Epidermolysis Bullosa
Epidermolysis Bullosa Dystrophica
Epidermolysis Bullosa Simplex
Epidermolysis Bullosa, Junctional
Epidermolysis Bullosa Acquisita
Collagène Type Vii
Plectine
Kératine-14
Peau
Pylore
Collagènes Non Fibrillaires
Intégrine Bêta4
Hémidesmosomes
Kératine-5
Kératines
Dermatoses Vésiculobulleuses
Kératinocytes
Collagènes Associés Aux Fibrilles
Généalogie
Desmosomes
Mutation Décalage Cadre Lecture
Membrane Basale
Codon Non-Sens
Protéines Filament Intermédiaire
Mutation
Maladies Génétiques De La Peau
Collagène
Peau Artificielle
Atrésie Intestinale
Auto-Antigène
Codon Terminateur
Pemphigoïde Bulleuse
Filaments Intermédiaires
Réticuline
Phénotype
Prurigo
Cornets
Laminine
Intégrine Alpha6 Bêta4
Mutation Ponctuelle
Mosaïcisme
Fatal Outcome
Microbial Collagenase
Exon
Intégrine Alpha6
Hétérozygote
Epidermis
Analyse Mutations Adn
Données Séquence Moléculaire
Molécules D'Adhérence Cellulaire
Acantholyse
Genetic Therapy
Maladies Rares
Séquence Nucléotidique
Microanalyse Par Sonde
Mutation Faux Sens
Maladies De La Bouche
Fibroblastes
Prurit
Diagnostic Prénatal
Hyperkératose
Family Health
Ichthyose Bulleuse De Siemens
Technique Anticorps Fluorescent
L'épidermolyse bulleuse (EB) est un groupe de troubles génétiques rares qui affectent la peau et les muqueuses. Ces maladies sont caractérisées par une fragilité cutanée extrême, entraînant des bulles et des plaies douloureuses à la suite de frottements ou de traumatismes même minimes.
Il existe plusieurs types d'EB, classés en fonction du niveau de décollement de la peau :
- EB simple : les bulles se forment dans l'épiderme (couche supérieure de la peau).
- EB jonctionnelle : les bulles se forment au niveau de la jonction dermo-épidermique (entre l'épiderme et le derme).
- EB dystrophique : les bulles se forment dans le derme (couche profonde de la peau).
Les symptômes varient en fonction du type et de la gravité de la maladie. Ils peuvent inclure des cloques et des plaies récurrentes, une cicatrisation anormale avec formation de cicatrices et de contractures, une perte de pigmentation cutanée, des ongles fragiles ou absents, et une inflammation chronique des muqueuses. Dans les cas graves, l'EB peut affecter d'autres organes, tels que le tractus gastro-intestinal, entraînant des complications potentiellement mortelles.
Le diagnostic de l'EB repose sur une combinaison d'examen clinique, d'anamnèse familiale et de tests génétiques. Actuellement, il n'existe pas de traitement curatif pour l'épidermolyse bulleuse. Le traitement est principalement axé sur les soins de la peau, la prévention des lésions et la prise en charge des complications associées à la maladie.
L'épidermolyse bulleuse dystrophique (EBD) est un groupe rare et héréditaire de désordres cutanés caractérisés par une fragilité cutanée excessive entraînant des bulles et des plaies spontanées ou à la suite d'un traumatisme mineur. Il s'agit d'une forme plus grave et généralement plus étendue de l'épidermolyse bulleuse que l'épidermolyse bulleuse simplicia (EBS).
L'EBD est causée par des mutations dans le gène COL7A1, qui code pour la chaîne alpha-1 de collagène de type VII, une protéine essentielle à l'ancrage de la membrane basale dermique-épidermique. Ces mutations entraînent une production réduite ou anormale de cette protéine, affaiblissant ainsi l'adhésion entre les couches de la peau et provoquant des bulles et des plaies.
Les symptômes de l'EBD peuvent varier en fonction de la gravité de la maladie et de la région du corps touchée. Les manifestations courantes comprennent :
1. Formation de bulles et de plaies spontanées ou à la suite d'un traumatisme mineur, en particulier sur les mains, les pieds, les coudes et les genoux.
2. Cicatrices excessives et contractures qui peuvent limiter la mobilité articulaire.
3. Déformations osseuses et pseudosyndactylie (fusion des doigts ou des orteils).
4. Érosions et infections cutanées récurrentes, ce qui peut entraîner une septicémie et d'autres complications potentiellement mortelles.
5. Perte de cheveux et ongles cassants.
6. Dysphagie (difficulté à avaler) due à des lésions dans la bouche et l'œsophage.
7. Anémie et carences nutritionnelles en raison d'une mauvaise absorption des nutriments.
Le traitement de l'EBD vise principalement à gérer les symptômes et à prévenir les complications. Les options thérapeutiques peuvent inclure :
1. Soins cutanés rigoureux pour prévenir les infections et favoriser la guérison des plaies.
2. Analgésiques et anti-inflammatoires pour soulager la douleur et l'inflammation.
3. Antibiotiques pour traiter et prévenir les infections cutanées.
4. Chirurgie reconstructive pour corriger les déformations osseuses et améliorer la fonction articulaire.
5. Nutrition entérale ou parentérale pour gérer la dysphagie et les carences nutritionnelles.
6. Thérapies complémentaires telles que la physiothérapie, l'ergothérapie et l'orthophonie pour améliorer la fonction physique et mentale.
7. Essais cliniques de médicaments expérimentaux ciblant les voies inflammatoires et immunitaires sous-jacentes à la maladie.
Épidermolyse bulleuse simplex (EBS) est une forme héréditaire de la peau fragile et blistering condition connue sous le nom d'épidermolyse bulleuse. Il s'agit d'une maladie génétique qui affecte les protéines qui assurent la cohésion entre les couches de la peau, entraînant ainsi une formation excessive de bulles et d'ampoules, en particulier dans les zones de friction ou de pression.
EBS est généralement causée par des mutations dans les gènes KRT5 ou KRT14, qui codent pour les chaînes keratine 5 et 14 respectivement. Ces protéines forment un réseau de fibres dans la couche basale de la peau, offrant résistance et intégrité mécanique à la peau. Les mutations dans ces gènes entraînent une instabilité du cytosquelette et une fragilité accrue de la peau, ce qui conduit à la formation de bulles et d'ampoules.
EBS se caractérise par des lésions cutanées récurrentes, souvent présentes dès la naissance ou apparaissant dans les premiers mois de vie. Les symptômes peuvent varier en gravité, allant de bulles mineures et de cloques à des ampoules plus graves et douloureuses. Les zones couramment touchées comprennent les mains, les pieds, les coudes, les genoux et le cuir chevelu. Les lésions cutanées peuvent s'infecter, entraînant des cicatrices, une contracture et une perte de fonction dans les cas graves.
Il existe plusieurs sous-types d'EBS, qui diffèrent par leurs manifestations cliniques et leur mode de transmission héréditaire. Les formes localisées d'EBS sont généralement moins graves et se limitent à certaines zones du corps, tandis que les formes généralisées peuvent affecter tout le corps et entraîner des complications plus graves.
Le traitement de l'EBS vise principalement à gérer les symptômes et à prévenir les complications. Cela peut inclure des soins de plaies appropriés, la gestion de la douleur et la prévention des infections. Dans certains cas, une intervention chirurgicale peut être nécessaire pour corriger les contractures ou les déformités.
La recherche sur l'EBS se poursuit, avec un intérêt particulier pour le développement de thérapies ciblées qui peuvent renforcer la structure et la fonction de la peau. Ces approches comprennent des stratégies visant à modifier les protéines du cytosquelette ou à remplacer les gènes défectueux par la thérapie génique.
La « Epidermolysis Bullosa, Junctional » (EBJ) est une forme rare et sévère d'épidermolyse bulleuse, une maladie génétique de la peau caractérisée par une fragilité cutanée extrême entraînant des bulles et des plaies douloureuses au moindre frottement ou traumatisme.
L'EBJ se distingue des autres formes d'épidermolyse bulleuse par la localisation de ces bulles, qui se forment au niveau des jonctions dermo-épidermiques (la jonction entre le derme et l'épiderme). Cette forme particulière est causée par des mutations dans les gènes responsables de la synthèse des protéines impliquées dans l'ancrage de la membrane basale, ce qui affaiblit considérablement l'adhésion entre le derme et l'épiderme.
Les symptômes de l'EBJ peuvent varier en fonction de sa sous-catégorie (Herlitz ou non-Herlitz), mais ils comprennent généralement des bulles et des plaies cutanées dès la naissance ou dans les premiers mois de vie, un risque élevé d'infections cutanées, une cicatrisation anormale avec formation de cicatrices atrophiques et/ou d'excroissances anormales (comme des doigts en « main de homard »), ainsi que des complications systémiques potentielles telles qu'une dénutrition, une anémie et des problèmes respiratoires.
Le traitement de l'EBJ est principalement symptomatique et vise à prévenir les traumatismes cutanés, assurer une bonne hygiène et cicatrisation des plaies, et gérer les complications associées. Dans certains cas, des traitements expérimentaux comme les thérapies géniques peuvent être envisagés pour tenter de corriger la base moléculaire de la maladie.
Épidermolyse bulleuse acquise (EBA) est une maladie rare et grave de la peau caractérisée par la formation de cloques et d'ampoules sur la peau et les muqueuses, souvent déclenchées par des traumatismes mineurs ou sans cause apparente. Contrairement à l'épidermolyse bulleuse héréditaire, qui est présente à la naissance ou se développe tôt dans l'enfance, l'EBA est généralement diagnostiquée chez les adultes, bien que des cas pédiatriques aient été signalés.
L'EBA est causée par des anomalies dans le fonctionnement du système immunitaire, entraînant une réaction auto-immune contre les protéines de la membrane basale, en particulier la collagène de type VII (COL7). Cette réaction provoque une inflammation et une dégradation de la jonction d'ancrage entre l'épiderme et le derme, entraînant des lésions cutanées et muqueuses.
Les symptômes courants de l'EBA comprennent:
1. Formation de cloques et d'ampoules sur la peau, en particulier sur les zones soumises à une friction ou à une pression accrues, telles que les mains, les pieds, les coudes et les genoux.
2. Cicatrisation lente et formation de cicatrices anormales après la guérison des plaies.
3. Blépharite (inflammation des paupières) et conjonctivite (inflammation de la membrane muqueuse recouvrant le blanc de l'œil), entraînant une sécheresse oculaire, une photophobie et une vision floue.
4. Sténose du tractus gastro-intestinal, provoquant des douleurs abdominales, des nausées, des vomissements, de la diarrhée et une perte de poids.
5. Atrophie vulvo-vaginale, entraînant une sécheresse vaginale, des rapports sexuels douloureux et une augmentation du risque d'infections urinaires.
Le diagnostic de l'EBA repose sur l'anamnèse, l'examen clinique, la biopsie cutanée et les tests immunologiques spécifiques. Le traitement de l'EBA vise à soulager les symptômes, à prévenir les complications et à améliorer la qualité de vie des patients. Les options thérapeutiques comprennent:
1. Corticostéroïdes topiques ou systémiques pour réduire l'inflammation cutanée.
2. Immunosuppresseurs, tels que l'azathioprine, la ciclosporine et le mycophénolate mofétil, pour moduler la réponse immunitaire.
3. Inhibiteurs de tyrosine kinase, comme l'imatinib et le nintédanib, pour inhiber la prolifération des cellules fibroblastiques.
4. Antagonistes des récepteurs de l'interféron gamma, tels que l'emapalumab, pour réguler l'activité immunitaire.
5. Thérapies ciblées, telles que les anticorps monoclonaux anti-IL-6 (tocilizumab) et anti-CD20 (rituximab), pour inhiber la réponse inflammatoire.
6. Chirurgie reconstructive pour corriger les déformations cutanées et fonctionnelles.
7. Soins de soutien, tels que l'hydratation cutanée, la protection solaire et l'évitement des facteurs déclenchants, pour améliorer le confort et la qualité de vie des patients.
Le collagène de type VII est un type spécifique de protéine qui joue un rôle crucial dans la structure et la fonction des tissus conjonctifs dans le corps humain. Plus précisément, il est un composant essentiel du complexe anchoring fibrillar (AF), qui assure l'ancrage de la membrane basale dermique à la membrane basale épidermique dans la jonction dermo-épidermique.
La protéine collagène de type VII est codée par le gène COL7A1 et se compose de trois chaînes polypeptidiques identiques, appelées alpha-1 (III) collagène de type VII. Ces chaînes s'assemblent pour former une molécule tridimensionnelle en forme de tours de guet, qui sont ensuite organisées en réseaux tridimensionnels pour former les fibres anchoring fibrils (AF).
Les AF sont ancrées dans la membrane basale dermique et s'étendent dans la membrane basale épidermique, où elles interagissent avec d'autres protéines de la matrice extracellulaire pour former un complexe anchoring. Ce complexe est essentiel pour maintenir l'intégrité structurelle et fonctionnelle de la jonction dermo-épidermique, ce qui est crucial pour la fonction barrière de la peau.
Les mutations du gène COL7A1 peuvent entraîner des maladies génétiques rares telles que le déficit en collagène de type VII, également connu sous le nom d'épidermolyse bulleuse acquise ou héréditaire. Ces conditions sont caractérisées par une fragilité excessive de la peau et des muqueuses, entraînant des bulles et des cicatrices.
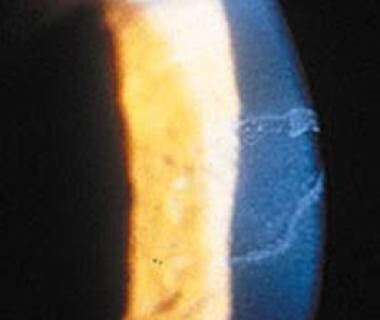
Une cloque est un type de lésion cutanée dans laquelle il y a accumulation de liquide sous la peau. Cela se produit lorsque les petits vaisseaux sanguins sous la peau sont endommagés, permettant au plasma de s'échapper et de s'accumuler dans l'espace situé entre la peau et le tissu sous-jacent. Les cloques peuvent être causées par une variété de facteurs, y compris les brûlures, les frottements excessifs, les éruptions cutanées, les infections ou certaines conditions médicales.
Les cloques sont généralement douloureuses et sensibles au toucher. Elles peuvent apparaître de différentes tailles, allant d'une petite bulle à une grande zone remplie de liquide. Dans la plupart des cas, il est recommandé de ne pas percer ou éclater une cloque, car cela peut entraîner une infection supplémentaire et ralentir le processus de guérison. Au lieu de cela, il est préférable de protéger la zone affectée en appliquant un pansement stérile et de consulter un professionnel de la santé si la cloque ne guérit pas ou s'il y a des signes d'infection, tels que rougeur, chaleur, gonflement ou pus.
La plectine est une protéine structurelle importante dans les cellules, en particulier dans le cytosquelette des muscles squelettiques. Elle joue un rôle crucial dans la liaison et l'organisation des filaments d'actine, qui sont des composants essentiels du cytosquelette. La plectine est également impliquée dans la formation et le maintien des jonctions communicantes entre les cellules musculaires squelettiques, ce qui permet la communication et la coordination entre ces cellules. Des mutations dans le gène de la plectine peuvent entraîner diverses maladies musculaires, y compris certaines formes de dystrophie musculaire.
La kératine-14 est une protéine structurelle spécifique qui appartient à la famille des kératines de filaments intermédiaires. Elle est principalement exprimée dans les cellules épithéliales stratifiées, en particulier dans les couches profondes de l'épiderme, où elle joue un rôle crucial dans le maintien de la structure et de la fonction de la peau.
Dans la peau, la kératine-14 s'associe à la kératine-5 pour former des hétérodimères qui forment ensuite des réseaux de filaments intermédiaires dans les cellules épithéliales. Ces réseaux aident à déterminer la forme et la fonction des cellules, ainsi qu'à fournir une résistance mécanique à l'épiderme.
Les mutations du gène KRT14, qui code pour la kératine-14, peuvent entraîner diverses maladies génétiques de la peau, telles que l'épidermolyse bulleuse et le psoriasis. Ces affections cutanées sont caractérisées par une fragilité accrue de la peau, des bulles et des lésions cutanées douloureuses.
En plus de son rôle dans la peau, la kératine-14 est également exprimée dans les follicules pileux, où elle contribue à la structure et à la fonction des cellules souches des follicules pileux. Les anomalies de la kératine-14 peuvent entraîner une alopécie ou une perte de cheveux.
En résumé, la kératine-14 est une protéine structurelle essentielle à la peau et aux follicules pileux, qui joue un rôle crucial dans le maintien de leur intégrité structurelle et fonctionnelle.
La peau est le plus grand organe du corps humain, servant de barrière physique entre l'intérieur du corps et son environnement extérieur. Elle a plusieurs fonctions importantes, y compris la protection contre les agents pathogènes, les dommages mécaniques, les variations de température et les rayons ultraviolets du soleil.
La peau est composée de trois couches principales : l'épiderme, le derme et l'hypoderme. L'épiderme est la couche externe, constituée principalement de cellules mortes qui sont constamment shed and replaced. The dermis, just below the epidermis, contains tough connective tissue, sweat glands, hair follicles, and blood vessels. The hypodermis is the deepest layer, composed of fat and connective tissue that provides padding and insulation for the body.
In addition to providing protection, the skin also plays a role in sensation through nerve endings that detect touch, temperature, and pain. It helps regulate body temperature through sweat glands that release perspiration to cool the body down when it's hot. Furthermore, the skin synthesizes vitamin D when exposed to sunlight.
Maintaining healthy skin is important for overall health and well-being. Proper care includes protecting it from excessive sun exposure, keeping it clean, moisturized, and nourished with essential nutrients.
Le pylore est la région musculaire située à la jonction entre l'estomac et le duodénum, qui est la première partie de l'intestin grêle. Il se compose d'un sphincter, un anneau musculaire qui régule le passage des aliments et des sécrétions gastriques dans le duodénum. Le pylore permet de contrôler la vidange de l'estomac en fonction de la quantité et de la composition du chyme (mélange semi-fluide d'aliments partiellement digérés, de sucs gastriques et d'enzymes) et des signaux nerveux et hormonaux reçus.
Le pylore joue un rôle important dans le processus de digestion en assurant une transition progressive entre les phases de broyage et de mélange effectuées par l'estomac et la phase d'absorption réalisée par l'intestin grêle. Une dysfonction du pylore peut entraîner des problèmes digestifs, tels que des nausées, des vomissements, une douleur abdominale et une malnutrition.
Les collagènes non fibrillaires sont un type de protéines structurelles qui jouent un rôle important dans la matrice extracellulaire des tissus conjonctifs. À la différence des collagènes fibrillaires, qui forment des fibres organisées et rigides, les collagènes non fibrillaires ont une structure plus désordonnée et sont souvent associés à d'autres protéines pour former des réseaux complexes.
Les collagènes non fibrillaires comprennent plusieurs sous-types, tels que le collagène de type IV, qui est un composant majeur de la membrane basale et joue un rôle important dans l'adhésion cellulaire et la régulation de la perméabilité des vaisseaux sanguins. Le collagène de type VII est également considéré comme non fibrillaire, car il forme des anchors fibres qui ancrent la membrane basale à la lame basale de la peau.
Les collagènes non fibrillaires sont impliqués dans une variété de processus biologiques, tels que la régulation de la migration cellulaire, la différenciation cellulaire et la morphogenèse des tissus. Des anomalies dans les gènes codant pour ces protéines peuvent entraîner des maladies génétiques graves, telles que l'épidermolyse bulleuse, une maladie de la peau caractérisée par une fragilité excessive de la peau et des muqueuses.
L'intégrine bêta-4, également connue sous le nom d'ITGB4, est un type de protéine qui appartient à la famille des intégrines. Les intégrines sont des récepteurs cellulaires transmembranaires qui jouent un rôle crucial dans l'adhésion cellulaire et la signalisation cellulaire.
L'intégrine bêta-4 est spécifiquement exprimée dans les cellules épithéliales et se lie à l'intégrine alpha-6 pour former un complexe hétérodimérique appelé très grand récepteur de la laminine (VLA-4 ou CD49d/CD29). Ce complexe est important pour l'adhésion des cellules épithéliales aux composants de la matrice extracellulaire, tels que la laminine et le collagène.
L'intégrine bêta-4 est ancrée dans la membrane cellulaire et s'étend profondément dans le cytoplasme, où elle se lie à des protéines structurelles telles que la filamine A et les kératines. Cette association permet de stabiliser l'ancrage des cellules épithéliales à la matrice extracellulaire et de participer à la régulation de processus tels que la différenciation cellulaire, la migration cellulaire et la croissance tumorale.
Des mutations dans le gène ITGB4 ont été associées à des maladies telles que l'épidermolyse bulleuse acquise, une forme d'épidermolyse bulleuse caractérisée par des bulles cutanées et muqueuses douloureuses. De plus, l'intégrine bêta-4 est souvent surexprimée dans divers types de cancer, ce qui peut contribuer à la progression tumorale en favorisant la migration cellulaire et l'angiogenèse.
Les hémidesmosomes sont des structures spécialisées situées à la surface basale des cellules épithéliales et des cellules myoépithéliales. Ils assurent l'ancrage de ces cellules à la membrane basale sous-jacente, ce qui permet une adhérence forte et stable entre l'épithélium et la matrice extracellulaire. Les hémidesmosomes sont composés de plusieurs protéines, y compris les intégrines alpha6beta4, les protéines BP230 (ou bullous pemphigoid antigen 1) et les protéines de la famille des plakins (par exemple, la plectine, la BP230 et la décorine). Les hémidesmosomes jouent un rôle crucial dans la préservation de l'intégrité structurale et fonctionnelle des épithéliums, en particulier dans les tissus qui subissent des forces mécaniques importantes, comme la peau et les muqueuses. Des anomalies dans la structure ou la composition des hémidesmosomes peuvent entraîner diverses affections cutanées et muqueuses, telles que le pemphigoid bulleux et l'épidermolyse bulleuse.
La kératine 5 est une protéine structurelle fibreuse qui fait partie de la famille des cytokératines. Elle est spécifiquement exprimée dans les cellules épithéliales stratifiées, en particulier dans l'épiderme et les muqueuses. Dans l'épiderme, elle est principalement trouvée dans le premier layer basal où elle forme des paires hétérodimériques avec la kératine 14. Ces deux protéines sont essentielles pour maintenir l'intégrité structurale et mécanique de l'épithélium, en particulier pendant les processus de division cellulaire et de différenciation. Les mutations dans le gène codant pour la kératine 5 peuvent entraîner des maladies génétiques telles que l'épidermolyse bulleuse, une affection caractérisée par une fragilité accrue de la peau et la formation de bulles.
Les kératines sont des protéines fibreuses qui composent la structure de divers tissus épithéliaux dans le corps humain. Elles sont particulièrement concentrées dans la couche externe de la peau, les cheveux et les ongles. Dans ces structures, les kératines forment des chaînes rigides qui leur confèrent une grande résistance à la traction et aux dommages mécaniques.
Dans la cornée de l'œil, qui est également constituée de cellules épithéliales, les kératines jouent un rôle important dans le maintien de la transparence et de la forme de cette structure essentielle à la vision. Les kératines sont synthétisées par des cellules spécialisées appelées kératinocytes.
Les mutations dans les gènes codant pour les différentes formes de kératine peuvent entraîner diverses affections cutanées, telles que le psoriasis, l'eczéma et certaines formes de dermatite. Des anomalies dans la structure des kératines peuvent également être associées à des maladies génétiques rares affectant les cheveux, la peau et les ongles, telles que la trichorrhée nodulaire ou le syndrome des ongles fragiles.
Un gène récessif est un type de gène qui doit être présent en double exemplaire (un hérité de chaque parent) pour qu'un trait ou une condition particulière se manifeste chez un individu. Dans l'espèce humaine, les caractéristiques contrôlées par des gènes récessifs ne s'expriment que si un individu hérite d'une copie de ce gène récessif de chaque parent.
Si un seul parent transmet le gène récessif, l'individu sera quand même porteur du trait récessif mais ne le manifestera pas, on l'appelle alors un porteur sain. Ce n'est que lorsque deux personnes qui sont toutes les deux porteuses d'un gène récessif ont un enfant qu'il y a une chance sur quatre (25%) à chaque grossesse pour que l'enfant hérite du trait récessif des deux parents et manifeste donc la caractéristique ou la maladie associée à ce gène.
Un exemple couramment cité de gène récessif est celui responsable de la drépanocytose, une maladie génétique affectant les globules rouges. Pour développer cette maladie, un individu doit hériter d'une copie du gène anormal de chaque parent. Si l'individu n'en hérite que d'une seule copie, il sera porteur sain du trait drépanocytaire mais ne développera pas la maladie.
Les dermatoses vésiculobulleuses sont un groupe de troubles cutanés caractérisés par la présence de cloques (vésicules) et de grandes bulles remplies de liquide sur la peau. Ces lésions cutanées se produisent lorsque la barrière protectrice de la peau est endommagée, entraînant une séparation anormale des couches supérieures et inférieures de la peau.
Ces affections peuvent être classées en deux catégories principales : les dermatoses vésiculobulleuses immunologiques et non immunologiques. Les dermatoses vésiculobulleuses immunologiques sont causées par des réactions anormales du système immunitaire, tandis que les dermatoses vésiculobulleuses non immunologiques sont dues à des causes génétiques, des irritants ou des maladies sous-jacentes.
Les exemples de dermatoses vésiculobulleuses incluent le pemphigus, la pemphigoïde bulleuse, le dermatite herpétiforme et l'épidermolyse bulleuse. Les symptômes peuvent varier en fonction du type spécifique de dermatose vésiculobulleuse, mais comprennent généralement des démangeaisons, une douleur cutanée, des rougeurs et l'apparition de cloques ou de bulles remplies de liquide. Le traitement dépend du type spécifique de dermatose vésiculobulleuse et peut inclure des médicaments topiques ou systémiques pour contrôler l'inflammation, des soins de la peau pour prévenir les infections et des mesures visant à éviter les déclencheurs connus.
Les kératinocytes sont les principales cellules constitutives de l'épiderme, la couche externe de la peau. Ils synthétisent la kératine, une protéine fibreuse qui confère à la peau sa résistance et son intégrité structurelle. Les kératinocytes subissent une différenciation progressive en migrant vers la surface de la peau, formant ainsi des couches de cellules cornées mortes qui assurent une barrière protectrice contre les agents pathogènes, les irritants et les pertes d'eau. Les kératinocytes jouent également un rôle crucial dans la réponse immunitaire cutanée en produisant divers facteurs chimiques qui régulent l'inflammation et aident à coordonner la défense de l'organisme contre les infections.
Les collagènes associés aux fibrilles sont des protéines structurelles qui s'associent à des fibrilles de collagène pour former des tissus conjonctifs dans le corps humain. Les fibrilles de collagène sont les principaux composants de la matrice extracellulaire et assurent la résistance mécanique, l'élasticité et la stabilité des tissus conjonctifs.
Les collagènes associés aux fibrilles comprennent plusieurs types de collagène, notamment les types IX, XII et XIV. Ces collagènes ont des domaines structurels qui leur permettent de s'associer à des fibrilles de collagène existantes, ce qui contribue à la régulation de la formation et du remodelage des fibrilles de collagène.
Les mutations dans les gènes codant pour ces collagènes associés aux fibrilles peuvent entraîner des maladies génétiques rares, telles que les myopathies et les ostéochondrodysplasies, qui affectent la structure et la fonction des tissus conjonctifs. Par exemple, une mutation dans le gène COL12A1, qui code pour le collagène de type XII, a été associée à une forme rare d'arthrite juvénile systémique.
En résumé, les collagènes associés aux fibrilles sont des protéines structurelles importantes qui jouent un rôle crucial dans la formation et le maintien de la matrice extracellulaire des tissus conjonctifs. Les mutations dans les gènes codant pour ces collagènes peuvent entraîner des maladies génétiques rares affectant la structure et la fonction des tissus conjonctifs.
En termes médicaux, la généalogie est l'étude systématique des antécédents familiaux et médicaux d'une personne ou d'une famille sur plusieurs générations. Elle vise à identifier les modèles de maladies héréditaires ou génétiques dans une famille, ce qui peut aider à évaluer le risque de développer certaines affections et à mettre en œuvre des stratégies de prévention et de dépistage appropriées.
Les professionnels de la santé utilisent souvent des arbres généalogiques pour représenter visuellement les relations familiales et les antécédents médicaux. Ces outils peuvent être particulièrement utiles dans la pratique clinique, en particulier lorsqu'il s'agit de maladies rares ou complexes qui ont tendance à se produire dans certaines familles en raison de facteurs génétiques sous-jacents.
En plus d'être un outil important pour la médecine préventive, la généalogie peut également fournir des informations précieuses sur l'histoire naturelle de diverses maladies et conditions, ce qui contribue à faire progresser notre compréhension globale de la pathogenèse et de la physiopathologie. Par conséquent, elle joue un rôle crucial dans la recherche médicale et les soins cliniques.
Les desmosomes sont des structures spécialisées dans la membrane plasmique qui servent de points d'ancrage entre les cellules épithéliales adjacentes. Ils aident à maintenir l'intégrité structurale et fonctionnelle des tissus en assurant une adhésion ferme entre les cellules. Les desmosomes sont composés de deux hémidésmosomes, chacun étant lié à la membrane plasmique d'une cellule différente.
Les desmosomes sont constitués de plusieurs protéines, y compris les cadhérines desmosomales, qui traversent la membrane plasmique et s'interdigitent avec les cadhérines desmosomales de la cellule adjacente pour former des jonctions intercellulaires. Les extrémités intracellulaires des cadhérines sont ancrées dans le cytosquelette via des protéines d'ancrage telles que les plakoglobines et les plakophilines.
Les desmosomes jouent un rôle crucial dans la résistance mécanique des tissus épithéliaux, en particulier ceux qui sont exposés à des forces de traction ou de cisaillement élevées, telles que la peau et le myocarde. Les mutations dans les gènes codant pour les protéines des desmosomes peuvent entraîner des maladies génétiques graves, telles que certaines formes d'épidermolyse bulleuse et de cardiomyopathie dilatée.
Une mutation décalage cadre de lecture (FCF, ou frameshift mutation en anglais) est un type de mutation génétique qui entraîne un décalage du cadre de lecture dans une région codante d'un gène. Cela se produit lorsqu'une ou plusieurs paires de bases sont insérées ou délétées dans l'ADN, ce qui modifie la séquence des codons suivants et perturbe la manière dont le code génétique est traduit en une chaîne d'acides aminés.
Dans un gène, les triplets de nucléotides (codons) spécifient l'ordre dans lequel les acides aminés doivent être assemblés pour former une protéine fonctionnelle. Un décalage du cadre de lecture entraîne la production d'une séquence d'acides aminés incorrecte et anormale, ce qui peut conduire à la synthèse d'une protéine non fonctionnelle ou tronquée.
Les mutations décalage cadre de lecture peuvent être causées par des erreurs de réplication de l'ADN, des dommages à l'ADN ou des événements d'insertion ou de délétion de séquences nucléotidiques. Elles sont souvent associées à des maladies génétiques graves et héréditaires, telles que certaines formes de cancer, la fibrose kystique, l'anémie falciforme et d'autres troubles liés aux gènes.
La membrane basale est une fine structure extracellulaire spécialisée, généralement constituée d'une matrice de fibres de collagène et de laminine, qui sépare et soutient les tissus ou les cellules adjacentes dans le corps. Elle sert souvent comme support structural pour les épithéliums et les endothéliums, régule la communication cellulaire et la migration, et joue un rôle crucial dans la cicatrisation des plaies et la pathogenèse de diverses maladies. La membrane basale est également connue sous le nom de membrane basale extracellulaire ou lame basale.
Les malformations de l'appareil digestif sont des anomalies congénitales qui affectent la structure et la fonction des organes du système digestif. Elles peuvent survenir à n'importe quel niveau, de la bouche à l'anus, et peuvent prendre diverses formes, selon l'organe concerné.
Voici quelques exemples de malformations de l'appareil digestif :
1. Fente labiale et palatine : Il s'agit d'une ouverture anormale dans le palais ou la lèvre supérieure, qui se forme pendant le développement du fœtus.
2. atrésie oesophagienne : C'est une malformation congénitale où l'oesophage ne se connecte pas à l'estomac.
3. Duodénum atresia : Il s'agit d'une malformation congénitale dans laquelle le duodénum, la première partie de l'intestin grêle, est obstrué ou mal formé.
4. Imperforation anale : C'est une malformation congénitale où l'anus ne se forme pas correctement, ce qui empêche les selles de quitter le corps.
5. Malrotation intestinale : Il s'agit d'une malformation congénitale dans laquelle les intestins ne tournent pas correctement pendant le développement du fœtus, ce qui peut entraîner une obstruction ou une torsion de l'intestin.
6. Hirsprung's disease : Il s'agit d'une malformation congénitale dans laquelle les muscles de la paroi intestinale ne se forment pas correctement, ce qui peut entraîner une obstruction ou un blocage des intestins.
Les malformations de l'appareil digestif peuvent être traitées par chirurgie, médicaments ou autres thérapies, selon la gravité et le type de la malformation.
Un codon non-sens, également connu sous le nom d'un codon stop ou d'un codon terminateur, est une séquence spécifique de trois nucléotides dans l'ARN messager (ARNm) qui conduit à la terminaison de la synthèse des protéines. Dans le code génétique, il y a 64 codons possibles, composés de combinaisons différentes d'un ensemble de quatre nucléotides (A, U, G et C). Parmi ces 64 codons, trois sont reconnus comme des codons non-sens : UAG (opale), UAA (ambre) et UGA (ocre). Lorsque la traduction ribosomique rencontre l'un de ces codons non-sens pendant le processus de synthèse des protéines, elle est interrompue, entraînant la libération prématurée de la chaîne polypeptidique et la formation d'une protéine tronquée ou non fonctionnelle.
Les mutations ponctuelles qui remplacent un codon de sens par un codon non-sens peuvent entraîner des maladies génétiques graves, car elles peuvent perturber la structure et la fonction des protéines essentielles au bon fonctionnement de l'organisme. Cependant, certains mécanismes cellulaires, tels que la relecture des codons non-sens ou la traduction des ARNm modifiés par des facteurs de correction de lecture, peuvent permettre de contourner les effets négatifs de ces mutations et de rétablir partiellement la production de protéines fonctionnelles.
Les protéines filamenteuses intermédiaires (IFP, ou en anglais, IFPs pour "Intermediate Filament Proteins") sont un type de protéines fibreuses qui forment des structures filamenteuses dans les cellules. Elles font partie du cytosquelette, le réseau de fibres et de tubules qui donne à la cellule sa forme et lui permet de se déplacer et de diviser.
Les IFP sont caractérisées par leur diamètre intermédiaire, entre celui des microtubules et des filaments d'actine. Elles sont constituées d'unités protéiques qui s'assemblent pour former des protofilaments, qui à leur tour s'associent pour former des filaments plus larges.
Il existe plusieurs types de IFP, chacun étant exprimé dans un type spécifique de cellule. Les cinq classes principales de IFP sont les kératines, les vimentines, les desmines, les neurofilaments et les lamines. Les kératines sont exprimées dans les épithéliums, les vimentines dans les cellules mésenchymateuses, les desmines dans les muscles squelettiques et cardiaques, les neurofilaments dans les neurones et les lamines dans la membrane nucléaire.
Les IFP jouent un rôle important dans la stabilité mécanique de la cellule, la protection contre les dommages physiques et la régulation des processus cellulaires tels que la division cellulaire et l'apoptose. Des anomalies dans les gènes codant pour les IFP peuvent entraîner des maladies telles que des dystrophies musculaires, des neuropathies et des cancers.
En génétique, une mutation est une modification permanente et héréditaire de la séquence nucléotidique d'un gène ou d'une région chromosomique. Elle peut entraîner des changements dans la structure et la fonction des protéines codées par ce gène, conduisant ainsi à une variété de phénotypes, allant de neutres (sans effet apparent) à délétères (causant des maladies génétiques). Les mutations peuvent être causées par des erreurs spontanées lors de la réplication de l'ADN, l'exposition à des agents mutagènes tels que les radiations ou certains produits chimiques, ou encore par des mécanismes de recombinaison génétique.
Il existe différents types de mutations, telles que les substitutions (remplacement d'un nucléotide par un autre), les délétions (suppression d'une ou plusieurs paires de bases) et les insertions (ajout d'une ou plusieurs paires de bases). Les conséquences des mutations sur la santé humaine peuvent être très variables, allant de maladies rares à des affections courantes telles que le cancer.
Les maladies génétiques de la peau sont des affections cutanées héréditaires causées par des mutations dans les gènes. Ces mutations peuvent être transmises des parents à l'enfant ou peuvent survenir spontanément pendant le développement de l'embryon. Les maladies génétiques de la peau affectent la structure, la fonction et l'apparence de la peau, entraînant souvent des symptômes cutanés visibles ainsi que d'autres complications systémiques.
Les exemples courants de maladies génétiques de la peau comprennent :
1. La neurofibromatose : une affection caractérisée par la croissance de tumeurs bénignes le long des nerfs, entraînant des lésions cutanées et des complications neurologiques.
2. Le xeroderma pigmentosum : un trouble rare qui rend la peau extrêmement sensible aux rayons UV, entraînant un risque élevé de cancer de la peau et d'autres dommages cutanés.
3. La maladie de Darier : une maladie héréditaire de la kératinisation qui provoque des plaques squameuses et des démangeaisons sur la peau, en particulier dans les zones humides et chaudes du corps.
4. L'ichtyose : un groupe de troubles génétiques qui affectent la différenciation des kératinocytes, entraînant une accumulation de squames et une peau sèche et écailleuse.
5. Le syndrome d'Ehlers-Danlos : un trouble du tissu conjonctif caractérisé par une peau fine, élastique et fragile, ainsi que des articulations hypermobiles et des complications vasculaires.
6. La maladie de Hurler : une maladie héréditaire rare du métabolisme des mucopolysaccharides qui affecte la peau, les os, les yeux, le cœur et d'autres organes.
7. Le syndrome de Marfan : un trouble génétique du tissu conjonctif qui affecte la croissance et le développement des os, des yeux, du cœur et des vaisseaux sanguins.
8. La neurofibromatose : un groupe de troubles héréditaires caractérisés par la formation de tumeurs bénignes sur les nerfs et la peau.
9. Le syndrome de Peutz-Jeghers : une maladie génétique rare qui se caractérise par des taches pigmentées sur la peau et les muqueuses, ainsi que des polypes intestinaux bénins.
10. La xeroderma pigmentosum : un trouble héréditaire rare de la réparation de l'ADN qui entraîne une sensibilité extrême aux rayons UV et un risque élevé de cancer de la peau.
Le collagène est une protéine structurelle abondante dans le corps humain, constituant environ un tiers des protéines totales. Il joue un rôle crucial dans la formation des structures de soutien et protectrices telles que la peau, les tendons, les ligaments, les os, les cartilages, les vaisseaux sanguins et les dents. Le collagène fournit force et souplesse à ces tissus en formant des fibres solides mais flexibles.
Il est synthétisé par divers types de cellules, y compris les fibroblastes, à partir d'acides aminés provenant de sources alimentaires ou du recyclage des propres protéines de l'organisme. Les trois acides aminés principaux utilisés dans la production de collagène sont la glycine, la proline et la hydroxyproline.
La structure unique du collagène, qui contient une grande quantité de résidus d'acide aminé hydroxyproline, lui confère sa rigidité et sa stabilité. Des anomalies dans la production ou la structure du collagène peuvent entraîner diverses maladies génétiques telles que l'ostéogenèse imparfaite (maladie des os de verre) et l'épidermolyse bulleuse (une forme grave de peau fragile).
La "peau artificielle" est un terme généralement utilisé pour décrire les matériaux synthétiques conçus pour imiter et remplacer temporairement ou peut-être même de manière permanente la fonction et la structure de la peau humaine. Ces matériaux peuvent être utilisés dans le traitement des brûlures graves, des ulcères de pression, des plaies chroniques et d'autres conditions où la peau a été endommagée ou perdue.
Les caractéristiques clés d'une peau artificielle comprennent généralement une barrière physique qui protège contre les infections, permet la perméabilité aux liquides et à l'oxygène tout en maintenant une humidité adéquate, et imite autant que possible les propriétés mécaniques de la peau réelle. Les exemples actuels de peaux artificielles comprennent des échafaudages biologiques dérivés de tissus comme le collagène ou l'élastine, ainsi que des matériaux synthétiques tels que les polymères et les hydrogels.
Cependant, il est important de noter qu'aucun matériau actuel ne peut pleinement reproduire toutes les fonctions complexes de la peau humaine, telles que la sensation tactile, la thermorégulation et l'immunité. La recherche dans ce domaine se poursuit activement dans le but d'améliorer ces matériaux et de développer des solutions plus efficaces pour aider à la guérison des plaies et au rétablissement de la fonction cutanée.
L'atrésie intestinale est une malformation congénitale dans laquelle il y a un manque complet ou partiel de continuité dans une partie du tube digestif, le plus souvent dans l'intestin grêle. Cela se produit lorsque la lumière intestinale ne parvient pas à se former correctement pendant le développement fœtal, entraînant une interruption de la circulation des aliments et des sécrétions dans le tube digestif.
Les types d'atrésie intestinale comprennent l'atrésie de l'iléon, l'atrésie du jéjunum et l'atrésie multiple, selon la partie de l'intestin grêle affectée. Dans certains cas, l'atrésie peut être associée à d'autres malformations congénitales, telles que les malrotations intestinales ou les fentes anales.
Les symptômes de l'atrésie intestinale peuvent inclure une distension abdominale, des vomissements bilieux, une absence de selles et un méconium épais et visqueux chez le nouveau-né. Le diagnostic est généralement posé par imagerie médicale, telle qu'une radiographie ou une échographie abdominales.
Le traitement de l'atrésie intestinale nécessite souvent une intervention chirurgicale pour rétablir la continuité du tube digestif. Dans certains cas, une résection de la partie affectée de l'intestin peut être nécessaire. Les nouveau-nés atteints d'atrésie intestinale peuvent également nécessiter une alimentation parentérale totale (TPN) pour fournir des nutriments jusqu'à ce que l'intestin soit capable de fonctionner normalement.
Les complications à long terme de l'atrésie intestinale peuvent inclure des problèmes de malabsorption, une dépendance à la TPN et un risque accru d'infections intestinales. Cependant, avec un traitement approprié et un suivi régulier, la plupart des enfants atteints d'atrésie intestinale peuvent mener une vie normale et active.
Un auto-antigène est une substance (généralement une protéine ou un polysaccharide) qui est présente dans l'organisme et qui peut déclencher une réponse immunitaire anormale chez certaines personnes. Dans des conditions normales, le système immunitaire ne réagit pas aux auto-antigènes car ils sont reconnus comme étant "propriétaires" de l'organisme.
Cependant, dans certaines situations, telles que lors d'une infection ou d'une maladie auto-immune, le système immunitaire peut commencer à produire des anticorps ou des cellules T qui attaquent les auto-antigènes, entraînant une inflammation et des dommages tissulaires.
Les maladies auto-immunes sont caractérisées par cette réponse anormale du système immunitaire contre ses propres tissus et organes. Les exemples de maladies auto-immunes comprennent la polyarthrite rhumatoïde, le lupus érythémateux disséminé, la sclérose en plaques, et le diabète sucré de type 1.
Un codon terminateur, également connu sous le nom de codon d'arrêt ou codon non-sens, est une séquence spécifique de trois nucléotides dans l'ARN messager (ARNm) qui signale la terminaison de la synthèse des protéines. Dans le code génétique, il n'y a trois codons terminaux : UAA (ou "ochre"), UAG ("ambre") et UGA ("opale" ou "tsiamis"). Lorsque les ribosomes atteignent ces codons au cours du processus de traduction, la synthèse des protéines s'arrête et l'enzyme peptidyl-transférase libère la nouvelle chaîne polypeptidique de l'ARN de transfert lié. Parfois, un ARNm peut contenir des codons terminaux prématurés en raison d'une mutation, ce qui entraîne la production de protéines tronquées et souvent non fonctionnelles.
La pemphigoïde bulleuse est une maladie auto-immune rare mais grave de la peau et des muqueuses. Elle se caractérise par la formation de bulles (vésicules) et de cloques (bulles) remplies de liquide clair sur la peau et, dans certains cas, à l'intérieur de la bouche, du nez, de la gorge ou des organes génitaux.
Cette maladie se produit lorsque le système immunitaire produit des anticorps qui attaquent les protéines de liaison des cellules épidermiques (desmigleine 1 et 3) situées dans la jonction dermo-épidermique. Cet attaque provoque une séparation anormale des couches de la peau, entraînant la formation de bulles.
Les symptômes de la pemphigoïde bulleuse apparaissent généralement progressivement et s'aggravent avec le temps. Ils peuvent inclure des démangeaisons sévères, l'apparition de petites bulles remplies de liquide clair sur la peau ou les muqueuses, et éventuellement une infection secondaire due à la rupture des bulles. Les zones les plus souvent touchées sont le tronc, les membres supérieurs et inférieurs, ainsi que les plis cutanés.
Le diagnostic de pemphigoïde bulleuse repose sur l'examen clinique, les antécédents médicaux du patient, des tests de laboratoire spécifiques (y compris des tests sanguins et des biopsies cutanées) et l'observation de certaines caractéristiques histopathologiques typiques.
Le traitement de la pemphigoïde bulleuse vise à contrôler les symptômes, prévenir les complications et maintenir une fonction normale des muqueuses. Les corticostéroïdes systémiques (comme la prednisone) sont généralement le traitement de première ligne pour cette maladie. D'autres médicaments immunosuppresseurs peuvent être utilisés en combinaison avec les corticostéroïdes ou comme alternative lorsque les corticostéroïdes ne sont pas tolérés ou ne fonctionnent pas suffisamment bien. Les soins de soutien, tels que l'hygiène cutanée appropriée et la prévention des infections, sont également importants pour assurer une prise en charge optimale de cette maladie.
Les filaments intermédiaires (FI) sont des structures protéiques intracellulaires qui forment un réseau dynamique dans la cellule. Ils jouent un rôle crucial dans la régulation de la forme et de la fonction cellulaire, ainsi que dans le processus de division cellulaire. Les FI sont principalement composés de deux types de protéines fibreuses : les lamins, qui se trouvent dans le nucléoplasme, et les cytoskelettons intermédiaires, qui se trouvent dans le cytoplasme.
Les lamines sont des protéines filamenteuses qui forment une couche sous la membrane nucléaire et fournissent une structure stable au noyau. Elles sont également impliquées dans la régulation de la transcription génétique, de la réplication de l'ADN et de la réparation de l'ADN.
Les cytoskelettons intermédiaires, quant à eux, comprennent des protéines telles que les kinésines, les dynéines, les moteurs myosines et les filaments d'actine. Ils sont responsables de la forme cellulaire, de la motilité cellulaire et du transport intracellulaire.
Les FI sont souvent altérés dans diverses maladies génétiques et neurodégénératives, telles que la dystrophie musculaire, la progéria et la maladie de Charcot-Marie-Tooth. Les mutations dans les gènes codant pour les protéines FI peuvent entraîner une instabilité structurelle, une dérégulation des processus cellulaires et finalement la mort cellulaire. Par conséquent, une meilleure compréhension de la structure et de la fonction des FI est essentielle pour élucider les mécanismes sous-jacents à ces maladies et développer de nouvelles stratégies thérapeutiques.
Je suis désolé, mais il semble y avoir une certaine confusion dans votre question. "Réticuline" n'est pas un terme médical communément utilisé. Cependant, la réticuline est une protéine qui peut être mentionnée dans des contextes scientifiques ou médico-scientifiques.
La réticuline est une protéine structurelle importante du cytosquelette et joue un rôle crucial dans la formation de la matrice extracellulaire. Elle est particulièrement abondante dans les tissus conjonctifs, nerveux et musculaires. Dans le système nerveux central, elle participe à la structure des gaines de myéline entourant les axones des neurones.
Si vous faisiez référence à un terme médical similaire ou associé, pouvez-vous s'il vous plaît me fournir plus d'informations ou clarifier votre question? Je serais heureux de vous aider davantage.
Le phénotype est le résultat observable de l'expression des gènes en interaction avec l'environnement et d'autres facteurs. Il s'agit essentiellement des manifestations physiques, biochimiques ou développementales d'un génotype particulier.
Dans un contexte médical, le phénotype peut se rapporter à n'importe quelle caractéristique mesurable ou observable résultant de l'interaction entre les gènes et l'environnement, y compris la couleur des yeux, la taille, le poids, certaines maladies ou conditions médicales, voire même la réponse à un traitement spécifique.
Il est important de noter que deux individus ayant le même génotype (c'est-à-dire la même séquence d'ADN) ne seront pas nécessairement identiques dans leur phénotype, car des facteurs environnementaux peuvent influencer l'expression des gènes. De même, des individus avec des génotypes différents peuvent partager certains traits phénotypiques en raison de similitudes dans leurs environnements ou dans d'autres facteurs non génétiques.
Prurigo est un terme général utilisé en dermatologie pour décrire des affections cutanées caractérisées par des lésions cutanées prurigineuses, qui sont intensément démangeaisons. Il existe plusieurs types de prurigo, dont le prurigo simplex acute (aigu), le prurigo simplex subacute (subaigu) et le prurigo nodularis.
Les lésions cutanées associées au prurigo sont souvent des papules ou des plaques surélevées, rouges ou brunes, qui peuvent être accompagnées de croûtes, d'écaillements ou de suintements si grattées. Les lésions peuvent apparaître sur n'importe quelle partie du corps, mais sont souvent localisées aux membres inférieurs et au tronc.
Le prurigo simplex acute est généralement déclenché par une irritation de la peau ou un eczéma et se caractérise par des lésions qui durent moins de six semaines. Le prurigo simplex subacute, en revanche, est caractérisé par des lésions persistant pendant plus de six semaines et peut être associé à une affection sous-jacente telle qu'une maladie rénale ou hépatique.
Le prurigo nodularis est le type le plus grave de prurigo, caractérisé par des lésions cutanées en forme de nœud qui sont très démangeantes et persistantes. Cette condition peut être difficile à traiter et peut entraîner des cicatrices et des changements de pigmentation de la peau.
Dans certains cas, le prurigo peut être associé à des troubles psychologiques tels que l'anxiété ou la dépression, qui peuvent aggraver les symptômes et rendre le traitement plus difficile.
En termes médicaux, les « cornets » se réfèrent aux cornets nasaux, qui sont des petites structures de tissus situées à l'intérieur de la cavité nasale. Ils aident à filtrer, humidifier et réchauffer l'air inspiré avant qu'il ne pénètre dans les poumons. Les cornets nasaux sont également responsables de la régulation du débit d'air dans les voies respiratoires supérieures et contribuent à la fonction immunitaire en piégeant les particules étrangères telles que la poussière, les allergènes et les micro-organismes. Il existe trois cornets nasaux de chaque côté du nez : le cornet inférieur, moyen et supérieur, chacun avec des tailles et des fonctions légèrement différentes.
La laminine est une protéine structurelle importante qui joue un rôle crucial dans la formation et le maintien des bases de la matrice extracellulaire (MEC) dans les tissus animaux. Elle contribue à créer un environnement cellulaire propice à l'adhérence, la migration, la différenciation et la prolifération cellulaires.
La laminine est composée de trois chaînes polypeptidiques différentes, appelées alpha, beta et gamma. Il existe plusieurs types isoformes de laminines, selon les combinaisons spécifiques de ces chaînes (par exemple, laminine-1, laminine-2, laminine-5, etc.). Chaque type de laminine possède des propriétés uniques et est exprimé dans différents tissus en fonction du stade de développement et des besoins fonctionnels.
Dans le contexte médical, la laminine a été étudiée pour ses potentielles implications thérapeutiques dans divers domaines, tels que la cicatrisation des plaies, la régénération tissulaire, la réparation nerveuse et la lutte contre le cancer. Cependant, ces recherches en sont encore à leurs balbutiements et nécessitent davantage d'études pour confirmer leur efficacité et leur sécurité.
L'intégrine alpha6beta4, également connue sous le nom de very late antigen-6 (VLA-6), est un type de protéine de la famille des intégrines qui se trouve à la surface des cellules. Elle est composée de deux chaînes polypeptidiques, l'alpha6 et la beta4, qui s'associent pour former un hétérodimère membranaire.
L'intégrine alpha6beta4 joue un rôle important dans la régulation de l'adhésion cellulaire, de la migration cellulaire et de la signalisation intracellulaire. Elle se lie spécifiquement à des ligands extracellulaires tels que la laminine-332, une protéine de la matrice extracellulaire, et participe ainsi à la formation et au maintien des jonctions hémidesmosomales entre les cellules épithéliales et la membrane basale.
Des anomalies dans l'expression ou la fonction de l'intégrine alpha6beta4 ont été associées à diverses pathologies, telles que le cancer du sein, la dermatite herpétiforme et certaines maladies génétiques rares affectant la peau et les muqueuses.
Un gène dominant est un type de gène qui, lorsqu'il est exprimé, sera manifestement exprimé dans l'organisme, que le gène ait deux copies (hétérozygote) ou deux copies identiques (homozygote). Cela signifie qu'une seule copie du gène dominant est suffisante pour provoquer une certaine apparence phénotypique ou une condition médicale, qui peut être bénigne ou pathologique.
Dans le contexte de la génétique mendélienne classique, un trait dominant est exprimé dans la progéniture même si l'autre copie du gène est normale (saine). Les traits dominants se manifestent généralement dans chaque génération et sont plus susceptibles d'être observés dans les familles affectées.
Un exemple bien connu de gène dominant est le gène de la névoid basocellulaire syndrome (NBS), également appelé syndrome du grain de beauté multiple, qui prédispose les individus à développer des cancers de la peau. Si un parent a ce trait et ne possède qu'une seule copie du gène NBS muté, chaque enfant aura 50% de chances d'hériter de cette copie mutée et donc de développer le syndrome.
Une mutation ponctuelle est un type spécifique de mutation génétique qui implique l'alteration d'une seule paire de bases dans une séquence d'ADN. Cela peut entraîner la substitution, l'insertion ou la délétion d'un nucléotide, ce qui peut à son tour modifier l'acide aminé codé par cette région particulière de l'ADN. Si cette modification se produit dans une région codante d'un gène (exon), cela peut entraîner la production d'une protéine anormale ou non fonctionnelle. Les mutations ponctuelles peuvent être héréditaires, transmises de parents à enfants, ou spontanées, survenant de novo dans un individu. Elles sont souvent associées à des maladies génétiques, certaines formes de cancer et au vieillissement.
Le mosaïcisme est un terme utilisé en génétique pour décrire la présence de deux ou plusieurs populations de cellules avec des génotypes différents dans un même individu. Cela se produit lorsqu'un événement mutationnel, comme une mutation génétique ou un réarrangement chromosomique, se produit après la fécondation et affecte seulement certaines cellules de l'organisme en développement.
Par exemple, dans le cas d'une mutation génétique, certaines cellules peuvent contenir la version mutée du gène, tandis que d'autres cellules peuvent contenir la version non-mutée du gène. Le mosaïcisme peut affecter n'importe quelle partie de l'organisme, y compris les tissus somatiques (corps) et germinaux (gamètes).
Le mosaïcisme peut être à l'origine de variations subtiles dans les caractéristiques physiques ou des différences plus importantes dans la fonction et la structure des organes. Dans certains cas, le mosaïcisme peut entraîner des troubles génétiques ou prédisposer une personne à certaines maladies. Cependant, dans d'autres cas, le mosaïcisme peut ne pas avoir de conséquences cliniquement significatives.
Un résultat fatal en médecine se réfère à un décès ou au fait de causer la mort. C'est un terme utilisé pour décrire un résultat particulièrement grave d'une maladie, d'un traumatisme ou d'une procédure médicale. Un résultat fatal peut être attendu dans certaines situations, comme dans le cas de maladies avancées et terminaux, ou peut être imprévu et survenir même avec un traitement approprié. Dans les deux cas, il s'agit d'un événement tragique qui a des implications importantes pour les patients, leurs familles et les professionnels de la santé qui les prennent en charge.
La collagénase microbienne est une enzyme produite par certains types de bactéries, principalement des genres Clostridium et Bacillus. Elle a la capacité de dégrader spécifiquement la molécule de collagène, qui est une protéine structurelle majeure trouvée dans les tissus conjonctifs des animaux, y compris les humains. La collagénase microbienne clive les liaisons peptidiques dans les régions triples hélicoïdales du collagène, entraînant ainsi sa dégradation et la destruction des tissus conjonctifs où il est présent.
Cette enzyme est souvent utilisée en médecine et en recherche biomédicale pour diverses applications, telles que la dissémination de tissus lors d'une greffe de peau ou d'un lambeau chirurgical, l'analyse structurale des protéines collagène et l'étude des processus pathologiques associés à la dégradation du collagène, comme dans le cas de maladies articulaires dégénératives ou de plaies chroniques.
Il est important de noter que la collagénase microbienne peut également contribuer à des infections graves et à la destruction tissulaire dans certains types d'infections bactériennes, comme le tétanos gazeux ou la gangrène gazeuse, qui sont causées par certaines espèces de Clostridium.
En génétique, un exon est une séquence d'ADN qui code pour une partie spécifique d'une protéine. Après la transcription de l'ADN en ARN messager (ARNm), les exons sont conservés et assemblés dans le processus de maturation de l'ARNm, tandis que les introns, qui sont les séquences non codantes, sont éliminés. Les exons forment ainsi la section codante finale de l'ARNm mature, qui est ensuite traduite en une chaîne polypeptidique lors de la synthèse des protéines.
En bref, un exon est une région d'un gène qui contribue à la séquence d'acides aminés d'une protéine après le traitement et l'assemblage de l'ARNm mature. Les mutations dans les exons peuvent entraîner des modifications dans la structure des protéines, ce qui peut conduire à des maladies génétiques ou à des changements phénotypiques.
L'intégrine alpha6, également connue sous le nom d'ITGA6, est un type de protéine de la famille des intégrines qui jouent un rôle crucial dans les processus cellulaires tels que l'adhésion cellulaire, la migration cellulaire et la signalisation cellulaire.
L'intégrine alpha6 se compose d'une chaîne alpha (alpha6) et d'une chaîne bêta (bêta1, bêta4 ou bêta7), ce qui donne lieu à différentes sous-unités fonctionnelles. Elle est largement exprimée dans divers tissus, y compris la peau, les poumons, le cœur et les vaisseaux sanguins.
L'intégrine alpha6 se lie spécifiquement aux laminines, qui sont des protéines de la matrice extracellulaire. Cette interaction est importante pour la formation et le maintien des jonctions entre les cellules épithéliales et endothéliales, ainsi que pour la migration et l'adhésion des cellules souches.
Des mutations dans le gène ITGA6 peuvent entraîner des maladies génétiques telles que le syndrome de Kindler, une maladie rare de la peau caractérisée par une inflammation cutanée, une fragilité et une cicatrisation anormale. De plus, l'intégrine alpha6 est également impliquée dans divers processus pathologiques tels que la progression du cancer, l'angiogenèse et la fibrose tissulaire.
Skin cream, également appelée crème hydratante, est un type de produit topique utilisé pour maintenir l'hydratation de la peau et prévenir la sécheresse. Elle contient une combinaison d'ingrédients tels que des humectants, des émollients et des occlusifs qui aident à piéger l'humidité dans la peau et à restaurer sa barrière cutanée.
Les humectants, comme la glycérine ou le sorbitol, attirent l'eau vers la surface de la peau, tandis que les émollients, tels que la lanoline ou les huiles végétales, adoucissent et assouplissent la peau en comblant les espaces entre les cellules cutanées. Les occlusifs, comme la vaseline ou la cire d'abeille, créent une barrière protectrice à la surface de la peau pour empêcher l'humidité de s'échapper.
Les crèmes pour la peau peuvent être formulées pour traiter une variété de problèmes de peau tels que l'eczéma, le psoriasis, l'acné ou les rides et ridules. Elles peuvent contenir des ingrédients actifs supplémentaires tels que des agents anti-inflammatoires, des antioxydants ou des agents exfoliants pour améliorer l'apparence et la santé de la peau.
Il est important de choisir une crème pour la peau adaptée à son type de peau et à ses besoins spécifiques, en évitant les ingrédients qui peuvent irriter ou aggraver les problèmes de peau existants. Il est également recommandé de faire un test épicutané avant d'utiliser une nouvelle crème pour la peau pour s'assurer qu'elle ne provoque pas de réactions allergiques.
En génétique, un hétérozygote est un individu qui possède deux allèles différents d'un même gène sur les deux chromosomes homologues. Cela signifie que l'individu a hérité d'un allèle particulier du gène en question de chacun de ses parents, et ces deux allèles peuvent être différents l'un de l'autre.
Dans le contexte de la génétique mendélienne classique, un hétérozygote est représenté par une notation avec une lettre majuscule suivie d'un signe plus (+) pour indiquer que cet individu est hétérozygote pour ce gène spécifique. Par exemple, dans le cas d'un gène avec deux allèles A et a, un hétérozygote serait noté Aa.
La présence d'hétérozygotie peut entraîner des phénotypes variés, en fonction du type de gène concerné et de la nature des allèles en présence. Dans certains cas, l'allèle dominant (généralement représenté par une lettre majuscule) détermine le phénotype, tandis que dans d'autres cas, les deux allèles peuvent contribuer au phénotype de manière égale ou interactive.
Il est important de noter qu'être hétérozygote pour certains gènes peut conférer des avantages ou des inconvénients en termes de santé, de résistance aux maladies et d'autres caractéristiques. Par exemple, l'hétérozygotie pour certaines mutations associées à la mucoviscidose (fibrose kystique) peut offrir une protection contre certaines bactéries nocives de l'appareil respiratoire.
L'épiderme est la couche externe et la plus fine de la peau. Il s'agit d'une structure stratifiée composée de cellules mortes et vivantes qui fournit une barrière protectrice contre les agents pathogènes, les substances chimiques et les dommages physiques. L'épiderme est principalement constitué de kératinocytes, qui produisent la kératine, une protéine fibreuse qui confère à la peau sa résistance et sa rigidité.
L'épiderme se compose de plusieurs couches : la couche basale, la couche spinuleuse, la couche granuleuse et la couche cornée. La couche basale est la plus profonde et contient des cellules souches qui peuvent se diviser et différencier en kératinocytes matures. Au fur et à mesure que les kératinocytes mûrissent, ils migrent vers la surface de la peau et traversent les différentes couches de l'épiderme.
Au cours de ce processus, les kératinocytes subissent des changements morphologiques et fonctionnels qui leur permettent de remplir leurs fonctions protectrices. Par exemple, dans la couche cornée, les kératinocytes sont complètement différenciés et remplis de kératine, ce qui les rend rigides et imperméables à l'eau.
En plus des kératinocytes, l'épiderme contient également d'autres types de cellules telles que les mélanocytes, qui produisent le pigment mélanine responsable de la couleur de la peau, et les cellules de Langerhans, qui jouent un rôle important dans le système immunitaire de la peau.
Des affections telles que l'eczéma, le psoriasis et le cancer de la peau peuvent affecter l'épiderme et perturber sa fonction protectrice.
Les auto-anticorps sont des anticorps produits par le système immunitaire qui ciblent et attaquent les propres cellules, tissus ou molécules d'un organisme. Normalement, le système immunitaire est capable de distinguer entre les substances étrangères (antigènes) et les composants du soi, et il ne produit pas de réponse immunitaire contre ces derniers.
Cependant, dans certaines conditions, telles que les maladies auto-immunes, le système immunitaire peut commencer à produire des auto-anticorps qui attaquent et détruisent les tissus sains. Les auto-anticorps peuvent être dirigés contre une grande variété de substances, y compris les protéines, les acides nucléiques, les lipides et les glucides.
La présence d'auto-anticorps dans le sang peut indiquer une maladie auto-immune sous-jacente ou une autre condition médicale. Les tests de dépistage des auto-anticorps sont souvent utilisés pour aider au diagnostic et à la surveillance des maladies auto-immunes telles que le lupus érythémateux disséminé, la polyarthrite rhumatoïde, la sclérodermie et la thyroïdite auto-immune.
Il est important de noter que la présence d'auto-anticorps ne signifie pas nécessairement qu'une personne a une maladie auto-immune ou va développer des symptômes associés à une telle maladie. Certains auto-anticorps peuvent être détectés chez des personnes en bonne santé, et d'autres conditions médicales peuvent également entraîner la production d'auto-anticorps. Par conséquent, les résultats des tests doivent être interprétés dans le contexte de l'histoire clinique et des symptômes du patient.
L'analyse des mutations de l'ADN est une méthode d'examen génétique qui consiste à rechercher des modifications (mutations) dans la séquence de l'acide désoxyribonucléique (ADN). L'ADN est le matériel génétique présent dans les cellules de tous les organismes vivants et contient les instructions pour le développement, la fonction et la reproduction des organismes.
Les mutations peuvent survenir spontanément ou être héritées des parents d'un individu. Elles peuvent entraîner des changements dans la structure et la fonction des protéines, ce qui peut à son tour entraîner une variété de conséquences, allant de mineures à graves.
L'analyse des mutations de l'ADN est utilisée dans un large éventail d'applications, y compris le diagnostic et le suivi des maladies génétiques, la détermination de la susceptibilité à certaines maladies, l'identification des auteurs de crimes, la recherche sur les maladies et le développement de médicaments.
Il existe différentes méthodes pour analyser les mutations de l'ADN, notamment la séquençage de nouvelle génération (NGS), la PCR en temps réel, la PCR quantitative et la Southern blotting. Le choix de la méthode dépend du type de mutation recherchée, de la complexité du test et des besoins du patient ou du chercheur.
Les données de séquence moléculaire se réfèrent aux informations génétiques ou protéomiques qui décrivent l'ordre des unités constitutives d'une molécule biologique spécifique. Dans le contexte de la génétique, cela peut inclure les séquences d'ADN ou d'ARN, qui sont composées d'une série de nucléotides (adénine, thymine, guanine et cytosine pour l'ADN; adénine, uracile, guanine et cytosine pour l'ARN). Dans le contexte de la protéomique, cela peut inclure la séquence d'acides aminés qui composent une protéine.
Ces données sont cruciales dans divers domaines de la recherche biologique et médicale, y compris la génétique, la biologie moléculaire, la médecine personnalisée, la pharmacologie et la pathologie. Elles peuvent aider à identifier des mutations ou des variations spécifiques qui peuvent être associées à des maladies particulières, à prédire la structure et la fonction des protéines, à développer de nouveaux médicaments ciblés, et à comprendre l'évolution et la diversité biologique.
Les technologies modernes telles que le séquençage de nouvelle génération (NGS) ont rendu possible l'acquisition rapide et économique de vastes quantités de données de séquence moléculaire, ce qui a révolutionné ces domaines de recherche. Cependant, l'interprétation et l'analyse de ces données restent un défi important, nécessitant des méthodes bioinformatiques sophistiquées et une expertise spécialisée.
Dans le domaine de la génétique, un individu homozygote est une personne qui hérite de deux allèles identiques pour un trait spécifique, ayant reçu ce même allèle de chacun de ses deux parents. Cela peut se traduire par l'expression d'un caractère particulier ou l'apparition d'une maladie génétique dans le cas où ces allèles sont défectueux.
On distingue deux types d'homozygotes : les homozygotes dominants et les homozygotes récessifs. Les homozygotes dominants présentent le phénotype lié au gène dominant, tandis que les homozygotes récessifs expriment le phénotype associé au gène récessif.
Par exemple, dans le cas de la drépanocytose, une maladie génétique héréditaire affectant l'hémoglobine des globules rouges, un individu homozygote pour cette affection possède deux allèles mutés du gène de l'hémoglobine et exprimera donc les symptômes de la maladie.
Les molécules d'adhésion cellulaire sont des protéines qui se trouvent à la surface des cellules et leur permettent de s'adhérer les unes aux autres ou à la matrice extracellulaire. Elles jouent un rôle crucial dans une variété de processus biologiques, tels que la communication intercellulaire, la migration cellulaire, la différenciation cellulaire et la régulation de la croissance cellulaire. Les molécules d'adhésion cellulaire peuvent être classées en plusieurs catégories, notamment les cadhérines, les immunoglobulines, les intégrines et les sélectines.
Les cadhérines sont des protéines transmembranaires qui médient l'adhésion homophilique entre les cellules, ce qui signifie qu'elles se lient préférentiellement à d'autres molécules de la même sous-classe. Les immunoglobulines sont des protéines transmembranaires qui médient l'adhésion hétérophilique entre les cellules, ce qui signifie qu'elles se lient à des molécules différentes de leur propre sous-classe.
Les intégrines sont des récepteurs transmembranaires qui médient l'adhésion des cellules à la matrice extracellulaire, tandis que les sélectines sont des protéines de surface cellulaire qui facilitent le contact initial et la reconnaissance entre les cellules.
Les molécules d'adhésion cellulaire peuvent être impliquées dans diverses pathologies, telles que l'inflammation, la tumorigénèse et la progression des tumeurs. Par conséquent, elles représentent des cibles thérapeutiques potentielles pour le développement de nouveaux traitements médicaux.
L'acantholysis est un terme médical qui décrit la séparation des cellules épithéliales (cellules qui recouvrent la surface du corps et tapissent les cavités internes) en raison de la dégénérescence ou de la perte de certaines structures de jonction entre ces cellules. Cette condition est souvent observée dans diverses affections cutanées, telles que le pemphigus et le herpès gestationis. L'acantholyse peut entraîner la formation de vésicules ou de bulles sur la peau, ce qui peut causer des démangeaisons, des douleurs et d'autres symptômes désagréables. Dans certains cas, l'acantholyse peut également affecter les muqueuses, telles que la bouche ou les organes génitaux. Le traitement de l'acantholyse dépend de la cause sous-jacente et peut inclure des médicaments topiques ou systémiques pour contrôler l'inflammation et favoriser la guérison.
La thérapie génétique est une forme avancée de médecine qui consiste à remplacer, manipuler ou inactiver des gènes spécifiques dans les cellules d'un patient pour traiter ou prévenir des maladies héréditaires ou acquises. Elle vise à corriger les défauts génétiques sous-jacents en introduisant des matériaux génétiques sains, tels que des gènes fonctionnels ou des acides nucléiques thérapeutiques, dans les cellules du patient.
Ces matériaux génétiques peuvent être délivrés directement aux cellules affectées par l'intermédiaire de vecteurs, tels que des virus inactivés ou des nanoparticules, qui permettent d'introduire les gènes thérapeutiques dans le génome ciblé. Une fois intégrés, ces nouveaux gènes peuvent aider à produire des protéines manquantes ou défectueuses, réguler l'expression de certains gènes, inhiber la production de protéines nocives ou même déclencher le processus de mort cellulaire programmée (apoptose) pour éliminer les cellules anormales.
Bien que la thérapie génétique offre des perspectives prometteuses dans le traitement de diverses affections, telles que les maladies génétiques rares, le cancer et certaines maladies infectieuses, elle est encore considérée comme une approche expérimentale et fait l'objet de recherches intensives pour évaluer son efficacité et sa sécurité à long terme.
Les maladies rares, également connues sous le nom de troubles rares ou de maladies orphelines, se réfèrent à des affections qui affectent un petit nombre de personnes dans la population. Selon l'Orphanet, le portail européen des maladies rares et des médicaments orphelins, une maladie est considérée comme rare en Europe lorsqu'elle affecte moins d'une personne sur 2 000.
Les maladies rares peuvent être causées par des mutations génétiques, des infections, des expositions environnementales ou encore rester sans cause identifiée. Elles peuvent toucher n'importe quel système organique du corps et se manifester par une grande variété de symptômes, allant de légers à graves, voire mortels.
Le diagnostic et le traitement des maladies rares sont souvent difficiles en raison de leur faible prévalence, ce qui peut entraîner un retard dans l'établissement d'un diagnostic correct et dans la mise en place d'un plan de traitement adapté. De plus, les ressources allouées à la recherche sur ces maladies sont souvent limitées, ce qui freine le développement de thérapies spécifiques et efficaces.
Il existe environ 7 000 maladies rares connues à ce jour, affectant près de 300 millions de personnes dans le monde. En raison de leur caractère rare et dispersé, la collaboration internationale entre médecins, chercheurs, patients et associations est essentielle pour améliorer la compréhension, le diagnostic et le traitement de ces affections.
Une séquence nucléotidique est l'ordre spécifique et linéaire d'une série de nucléotides dans une molécule d'acide nucléique, comme l'ADN ou l'ARN. Chaque nucléotide se compose d'un sucre (désoxyribose dans le cas de l'ADN et ribose dans le cas de l'ARN), d'un groupe phosphate et d'une base azotée. Les bases azotées peuvent être adénine (A), guanine (G), cytosine (C) et thymine (T) dans l'ADN, tandis que dans l'ARN, la thymine est remplacée par l'uracile (U).
La séquence nucléotidique d'une molécule d'ADN ou d'ARN contient des informations génétiques cruciales qui déterminent les caractéristiques et les fonctions de tous les organismes vivants. La décodage de ces séquences, appelée génomique, est essentiel pour comprendre la biologie moléculaire, la médecine et la recherche biologique en général.
Une mutation « faux sens » (ou missense mutation) est un type de mutation génétique où une seule paire de bases dans l'ADN est modifiée, ce qui entraîne le remplacement d'un acide aminé par un autre dans la protéine codée par ce gène. Cela peut altérer la fonction, la structure ou la stabilité de la protéine, dépendant de la position et de l'importance de l'acide aminé remplacé. Dans certains cas, ces mutations peuvent entraîner des maladies génétiques ou prédisposer à certaines conditions médicales. Toutefois, il est important de noter que toutes les mutations faux sens ne sont pas nécessairement pathogènes et que leur impact sur la santé dépend du contexte dans lequel elles se produisent.
Les « Maladies De La Bouche » est un terme général qui se réfère à diverses affections et conditions médicales qui affectent la santé bucco-dentaire. Cela peut inclure des infections, des maladies inflammatoires, des tumeurs bénignes ou malignes, des lésions traumatiques, des désordres auto-immuns, et d'autres affections qui peuvent affecter les structures orales telles que les gencives, les dents, la langue, le palais, les lèvres, les joues, et le plancher de la bouche.
Les maladies courantes de la bouche comprennent la carie dentaire, la gingivite (inflammation des gencives), la parodontite (maladie des gencives sévère qui peut entraîner une perte osseuse et dentaire), l'aphte (petites ulcérations douloureuses de la muqueuse buccale), le lichen plan buccal (une éruption cutanée chronique et souvent récurrente de la muqueuse buccale), et le cancer de la bouche.
Les facteurs de risque pour les maladies de la bouche peuvent inclure une mauvaise hygiène bucco-dentaire, le tabagisme, la consommation excessive d'alcool, une mauvaise alimentation, le stress, et certaines affections systémiques telles que le diabète. Le traitement dépend de la maladie spécifique et peut inclure des antibiotiques, des antifongiques, des corticostéroïdes, des médicaments immunosuppresseurs, une chirurgie, ou une combinaison de ces options.
Les fibroblastes sont des cellules présentes dans les tissus conjonctifs de l'organisme, qui produisent et sécrètent des molécules structurelles telles que le collagène et l'élastine. Ces protéines assurent la cohésion, la résistance et l'élasticité des tissus conjonctifs, qui constituent une grande partie de notre organisme et ont pour rôle de relier, soutenir et protéger les autres tissus et organes.
Les fibroblastes jouent également un rôle important dans la cicatrisation des plaies en synthétisant et déposant du collagène et d'autres composants de la matrice extracellulaire, ce qui permet de combler la zone lésée et de rétablir l'intégrité du tissu.
En plus de leur activité structurelle, les fibroblastes sont également capables de sécréter des facteurs de croissance, des cytokines et d'autres molécules de signalisation qui influencent le comportement des cellules voisines et participent à la régulation des processus inflammatoires et immunitaires.
Dans certaines circonstances pathologiques, comme en cas de cicatrices excessives ou de fibroses, les fibroblastes peuvent devenir hyperactifs et produire une quantité excessive de collagène et d'autres protéines, entraînant une altération de la fonction des tissus concernés.
Le prurit est un symptôme commun dans le domaine médical qui se réfère à une sensation désagréable et irritante de démangeaison sur la peau. Cette sensation provoque souvent l'envie de se gratter, ce qui peut parfois aggraver les démangeaisons ou même endommager la peau. Le prurit peut être localisé à une petite zone du corps ou généralisé, affectant de larges étendues de peau.
Les causes du prurit sont variées et peuvent inclure des affections dermatologiques telles que l'eczéma, le psoriasis, la dermatite séborrhéique ou l'urticaire. Des maladies systémiques comme le diabète, l'insuffisance rénale chronique, les troubles hépatiques (y compris la cholestase) et certains cancers peuvent également causer des démangeaisons. De plus, certaines infections, réactions médicamenteuses, irritants chimiques, stress émotionnel et changements hormonaux peuvent aussi être à l'origine de ce symptôme.
Le traitement du prurit dépendra de la cause sous-jacente. Il peut inclure des crèmes ou pommades topiques pour hydrater la peau et réduire l'inflammation, des antihistaminiques oraux pour contrôler les démangeaisons sévères, des corticostéroïdes systémiques pour traiter certaines affections sous-jacentes ou d'autres thérapies spécifiques à la cause identifiée.
Le diagnostic prénatal (DPN) est un ensemble de techniques médicales utilisées pour déterminer la présence ou l'absence de maladies, d'anomalies congénitales ou de conditions génétiques dans le fœtus avant sa naissance. Il s'agit d'un processus invasif ou non invasif qui permet aux médecins d'identifier précocement les problèmes de santé potentiels du fœtus, offrant ainsi la possibilité aux parents et aux prestataires de soins de prendre des décisions éclairées sur la poursuite ou non de la grossesse, la planification des soins prénatals et postnatals, et la préparation à l'accueil d'un enfant malade.
Les méthodes de diagnostic prénatal comprennent les échographies, les analyses sanguines maternelles, les tests invasifs tels que l'amniocentèse et la biopsie des villosités choriales, ainsi que les examens génétiques préimplantatoires dans le cadre d'une fécondation in vitro. Ces procédures permettent de rechercher des anomalies chromosomiques, des mutations génétiques spécifiques et d'autres problèmes structurels ou fonctionnels du fœtus.
Il est important de noter que le diagnostic prénatal soulève des questions éthiques complexes et controversées concernant l'eugénisme, la sélection des embryons et les droits des personnes handicapées. Les professionnels de santé doivent donc veiller à fournir des informations complètes, impartiales et sensibles sur les avantages, les risques et les implications potentielles des tests de diagnostic prénatal, afin que les parents puissent prendre une décision éclairée et en accord avec leurs valeurs personnelles et leur contexte culturel.
Le dépistage néonatal est un processus systématique de détection précoce, à grande échelle et généralisée, de certaines conditions médicales congénitales ou acquises à la naissance chez les nouveau-nés. Il est réalisé en prenant des échantillons de sang, d'urine ou d'autres tissus peu après la naissance, puis en analysant ces échantillons à l'aide de divers tests de laboratoire.
Le dépistage néonatal vise à identifier rapidement les nouveau-nés qui présentent un risque accru de développer des problèmes de santé graves et potentiellement évitables, tels que les troubles métaboliques héréditaires, les maladies du sang, les déficits hormonaux et d'autres affections congénitales. Une détection précoce permet une intervention thérapeutique rapide, ce qui peut améliorer considérablement les résultats pour la santé des nourrissons concernés, réduire la morbidité et la mortalité, et améliorer leur qualité de vie globale.
Les programmes de dépistage néonatal sont généralement mis en œuvre par les autorités sanitaires publiques ou les établissements de santé, et ils sont recommandés dans de nombreux pays développés pour tous les nouveau-nés à moins que des contre-indications médicales ne soient présentes. Les conditions ciblées par le dépistage néonatal peuvent varier selon les pays et les régions en fonction des ressources disponibles, des priorités de santé publique et des prévalences locales des différentes affections.
La santé familiale est un domaine de la médecine qui se concentre sur la santé et le bien-être des membres d'une famille pris dans leur ensemble, ainsi que sur les facteurs sociaux, économiques et environnementaux qui peuvent influencer leur santé. Elle met l'accent sur la prévention des maladies et des blessures, la promotion de la santé et le traitement des problèmes de santé existants.
La santé familiale peut inclure des soins primaires pour tous les membres de la famille, ainsi que des services spécialisés pour les membres qui ont des besoins de santé spécifiques. Les prestataires de soins de santé familiaux peuvent offrir une gamme de services, y compris des examens physiques, des vaccinations, des contrôles de la tension artérielle et du cholestérol, des dépistages de maladies chroniques telles que le diabète et l'hypertension, des soins de maternité et de nouveau-né, et des services de santé mentale.
La santé familiale peut également inclure des services d'éducation et de counseling pour aider les familles à adopter des modes de vie sains, tels que une alimentation équilibrée, l'exercice régulier, la gestion du stress et la prévention du tabagisme et de la consommation excessive d'alcool.
En outre, les prestataires de soins de santé familiaux peuvent travailler en étroite collaboration avec d'autres professionnels de la santé, tels que des spécialistes, des infirmières praticiennes, des travailleurs sociaux et des thérapeutes, pour fournir des soins coordonnés et complets à tous les membres de la famille.
Dans l'ensemble, la santé familiale vise à promouvoir la santé et le bien-être des familles dans leur ensemble, en prenant en compte les facteurs qui influencent leur santé et en travaillant avec eux pour atteindre leurs objectifs de santé.
L'ichthyose bulleuse de Siemens est une forme rare d'ichtyose, une maladie génétique de la peau. Cette condition est caractérisée par la présence de vésicules (petites bulles) et d'écailles sur la peau. Les symptômes apparaissent généralement à la naissance ou dans les premiers mois de vie.
Les vésicules sont souvent déclenchées par des frottements, des changements de température ou une humidité élevée. Elles peuvent se rompre et former des croûtes, ce qui peut entraîner des cicatrices permanentes. Les zones les plus touchées sont souvent les membres, le tronc et la tête.
L'ichthyose bulleuse de Siemens est causée par une mutation du gène KRT5 ou KRT14, qui code pour les protéines keratine 5 et keratine 14. Ces protéines sont importantes pour la structure et la fonction des cellules de la peau. La mutation de ces gènes entraîne une production anormale de ces protéines, ce qui affaiblit les couches supérieures de la peau et rend vulnérable à la formation de vésicules.
Le traitement de l'ichthyose bulleuse de Siemens est principalement symptomatique et peut inclure des crèmes hydratantes, des antibiotiques pour les infections cutanées et des antiseptiques pour prévenir les infections. Dans certains cas, une transplantation de cellules souches peut être envisagée.
La technique des anticorps fluorescents, également connue sous le nom d'immunofluorescence, est une méthode de laboratoire utilisée en médecine et en biologie pour détecter et localiser les antigènes spécifiques dans des échantillons tels que des tissus, des cellules ou des fluides corporels. Cette technique implique l'utilisation d'anticorps marqués avec des colorants fluorescents, tels que la FITC (fluorescéine isothiocyanate) ou le TRITC (tétraméthylrhodamine isothiocyanate).
Les anticorps sont des protéines produites par le système immunitaire qui reconnaissent et se lient spécifiquement à des molécules étrangères, appelées antigènes. Dans la technique des anticorps fluorescents, les anticorps marqués sont incubés avec l'échantillon d'intérêt, ce qui permet aux anticorps de se lier aux antigènes correspondants. Ensuite, l'échantillon est examiné sous un microscope à fluorescence, qui utilise une lumière excitatrice pour activer les colorants fluorescents et produire une image lumineuse des sites d'antigène marqués.
Cette technique est largement utilisée en recherche et en médecine diagnostique pour détecter la présence et la distribution d'un large éventail d'antigènes, y compris les protéines, les sucres et les lipides. Elle peut être utilisée pour diagnostiquer une variété de maladies, telles que les infections bactériennes ou virales, les maladies auto-immunes et le cancer.
 Frédérique Vidal
Frédérique Vidal Silvestrelli Maurizio IFCE
Silvestrelli Maurizio IFCE DeCS
DeCS