Syndrome De Bardet-Biedl
Group II Chaperonins
Syndrome De Laurence-Moon
Cils Vibratiles
Polydactylie
Alstrom Syndrome
Malformations Du Système Stomatognathique
Protéines Associées Tubuline
Newfoundland and Labrador
Chaperonines
Généalogie
Protéines
Dégénérescence De La Rétine
Mutation
Malformations Multiples
Phénotype
Hétérogénéité Génétique
Rétinite Pigmentaire
Danio Zébré
Analyse Mutations Adn
Facteurs D'Adp-Ribosylation
Genetic Testing
Protéines Zebrafish
Cartographie Chromosomique
Chaperon Moléculaire
Obésité
Hypogonadisme
Encyclopedias as Topic
Médicament Orphelin
Maladies Rares
Canal Membranaire
Porte Canal Membranaire
Canaux Calciques
Le syndrome de Bardet-Biedl est un trouble génétique rare caractérisé par une combinaison de symptômes, notamment une obésité abdominale, des polydactylies (doigts ou orteils supplémentaires), une rétinite pigmentaire (une dégénérescence progressive de la rétine entraînant une perte de vision), des anomalies génitales et/ou rénales, ainsi que des retards intellectuels et développementaux. Ce syndrome est hérité selon un mode autosomique récessif, ce qui signifie qu'un individu doit hériter de deux copies du gène muté (une de chaque parent) pour développer la maladie. Il existe plusieurs gènes différents qui peuvent être impliqués dans le syndrome de Bardet-Biedl, et les symptômes peuvent varier en fonction du gène spécifique qui est muté. Le traitement du syndrome de Bardet-Biedl se concentre généralement sur la gestion des symptômes individuels et peut inclure une thérapie physique et occupante, une éducation spécialisée, des médicaments pour contrôler le poids et d'autres problèmes de santé associés, et une surveillance régulière par un médecin.
Les chaperonines de classe II sont des protéines moléculaires qui aident à la folding et l'assemblage d'autres protéines dans les cellules. Ils forment un complexe multiprotéique composé de deux anneaux stackés l'un sur l'autre, formant une cage cylindrique creuse. Cette structure permet d'accueillir des polypeptides dépliés à l'intérieur et de faciliter leur repliement en protéines fonctionnelles.
Les chaperonines de classe II sont également appelées chaperonines de type CPN60 (HSP60) et CPN10 (HSP10). Elles se trouvent principalement dans les mitochondries et les chloroplastes des cellules eucaryotes, ainsi que dans certaines bactéries. Les chaperonines de classe II sont essentielles pour la survie cellulaire, car elles assistent le repliement d'un grand nombre de protéines impliquées dans divers processus cellulaires tels que la respiration et la photosynthèse.
Les chaperonines de classe II fonctionnent en collaboration avec des co-chaperonines qui régulent leur activité et facilitent le transfert des polypeptides entre l'intérieur et l'extérieur de la cage protéique. Les co-chaperonines sont souvent associées aux anneaux externes des chaperonines de classe II et se lient aux polypeptides partiellement pliés pour les libérer une fois le repliement terminé.
Dans l'ensemble, les chaperonines de classe II sont des protéines cruciales qui assurent la stabilité et la fonctionnalité des autres protéines dans la cellule en facilitant leur repliement correct.
Le syndrome de Laurence-Moon est un trouble génétique rare caractérisé par une combinaison de symptômes comprenant une neuropathie sensorielle et moteur, une atrophie optique, des polydactylies (doigts ou orteils supplémentaires) et une insuffisance rénale. Il est souvent associé à une obésité précoce et à un retard mental léger. Ce syndrome est hérité de manière autosomique récessive, ce qui signifie que les deux copies du gène doivent être altérées pour que la maladie se manifeste. Il a été nommé d'après les médecins britanniques qui l'ont décrit pour la première fois en 1866, Frederick Parkes Weber et John Zachariah Laurence, ainsi que le médecin irlandais Robert Charles Moon.
Je suis désolé, mais la phrase que vous avez demandée ne correspond pas à une définition médicale standard. Les cils sont de petits poils qui poussent sur la bordure des paupières supérieures et inférieures des yeux chez les humains et de nombreux animaux. Le terme «vibratile» ne fait généralement pas partie de la description ou de la définition des cils dans un contexte médical.
Si vous cherchiez une information sur une condition oculaire spécifique ou un phénomène lié aux cils, veuillez fournir plus de détails et je ferai de mon mieux pour vous fournir une réponse appropriée.
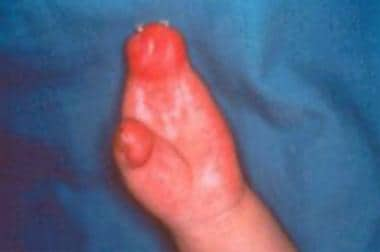
La polydactylie est un terme utilisé en médecine et en génétique pour décrire une anomalie congénitale caractérisée par la présence d'un ou plusieurs doigts ou orteils supplémentaires sur les mains ou les pieds. Ces doigts ou orteils surnuméraires peuvent varier en taille, en fonctionnalité et en positionnement. Ils peuvent être complètement développés avec des os, des muscles, des nerfs et des vaisseaux sanguins, ou ils peuvent être sous-développés, ressemblant à des petites bosses de peau.
La polydactylie est généralement héréditaire et peut être associée à certaines maladies génétiques ou syndromiques. Cependant, dans la plupart des cas, elle se produit isolément, sans autres anomalies associées. Le traitement de la polydactylie dépend de sa sévérité et de son impact fonctionnel. Dans certains cas, les doigts ou orteils supplémentaires peuvent être enlevés chirurgicalement pour des raisons esthétiques ou fonctionnelles.
Le syndrome d'Alström est une maladie génétique rare et héréditaire qui affecte plusieurs systèmes du corps. Il est causé par des mutations dans le gène ALMS1. Cette maladie se caractérise par une variété de symptômes, notamment une perte de vision précoce due à une neuropathie optique, une surdité progressive, une obésité, un diabète insulino-dépendant, des anomalies cardiaques et rénales, ainsi qu'une insuffisance hépatique. Les personnes atteintes de cette maladie peuvent également présenter des problèmes respiratoires, une faiblesse musculaire et un retard de développement.
Les symptômes du syndrome d'Alström apparaissent généralement pendant la petite enfance et s'aggravent progressivement avec le temps. Il n'existe actuellement aucun traitement curatif pour cette maladie, mais une prise en charge multidisciplinaire peut aider à gérer les symptômes et à améliorer la qualité de vie des personnes atteintes.
Le syndrome d'Alström est héréditaire et se transmet selon un mode autosomique récessif, ce qui signifie que les deux copies du gène doivent être altérées pour que la maladie se manifeste. Les personnes qui ne présentent qu'une copie altérée du gène sont des porteurs sains et ne développent pas la maladie, mais peuvent transmettre la mutation à leur progéniture.
Un syndrome, dans le contexte médical, est un ensemble de symptômes ou de signes cliniques qui, considérés dans leur globalité, suggèrent l'existence d'une pathologie spécifique ou d'un état anormal dans le fonctionnement de l'organisme. Il s'agit essentiellement d'un ensemble de manifestations cliniques qui sont associées à une cause sous-jacente commune, qu'elle soit connue ou inconnue.
Un syndrome n'est pas une maladie en soi, mais plutôt un regroupement de signes et symptômes qui peuvent être liés à différentes affections médicales. Par exemple, le syndrome métabolique est un ensemble de facteurs de risque qui augmentent la probabilité de développer des maladies cardiovasculaires et du diabète de type 2. Ces facteurs comprennent l'obésité abdominale, l'hypertension artérielle, l'hyperglycémie à jeun et les taux élevés de triglycérides et de faibles taux de HDL-cholestérol.
La définition d'un syndrome peut évoluer avec le temps, alors que la compréhension des mécanismes sous-jacents s'améliore grâce aux recherches médicales et scientifiques. Certains syndromes peuvent être nommés d'après les professionnels de la santé qui ont contribué à leur identification ou à leur description, comme le syndrome de Down (trisomie 21) ou le syndrome de Klinefelter (XXY).
Il est important de noter que la présence d'un syndrome ne permet pas toujours d'établir un diagnostic définitif, car plusieurs affections médicales peuvent partager des symptômes similaires. Cependant, l'identification d'un syndrome peut aider les professionnels de la santé à orienter le diagnostic et le traitement vers des causes probables ou à fournir des informations sur le pronostic et la prise en charge globale du patient.
Les malformations du système stomatognathique se réfèrent à un large éventail d'anomalies congénitales qui affectent les structures et les fonctions du système stomatognathique. Ce système comprend la cavité buccale, les dents, les mâchoires, les muscles de la mastication, les articulations temporo-mandibulaires, et les nerfs associés.
Les malformations peuvent varier en gravité et en complexité. Elles peuvent inclure des anomalies telles que des fentes labiales et palatines (becs-de-lièvre), des dents supplémentaires ou manquantes, des mâchoires mal alignées ou de taille anormale, des muscles de la mastication sous-développés, et des articulations temporo-mandibulaires anormales.
Ces malformations peuvent entraîner une variété de problèmes fonctionnels, tels que des difficultés à manger, à parler et à avaler, ainsi que des problèmes esthétiques. Elles peuvent également être associées à d'autres anomalies congénitales ou syndromiques.
Le traitement dépend de la nature et de la gravité de la malformation et peut inclure une combinaison de chirurgie, d'orthodontie, de thérapie de réadaptation et de soins dentaires spécialisés.
La consanguinité, en termes médicaux, se réfère à la pratique d'avoir des relations sexuelles ou de se marier entre individus qui sont apparentés de près, ce qui peut augmenter la probabilité de naissance d'enfants avec des anomalies génétiques et certaines maladies héréditaires. Cela est dû au fait que les gènes récessifs nocifs ont une plus grande chance d'être présents dans les deux copies du gène (une copie de chaque parent) chez les enfants issus de ces unions, ce qui peut entraîner leur expression et la manifestation de problèmes de santé.
Plus le taux de consanguinité est élevé (c'est-à-dire, plus les individus sont étroitement liés), plus le risque de maladies génétiques récessives est grand chez leur progéniture. Des mariages entre cousins germains, par exemple, présentent un risque légèrement accru de certains problèmes de santé congénitaux, mais ce risque n'est généralement pas aussi élevé que dans le cas d'unions entre frères et sœurs ou parents plus proches.
Il est important de noter que la consanguinité existe depuis longtemps dans l'histoire humaine pour des raisons sociales, culturelles et géographiques. Cependant, les professionnels de la santé doivent être conscients de ce risque accru de problèmes de santé génétiques lorsqu'ils traitent des patients issus de ces unions consanguines et offrir des conseils appropriés en matière de planification familiale et de dépistage prénatal.
Les protéines associées à la tubuline sont un groupe divers de protéines qui interagissent et se lient à la tubuline, le composant principal des microtubules. Les microtubules sont des structures cylindriques dynamiques qui jouent un rôle crucial dans la division cellulaire, le transport intracellulaire, la forme et la motilité cellulaires, ainsi que dans d'autres processus cellulaires importants.
Les protéines associées à la tubuline peuvent être classées en plusieurs catégories fonctionnelles, notamment:
1. Protéines de liaison aux microtubules: Ces protéines se lient directement aux microtubules et régulent leur dynamique et leur stabilité. Elles comprennent des protéines motrices telles que les kinésines et les dynéines, qui déplacent des cargaisons le long des microtubules, ainsi que des protéines structurales comme les MAPs (protéines associées aux microtubules) et les TACCs (transforming acidic coiled-coil proteins).
2. Protéines de régulation: Ces protéines modulent la dynamique des microtubules en interagissant avec des facteurs de croissance et d'assemblage des microtubules, tels que les GTPases de la tubuline et les protéines de chélation du GTP. Elles comprennent des protéines comme les Stathmins, les TOGs (tumor overexpressed genes) et les CLASPs (cytoplasmic linker associated proteins).
3. Protéines d'ancrage: Ces protéines ancrent les microtubules aux structures cellulaires, telles que le cortex cellulaire, les membranes organelles et les chromosomes. Elles comprennent des protéines comme les NuMAs (nuclear mitotic apparatus proteins) et les dyneines.
4. Protéines de modification post-traductionnelle: Ces protéines modifient les microtubules en ajoutant ou en éliminant des groupes chimiques, tels que l'acétylation, la tyrosination et le polyglutamylation. Elles comprennent des protéines comme les HDACs (histone déacétylases) et les TTLs (tubuline tyrosine ligases).
Les protéines impliquées dans l'interaction avec les microtubules jouent un rôle crucial dans la régulation de leur dynamique, de leur organisation spatiale et de leurs fonctions cellulaires. Les perturbations de ces interactions peuvent entraîner des défauts dans la division cellulaire, le transport intracellulaire et l'organisation du cytosquelette, ce qui peut conduire au développement de diverses pathologies, telles que les maladies neurodégénératives, le cancer et les anomalies congénitales.
Je suis désolé, mais il semble y avoir une certaine confusion dans votre demande. Newfoundland and Labrador est en fait une province du Canada et non un terme médical ou une condition médicale. Il n'y a pas de définition médicale associée à Newfoundland and Labrador.
Newfoundland and Labrador est située dans la partie orientale du Canada et est composée de l'île de Newfoundland et de la région continentale de Labrador. C'est une destination touristique populaire avec sa beauté naturelle, sa riche histoire et sa culture unique.
Chaperonines sont des protéines complexes qui aident à plier d'autres protéines dans les cellules. Ils forment une cage protéique qui agit comme un moule pour la protéine naissante, aidant ainsi à assurer que la protéine se plie correctement dans sa structure tridimensionnelle appropriée et fonctionnelle. Ce processus est crucial pour la fonction et la stabilité des protéines, et les chaperonines jouent donc un rôle important dans la prévention de l'agrégation des protéines et dans la protection de la cellule contre le stress proteotoxique. Les chaperonines sont présentes dans tous les domaines de la vie et sont particulièrement importantes dans les environnements à haute température ou à forte teneur en protéines, comme les bactéries thermophiles et les mitochondries.
En termes médicaux, la généalogie est l'étude systématique des antécédents familiaux et médicaux d'une personne ou d'une famille sur plusieurs générations. Elle vise à identifier les modèles de maladies héréditaires ou génétiques dans une famille, ce qui peut aider à évaluer le risque de développer certaines affections et à mettre en œuvre des stratégies de prévention et de dépistage appropriées.
Les professionnels de la santé utilisent souvent des arbres généalogiques pour représenter visuellement les relations familiales et les antécédents médicaux. Ces outils peuvent être particulièrement utiles dans la pratique clinique, en particulier lorsqu'il s'agit de maladies rares ou complexes qui ont tendance à se produire dans certaines familles en raison de facteurs génétiques sous-jacents.
En plus d'être un outil important pour la médecine préventive, la généalogie peut également fournir des informations précieuses sur l'histoire naturelle de diverses maladies et conditions, ce qui contribue à faire progresser notre compréhension globale de la pathogenèse et de la physiopathologie. Par conséquent, elle joue un rôle crucial dans la recherche médicale et les soins cliniques.
Un gène récessif est un type de gène qui doit être présent en double exemplaire (un hérité de chaque parent) pour qu'un trait ou une condition particulière se manifeste chez un individu. Dans l'espèce humaine, les caractéristiques contrôlées par des gènes récessifs ne s'expriment que si un individu hérite d'une copie de ce gène récessif de chaque parent.
Si un seul parent transmet le gène récessif, l'individu sera quand même porteur du trait récessif mais ne le manifestera pas, on l'appelle alors un porteur sain. Ce n'est que lorsque deux personnes qui sont toutes les deux porteuses d'un gène récessif ont un enfant qu'il y a une chance sur quatre (25%) à chaque grossesse pour que l'enfant hérite du trait récessif des deux parents et manifeste donc la caractéristique ou la maladie associée à ce gène.
Un exemple couramment cité de gène récessif est celui responsable de la drépanocytose, une maladie génétique affectant les globules rouges. Pour développer cette maladie, un individu doit hériter d'une copie du gène anormal de chaque parent. Si l'individu n'en hérite que d'une seule copie, il sera porteur sain du trait drépanocytaire mais ne développera pas la maladie.
En médecine et en biologie, les protéines sont des macromolécules essentielles constituées de chaînes d'acides aminés liés ensemble par des liaisons peptidiques. Elles jouent un rôle crucial dans la régulation et le fonctionnement de presque tous les processus biologiques dans les organismes vivants.
Les protéines ont une grande variété de fonctions structurales, régulatrices, enzymatiques, immunitaires, transport et signalisation dans l'organisme. Leur structure tridimensionnelle spécifique détermine leur fonction particulière. Les protéines peuvent être composées de plusieurs types différents d'acides aminés et varier considérablement en taille, allant de petites chaînes de quelques acides aminés à de longues chaînes contenant des milliers d'unités.
Les protéines sont synthétisées dans les cellules à partir de gènes qui codent pour des séquences spécifiques d'acides aminés. Des anomalies dans la structure ou la fonction des protéines peuvent entraîner diverses maladies, y compris des maladies génétiques et des troubles dégénératifs. Par conséquent, une compréhension approfondie de la structure, de la fonction et du métabolisme des protéines est essentielle pour diagnostiquer et traiter ces affections.
La dégénérescence rétinienne se réfère à un groupe de maladies oculaires caractérisées par la détérioration et la mort des cellules de la rétine. La rétine est une couche fine de tissu sensible à la lumière située à l'arrière de l'œil qui joue un rôle crucial dans le processus visuel en convertissant les stimuli lumineux en signaux électriques transmis au cerveau via le nerf optique.
Il existe deux types principaux de dégénérescence rétinienne : sèche et humide. La dégénérescence rétinienne sèche est la forme la plus courante et se caractérise par l'accumulation de dépôts jaunâtres appelés drusen sous la rétine, ce qui entraîne une atrophie progressive des cellules rétiniennes. La dégénérescence rétinienne humide est moins fréquente mais plus agressive ; elle se caractérise par la croissance anormale de nouveaux vaisseaux sanguins sous la rétine, qui peuvent fuir et provoquer un œdème maculaire, une hémorragie et une cicatrisation, entraînant une perte de vision sévère.
Les facteurs de risque de dégénérescence rétinienne comprennent l'âge avancé, le tabagisme, l'hypertension artérielle, l'obésité et les antécédents familiaux de la maladie. Actuellement, il n'existe aucun traitement curatif pour la dégénérescence rétinienne sèche, bien que des suppléments nutritionnels et des lunettes à faible intensité lumineuse puissent ralentir sa progression. Le traitement de la dégénérescence rétinienne humide implique généralement l'utilisation d'agents anti-VEGF, qui inhibent la croissance des vaisseaux sanguins anormaux, ainsi que la photocoagulation au laser et la thérapie par radiothérapie.
En génétique, une mutation est une modification permanente et héréditaire de la séquence nucléotidique d'un gène ou d'une région chromosomique. Elle peut entraîner des changements dans la structure et la fonction des protéines codées par ce gène, conduisant ainsi à une variété de phénotypes, allant de neutres (sans effet apparent) à délétères (causant des maladies génétiques). Les mutations peuvent être causées par des erreurs spontanées lors de la réplication de l'ADN, l'exposition à des agents mutagènes tels que les radiations ou certains produits chimiques, ou encore par des mécanismes de recombinaison génétique.
Il existe différents types de mutations, telles que les substitutions (remplacement d'un nucléotide par un autre), les délétions (suppression d'une ou plusieurs paires de bases) et les insertions (ajout d'une ou plusieurs paires de bases). Les conséquences des mutations sur la santé humaine peuvent être très variables, allant de maladies rares à des affections courantes telles que le cancer.
Les malformations multiples, également connues sous le nom de malformations congénitales multiples, se réfèrent à la présence de deux ou plusieurs anomalies congénitales affectant différents organes ou systèmes du corps. Ces anomalies sont présentes dès la naissance et peuvent être causées par des facteurs génétiques, environnementaux ou une combinaison des deux.
Les malformations multiples peuvent affecter n'importe quelle partie du corps et peuvent varier en gravité, allant de légères à graves. Elles peuvent également affecter la fonctionnalité des organes touchés et dans les cas les plus sévères, peuvent être fatales.
Les exemples courants de malformations multiples comprennent le syndrome de Down (trisomie 21), qui est caractérisé par un retard mental, une apparence faciale distinctive et souvent d'autres anomalies telles que des problèmes cardiaques congénitaux ; le syndrome de Di George, qui affecte la croissance et le développement et peut causer des problèmes cardiaques, immunitaires et de développement du cerveau ; et le spina bifida, une anomalie de la colonne vertébrale qui peut causer des problèmes de mouvement et de sensation dans les jambes.
Le diagnostic et le traitement des malformations multiples dépendent du type et de la gravité des anomalies présentes. Les soins peuvent inclure une combinaison de chirurgie, de médicaments, de thérapies et de soutien de développement pour aider à gérer les symptômes et améliorer la qualité de vie de l'enfant affecté.
Dans le domaine de la génétique, un individu homozygote est une personne qui hérite de deux allèles identiques pour un trait spécifique, ayant reçu ce même allèle de chacun de ses deux parents. Cela peut se traduire par l'expression d'un caractère particulier ou l'apparition d'une maladie génétique dans le cas où ces allèles sont défectueux.
On distingue deux types d'homozygotes : les homozygotes dominants et les homozygotes récessifs. Les homozygotes dominants présentent le phénotype lié au gène dominant, tandis que les homozygotes récessifs expriment le phénotype associé au gène récessif.
Par exemple, dans le cas de la drépanocytose, une maladie génétique héréditaire affectant l'hémoglobine des globules rouges, un individu homozygote pour cette affection possède deux allèles mutés du gène de l'hémoglobine et exprimera donc les symptômes de la maladie.
Le phénotype est le résultat observable de l'expression des gènes en interaction avec l'environnement et d'autres facteurs. Il s'agit essentiellement des manifestations physiques, biochimiques ou développementales d'un génotype particulier.
Dans un contexte médical, le phénotype peut se rapporter à n'importe quelle caractéristique mesurable ou observable résultant de l'interaction entre les gènes et l'environnement, y compris la couleur des yeux, la taille, le poids, certaines maladies ou conditions médicales, voire même la réponse à un traitement spécifique.
Il est important de noter que deux individus ayant le même génotype (c'est-à-dire la même séquence d'ADN) ne seront pas nécessairement identiques dans leur phénotype, car des facteurs environnementaux peuvent influencer l'expression des gènes. De même, des individus avec des génotypes différents peuvent partager certains traits phénotypiques en raison de similitudes dans leurs environnements ou dans d'autres facteurs non génétiques.
L'hétérogénéité génétique est un terme utilisé en génétique pour décrire la situation où différents gènes ou différentes mutations dans le même gène peuvent conduire à des phénotypes similaires ou identiques. Cela signifie qu'il existe plusieurs façons dont une maladie particulière peut se manifester, en fonction de la manière dont les gènes sont hérités ou altérés.
L'hétérogénéité génétique peut être classée en deux types : l'hétérogénéité génétique d'un gène (ou hétérogénéité algique) et l'hétérogénéité génétique intergénique (ou hétérogénéité non algique).
L'hétérogénéité génétique d'un gène se produit lorsque différentes mutations dans le même gène entraînent la même maladie. Par exemple, il existe de nombreuses mutations différentes dans le gène CFTR qui peuvent causer la mucoviscidose, une maladie génétique grave qui affecte principalement les poumons et le système digestif.
D'autre part, l'hétérogénéité génétique intergénique se produit lorsque des mutations dans différents gènes entraînent la même maladie. Par exemple, il existe plusieurs types de cancer du sein qui peuvent être causés par des mutations dans différents gènes, tels que BRCA1, BRCA2, P53 et autres.
L'hétérogénéité génétique peut compliquer le diagnostic et le traitement des maladies génétiques, car il peut être difficile de déterminer quelle mutation ou quel gène est à l'origine de la maladie chez un patient donné. Cependant, la compréhension de l'hétérogénéité génétique peut également fournir des informations importantes sur les mécanismes sous-jacents des maladies et aider au développement de nouveaux traitements personnalisés pour les patients atteints de maladies rares ou complexes.
La rétinite pigmentaire (RP) est un groupe de maladies génétiques oculaires qui affectent la rétine, une membrane nerveuse située à l'arrière de l'œil qui détecte la lumière et envoie des signaux au cerveau via le nerf optique. Ces signaux sont interprétés par le cerveau comme des images visuelles.
Dans la rétinite pigmentaire, les cellules photoréceptrices de la rétine, appelées bâtonnets et cônes, se détériorent progressivement. Les bâtonnets sont principalement responsables de la vision nocturne et périphérique, tandis que les cônes sont responsables de la vision centrale et des couleurs. Lorsque ces cellules dégénèrent, cela entraîne une perte de vision progressive, commençant souvent par la vision nocturne et la vision périphérique (également appelée vision tunnel).
La progression de la maladie varie considérablement d'une personne à l'autre. Chez certaines personnes, la perte de vision peut progresser relativement lentement sur plusieurs décennies, tandis que chez d'autres, la perte de vision peut être plus rapide et sévère. Dans les cas graves, la rétinite pigmentaire peut entraîner une cécité complète.
Actuellement, il n'existe pas de traitement curatif pour la rétinite pigmentaire. Cependant, des recherches sont en cours pour développer des thérapies géniques et d'autres approches visant à ralentir ou arrêter la progression de la maladie.
Le Danio Zébré, également connu sous le nom de Danio rerio, est un petit poisson d'eau douce souvent utilisé en recherche biomédicale comme modèle animal. Il est originaire d'Asie du Sud et d'Asie du Sud-Est. Bien que ce ne soit pas une espèce animale couramment utilisée dans la médecine humaine, il est largement employé dans les études de développement, de génétique et de toxicologie en raison de sa facilité de maintenance en laboratoire, de son cycle de vie court, de sa transparence à certains stades de développement, et de la possibilité de modifier facilement son génome.
Cependant, il est important de noter que cette réponse concerne l'utilisation du Danio Zébré dans le contexte de la recherche biologique et non pas dans un sens médical direct.
L'analyse des mutations de l'ADN est une méthode d'examen génétique qui consiste à rechercher des modifications (mutations) dans la séquence de l'acide désoxyribonucléique (ADN). L'ADN est le matériel génétique présent dans les cellules de tous les organismes vivants et contient les instructions pour le développement, la fonction et la reproduction des organismes.
Les mutations peuvent survenir spontanément ou être héritées des parents d'un individu. Elles peuvent entraîner des changements dans la structure et la fonction des protéines, ce qui peut à son tour entraîner une variété de conséquences, allant de mineures à graves.
L'analyse des mutations de l'ADN est utilisée dans un large éventail d'applications, y compris le diagnostic et le suivi des maladies génétiques, la détermination de la susceptibilité à certaines maladies, l'identification des auteurs de crimes, la recherche sur les maladies et le développement de médicaments.
Il existe différentes méthodes pour analyser les mutations de l'ADN, notamment la séquençage de nouvelle génération (NGS), la PCR en temps réel, la PCR quantitative et la Southern blotting. Le choix de la méthode dépend du type de mutation recherchée, de la complexité du test et des besoins du patient ou du chercheur.
Les facteurs d'ADP-ribosylation sont des enzymes qui catalysent l'ajout d'un ou plusieurs groupes ADP-ribose à une protéine cible. Ce processus, appelé ADP-ribosylation, joue un rôle crucial dans divers processus cellulaires tels que la réparation de l'ADN, le contrôle de la transcription, la régulation du cycle cellulaire et la réponse au stress oxydatif.
Il existe deux types principaux de facteurs d'ADP-ribosylation : les poly(ADP-ribose) polymérases (PARPs) et les mono(ADP-ribose) transférases (MARTs). Les PARPs sont responsables de l'ajout de chaînes de poly(ADP-ribose) à des protéines cibles, tandis que les MARTs ajoutent un seul groupe ADP-ribose.
Les facteurs d'ADP-ribosylation sont également importants dans la réponse cellulaire au dommage de l'ADN. Lorsque l'ADN est endommagé, les PARPs sont activées et catalysent l'ajout de chaînes de poly(ADP-ribose) sur des protéines impliquées dans la réparation de l'ADN. Cela permet de recruter d'autres protéines pour réparer l'ADN endommagé et aide à maintenir la stabilité génomique.
Cependant, une activation excessive des facteurs d'ADP-ribosylation peut entraîner une déplétion de NAD+, un cofacteur essentiel pour de nombreuses réactions métaboliques dans la cellule. Cela peut entraîner une altération de la fonction mitochondriale et finalement entraîner une mort cellulaire programmée ou apoptose.
Des études ont montré que les facteurs d'ADP-ribosylation sont également impliqués dans divers processus pathologiques, tels que l'inflammation, le stress oxydatif et la neurodégénération. Par conséquent, les inhibiteurs des facteurs d'ADP-ribosylation sont actuellement étudiés comme thérapies potentielles pour diverses maladies, telles que le cancer, les maladies neurodégénératives et les maladies cardiovasculaires.
Le test génétique est un type d'examen diagnostique qui consiste à analyser les gènes d'un individu dans le but d'identifier des modifications ou des variations dans l'ADN qui peuvent être associées à un risque accru de développer une maladie héréditaire ou d'autres conditions médicales. Les tests génétiques peuvent également être utilisés pour déterminer la susceptibilité d'un individu à répondre à certains traitements médicaux ou pour établir des relations familiales.
Les tests génétiques peuvent impliquer l'analyse de l'ADN, de l'ARN ou des protéines, et peuvent être effectués sur des échantillons de sang, de salive, de cheveux ou d'autres tissus. Les résultats du test génétique peuvent aider les médecins à poser un diagnostic, à prévoir le risque de développer une maladie à l'avenir, à déterminer le meilleur traitement pour une maladie existante ou à fournir des conseils en matière de planification familiale.
Il est important de noter que les tests génétiques peuvent avoir des implications émotionnelles et sociales importantes pour les individus et leur famille, et qu'il est donc essentiel de recevoir un counseling génétique avant et après le test pour comprendre pleinement les résultats et les conséquences potentielles.
Je suis désolé, mais la requête « Protéines Zebrafish » est un peu imprécise. Le terme « protéines » fait référence à des molécules complexes présentes dans toutes les cellules vivantes, tandis que « Zebrafish » est le nom commun du poisson-zèbre, qui est souvent utilisé comme organisme modèle en biologie et en médecine.
Si vous cherchez des informations sur les protéines spécifiques du poisson-zèbre, il faudrait préciser laquelle pour que je puisse vous fournir une définition médicale ou scientifique exacte. Le poisson-zèbre a un génome séquencé et bien annoté, ce qui permet d'identifier et d'étudier les protéines spécifiques qu'il exprime.
Cependant, sans plus de précision, il m'est difficile de vous donner une définition médicale pertinente des « protéines Zebrafish ». Pouvez-vous me fournir plus d'informations sur le type de protéine qui vous intéresse ?
La cartographie chromosomique est une discipline de la génétique qui consiste à déterminer l'emplacement et l'ordre relatif des gènes et des marqueurs moléculaires sur les chromosomes. Cette technique utilise généralement des méthodes de laboratoire pour analyser l'ADN, comme la polymerase chain reaction (PCR) et la Southern blotting, ainsi que des outils d'informatique pour visualiser et interpréter les données.
La cartographie chromosomique est un outil important dans la recherche génétique, car elle permet aux scientifiques de comprendre comment les gènes sont organisés sur les chromosomes et comment ils interagissent entre eux. Cela peut aider à identifier les gènes responsables de certaines maladies héréditaires et à développer des traitements pour ces conditions.
Il existe deux types de cartographie chromosomique : la cartographie physique et la cartographie génétique. La cartographie physique consiste à déterminer l'emplacement exact d'un gène ou d'un marqueur sur un chromosome en termes de distance physique, exprimée en nucléotides. La cartographie génétique, quant à elle, consiste à déterminer l'ordre relatif des gènes et des marqueurs sur un chromosome en fonction de la fréquence de recombinaison entre eux lors de la méiose.
En résumé, la cartographie chromosomique est une technique utilisée pour déterminer l'emplacement et l'ordre relatif des gènes et des marqueurs moléculaires sur les chromosomes, ce qui permet aux scientifiques de mieux comprendre comment les gènes sont organisés et interagissent entre eux.
Un chaperon moléculaire est une protéine qui assiste et régule le pliage et l'assemblage corrects d'autres protéines dans les cellules vivantes. Ils aident à prévenir l'agrégation des protéines mal pliées et favorisent leur dégradation si un repliage correct ne peut être accompli. Les chaperons moléculaires jouent donc un rôle crucial dans la protection de l'intégrité du proteome cellulaire et dans la prévention des maladies liées à des protéines mal pliées, telles que les maladies neurodégénératives.
Les chaperons moléculaires sont souvent classés en fonction de leur taille et de leur structure. Les plus étudiés comprennent les chaperonnes de la famille des chocs thermiques (HSP), comme Hsp60, Hsp70 et Hsp90. Ces protéines sont hautement conservées dans divers organismes vivants, ce qui témoigne de leur importance fonctionnelle cruciale.
Les chaperons moléculaires peuvent se lier aux protéines clientes à différents stades du processus de pliage, depuis la traduction des ARNm jusqu'à l'assemblage et au transport des complexes multiprotéiques vers leurs destinations intracellulaires. Leur activité est généralement régulée par des changements dans les interactions protéine-protéine, la modification post-traductionnelle et l'énergie fournie par l'hydrolyse de l'ATP.
Dans l'ensemble, les chaperons moléculaires sont essentiels pour maintenir l'homéostasie protéique et prévenir le stress cellulaire associé aux protéines mal pliées. Leur dysfonctionnement a été lié à diverses affections pathologiques, notamment les maladies neurodégénératives, la fibrose kystique, le cancer et les infections virales.
L'obésité est une accumulation anormale ou excessive de graisse corporelle qui présente un risque pour la santé. Elle est généralement définie en termes d'indice de masse corporelle (IMC), qui est une mesure de la proportion de poids corporel due à la graisse. Un IMC de 30 ou plus est considéré comme obèse, selon l'Organisation mondiale de la santé (OMS).
L'obésité peut entraîner divers problèmes de santé graves, notamment des maladies cardiovasculaires, le diabète de type 2, l'apnée du sommeil, les maladies articulaires dégénératives et certains types de cancer. Elle résulte généralement d'une combinaison de facteurs, tels qu'une alimentation déséquilibrée, un manque d'activité physique, une prédisposition génétique et des facteurs environnementaux et psychologiques.
Il est important de noter que l'obésité n'est pas simplement une question de choix personnel ou de manque de volonté, mais plutôt le résultat d'un ensemble complexe de facteurs interagissant les uns avec les autres. De nombreuses personnes obèses ont des difficultés à maintenir une perte de poids durable en raison de facteurs biologiques et environnementaux qui rendent difficile la modification des habitudes alimentaires et de l'activité physique.
L'hypogonadisme est un trouble endocrinien qui se caractérise par une production insuffisante d'hormones sexuelles en raison d'une anomalie des gonades, c'est-à-dire les testicules chez l'homme et les ovaires chez la femme. Chez l'homme, cela peut entraîner une diminution de la production de spermatozoïdes et de testostérone, ce qui peut se traduire par des symptômes tels que des troubles de la fonction sexuelle, une baisse de la libido, une fatigue, une perte de masse musculaire, une ostéoporose et des bouffées de chaleur. Chez la femme, cela peut entraîner une absence ou un retard des règles, une stérilité, des bouffées de chaleur et une sécheresse vaginale.
L'hypogonadisme peut être congénital ou acquis, et il existe plusieurs causes possibles, notamment des anomalies génétiques, des lésions cérébrales, des tumeurs, des infections, des troubles systémiques tels que le diabète et l'insuffisance rénale, ainsi que certains médicaments. Le diagnostic de l'hypogonadisme repose sur les antécédents médicaux, un examen physique, des analyses de sang pour mesurer les niveaux d'hormones sexuelles et d'autres tests complémentaires si nécessaire. Le traitement dépend de la cause sous-jacente et peut inclure un remplacement hormonal, une fertilité assistée ou d'autres interventions thérapeutiques.
Je suis désolé, "Encyclopedias as Topic" n'est pas une définition médicale. Il s'agit plutôt d'une catégorie de sujets dans la classification MeSH (Medical Subject Headings) utilisée pour indexer les articles de bibliographie en médecine et en sciences de la santé. Cette catégorie comprend des encyclopédies médicales générales ou spécialisées, des dictionnaires médicaux, des manuels médicaux et d'autres ressources similaires. Cependant, il ne s'agit pas d'une définition médicale à proprement parler.
Un médicament orphelin est un médicament ou une substance biologique développé(e) spécifiquement pour le traitement, la prévention ou le diagnostic d'une maladie rare, c'est-à-dire qui affecte moins de 200 000 personnes aux États-Unis ou moins de 1 personne sur 2 000 dans l'Union européenne.
En raison du faible nombre de patients atteints de ces maladies rares, les sociétés pharmaceutiques peuvent être réticentes à investir dans le développement et la commercialisation de médicaments orphelins en raison des coûts élevés et des risques financiers associés. Pour encourager la recherche et le développement de ces médicaments, les gouvernements offrent souvent des incitations telles que des crédits d'impôt, des exonérations de frais réglementaires et une période d'exclusivité commerciale prolongée après l'approbation du médicament.
Les médicaments orphelins peuvent être approuvés pour un usage général ou pour une utilisation compassionnelle, ce qui permet aux médecins de prescrire des médicaments non approuvés pour traiter des patients gravement malades atteints de maladies rares lorsqu'il n'existe aucune autre option thérapeutique satisfaisante.
Les maladies rares, également connues sous le nom de troubles rares ou de maladies orphelines, se réfèrent à des affections qui affectent un petit nombre de personnes dans la population. Selon l'Orphanet, le portail européen des maladies rares et des médicaments orphelins, une maladie est considérée comme rare en Europe lorsqu'elle affecte moins d'une personne sur 2 000.
Les maladies rares peuvent être causées par des mutations génétiques, des infections, des expositions environnementales ou encore rester sans cause identifiée. Elles peuvent toucher n'importe quel système organique du corps et se manifester par une grande variété de symptômes, allant de légers à graves, voire mortels.
Le diagnostic et le traitement des maladies rares sont souvent difficiles en raison de leur faible prévalence, ce qui peut entraîner un retard dans l'établissement d'un diagnostic correct et dans la mise en place d'un plan de traitement adapté. De plus, les ressources allouées à la recherche sur ces maladies sont souvent limitées, ce qui freine le développement de thérapies spécifiques et efficaces.
Il existe environ 7 000 maladies rares connues à ce jour, affectant près de 300 millions de personnes dans le monde. En raison de leur caractère rare et dispersé, la collaboration internationale entre médecins, chercheurs, patients et associations est essentielle pour améliorer la compréhension, le diagnostic et le traitement de ces affections.
Le canal membranaire, également connu sous le nom de canal de Nérée ou aqueduc de Sylvius, est un petit conduit présent dans l'anatomie humaine. Il s'agit d'une structure tubulaire située dans le cerveau qui connecte l'oreille interne au quatrième ventricule, un espace rempli de liquide céphalo-rachidien (LCR) dans le tronc cérébral.
Le canal membranaire a une fonction cruciale dans la protection et la régulation du système auditif interne. Il permet au liquide endolymphatique, qui remplit la cochlée et les canaux semi-circulaires de l'oreille interne, de circuler et de communiquer avec le quatrième ventricule et le reste du système nerveux central.
Ce canal peut être affecté par certaines affections médicales, telles que la maladie de Ménière ou les tumeurs du nerf vestibulaire (schwannome vestibulaire), ce qui peut entraîner des symptômes auditifs et d'équilibre. Des procédures chirurgicales peuvent être nécessaires pour traiter ces conditions, telles que la décompression du canal membranaire ou l'ablation de tumeurs.
Les contraceptifs oraux hormonaux, également connus sous le nom de « pilules contraceptives », sont des formulations pharmaceutiques contenant des hormones synthétiques qui imitent l'action des œstrogènes et de la progestérone dans l'organisme. Ils sont conçus pour interférer avec le processus naturel de reproduction en empêchant l'ovulation, en épaississant la glaire cervicale pour rendre plus difficile la traversée des spermatozoïdes ou en modifiant la muqueuse utérine pour prévenir l'implantation d'un ovule fécondé.
Les contraceptifs oraux hormonaux se présentent généralement sous deux formes : les combinaisons œstro-progestatives et les progestatifs seuls (mini-pilules). Les combinaisons œstro-progestatives contiennent à la fois un dérivé d'œstrogène et un dérivé de progestérone, tandis que les mini-pilules ne contiennent qu'un progestatif. Chaque type a ses avantages et ses inconvénients, et le choix du contraceptif oral hormonal dépend souvent des préférences personnelles, des antécédents médicaux et des besoins de chaque individu.
L'utilisation régulière et correcte des contraceptifs oraux hormonaux est globalement sûre et efficace pour prévenir la grossesse, avec un taux d'échec inférieur à 1 % lorsqu'ils sont utilisés de manière optimale. Cependant, il convient de noter que les contraceptifs oraux hormonaux ne protègent pas contre les infections sexuellement transmissibles (IST) et doivent être associés à des méthodes de barrière, telles que le préservatif, pour une protection optimale contre les IST.
Comme tout médicament, les contraceptifs oraux hormonaux peuvent entraîner des effets secondaires et des risques potentiels. Les effets indésirables courants comprennent des saignements irréguliers, des nausées, des maux de tête, des seins douloureux ou enflés, une prise de poids et une humeur changeante. Dans de rares cas, les contraceptifs oraux hormonaux peuvent augmenter le risque de thromboembolie veineuse (TEV), c'est-à-dire la formation d'un caillot sanguin dans une veine, qui peut entraîner des complications graves telles que des embolies pulmonaires ou des accidents vasculaires cérébraux. Les femmes présentant des facteurs de risque de TEV, tels qu'une histoire personnelle ou familiale de thrombose veineuse, un âge avancé, une obésité morbide, une hypertension artérielle sévère, un tabagisme ou une fibrillation auriculaire, doivent faire l'objet d'une évaluation attentive avant de commencer à prendre des contraceptifs oraux hormonaux.
Les contraceptifs oraux combinés (COC) sont disponibles sous différentes formulations, avec des dosages et des types d'hormones variables. Les COC les plus couramment prescrits contiennent de l'éthinylestradiol à une dose comprise entre 20 et 35 microgrammes et un progestatif, tel que le lévonorgestrel, le norgestimate, le désogestrel, la drospirénone ou le gestodène. Les COC contenant de faibles doses d'éthinylestradiol (20 microgrammes) sont associés à un risque réduit de TEV par rapport aux COC contenant des doses plus élevées (35 microgrammes). Cependant, les COC contenant des progestatifs de troisième et quatrième générations, tels que la drospirénone et le désogestrel, peuvent être associés à un risque accru de TEV par rapport aux COC contenant des progestatifs de deuxième génération, tels que le lévonorgestrel.
Les contraceptifs oraux progestatifs (POP) ne contiennent qu'une seule hormone, un progestatif, et sont disponibles sous différentes formulations. Les POP les plus couramment prescrits contiennent du lévonorgestrel, de la noréthistérone ou de la médroxyprogestérone acétate. Les POP sont associés à un risque réduit de TEV par rapport aux COC, en particulier chez les femmes présentant des facteurs de risque tels que l'obésité, le tabagisme et l'âge avancé. Cependant, les POP peuvent ne pas être aussi efficaces que les COC pour prévenir la grossesse, en particulier chez les femmes présentant un cycle menstruel irrégulier ou une masse corporelle élevée.
Les contraceptifs oraux d'urgence (COU) sont des médicaments qui peuvent être utilisés après un rapport sexuel non protégé pour prévenir une grossesse non désirée. Les deux types de COU les plus couramment utilisés sont la levonorgestrel et l'acétate d'ulipristal. La levonorgestrel est disponible en vente libre aux États-Unis pour les femmes âgées de 15 ans et plus, tandis que l'acétate d'ulipristal nécessite une prescription médicale. Les COU sont généralement considérés comme sûrs et efficaces, mais ils peuvent ne pas être aussi efficaces que les contraceptifs oraux réguliers pour prévenir la grossesse.
Les contraceptifs oraux peuvent également avoir des avantages pour la santé en dehors de la planification familiale. Par exemple, certaines études ont suggéré que les COC peuvent réduire le risque de cancer de l'ovaire et de l'endomètre, tandis que d'autres études ont montré qu'ils peuvent améliorer les symptômes du syndrome des ovaires polykystiques (SOPK) et de l'acné. Cependant, il est important de noter que les contraceptifs oraux ne sont pas sans risques et qu'ils peuvent augmenter le risque de certaines maladies, telles que les accidents vasculaires cérébraux, les caillots sanguins et les maladies du foie.
En conclusion, les contraceptifs oraux sont des médicaments qui peuvent être utilisés pour prévenir une grossesse non désirée. Ils sont disponibles sous différentes formes, telles que les pilules, les patchs et les anneaux vaginaux, et ils fonctionnent en empêchant l'ovulation, en épaississant la glaire cervicale ou en modifiant l'endomètre. Les contraceptifs oraux peuvent également avoir des avantages pour la santé en dehors de la planification familiale, mais ils ne sont pas sans risques et doivent être utilisés avec prudence. Il est important de consulter un professionnel de la santé avant de commencer à utiliser des contraceptifs oraux pour déterminer le type le plus approprié et pour discuter des risques et des avantages.
Le terme "porte canal membranaire" est utilisé en anatomie et en physiologie pour décrire une structure fine et tubulaire qui traverse la membrane cellulaire, permettant ainsi le passage de certaines substances telles que les ions ou les molécules hydrophiles. Ces canaux sont composés de protéines et jouent un rôle crucial dans la régulation des échanges ioniques et moléculaires entre le milieu extracellulaire et le cytoplasme.
Les portes canal membranaires peuvent être spécifiques à certains types d'ions ou de molécules, ce qui signifie qu'ils ne permettent que le passage de certaines substances tout en empêchant celui des autres. Par exemple, les canaux sodium sont sélectivement perméables aux ions sodium, tandis que les canaux potassium sont perméables aux ions potassium.
Les portes canal membranaires peuvent être régulés de différentes manières, notamment par des changements de tension membranaire, des modifications post-traductionnelles des protéines qui composent les canaux, ou encore par l'action de messagers chimiques tels que les neurotransmetteurs. Ces mécanismes permettent de contrôler finement le flux d'ions et de molécules à travers la membrane cellulaire, ce qui est essentiel pour de nombreux processus physiologiques tels que la transmission nerveuse, la contraction musculaire, ou encore la régulation du volume cellulaire.
Les canaux calciques sont des protéines qui forment des pores dans la membrane cellulaire, permettant au calcium d'entrer et de sortir des cellules. Ils jouent un rôle crucial dans la régulation de divers processus cellulaires tels que la contraction musculaire, la libération de neurotransmetteurs et la signalisation cellulaire.
Il existe différents types de canaux calciques, chacun ayant des propriétés spécifiques et des rôles distincts dans l'organisme. Certains sont sensibles au voltage et s'ouvrent en réponse à un changement du potentiel électrique de la membrane cellulaire, tandis que d'autres sont activés par des messagers intracellulaires tels que l'IP3 (inositol trisphosphate) ou le calcium lui-même.
Les canaux calciques peuvent être affectés dans certaines maladies, telles que les maladies cardiovasculaires, l'hypertension artérielle et certaines formes d'épilepsie. Des médicaments appelés bloqueurs des canaux calciques sont souvent utilisés pour traiter ces conditions en régulant la quantité de calcium qui entre dans les cellules.
Il est important de noter que l'équilibre du calcium intracellulaire est essentiel au fonctionnement normal des cellules, et que des perturbations de ce processus peuvent entraîner des conséquences néfastes pour la santé.
Les "Acid Sensing Ion Channels" (ASIC) sont des canaux ioniques activés par le pH qui s'ouvrent en réponse à une acidification du milieu extracellulaire. Ils sont largement distribués dans le système nerveux central et périphérique des mammifères et jouent un rôle important dans la détection de la douleur, la plasticité synaptique et la régulation de l'excitabilité neuronale.
Les canaux ASIC sont constitués de sous-unités protéiques qui s'assemblent pour former des homo ou hétéotrimères. Il existe au moins six sous-unités différentes (ASIC1a, ASIC1b, ASIC2a, ASIC2b, ASIC3 et ASIC4) qui peuvent se combiner pour former divers types de canaux avec des propriétés pharmacologiques et fonctionnelles distinctes.
L'activation des canaux ASIC entraîne une entrée de cations (principalement Na+) dans la cellule, ce qui peut dépolariser la membrane et potentialiser l'activité neuronale. L'acidification du milieu extracellulaire peut se produire dans divers contextes physiologiques et pathologiques, tels que l'ischémie, l'inflammation, l'hypoxie et la douleur.
Les canaux ASIC sont donc des cibles thérapeutiques potentielles pour le traitement de diverses affections neurologiques et dolorifiques, telles que les migraines, les neuropathies périphériques, l'épilepsie et la douleur chronique.
 Syndrome de Bardet-Biedl
Syndrome de Bardet-Biedl
Syndrome de Prader-Willi
Georges Bardet
Bardet
Rétinite pigmentaire
Syndrome de Borjeson-Forssman-Lehmann
Jean Bardet (éditeur)
BBS
Syndrome de McKusick Kaufman
Habiba Bouhamed Chaabouni
Polykystose rénale autosomique récessive
Transmission autosomique récessive
Atrésie vaginale
Syndrome de Pallister-Hall
Syndrome de Laurence-Moon
CIM-10 Chapitre 17 : Malformations congénitales et anomalies chromosomiques
Liste de maladies rares
Syndrome de Bardet-Biedl - Wikipedia
 syndrome de Bardet Biedl | 3 Points Important
syndrome de Bardet Biedl | 3 Points Important
 Définition | Endogamie
Définition | Endogamie
 L'association BARDET BIEDL SYNDROME - Facile2Soutenir
L'association BARDET BIEDL SYNDROME - Facile2Soutenir
 La Maladie de Hirschsprung (HSCR) - Risques, Traitements, Symptômes
La Maladie de Hirschsprung (HSCR) - Risques, Traitements, Symptômes
 Veille bibliographique
Veille bibliographique
 Centre de Référence Constitutif des Anomalies du Développement et Syndromes Malformatifs de L'Ouest
Centre de Référence Constitutif des Anomalies du Développement et Syndromes Malformatifs de L'Ouest
 Archives des Laser Game Evolution - Laser Game Entreprise
Archives des Laser Game Evolution - Laser Game Entreprise
 AtlasRLeye - Dystrophies Maculaires
AtlasRLeye - Dystrophies Maculaires
 Sumycin Pharmacie En Ligne La Moins Chere | costreview.com | PACS
Sumycin Pharmacie En Ligne La Moins Chere | costreview.com | PACS
 16 novembre 2017
16 novembre 2017
 Syndrome de Senior-Loken • Retina Suisse
Syndrome de Senior-Loken • Retina Suisse
 39ème Congrès de la Société Française d'Endocrinologie - du 4 au 7 octobre 2023, Marseille, Palais du Pharo | Programme du...
39ème Congrès de la Société Française d'Endocrinologie - du 4 au 7 octobre 2023, Marseille, Palais du Pharo | Programme du...
 Malformations congénitales des membres - Pédiatrie - Édition professionnelle du Manuel MSD
Malformations congénitales des membres - Pédiatrie - Édition professionnelle du Manuel MSD
SYNDROME DE BAINBRIDGE-ROPERS - Page 2 - Le Forum maladies rares
 Identifier les maladies rares grâce à l'intelligence artificielle - Plus Magazine
Identifier les maladies rares grâce à l'intelligence artificielle - Plus Magazine
Association BARDET-BIEDL France
 Rare Diabetes | The Euro-WABB projectWolfram
Rare Diabetes | The Euro-WABB projectWolfram
 S'informer sur les maladies et leurs conséquences | Tous à l'école
S'informer sur les maladies et leurs conséquences | Tous à l'école
Association BARDET-BIEDL France
Actualités Scientifiques - Médicales: #thelancet #exclusif #fenfluramine #épilepsie Chlorhydrate de fenfluramine pour le...
Actualités Scientifiques - Médicales: Canalisation de la glycémie: variations dans les canaux ioniques et perturbation de l...
Je fais un don | Association BARDET-BIEDL Syndrome
 février, 2009 | MyPharma Editions - Part 4
février, 2009 | MyPharma Editions - Part 4
 Cystic kidney diseases in children - EM consulte
Cystic kidney diseases in children - EM consulte
Association BARDET-BIEDL France
 Les malformations congénitales - Diagnostic Anténatal et Devenir - Tome 3 - Alain Couture - Broché - SAURAMPS MEDICAL -...
Les malformations congénitales - Diagnostic Anténatal et Devenir - Tome 3 - Alain Couture - Broché - SAURAMPS MEDICAL -...
 DeCS
DeCS
NeuroDegen - ITI NeuroStra - Le système nerveux de l'adaptation aux pathologies - Université de Strasbourg
 École secondaire Augustin-Norbert Morin - Derniers articles Valeurs
École secondaire Augustin-Norbert Morin - Derniers articles Valeurs
 Obésité précoce : une perte de poids radicale et durable sous setmélanotide
Obésité précoce : une perte de poids radicale et durable sous setmélanotide
Association BARDET-BIEDL France
WIF FRANCE - Facile2Soutenir
LES BEBES BUGS - Facile2Soutenir
Actualités Scientifiques - Médicales: #mers-cov #ribavirine #interferon Ribavirine et interferon alfa-2a pour le traitement du...
Actualités Scientifiques - Médicales: #thelancetinfectiousdiseases #éditorial #monkeypox #épidémie Suivi de l'épidémie de...
Association Bardet-Biedl1
- jeg går PAR COURRIER Cliquez sur le lien ci-dessous, imprimez le document puis une fois complété, adressez le, accompagné de votre règlement, à l'adresse suivante : Association Bardet-Biedl chez M. Grégory BOUETEL 5 rue du petit prince 49610 MURS-ERIGNE. (bardet-biedl.com)
L'association3
- Grâce à eux, nous avons pu reverser à l'association Bardet Bield Syndrome, 3% de notre chiffre d'affaires. (lasergame-entreprise.com)
- N'hésitez pas à prendre contact avec notre trésorier jeg går NOUS SOUTENIR GRACE A VOS ACHATS SUR INTERNET L'association est inscrite sur le site facile2soutenir.fr Facile2soutenir permet de générer des dons pour notre cause grâce à nos achats en ligne. (bardet-biedl.com)
- Votre engagement auprès de l'Association BARDET-BIEDL peut aussi prendre la forme d'un don ponctuel. (bardet-biedl.com)
Maladie6
- Le Syndrome de Bardet Biedl est une maladie génétique rare qui affecte de nombreux systèmes du corps. (12steplife.org)
- Le Syndrome de Bardet Biedl (BBS) est une colopathie non motte-le multisystémique principalement caractérisée par la dystrophie rétinienne des cônes-bâtonnets, l'obésité et ses complications, la polydactylie post-axiale, l'insuffisance hypogonadotrope-hypogonadisme ou les malformations génitales-urinaires, et les malformations rénales ou maladie parenchymateuse rénale. (12steplife.org)
- VarCoPP pourrait ainsi étudier et fournir des suggestions de combinaisons diagnostiques pour toute maladie pour laquelle des gènes causaux sont inconnus ou connus comme, par exemple, les centaines de gènes d'autisme ou d'épilepsie ou les 20 gènes du Syndrome malformatif de Bardet-Biedl, une maladie rare où les combinaisons de variants sont très présentes, détaille l'ULB. (plusmagazine.be)
- Le mot syndrome désigne une maladie dont les symptômes se présentent de façon constante et dont la cause n'est pas élucidée. (euro-wabb.org)
- Le syndrome de Wolfram est une maladie récessive autosomique, ce qui signifie qu'il est transmis par les deux parents, porteur chacun d'une copie anormale du gène responsable du syndrome et d'une copie normale. (euro-wabb.org)
- Dans le syndrome de Dravet, la fenfluramine a produit une diminution significativement plus importante de fréquence des convulsions en comparaison du placebo et était bien tolérée de manière générale, avec notamment aucune maladie des valvules cardiaques ni d'hypertension artérielle pulmonaire détectées. (actuscimed.com)
Laurence Moon2
- Syndrome de Laurence Moon Bardet Bield. (wikipedia.org)
- Au cours du temps, les syndromes de Laurence Moon et de Bardet-Biedl ont été soit séparés, soit regroupés. (wikipedia.org)
Produisent2
- La plupart des hommes atteints de Syndrome de Bardet Biedl produisent des quantités réduites d'hormones sexuelles (hypogonadisme) et sont habituellement incapables de concevoir des enfants biologiques (infertiles). (12steplife.org)
- Les anomalies congénitales des membres ont des causes multiples et se produisent souvent comme une composante de divers syndromes congénitaux. (msdmanuals.com)
Perte4
- Syndrome de McKusick Kaufman : Ce syndrome associant hydrohematocolpos, polydactylie et cardiopathie congénitale est en rapport avec une mutation du gène MKKS impliqué dans certains types de syndrome de Bardet-Biedl Syndrome d'Alström qui associe rétinite pigmentaire, obésité, perte d'audition progressive, cardiomyopathie, insulino-résistance et retard de développement. (wikipedia.org)
- La perte de vision est l'une des principales caractéristiques du Syndrome de Bardet Biedl. (12steplife.org)
- Des caractéristiques faciales distinctives, des anomalies dentaires, des doigts ou des orteils anormalement courts ou fusionnés et une perte partielle ou complète du sens de l'odeur (anosmie) ont aussi été signalés chez certaines personnes atteintes du Syndrome de Bardet Biedl. (12steplife.org)
- Des anomalies supplémentaires peuvent être également visibles : la perte auditive neurosensorielle (syndrome de Waardenburg-Shah), une déficience intellectuelle (syndrome de Mowat-Wilson), une hypoventilation alvéolaire centrale (syndrome de Haddad), des anomalies des membres (syndrome de Bardet-Biedl), un cancer médullaire de la thyroide (néoplasie endocrinienne multiple type 2B) ou encore des anomalies chromosomiques (syndrome de Down). (passeportsante.net)
Atteints3
- Le but de la présente étude était d'évaluer l'efficacité et l'innocuité de la fenfluramine chez des patients atteints du syndrome de Dravet. (actuscimed.com)
- Dans cette étude clinique randomisée, en double aveugle, contrôlée par placebo, nous avons recruté des enfants et des jeunes adultes atteints par le syndrome de Dravet. (actuscimed.com)
- La fenfluramine pourrait donc représenter une nouvelle option de traitement chez les patients atteints du syndrome de Dravet. (actuscimed.com)
Anomalies4
- Syndrome de Bardet-Biedl Mise en garde médicale modifier - modifier le code - voir Wikidata (aide) Le syndrome de Bardet-Biedl est un syndrome qui associe une rétinite pigmentaire (toujours), une obésité (souvent), une polydactylie post axiale (souvent), un hypogonadisme hypogonadotrope chez les garçons (parfois), des malformations génitales complexes chez les filles (parfois), des anomalies rénales (souvent) responsables de la mortalité de ce syndrome. (wikipedia.org)
- D'autres signes et symptômes importants du Syndrome de Bardet Biedl incluent la présence de doigts ou d'orteils supplémentaires (polydactylie), une déficience intellectuelle ou des problèmes d'apprentissage, et des anomalies des organes génitaux. (12steplife.org)
- De nombreuses personnes atteintes de Syndrome de Bardet Biedl présentent également des anomalies rénales, qui peuvent être graves ou mettant en jeu le pronostic vital. (12steplife.org)
- Les causes les plus fréquentes d'amputation congénitale des membres sont des anomalies des tissus mous et/ou vasculaires, telles qu'un déficit des membres lié à une bride amniotique, un syndrome dans lequel des brides libres d'amnion s'enchevêtrent ou se fondent avec le tissu fœtal. (msdmanuals.com)
Responsable du syndrome1
- Seul le séquençage de la mutation M390R responsable du syndrome de Bardet-Biedl de type 1 était disponible en 2005. (wikipedia.org)
D'autres2
- Plusieurs mutations impliquant plus de 18 différents gènes sont responsables de ce syndrome : D'autres gènes sont impliquées, dont le CEP290. (wikipedia.org)
- L'apparition de trouble visuel en rapport avec la choriorétinite permet le diagnostic, encore que ce syndrome soit le deuxième plus mal diagnostiqué, parmi les Rétinites Pigmentaires syndromiques (associant d'autres symptômes). (wikipedia.org)
Forme2
- Dans la forme la plus grave de syndrome (déficit) de radius, le radius est absent, comme chez ce patient. (msdmanuals.com)
- Le syndrome de Dravet est une forme rare d'encéphalopathie épileptique et développementale résistante aux traitements, caractérisée par des convulsions multiples, fréquentes, et invalidantes. (actuscimed.com)
Diagnostic2
- Un diagnostic différentiel distingue le SLSN du syndrome de Bardet-Biedl. (retina.ch)
- Bonjour je suis la maman de Louise 2 ans edemi qui a été diagnostic en janvier 2021 du syndrome de bainbridge ropers, elle se déplace à l'aide de jeux, problèmes de sommeil elle mange des morceaux, elle ne parle pas ne nous montre pas se qu'elle veut , n'aime pas être dans les bras Trouble autistique elle a peur de la foule crise de convulsions. (maladiesraresinfo.org)
Variants1
- Dans un papier récent, nous avons montré dans une cohorte de patients avec un syndrome d'Alport lié à l'X, que seuls 60% des variants introniques profonds identifiés chez les patients, et dont l'effet sur l'épissage avait été prouvé, étaient décrit comme altérant probablement l'épissage par les scores de prédictions. (mycongressonline.net)
Polydactylie1
- Ce syndrome ressemble au syndrome de Bardet Biedl mais il n'existe pas de polydactylie et les personnes vont développer une paralysie spastique. (wikipedia.org)
Plupart1
- La plupart des personnes atteintes de Syndrome de Bardet Biedl développent également une vision centrale floue (mauvaise acuité visuelle) et deviennent légalement aveugles à l'adolescence ou au début de l'âge adulte. (12steplife.org)
Rare1
- An EU Rare Diseases Registry for Wolfram syndrome, Alström syndrome, Bardet-Biedl syndrome and other rare diabetes syndromes. (euro-wabb.org)
Adultes1
- J'aimerai partager avec des parents d'enfants ou adultes touchés par ce syndrome afin d'échanger sur les différentes méthodes adoptées et adaptées à chacun. (maladiesraresinfo.org)
Pigmentaire2
- Syndrome de Bardet-Biedl Mise en garde médicale modifier - modifier le code - voir Wikidata (aide) Le syndrome de Bardet-Biedl est un syndrome qui associe une rétinite pigmentaire (toujours), une obésité (souvent), une polydactylie post axiale (souvent), un hypogonadisme hypogonadotrope chez les garçons (parfois), des malformations génitales complexes chez les filles (parfois), des anomalies rénales (souvent) responsables de la mortalité de ce syndrome. (wikipedia.org)
- Syndrome de McKusick Kaufman : Ce syndrome associant hydrohematocolpos, polydactylie et cardiopathie congénitale est en rapport avec une mutation du gène MKKS impliqué dans certains types de syndrome de Bardet-Biedl Syndrome d'Alström qui associe rétinite pigmentaire, obésité, perte d'audition progressive, cardiomyopathie, insulino-résistance et retard de développement. (wikipedia.org)
Responsables1
- Identifier les gènes responsables du syndrome de Bardet-Biedl (BBS). (bardet-biedl.com)
Personnes1
- De nombreuses personnes atteintes de Syndrome de Bardet Biedl présentent également des anomalies rénales, qui peuvent être graves ou mettant en jeu le pronostic vital. (12steplife.org)
Disponible1
- Seul le séquençage de la mutation M390R responsable du syndrome de Bardet-Biedl de type 1 était disponible en 2005. (wikipedia.org)
Souvent1
- Souvent, il peut exister aussi des aussi reflux vésico-urétraux et un syndrome polyurie/polydipsie. (wikipedia.org)