Comparative Genomic Hybridization
Hybridation Acide Nucléique
Aberrations Chromosomiques
Dosage Génique
Hybridation In Situ Fluorescence
Séquençage Par Oligonucléotides En Batterie
DNA Copy Number Variations
Chromosomes Artificiels De Bactérie
Amplification Génique
Génome Humain
Chromosome X Humain
Cartographie Chromosomique
Aneuploïdie
Chromosomes Humains De La Paire 20
Analyse Cytogénétique
Hybridation In Situ
Cytogénétique
Chromosomes Humains De La Paire 18
Chromosomes Humains De La Paire 1
Analyse Expression Gène
Maladies Chromosomiques
Détermination Séquence Adn
Bande Chromosomique
Chromosomes Humains De La Paire 13
Intellectual Disability
Délétion Génique
Instabilité Génomique
Perte Hétérozygotie
Chromosome Duplication
Chromosomes Humains De La Paire 17
Génome
Chromosomes Humains De La Paire 11
Caryotypage Spectral
Chromosomes Humains De La Paire 19
Duplication Génique
Chromosomes Humains De La Paire 13
Chromosomes Humains De La Paire 16
Données Séquence Moléculaire
Séquence Nucléotidique
Réaction Polymérisation En Chaîne
Chromosomes Humains De La Paire 14
Chromosomes Humains De La Paire 17
Analyse Par Microarray
Chromosomes Humains De La Paire 12
Chromosomes Humains De La Paire 16
Déséquilibre Allélique
Chromosome Breakpoints
Analyse D'Aggrégats
Chromosomes Humains De La Paire 15
Malformations Multiples
Polar Bodies
Chromosomes Humains De La Paire 12
Inclusion Paraffine
Chromosomes Humains De La Paire 22
Phénotype
Peinture Chromosomique
Cassure Chromosomique
Segmental Duplications, Genomic
Régulation Expression Génique Tumorale
Chromosomes Humains De La Paire 18
Instabilité Chromosomique
Karyotype
Ilots Génomiques
Techniques Amplification Acides Nucléiques
Gènes Tumoraux
Délétion Séquentielle
Microsatellites
Translocation, Genetic
Diagnostic Préimplantatoire
Chromosomes Humains De La Paire 15
Mutation
Adn
Evolution, Molecular
Spécificité Espèce
Chromosomes Humains De La Paire 14
Chromosome X Humain
Kératoacanthome
Sondes Adn
Famille Multigénique
Pronostic
Chromosomes Artificiels De Bactériophage P1
Algorithme
Chromosomes
Modèle Génétique
Hybridation Génétique
Arn Messager
Southern-Blot
Formaldéhyde
Genetic Testing
Fixateurs
Carcinome
Marqueur Génétique
Analyse Mutations Adn
Réaction Polymérisation En Chaîne Par Transcriptase Inverse
Reproductibilité Des Résultats
Bioinformatique
Léiomyosarcome
Immunohistochimie
Réarrangement Gène
Chromosomes Humains De La Paire 19
Lymphoma, Large B-Cell, Diffuse
Faciès
Gène Suppresseur Tumeur
Diagnostic Prénatal
Génotype
Histiocytome Fibreux Bénin
Cartographie Physique Des Chromosomes
Alignement Séquences
Carcinoma in Situ
Oncogène
Troubles Du Développement
Ependymoma
Logiciel
Sondes Oligonucléotides
Chromosomes Humains De La Paire 10
Séquence Des Acides Aminés
Astrocytome
Fusion De Gènes
Genomic Structural Variation
Polymorphisme Simple Nucléotide
Cellules Cancéreuses En Culture
Isochromosomes
Métaphase
Clonage Moléculaire
Protéines Tumorales
Carcinome Canalaire Du Sein
Sensibilité Et Spécificité (
Disease Progression
Retard Mental Lié
Médulloblastome
Tumeurs Du Cerveau
Amorces Adn
Dermatofibrosarcome
Marqueur Biologique Tumeur
Expression Génique
Traitement Image Assisté Ordinateur
Généalogie
Monosomie
Neoplasms, Plasma Cell
Exon
Oligodendrogliome
Adn Complémentaire
Tumeurs
Hernie Diaphragmatique
Malformations Crâniofaciales
Mélanome
Trouble Autistique
Genetic Association Studies
Hétérogénéité Génétique
Analyse Par Puce
Lymphome B
Synténie
Banque Génome
Fixation Tissulaire
Coupe
Tumeurs Sus-Tentorielles
Apraxies
Carcinomes
Clostridium Botulinum Type B
Gène P53
Transcription Génétique
Paragangliome
Gène Myc
Méthylation Adn
Grossesse
Genetic Loci
Tumeurs De L'Ovaire
Gène Dupliqué
Diploïdie
Maladies Génétiques Congénitales
Techniques Génétiques
Tumeurs Du Cervelet
Mésothéliome
La hybridation génomique comparative (CGH) est une technique de laboratoire utilisée pour comparer les différences dans le contenu en ADN entre deux échantillons, généralement le DNA d'un patient et un échantillon de référence. Cette méthode permet la détection des variations du nombre de copies (gains ou pertes) de grandes régions chromosomiques entre les deux échantillons.
Le processus implique l'étiquetage de deux échantillons d'ADN avec des fluorophores différents, créant ainsi une sonde "marquée". Les deux échantillons sont ensuite mélangés et hybridés à un seul ensemble de chromosomes métaphasiques ou à une matrice artificielle contenant tout le génome. En analysant l'intensité des signaux fluorescents, on peut déterminer les variations du nombre de copies dans les régions chromosomiques entre les deux échantillons initiaux.
La CGH est une technique sensible et utile pour le diagnostic et la recherche en génétique humaine, en particulier dans l'identification des anomalies chromosomiques submicroscopiques associées à diverses affections, telles que les troubles du développement, les maladies congénitales, les cancers et les maladies mentales.
L'hybridation d'acides nucléiques est un processus dans lequel deux molécules d'acides nucléiques, généralement une molécule d'ADN et une molécule d'ARN ou deux molécules d'ADN complémentaires, s'apparient de manière spécifique par des interactions hydrogène entre leurs bases nucléotidiques correspondantes. Ce processus est largement utilisé en biologie moléculaire et en génétique pour identifier, localiser et manipuler des séquences d'ADN ou d'ARN spécifiques.
L'hybridation a lieu lorsque les deux brins d'acides nucléiques sont mélangés et portés à des températures et des concentrations de sel optimales pour permettre la formation de paires de bases complémentaires. Les conditions d'hybridation doivent être soigneusement contrôlées pour assurer la spécificité et la stabilité de l'appariement des bases.
L'hybridation d'acides nucléiques est une technique sensible et fiable qui peut être utilisée pour détecter la présence de séquences d'ADN ou d'ARN spécifiques dans un échantillon, pour mesurer l'abondance relative de ces séquences, et pour analyser les relations évolutives entre différentes espèces ou populations. Elle est largement utilisée dans la recherche en génétique, en médecine, en biologie moléculaire, en agriculture et dans d'autres domaines où l'identification et l'analyse de séquences d'acides nucléiques sont importantes.
Les aberrations chromosomiques sont des anomalies dans la structure, le nombre ou l'arrangement des chromosomes dans une cellule. Ces anomalies peuvent entraîner une variété de conséquences sur la santé, allant de légères à graves.
Les aberrations chromosomiques peuvent être héréditaires ou spontanées et peuvent affecter n'importe quel chromosome. Les types courants d'aberrations chromosomiques comprennent :
1. Aneuploïdie : Il s'agit d'une anomalie du nombre de chromosomes, dans laquelle il y a soit un chromosome supplémentaire (trisomie), soit un chromosome manquant (monosomie). Un exemple courant est la trisomie 21, qui est associée au syndrome de Down.
2. Translocation : Il s'agit d'un réarrangement des morceaux de chromosomes entre eux. Les translocations peuvent être équilibrées (aucun matériel génétique n'est gagné ou perdu) ou déséquilibrées (le matériel génétique est gagné ou perdu).
3. Déletion : Il s'agit d'une perte de partie d'un chromosome. Les délétions peuvent entraîner une variété de problèmes de santé, selon la taille et l'emplacement du morceau manquant.
4. Inversion : Il s'agit d'un renversement de section d'un chromosome. Les inversions peuvent être associées à des problèmes de fertilité ou à un risque accru de malformations congénitales chez les enfants.
5. Duplication : Il s'agit d'une copie supplémentaire d'une partie d'un chromosome. Les duplications peuvent entraîner une variété de problèmes de santé, selon la taille et l'emplacement du morceau supplémentaire.
Les anomalies chromosomiques peuvent être causées par des erreurs lors de la division cellulaire ou par des mutations génétiques héréditaires. Certaines anomalies chromosomiques sont associées à un risque accru de maladies génétiques, tandis que d'autres n'ont aucun impact sur la santé. Les tests génétiques peuvent être utilisés pour détecter les anomalies chromosomiques et évaluer le risque de maladies génétiques.
Un dosage génique, également connu sous le nom de test de dosage génique ou dosage quantitatif d'acide nucléique, est un type de test de laboratoire utilisé pour déterminer la quantité ou l'expression relative d'un gène ou d'un ARN spécifique dans un échantillon donné. Ce type de test peut être utilisé à des fins diagnostiques pour aider à identifier les maladies génétiques, les troubles chromosomiques et certains types de cancer. Il peut également être utilisé à des fins de recherche pour étudier l'expression des gènes dans différents tissus ou à différentes étapes du développement.
Le dosage génique implique généralement l'amplification de l'acide nucléique cible à l'aide d'une technique de PCR (polymerase chain reaction) ou d'autres méthodes d'amplification, suivie de la détection et de la quantification du produit amplifié. Les résultats peuvent être exprimés en termes de nombre de copies du gène ou de l'ARN par unité de volume d'échantillon ou en termes relatifs par rapport à un échantillon de référence.
Il est important de noter que le dosage génique ne doit pas être confondu avec le séquençage de l'ADN, qui est utilisé pour déterminer la séquence exacte des nucléotides dans une région spécifique de l'ADN. Alors que le séquençage de l'ADN peut fournir des informations sur les variations génétiques spécifiques, le dosage génique permet de déterminer la quantité relative d'un gène ou d'un ARN donné dans un échantillon.
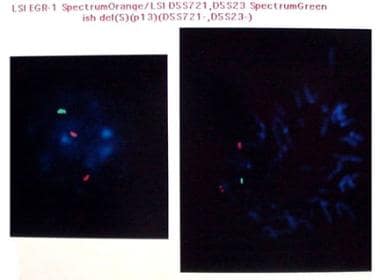
La fluorescence in situ hybride (FISH) est une technique de biologie moléculaire utilisée pour détecter et localiser des séquences d'ADN spécifiques dans des cellules ou des tissus préservés. Cette méthode consiste à faire réagir des sondes d'ADN marquées avec des fluorophores spécifiques, qui se lient de manière complémentaire aux séquences d'intérêt sur les chromosomes ou l'ARN dans les cellules préparées.
Dans le cas particulier de l'hybridation in situ fluorescente (FISH), les sondes sont appliquées directement sur des échantillons de tissus ou de cellules fixés et préparés, qui sont ensuite exposés à des températures et à une humidité contrôlées pour favoriser la liaison des sondes aux cibles. Les échantillons sont ensuite examinés au microscope à fluorescence, ce qui permet de visualiser les signaux fluorescents émis par les sondes liées et donc de localiser les séquences d'ADN ou d'ARN d'intérêt dans le contexte des structures cellulaires et tissulaires.
La FISH est largement utilisée en recherche et en médecine diagnostique pour détecter des anomalies chromosomiques, des réarrangements génétiques, des mutations spécifiques ou des modifications de l'expression génique dans divers contextes cliniques, tels que le cancer, les maladies génétiques et les infections virales.
Le terme «séquençage par oligonucléotides en batterie» ne semble pas être une expression ou un concept reconnu dans le domaine de la médecine ou de la biologie moléculaire. Il est possible que vous ayez fait une erreur ou que ce terme spécifique soit utilisé dans un contexte particulier et restreint qui m'est inconnu.
Le séquençage d'oligonucléotides, cependant, est une technique de biologie moléculaire permettant de déterminer l'ordre des nucléotides dans une chaîne d'acide nucléique (ADN ou ARN). Cette méthode implique généralement l'utilisation de petits oligonucléotides marqués comme sondes pour identifier et séquencer des régions spécifiques du brin d'acide nucléique.
Si vous cherchiez une définition pour un terme similaire ou lié, veuillez me fournir plus de détails afin que je puisse vous aider au mieux.
Les variations du nombre de copies d'ADN (VNCAs - Variations of Number of Copies of DNA en anglais) correspondent à des écarts dans la quantité de matériel génétique présent dans le génome d'un individu, par rapport à une population de référence. Contrairement aux mutations ponctuelles qui affectent un seul nucléotide, les VNCAs concernent des segments d'ADN de taille variable, allant de quelques centaines de paires de bases à plusieurs méga-bases.
Ces variations peuvent entraîner des conséquences fonctionnelles sur l'expression et la régulation des gènes, ainsi que sur la structure des chromosomes. Elles sont souvent associées à une prédisposition à certaines maladies génétiques, telles que les troubles du spectre autistique, les maladies neurodégénératives, les cancers et d'autres affections complexes. Les VNCAs peuvent être héritées ou acquises au cours de la vie, par exemple en raison d'erreurs lors de la réplication de l'ADN ou de mécanismes de réparation défectueux.
Les techniques de cytogénétique moléculaire, telles que l'hybridation in situ à fluorescence (FISH) et les puces à ADN à haute densité, sont couramment utilisées pour détecter et caractériser ces variations du nombre de copies d'ADN.
Une délétion chromosomique est un type d'anomalie chromosomique qui se produit lorsqu'une partie d'un chromosome est manquante ou absente. Cela se produit lorsque des segments du chromosome se cassent et que les morceaux perdus ne sont pas correctement réintégrés. Les délétions chromosomiques peuvent être héréditaires ou spontanées, et leur taille et leur emplacement varient considérablement.
Les conséquences d'une délétion chromosomique dépendent de la taille et de l'emplacement de la région déléguée. Les petites délétions peuvent ne provoquer aucun symptôme, tandis que les grandes délétions peuvent entraîner des anomalies congénitales graves, un retard mental et d'autres problèmes de santé.
Les délétions chromosomiques peuvent être détectées avant la naissance par le biais de tests prénataux tels que l'amniocentèse ou le prélèvement de villosités choriales. Les nouveau-nés atteints d'une délétion chromosomique peuvent présenter des caractéristiques physiques uniques, telles qu'un visage allongé, une petite tête, des yeux largement séparés et des oreilles bas situées.
Le traitement d'une délétion chromosomique dépend de la gravité des symptômes et peut inclure une thérapie physique, une thérapie occupationnelle, une éducation spécialisée et d'autres interventions de soutien. Dans certains cas, les personnes atteintes d'une délétion chromosomique peuvent mener une vie relativement normale avec un traitement et un soutien appropriés.
Les chromosomes artificiels de bactéries sont des vecteurs d'ADN artificiels créés en laboratoire qui peuvent être utilisés pour transférer des gènes ou des segments d'ADN spécifiques dans des bactéries hôtes. Ils sont souvent fabriqués en prenant un plasmide, une petite molécule d'ADN circulaire autoreproductible trouvée dans de nombreuses bactéries, et en y insérant des gènes ou des segments d'ADN d'intérêt.
Ces chromosomes artificiels peuvent être utilisés pour étudier la fonction des gènes, produire des protéines recombinantes à grande échelle, ou créer des organismes génétiquement modifiés avec des propriétés améliorées. Ils sont un outil important en biologie moléculaire et en biotechnologie.
Cependant, il est important de noter que le terme "chromosome artificiel" peut être trompeur, car ces structures ne sont pas vraiment des chromosomes au sens où les cellules eucaryotes les définissent. Elles n'ont pas la même structure complexe et ne se comportent pas de la même manière pendant la division cellulaire.
L'amplification génétique est un processus de laboratoire qui permet de copier et de multiplier des segments spécifiques d'ADN à des fins d'analyse. Ce procédé est couramment utilisé en médecine légale, dans le diagnostic et la recherche médicale pour détecter et analyser des gènes ou des séquences d'ADN spécifiques associés à des maladies héréditaires, des mutations ou des marqueurs génétiques.
La technique la plus courante d'amplification génétique est la réaction en chaîne par polymérase (PCR), qui permet de copier rapidement et avec une grande précision des millions à des milliards de copies d'un segment d'ADN spécifique. Cette méthode est basée sur l'utilisation d'enzymes, de primers et de nucléotides pour amplifier la séquence d'intérêt.
L'amplification génétique a révolutionné le domaine de la médecine moléculaire en permettant une analyse plus sensible, spécifique et rapide des gènes et des mutations associées à diverses maladies et affections. Elle est également utilisée dans les tests de paternité, l'identification de victimes dans des scènes de crime, la détection d'agents pathogènes et la recherche en génétique évolutive.
Un caryotype est une représentation standardisée de l'ensemble des chromosomes d'une cellule, organisme ou espèce donnée. Il s'agit d'un outil diagnostique important en génétique médicale pour identifier d'éventuelles anomalies chromosomiques.
Un caryotype humain typique se compose de 46 chromosomes, répartis en 23 paires. Chaque paire est constituée d'un chromosome d'origine maternelle et d'un chromosome d'origine paternelle, à l'exception des chromosomes sexuels X et Y. Les femmes ont deux chromosomes X (XX), tandis que les hommes en ont un X et un Y (XY).
Pour réaliser un caryotype, on prélève généralement des cellules du sang ou des tissus. Ensuite, ces cellules sont cultivées en laboratoire pour parvenir à la phase de division cellulaire appelée métaphase. À ce stade, les chromosomes sont le plus condensés et donc les plus faciles à visualiser.
Les chromosomes sont ensuite colorés avec des teintures spécifiques qui permettent de distinguer visuellement chaque paire. Ils sont ensuite disposés en fonction de leur taille, du centromère (point de jonction entre les bras courts et longs) et des bandes caractéristiques propres à chaque chromosome.
Un caryotype anormal peut révéler divers types d'anomalies chromosomiques, telles que des délétions, des duplications, des translocations ou des inversions partielles ou totales de certains segments chromosomiques. Ces anomalies peuvent être responsables de maladies génétiques, de retards de développement, d'anomalies congénitales et d'autres problèmes de santé.
ADN tumoral, également connu sous le nom d'ADN circulant tumoral (ctDNA), fait référence à des fragments d'ADN qui sont libérés dans le sang lorsque les cellules cancéreuses meurent. Contrairement à l'ADN normal, qui est stable et se trouve principalement dans les noyaux des cellules, l'ADN tumoral est présent dans le sérum ou le plasma sanguin.
L'analyse de l'ADN tumoral peut fournir des informations importantes sur la composition génétique d'une tumeur cancéreuse, y compris les mutations spécifiques qui peuvent être présentes. Cette information peut être utilisée pour diagnostiquer le cancer, prédire la réponse au traitement et surveiller la maladie au fil du temps.
L'analyse de l'ADN tumoral peut être effectuée en prélevant un échantillon de sang, ce qui est moins invasif que les biopsies traditionnelles des tissus. Cependant, il convient de noter que la quantité d'ADN tumoral dans le sang peut varier considérablement d'une personne à l'autre et dépendre de facteurs tels que la taille de la tumeur et son stade.
En résumé, l'ADN tumoral est un type d'ADN qui est libéré dans le sang lorsque les cellules cancéreuses meurent. L'analyse de l'ADN tumoral peut fournir des informations importantes sur la composition génétique d'une tumeur et être utilisée pour diagnostiquer, prédire la réponse au traitement et surveiller le cancer.
Le génome humain est la totalité de l'information génétique héréditaire d'un être humain, encodée dans l'ADN. Il est contenu dans chaque cellule du corps, à l'exception des spermatozoïdes et des ovules, qui ne contiennent que la moitié du génome. Le génome humain est organisé en 23 paires de chromosomes, y compris les chromosomes sexuels X et Y, contenant environ 3 milliards de paires de bases d'ADN. Il comprend tous les gènes (environ 20 000 à 25 000) qui déterminent les caractéristiques physiques et les traits héréditaires, ainsi que des segments d'ADN qui ne codent pas de protéines mais qui peuvent réguler l'expression des gènes ou avoir d'autres fonctions importantes. L'étude du génome humain est appelée génomique et aide à comprendre les maladies, les traits héréditaires, les relations évolutives et la diversité biologique entre les populations humaines.
Le chromosome X humain est l'un des deux chromosomes sexuels chez les humains, l'autre étant le chromosome Y. Les humains ont normalement 46 chromosomes répartis en 23 paires, dont une paire de chromosomes sexuels. La plupart des femmes ont deux chromosomes X (XX), et la plupart des hommes ont un chromosome X et un chromosome Y (XY).
Le chromosome X est beaucoup plus grand que le chromosome Y et contient environ 155 millions de paires de bases, ce qui représente environ 5% du génome humain. Il code pour environ 1 098 protéines. Le chromosome X contient un certain nombre de gènes importants liés à la fonction cérébrale, à l'immunité et aux maladies génétiques.
Les femmes ont deux copies actives du chromosome X dans la plupart des cellules de leur corps, ce qui est appelé dosage génique. Cependant, les hommes n'ont qu'une seule copie active du chromosome X. Pour équilibrer le niveau d'expression des gènes sur le chromosome X chez les hommes, certains gènes sur le chromosome X sont soumis à une inactivation de l'X ou à la méthylation de l'ADN, ce qui entraîne leur silenciation.
Des mutations dans les gènes du chromosome X peuvent entraîner des maladies génétiques liées au sexe, telles que la dystrophie musculaire de Duchenne, le syndrome de l'X fragile et la maladie de Hunter. Les femmes qui sont porteuses d'une mutation sur un gène du chromosome X ont un risque accru de transmettre cette mutation à leurs enfants.
La cartographie chromosomique est une discipline de la génétique qui consiste à déterminer l'emplacement et l'ordre relatif des gènes et des marqueurs moléculaires sur les chromosomes. Cette technique utilise généralement des méthodes de laboratoire pour analyser l'ADN, comme la polymerase chain reaction (PCR) et la Southern blotting, ainsi que des outils d'informatique pour visualiser et interpréter les données.
La cartographie chromosomique est un outil important dans la recherche génétique, car elle permet aux scientifiques de comprendre comment les gènes sont organisés sur les chromosomes et comment ils interagissent entre eux. Cela peut aider à identifier les gènes responsables de certaines maladies héréditaires et à développer des traitements pour ces conditions.
Il existe deux types de cartographie chromosomique : la cartographie physique et la cartographie génétique. La cartographie physique consiste à déterminer l'emplacement exact d'un gène ou d'un marqueur sur un chromosome en termes de distance physique, exprimée en nucléotides. La cartographie génétique, quant à elle, consiste à déterminer l'ordre relatif des gènes et des marqueurs sur un chromosome en fonction de la fréquence de recombinaison entre eux lors de la méiose.
En résumé, la cartographie chromosomique est une technique utilisée pour déterminer l'emplacement et l'ordre relatif des gènes et des marqueurs moléculaires sur les chromosomes, ce qui permet aux scientifiques de mieux comprendre comment les gènes sont organisés et interagissent entre eux.
La génomique est le domaine de la biologie qui traite de l'étude complète des gènes, y compris leur structure, fonction, interaction et évolution, ainsi que des cartes d'expression des gènes au niveau populationnel. Elle implique l'analyse du génome, qui est l'ensemble complet de l'information génétique héréditaire d'un individu, organisée dans 23 paires de chromosomes humains et le chromosome X ou Y supplémentaire, contenant environ 20 000 à 25 000 gènes.
La génomique utilise des techniques de pointe telles que la séquençage de nouvelle génération pour étudier non seulement les gènes codants pour des protéines, mais aussi d'autres éléments fonctionnels du génome, tels que les ARN non codants, les éléments régulateurs et les séquences répétitives.
Les applications de la génomique comprennent la médecine personnalisée, qui consiste à utiliser des informations sur les variations génétiques d'un individu pour prédire sa susceptibilité aux maladies et optimiser son traitement ; la découverte de nouveaux médicaments et cibles thérapeutiques ; et la compréhension de l'évolution, de la diversité et de la pathogenèse des organismes.
Aneuploïdie est un terme utilisé en génétique pour décrire une condition dans laquelle il y a un changement dans le nombre normal de chromosomes dans une cellule. Les chromosomes sont des structures situées dans le noyau des cellules qui contiennent les gènes, qui sont les unités de base de l'hérédité.
Dans une cellule humaine normale, il y a 23 paires de chromosomes, pour un total de 46 chromosomes. L'aneuploïdie se produit lorsqu'il y a soit une absence (monosomie) ou un excès (trisomie) d'un chromosome dans une de ces paires.
Les aneuploïdies peuvent survenir spontanément en raison d'erreurs qui se produisent pendant la division cellulaire, ou elles peuvent être héritées d'un parent atteint d'une condition chromosomique particulière. Les aneuploïdies peuvent entraîner une variété de problèmes de santé, en fonction du chromosome concerné et de la quantité de matériel génétique supplémentaire ou manquant.
Par exemple, la trisomie 21, également connue sous le nom de syndrome de Down, est une aneuploïdie courante qui se produit lorsqu'il y a un chromosome 21 supplémentaire, entraînant un total de 47 chromosomes. Cette condition est associée à des caractéristiques physiques et développementales spécifiques, telles qu'un visage plat, des yeux inclinés vers le haut, une petite tête et des retards de développement intellectuel et moteur.
D'autres exemples d'aneuploïdies comprennent la trisomie 18 (syndrome d'Edwards) et la trisomie 13 (syndrome de Patau), qui sont toutes deux associées à des anomalies graves et souvent fatales. La monosomie X, ou syndrome de Turner, est une aneuploïdie qui se produit lorsqu'il n'y a qu'un seul chromosome X présent dans les cellules du corps, entraînant des caractéristiques physiques et développementales uniques.
Dans l'ensemble, les aneuploïdies peuvent avoir des conséquences graves sur la santé et le développement d'un individu, et peuvent être détectées avant la naissance par le biais de tests prénataux tels que l'amniocentèse ou le prélèvement de villosités choriales.
Les chromosomes humains de la paire 2
L'analyse cytogénétique est une méthode de laboratoire utilisée pour examiner les chromosomes et les structures chromosomiques dans les cellules d'un individu. Cette technique est couramment utilisée en médecine et en biologie pour diagnostiquer des affections telles que les anomalies chromosomiques congénitales, les cancers et les troubles héréditaires.
L'analyse cytogénétique implique généralement la culture de cellules d'un échantillon de tissu ou de sang, suivie du blocage de la division cellulaire au bon moment pour permettre l'observation des chromosomes. Les chromosomes sont ensuite colorés et examinés au microscope pour identifier tout schéma anormal dans leur nombre, forme ou structure.
Les résultats de l'analyse cytogénétique peuvent aider à confirmer un diagnostic, à évaluer la gravité d'une maladie, à surveiller la réponse au traitement et à fournir des informations sur le risque de transmission d'un trouble génétique à la progéniture.
Les exemples courants d'anomalies chromosomiques détectées par l'analyse cytogénétique comprennent la trisomie 21 (syndrome de Down), la trisomie 18 (syndrome d'Edwards) et la monosomie X (syndrome de Turner). Dans le cancer, les anomalies chromosomiques peuvent inclure des translocations, des délétions ou des amplifications qui contribuent au développement et à la progression de la maladie.
L'hybridation in situ (HIS) est une technique de biologie moléculaire utilisée en histopathologie et en cytogénétique pour localiser et identifier spécifiquement des séquences d'ARN ou d'ADN dans des tissus ou des cellules. Cette méthode consiste à introduire un fragment d'ADN ou d'ARN marqué (probe) dans des sections de tissus préalablement traités et fixés, puis à détecter l'hybridation entre la sonde et les séquences cibles par différentes méthodes de détection.
La hybridation in situ est souvent utilisée pour étudier l'expression génique au niveau cellulaire et subcellulaire dans des tissus normaux ou pathologiques, ce qui permet d'identifier la distribution et l'abondance relative des gènes d'intérêt. Elle peut également être utilisée en combinaison avec d'autres techniques pour caractériser les réarrangements chromosomiques et les mutations génétiques dans des cellules cancéreuses ou autres maladies liées à des altérations génétiques.
Il existe plusieurs types d'hybridation in situ, y compris l'hybridation in situ standard (FISH), l'hybridation in situ en chromosome entier (EISH), et l'hybridation in situ avec amplification par réaction en chaîne de la polymérase (PCR-ISH). Chacune de ces méthodes a ses avantages et ses limites, et elles sont utilisées dans différents contextes pour répondre à des questions spécifiques en recherche biomédicale.
La cytogénétique est une sous-spécialité de la génétique qui s'intéresse à l'étude des chromosomes et de leur impact sur la santé, les maladies et les caractéristiques héréditaires. Elle implique l'analyse des structures et des comportements chromosomiques, y compris les anomalies numériques ou structurelles, dans les cellules humaines. Les techniques cytogénétiques comprennent la coloration des chromosomes pour identifier leur forme et leur taille (coloration de Giemsa ou bandage), l'hybridation in situ en fluorescence (FISH) pour détecter et localiser des séquences d'ADN spécifiques, et le séquençage de nouvelle génération pour analyser les variations du nombre de copies et les réarrangements chromosomiques à grande échelle. La cytogénétique est utilisée dans le diagnostic prénatal, le dépistage des cancers et d'autres maladies génétiques, ainsi que dans la recherche sur l'évolution et la fonction des gènes.
Les chromosomes humains de la paire 18, également connus sous le nom de chromosomes 18, sont des structures composées de ADN et protéines trouvés dans le noyau de chaque cellule du corps humain. Les humains ont normalement 23 paires de chromosomes, pour un total de 46 chromosomes, dans chaque cellule. Les chromosomes 18 sont la dix-huitième paire de ces chromosomes, et chaque personne hérite d'une copie de ce chromosome de chacun de ses parents.
Les chromosomes 18 sont des structures relativement grandes, représentant environ 2 à 2,5 % du total de l'ADN dans une cellule humaine. Ils contiennent des milliers de gènes qui fournissent des instructions pour la production de protéines et d'autres produits fonctionnels nécessaires au développement, à la fonction et à la survie de l'organisme.
Les anomalies chromosomiques des chromosomes 18 peuvent entraîner diverses conditions médicales. Par exemple, une trisomie 18, dans laquelle il y a trois copies du chromosome 18 au lieu des deux copies normales, est associée à une condition appelée syndrome d'Edwards. Cette condition est caractérisée par un ensemble de symptômes et de traits physiques distincts, notamment un retard de développement, des anomalies faciales, des problèmes cardiaques et d'autres anomalies congénitales.
D'autre part, une monosomie 18, dans laquelle il n'y a qu'une seule copie du chromosome 18 au lieu des deux copies normales, est associée à une condition appelée syndrome de Jacobsen. Cette condition est également caractérisée par un ensemble de symptômes et de traits physiques distincts, notamment un retard de développement, des anomalies faciales, des problèmes cardiaques et d'autres anomalies congénitales.
Dans l'ensemble, les chromosomes 18 jouent un rôle crucial dans le développement et la fonction normaux de l'organisme. Les anomalies chromosomiques de ces chromosomes peuvent entraîner divers problèmes de santé et des déficiences du développement.
Les chromosomes humains de la paire 1, également connus sous le nom de chromosome 1, sont les plus grands chromosomes humains en termes de longueur et de nombre de gènes. Ils font partie des 23 paires de chromosomes humains et chacun d'entre eux contient environ 2 800 gènes, ce qui représente environ 8% du total des gènes du génome humain.
Les chromosomes de la paire 1 sont présents dans toutes les cellules du corps à l'exception des gamètes (spermatozoïdes et ovules), qui ne contiennent que 23 chromosomes individuels. Pendant la fécondation, lorsque les gamètes mâles et femelles se rencontrent, leurs chromosomes s'associent pour former le zygote, qui contient 46 chromosomes au total, répartis en 23 paires.
Les chromosomes de la paire 1 sont cruciaux pour une variété de fonctions cellulaires et peuvent être associés à un certain nombre de maladies génétiques lorsqu'ils présentent des anomalies structurelles ou fonctionnelles. Par exemple, certaines conditions telles que la microdélétion 1p36, la délétion 1q21.1 et l'anomalie du chromosome 1q44 peuvent être causées par des réarrangements structurels spécifiques de ce chromosome.
Les recherches sur les chromosomes humains de la paire 1 se poursuivent activement, car une meilleure compréhension de leur fonction et de leur régulation peut fournir des informations importantes sur le développement et les maladies humaines.
L'analyse de l'expression des gènes est une méthode de recherche qui mesure la quantité relative d'un ARN messager (ARNm) spécifique produit par un gène dans un échantillon donné. Cette analyse permet aux chercheurs d'étudier l'activité des gènes et de comprendre comment ils fonctionnent ensemble pour réguler les processus cellulaires et les voies métaboliques.
L'analyse de l'expression des gènes peut être effectuée en utilisant plusieurs techniques, y compris la microarray, la PCR quantitative en temps réel (qPCR), et le séquençage de l'ARN. Ces méthodes permettent de mesurer les niveaux d'expression des gènes à grande échelle, ce qui peut aider à identifier les différences d'expression entre des échantillons normaux et malades, ou entre des cellules avant et après un traitement.
L'analyse de l'expression des gènes est utilisée dans divers domaines de la recherche biomédicale, y compris la génétique, la biologie moléculaire, la pharmacologie, et la médecine translationnelle. Elle peut fournir des informations importantes sur les mécanismes sous-jacents à une maladie, aider au diagnostic précoce et à la surveillance de l'évolution de la maladie, et contribuer au développement de nouveaux traitements ciblés.
Les maladies chromosomiques sont des troubles médicaux causés par des anomalies dans le nombre ou la structure des chromosomes. Les chromosomes sont des structures situées dans le noyau des cellules qui contiennent nos gènes, les unités de base de l'hérédité. Normalement, chaque cellule humaine a 46 chromosomes répartis en 23 paires, sauf les spermatozoïdes et les ovules qui n'en ont qu'une seule de chaque.
Les maladies chromosomiques peuvent résulter d'une absence (délétion), d'un surplus (duplication) ou d'une mauvaise position (translocation) d'un segment chromosomique, ou encore d'un nombre anormal de chromosomes. Par exemple, la trisomie 21, également connue sous le nom de syndrome de Down, est une maladie chromosomique courante causée par la présence d'un chromosome supplémentaire à la paire 21, ce qui donne un total de 47 chromosomes.
Ces anomalies chromosomiques peuvent se produire pendant la formation des ovules ou des spermatozoïdes (méiose) ou pendant le développement embryonnaire (segmentation). Elles peuvent entraîner une grande variété de symptômes, selon la région du chromosome affectée et l'ampleur de l'anomalie.
Les maladies chromosomiques comprennent des affections bien connues telles que le syndrome de Down, le syndrome d'Edwards (trisomie 18), le syndrome de Patau (trisomie 13), la syndactylie (doigts ou orteils collés ensemble) et le Turner et le syndrome de Klinefelter. Ces maladies peuvent entraîner une variété de problèmes de santé, notamment des anomalies physiques, des retards de développement, des déficiences intellectuelles et des problèmes de croissance.
Le génome bactérien se réfère à l'ensemble complet de matériel génétique présent dans une bactérie. Il est composé d'une unique molécule circulaire d'ADN (appelée chromosome bactérien) qui contient tous les gènes nécessaires à la croissance, au développement et à la survie de la bactérie. Le génome bactérien peut également contenir des plasmides, qui sont des petites molécules d'ADN extrachromosomiques qui peuvent porter des gènes supplémentaires tels que ceux codant pour la résistance aux antibiotiques. La taille du génome bactérien varie considérablement selon les espèces, allant de quelques centaines de milliers à plusieurs millions de paires de bases. L'étude du génome bactérien permet de comprendre les mécanismes moléculaires et cellulaires des bactéries, ce qui aide à développer des stratégies pour combattre les maladies infectieuses et à exploiter les bactéries dans des applications industrielles et médicales utiles.
La détermination de la séquence d'ADN est un processus de laboratoire qui consiste à déterminer l'ordre des nucléotides dans une molécule d'ADN. Les nucléotides sont les unités de base qui composent l'ADN, et chacun d'entre eux contient un des quatre composants différents appelés bases : adénine (A), guanine (G), cytosine (C) et thymine (T). La séquence spécifique de ces bases dans une molécule d'ADN fournit les instructions génétiques qui déterminent les caractéristiques héréditaires d'un organisme.
La détermination de la séquence d'ADN est généralement effectuée en utilisant des méthodes de séquençage de nouvelle génération (NGS), telles que le séquençage Illumina ou le séquençage Ion Torrent. Ces méthodes permettent de déterminer rapidement et à moindre coût la séquence d'un grand nombre de molécules d'ADN en parallèle, ce qui les rend utiles pour une variété d'applications, y compris l'identification des variations génétiques associées à des maladies humaines, la surveillance des agents pathogènes et la recherche biologique fondamentale.
Il est important de noter que la détermination de la séquence d'ADN ne fournit qu'une partie de l'information génétique d'un organisme. Pour comprendre pleinement les effets fonctionnels des variations génétiques, il est souvent nécessaire d'effectuer d'autres types d'analyses, tels que la détermination de l'expression des gènes et la caractérisation des interactions protéine-protéine.
Une bande chromosomique est une section ou une région spécifique d'un chromosome qui a été identifiée et caractérisée par sa position, sa taille et son motif de bandes sombres et pâles lorsqu'il est visualisé au microscope à l'aide de techniques de coloration spéciales.
Les bandes chromosomiques sont un outil important en cytogénétique, qui est la branche de la génétique qui étudie les chromosomes et leur rôle dans l'hérédité et les maladies humaines. Les motifs de bandes caractéristiques permettent aux chercheurs d'identifier des anomalies chromosomiques spécifiques, telles que des délétions, des duplications ou des translocations, qui peuvent être associées à des troubles génétiques ou à des maladies.
Les bandes chromosomiques sont généralement nommées en fonction de leur position relative sur le bras court (p) ou le bras long (q) d'un chromosome particulier. Par exemple, la bande 12p13.1 est située dans la région 13.1 du bras court du chromosome 12. Les bandes sont également classées en fonction de leur largeur relative, allant des plus larges (bandes 1 à 5) aux plus étroites (bandes 6 à 10).
En résumé, les bandes chromosomiques sont des marqueurs visuels importants utilisés pour identifier et caractériser les anomalies chromosomiques associées à divers troubles génétiques et maladies.
Les chromosomes humains de la paire 13, également connus sous le nom de chromosomes 13, sont une partie importante du matériel génétique d'un être humain. Ils font partie des 23 paires de chromosomes contenues dans chaque cellule du corps humain, à l'exception des spermatozoïdes et des ovules qui n'en contiennent que 22 paires.
Chaque chromosome 13 est constitué d'une longue molécule d'ADN enroulée autour de protéines histones, formant une structure en forme de X appelée chromatine. Les chromosomes 13 sont classés comme des chromosomes acrocentriques, ce qui signifie qu'ils ont un centromère situé près d'un extrémité et un petit bras court (p) ainsi qu'un grand bras long (q).
Les chromosomes humains de la paire 13 contiennent environ 114 millions de paires de bases d'ADN, ce qui représente environ 3,5% du total des gènes du génome humain. Ils sont responsables de la production de certaines protéines importantes pour le développement et le fonctionnement normaux de l'organisme.
Les anomalies chromosomiques impliquant les chromosomes 13 peuvent entraîner des troubles génétiques graves, tels que la trisomie 13 ou syndrome de Patau, qui se caractérise par une copie supplémentaire du chromosome 13. Cette condition est associée à un certain nombre d'anomalies congénitales et de retards de développement, ainsi qu'à une espérance de vie considérablement réduite.
L'intelligence désigne les capacités d'une personne à apprendre, à raisonner, à résoudre des problèmes, à faire preuve de jugement et de pensée abstraite. Un handicap intellectuel, également connu sous le nom de déficience intellectuelle ou retard mental, est un trouble du développement qui affecte ces capacités intellectuelles et la capacité d'une personne à fonctionner de manière indépendante dans la vie quotidienne.
Il est généralement diagnostiqué avant l'âge de 18 ans et peut varier de léger à sévère. Les personnes atteintes de handicap intellectuel peuvent avoir des difficultés à acquérir et à appliquer de nouvelles connaissances, à communiquer efficacement, à prendre soin d'elles-mêmes, à établir des relations sociales et à faire face aux situations stressantes.
Les causes du handicap intellectuel peuvent être génétiques, environnementales ou résulter de complications pendant la grossesse ou la naissance. Il est important de noter que les personnes atteintes de handicap intellectuel ont des capacités et des besoins uniques, et qu'un diagnostic précoce et une intervention appropriée peuvent améliorer considérablement leur qualité de vie et leurs perspectives d'avenir.
La délétion génique est un type d'anomalie chromosomique où une partie du chromosome est manquante ou absente. Cela se produit lorsque une certaine séquence d'ADN, qui contient généralement des gènes, est supprimée au cours du processus de réplication de l'ADN ou de la division cellulaire.
Cette délétion peut entraîner la perte de fonction de uno ou plusieurs gènes, en fonction de la taille et de l'emplacement de la délétion. Les conséquences de cette perte de fonction peuvent varier considérablement, allant d'aucun effet notable à des anomalies graves qui peuvent affecter le développement et la santé de l'individu.
Les délétions géniques peuvent être héréditaires ou spontanées (de novo), et peuvent survenir dans n'importe quel chromosome. Elles sont souvent associées à des troubles génétiques spécifiques, tels que la syndrome de cri du chat, le syndrome de Williams-Beuren, et le syndrome de délétion 22q11.2.
Le diagnostic d'une délétion génique peut être établi par l'analyse cytogénétique ou moléculaire, qui permettent de détecter les anomalies chromosomiques et génétiques spécifiques. Le traitement et la prise en charge d'une délétion génique dépendent du type et de la gravité des symptômes associés à la perte de fonction des gènes affectés.
L'instabilité génomique est un terme utilisé en génétique et en oncologie pour décrire une condition dans laquelle il y a une augmentation anormale du taux de mutations dans l'ADN d'une cellule ou d'un organisme. Cela peut entraîner des changements dans la structure, la fonction et le nombre de gènes, ce qui peut conduire au développement de maladies, en particulier les cancers.
L'instabilité génomique peut être classée en deux types principaux : l'instabilité génomique par microsatellites (IGM) et l'instabilité chromosomique. L'IGM est caractérisée par des modifications dans la répétition de séquences nucléotidiques courtes dans l'ADN, tandis que l'instabilité chromosomique se réfère à des anomalies structurelles et numériques des chromosomes.
L'instabilité génomique peut être causée par une variété de facteurs, y compris les défauts dans les mécanismes de réparation de l'ADN, l'exposition à des agents mutagènes tels que les radiations et les produits chimiques, et certaines maladies héréditaires. Les personnes atteintes d'instabilité génomique peuvent présenter un risque accru de développer certains types de cancer, en particulier ceux qui se développent dans le côlon, l'endomètre, l'estomac et les ovaires.
Le diagnostic et le traitement de l'instabilité génomique peuvent être complexes et nécessitent souvent une évaluation multidisciplinaire par des spécialistes en oncologie, en génétique et en médecine de laboratoire. Les options de traitement peuvent inclure la chirurgie, la radiothérapie, la chimiothérapie et les thérapies ciblées qui visent à réparer ou à prévenir les dommages à l'ADN.
La perte d'hétérozygotie (LOH), également appelée "perte de hétérozygosité" ou "déficience de l'hétérozygotie", est un phénomène dans lequel une cellule ou un organisme perd un allèle fonctionnel sur un locus chromosomique particulier. Dans un individu hétérozygote pour un gène donné, qui possède donc deux allèles différents de ce gène (par exemple, A et a), la perte d'hétérozygotie entraîne l'acquisition d'une homozygosité complète pour ce locus (par exemple, AA ou aa).
Ce phénomène peut survenir par plusieurs mécanismes, notamment la conversion de gènes, la recombinaison inégale et la perte de chromosomes entiers ou de segments chromosomiques. La perte d'hétérozygotie est souvent observée dans les cellules cancéreuses et peut contribuer au développement et à la progression du cancer en désactivant des gènes suppresseurs de tumeurs ou en activant des oncogènes.
En médecine, l'analyse de la perte d'hétérozygotie est utilisée comme un outil pour identifier les altérations génétiques dans diverses maladies, y compris les cancers et certaines maladies héréditaires.
La duplication chromosomique est un type d'anomalie chromosomique où une partie ou la totalité d'un chromosome est dupliquée, ce qui entraîne une quantité excessive de matériel génétique à partir de ce chromosome. Cela peut se produire de différentes manières, soit en ayant un morceau supplémentaire du même chromosome (duplication interstitielle), soit en ayant une copie entière supplémentaire d'un chromosome (duplication en tandem).
Les duplications chromosomiques peuvent être héritées ou spontanées et peuvent entraîner divers effets sur la santé, selon la taille et l'emplacement de la duplication. Les petites duplications peuvent ne pas causer de problèmes de santé significatifs, tandis que les grandes duplications peuvent entraîner des anomalies congénitales, des retards de développement, des déficiences intellectuelles et d'autres problèmes de santé.
Le diagnostic d'une duplication chromosomique peut être posé par analyse cytogénétique ou par tests moléculaires tels que l'hybridation in situ fluorescente (FISH) ou le séquençage de nouvelle génération (NGS). Le traitement dépendra des symptômes spécifiques présentés par la personne affectée et peut inclure une thérapie de soutien, une intervention chirurgicale, une thérapie médicamenteuse ou une combinaison de ces options.
Les chromosomes humains de la paire 17, également connus sous le nom de chromosomes 17, sont une partie importante du matériel génétique d'un être humain. Les chromosomes sont des structures en forme de bâtonnet dans le noyau des cellules qui contiennent des gènes, qui sont les unités de base de l'hérédité.
Chaque personne a 23 paires de chromosomes, pour un total de 46 chromosomes, dans chaque cellule de leur corps, sauf les cellules reproductives (spermatozoïdes et ovules), qui ne contiennent qu'une seule copie de chaque chromosome. Les chromosomes 17 sont la quatorzième paire de chromosomes dans l'ensemble des 23 paires.
Les chromosomes 17 sont relativement grands et contiennent environ 800 millions de paires de bases, ce qui représente environ 6 à 7 % du génome humain total. Ils contiennent entre 1 500 et 1 600 gènes, qui sont responsables de la production de protéines importantes pour diverses fonctions corporelles, telles que la réparation de l'ADN, le métabolisme des lipides et des glucides, la signalisation cellulaire, la division cellulaire et la différenciation cellulaire.
Les chromosomes 17 sont également associés à plusieurs maladies génétiques rares et courantes, telles que le syndrome de Li-Fraumeni, qui est un trouble héréditaire du cancer, et la neuropathie sensitive héréditaire de type 1, qui est une maladie neurologique héréditaire. Les mutations dans certains gènes situés sur les chromosomes 17 peuvent également augmenter le risque de développer des cancers tels que le cancer du sein, du poumon et du côlon.
En résumé, les chromosomes humains 17 sont importants pour la santé humaine car ils contiennent des gènes responsables de diverses fonctions corporelles et sont associés à plusieurs maladies génétiques courantes et rares. Les mutations dans certains gènes situés sur les chromosomes 17 peuvent également augmenter le risque de développer certains cancers.
Le génome est la totalité de l'information génétique héréditaire d'un organisme encodée dans ses acides nucléiques (ADN et ARN). Il comprend tous les gènes, ainsi que l'ensemble des séquences non codantes. Chez les humains, il est composé de près de 3 milliards de paires de bases d'ADN, réparties sur 23 paires de chromosomes dans le noyau de chaque cellule somatique. Le génome contient toutes les instructions nécessaires pour construire et maintenir un organisme, y compris les informations sur la structure et la fonction des protéines, la régulation de l'expression des gènes et les mécanismes de réparation de l'ADN. L'étude du génome est appelée génomique.
Les chromosomes humains de la paire 11, également connus sous le nom de chromosomes 11, sont une partie importante du matériel génétique d'un être humain. Chaque personne a deux chromosomes 11, une copie héritée de chaque parent. Les chromosomes 11 sont des structures en forme de bâtonnet qui se trouvent dans le noyau de chaque cellule du corps et contiennent des milliers de gènes responsables de la détermination de nombreuses caractéristiques physiques et fonctionnelles d'un individu.
Les chromosomes 11 sont les quatrièmes plus grands chromosomes humains, mesurant environ 220 nanomètres de longueur. Ils contiennent environ 135 millions de paires de bases d'ADN et représentent environ 4 à 4,5 % du génome humain total.
Les gènes situés sur les chromosomes 11 sont responsables de la régulation de divers processus physiologiques tels que le développement et la fonction des systèmes nerveux central et périphérique, la croissance et le développement des os et des muscles, la production d'hormones et l'homéostasie métabolique.
Des anomalies chromosomiques sur les chromosomes 11 peuvent entraîner diverses conditions médicales telles que le syndrome de Williams-Beuren, le syndrome de WAGR, la neurofibromatose de type 1 et certaines formes de cancer. Par conséquent, il est important de comprendre les caractéristiques des chromosomes humains de la paire 11 pour mieux comprendre les causes sous-jacentes de ces conditions et développer des stratégies thérapeutiques appropriées.
Le caryotypage spectral est une technique de cytogénétique avancée qui permet l'analyse détaillée de l'ensemble du génome chromosomique d'un individu. Cette méthode combine la technologie de marquage fluorescent des chromosomes (FISH) et le caryotypage conventionnel pour fournir une représentation visuelle et détaillée de l'arrangement, du nombre et de la structure des chromosomes d'un échantillon.
Dans le caryotypage spectral, chaque paire de chromosomes est teintée avec des colorants fluorescents spécifiques qui émettent des spectres de couleurs uniques lorsqu'ils sont exposés à une lumière excitatrice. Cette technique permet non seulement d'identifier et de localiser les anomalies chromosomiques, telles que les délétions, les duplications et les translocations, mais aussi de quantifier l'ampleur de ces altérations.
Le caryotypage spectral est particulièrement utile dans le diagnostic et la surveillance des troubles génétiques, tels que les syndromes de Down, de Turner et de Klinefelter, ainsi que dans l'évaluation de la réponse au traitement dans les cas de cancer. Cette technique offre une résolution plus élevée et une précision accrue par rapport aux méthodes traditionnelles de caryotypage, ce qui en fait un outil essentiel pour les chercheurs et les cliniciens travaillant dans le domaine de la génétique humaine.
Les chromosomes humains de la paire 19, également connus sous le nom de chromosomes 19, sont l'une des 23 paires de chromosomes présentes dans les cellules humaines. Chaque personne hérite d'une copie de chaque chromosome de chaque parent, ce qui signifie que nous avons deux copies du chromosome 19 en tout.
Le chromosome 19 est l'un des plus grands chromosomes humains et contient un grand nombre de gènes, estimés à environ 1 400. Il code pour une variété de protéines et de molécules impliquées dans divers processus biologiques, tels que le métabolisme, la réponse immunitaire, la fonction nerveuse et la croissance cellulaire.
Des variations dans les gènes situés sur le chromosome 19 ont été associées à un certain nombre de maladies génétiques, notamment la maladie d'Alzheimer, la thrombose veineuse profonde, la sclérose latérale amyotrophique et la surdité neurosensorielle. Les chercheurs continuent d'étudier le chromosome 19 pour comprendre son rôle dans la santé humaine et les maladies.
La duplication génique est un phénomène dans lequel une section spécifique d'un chromosome, comprenant souvent un ou plusieurs gènes, est copiée et insérée à une autre partie du même chromosome ou sur un autre chromosome. Cela entraîne la présence de deux copies ou plus du même gène dans le génome.
Ces duplications peuvent être détectées lors des tests génétiques et sont souvent classées en fonction de leur taille et de la quantité de matériel génétique qu'elles contiennent. Elles peuvent varier d'une petite duplication impliquant quelques centaines de paires de bases à une grande duplication couvrant plusieurs milliers de paires de bases.
Les duplications géniques peuvent être héritées ou se produire spontanément en raison d'erreurs lors de la division cellulaire. Elles sont souvent associées à des conditions génétiques et à des maladies, telles que les troubles neurodéveloppementaux, les maladies cardiaques congénitales et certains types de cancer. Cependant, il est important de noter que toutes les duplications géniques ne causent pas nécessairement une maladie, car la fonction des gènes peut être redondante ou compensée par d'autres facteurs.
Les chromosomes humains de la paire 13, également connus sous le nom de chromosomes 13, sont une partie importante du matériel génétique d'un être humain. Ils font partie des 23 paires de chromosomes contenues dans chaque cellule du corps humain, à l'exception des spermatozoïdes et des ovules qui n'en contiennent que 22 paires.
Chaque chromosome 13 est constitué d'une longue molécule d'ADN enroulée autour de protéines histones, formant une structure en forme de X appelée chromatine. Les chromosomes 13 sont classés comme des chromosomes acrocentriques, ce qui signifie qu'ils ont un centromère situé près d'un extrémité et un petit bras court (p) ainsi qu'un grand bras long (q).
Les chromosomes humains de la paire 13 contiennent environ 114 millions de paires de bases d'ADN, ce qui représente environ 3,5% du total des gènes du génome humain. Ils sont responsables de la production de certaines protéines importantes pour le développement et le fonctionnement normaux de l'organisme.
Les anomalies chromosomiques impliquant les chromosomes 13 peuvent entraîner des troubles génétiques graves, tels que la trisomie 13 ou syndrome de Patau, qui se caractérise par une copie supplémentaire du chromosome 13. Cette condition est associée à un certain nombre d'anomalies congénitales et de retards de développement, ainsi qu'à une espérance de vie considérablement réduite.
Les chromosomes humains de la paire 16, également connus sous le nom de chromosomes 16, sont une partie importante du matériel génétique d'un être humain. Chaque personne a deux chromosomes 16, une copie héritée de chaque parent. Les chromosomes 16 sont des structures en forme de bâtonnet qui se trouvent dans le noyau de chaque cellule du corps et contiennent des milliers de gènes responsables de la détermination de nombreuses caractéristiques physiques et fonctionnelles d'un individu.
Les chromosomes 16 sont l'une des 23 paires de chromosomes humains, ce qui signifie qu'il y a en tout 46 chromosomes dans chaque cellule du corps humain (à l'exception des spermatozoïdes et des ovules, qui n'en contiennent que 23). Les chromosomes 16 sont relativement grands et se situent au milieu de la gamme de taille des chromosomes humains.
Les gènes contenus dans les chromosomes 16 jouent un rôle important dans divers processus biologiques, notamment le développement du cerveau, le métabolisme des lipides et des glucides, la réponse immunitaire et la régulation de l'expression génétique. Les mutations dans ces gènes peuvent entraîner un certain nombre de maladies génétiques rares, telles que la neurofibromatose de type 1, le syndrome de Prader-Willi et le syndrome d'Angelman.
En résumé, les chromosomes humains de la paire 16 sont des structures en forme de bâtonnet qui se trouvent dans le noyau de chaque cellule du corps humain et contiennent des milliers de gènes responsables de nombreuses caractéristiques physiques et fonctionnelles d'un individu. Les mutations dans ces gènes peuvent entraîner un certain nombre de maladies génétiques rares.
Les données de séquence moléculaire se réfèrent aux informations génétiques ou protéomiques qui décrivent l'ordre des unités constitutives d'une molécule biologique spécifique. Dans le contexte de la génétique, cela peut inclure les séquences d'ADN ou d'ARN, qui sont composées d'une série de nucléotides (adénine, thymine, guanine et cytosine pour l'ADN; adénine, uracile, guanine et cytosine pour l'ARN). Dans le contexte de la protéomique, cela peut inclure la séquence d'acides aminés qui composent une protéine.
Ces données sont cruciales dans divers domaines de la recherche biologique et médicale, y compris la génétique, la biologie moléculaire, la médecine personnalisée, la pharmacologie et la pathologie. Elles peuvent aider à identifier des mutations ou des variations spécifiques qui peuvent être associées à des maladies particulières, à prédire la structure et la fonction des protéines, à développer de nouveaux médicaments ciblés, et à comprendre l'évolution et la diversité biologique.
Les technologies modernes telles que le séquençage de nouvelle génération (NGS) ont rendu possible l'acquisition rapide et économique de vastes quantités de données de séquence moléculaire, ce qui a révolutionné ces domaines de recherche. Cependant, l'interprétation et l'analyse de ces données restent un défi important, nécessitant des méthodes bioinformatiques sophistiquées et une expertise spécialisée.
La phylogénie est une discipline scientifique qui étudie et reconstruit l'histoire évolutive des espèces ou groupes d'organismes vivants, en se basant sur leurs caractères biologiques partagés. Elle vise à déterminer les relations de parenté entre ces différents taxons (unités systématiques) et à établir leur arbre évolutif, appelé également phylogramme ou cladogramme.
Dans un contexte médical, la phylogénie peut être utilisée pour comprendre l'évolution des agents pathogènes, tels que les virus, bactéries ou parasites. Cette approche permet de mieux appréhender leur diversité génétique, l'origine et la diffusion des épidémies, ainsi que d'identifier les facteurs responsables de leur virulence ou résistance aux traitements. En conséquence, elle contribue au développement de stratégies préventives et thérapeutiques plus efficaces contre les maladies infectieuses.
Une séquence nucléotidique est l'ordre spécifique et linéaire d'une série de nucléotides dans une molécule d'acide nucléique, comme l'ADN ou l'ARN. Chaque nucléotide se compose d'un sucre (désoxyribose dans le cas de l'ADN et ribose dans le cas de l'ARN), d'un groupe phosphate et d'une base azotée. Les bases azotées peuvent être adénine (A), guanine (G), cytosine (C) et thymine (T) dans l'ADN, tandis que dans l'ARN, la thymine est remplacée par l'uracile (U).
La séquence nucléotidique d'une molécule d'ADN ou d'ARN contient des informations génétiques cruciales qui déterminent les caractéristiques et les fonctions de tous les organismes vivants. La décodage de ces séquences, appelée génomique, est essentiel pour comprendre la biologie moléculaire, la médecine et la recherche biologique en général.
La réaction de polymérisation en chaîne est un processus chimique au cours duquel des molécules de monomères réagissent ensemble pour former de longues chaînes de polymères. Ce type de réaction se caractérise par une vitesse de réaction rapide et une exothermie, ce qui signifie qu'elle dégage de la chaleur.
Dans le contexte médical, les réactions de polymérisation en chaîne sont importantes dans la production de matériaux biomédicaux tels que les implants et les dispositifs médicaux. Par exemple, certains types de plastiques et de résines utilisés dans les équipements médicaux sont produits par polymérisation en chaîne.
Cependant, il est important de noter que certaines réactions de polymérisation en chaîne peuvent également être impliquées dans des processus pathologiques, tels que la formation de plaques amyloïdes dans les maladies neurodégénératives telles que la maladie d'Alzheimer. Dans ces cas, les protéines se polymérisent en chaînes anormales qui s'accumulent et endommagent les tissus cérébraux.
Les chromosomes humains de la paire 14, également connus sous le nom de chromosomes 14, sont des structures composées de ADN et protéines qui contiennent des gènes et se trouvent dans le noyau de chaque cellule du corps. Les chromosomes 14 sont une paire de chromosomes homologues, ce qui signifie qu'ils ont la même taille, la même forme et contiennent des gènes similaires aux mêmes emplacements le long de la chromosome.
Chaque personne a 23 paires de chromosomes dans chaque cellule de leur corps, pour un total de 46 chromosomes. Les chromosomes 14 sont la 14ème paire de ces chromosomes, et ils sont numérotés de 14 à 14, ce qui signifie qu'ils sont présents deux fois dans chaque cellule du corps.
Les chromosomes 14 contiennent des centaines de gènes qui fournissent des instructions pour la production de protéines et d'autres produits génétiques importants pour le fonctionnement normal du corps. Les mutations dans ces gènes peuvent entraîner diverses maladies et conditions, telles que les troubles neurodégénératifs, les cancers et les maladies héréditaires.
En résumé, les chromosomes humains de la paire 14 sont des structures composées d'ADN et de protéines qui contiennent des gènes importants pour le fonctionnement normal du corps. Les mutations dans ces gènes peuvent entraîner diverses maladies et conditions.
Les chromosomes humains de la paire 17, également connus sous le nom de chromosomes 17, sont une partie importante du matériel génétique d'un être humain. Les chromosomes sont des structures en forme de bâtonnet dans le noyau des cellules qui contiennent des gènes, qui sont les unités de base de l'hérédité.
Chaque personne a 23 paires de chromosomes, pour un total de 46 chromosomes, dans chaque cellule de leur corps, sauf les cellules reproductives (spermatozoïdes et ovules), qui ne contiennent qu'une seule copie de chaque chromosome. Les chromosomes 17 sont la quatorzième paire de chromosomes dans l'ensemble des 23 paires.
Les chromosomes 17 sont relativement grands et contiennent environ 800 millions de paires de bases, ce qui représente environ 6 à 7 % du génome humain total. Ils contiennent entre 1 500 et 1 600 gènes, qui sont responsables de la production de protéines importantes pour diverses fonctions corporelles, telles que la réparation de l'ADN, le métabolisme des lipides et des glucides, la signalisation cellulaire, la division cellulaire et la différenciation cellulaire.
Les chromosomes 17 sont également associés à plusieurs maladies génétiques rares et courantes, telles que le syndrome de Li-Fraumeni, qui est un trouble héréditaire du cancer, et la neuropathie sensitive héréditaire de type 1, qui est une maladie neurologique héréditaire. Les mutations dans certains gènes situés sur les chromosomes 17 peuvent également augmenter le risque de développer des cancers tels que le cancer du sein, du poumon et du côlon.
En résumé, les chromosomes humains 17 sont importants pour la santé humaine car ils contiennent des gènes responsables de diverses fonctions corporelles et sont associés à plusieurs maladies génétiques courantes et rares. Les mutations dans certains gènes situés sur les chromosomes 17 peuvent également augmenter le risque de développer certains cancers.
L'analyse par microarray est une technique de laboratoire utilisée pour mesurer l'expression simultanée de milliers de gènes dans un échantillon donné. Cette méthode implique l'utilisation d'une puce à ADN, qui contient des centaines de milliers de petits fragments d'ADN, appelés sondes, disposés sur une surface solide.
Dans le processus d'analyse, l'ARNm (un précurseur de l'ARN messager) est extrait de l'échantillon et converti en ADN complémentaire (ADNc). Cet ADNc est ensuite étiqueté avec une molécule fluorescente et hybridé à la puce à ADN. Les sondes sur la puce qui correspondent aux séquences d'ARNm présentes dans l'échantillon s'hybrideront avec l'ADNc étiqueté, créant des signaux fluorescents détectables.
En analysant les intensités de ces signaux fluorescents, les chercheurs peuvent déterminer quels gènes sont surexprimés ou sous-exprimés dans l'échantillon, ce qui peut fournir des informations importantes sur les voies moléculaires et les processus cellulaires impliqués dans une maladie ou un état physiologique particulier.
L'analyse par microarray est largement utilisée en recherche biomédicale pour l'étude de diverses affections, telles que le cancer, les maladies cardiovasculaires et neurologiques, ainsi que pour la découverte de nouveaux biomarqueurs et cibles thérapeutiques.
Les chromosomes humains de la paire 12, également connus sous le nom de chromosomes 12, sont des structures composées de ADN et des protéines appelées histones. Ils font partie d'un ensemble de 46 chromosomes dans chaque cellule humaine et se trouvent dans chaque noyau cellulaire.
Chaque personne a deux chromosomes 12, une héritée de chaque parent, ce qui signifie qu'il y a 23 paires de chromosomes au total dans le génome humain. Les chromosomes 12 sont subdivisés en plusieurs régions et bandes, chacune contenant des gènes spécifiques qui codent pour des protéines importantes pour le fonctionnement normal du corps.
Les mutations ou les variations dans les gènes situés sur les chromosomes 12 peuvent être associées à certaines maladies génétiques, telles que la neurofibromatose de type 1, le syndrome de Wilms et l'anémie de Fanconi. Des études continues sont en cours pour mieux comprendre les fonctions des gènes situés sur les chromosomes 12 et leur rôle dans le développement et la progression des maladies humaines.
Les chromosomes humains de la paire 16, également connus sous le nom de chromosomes 16, sont une partie importante du matériel génétique d'un être humain. Chaque personne a deux chromosomes 16, une copie héritée de chaque parent. Les chromosomes 16 sont des structures en forme de bâtonnet qui se trouvent dans le noyau de chaque cellule du corps et contiennent des milliers de gènes responsables de la détermination de nombreuses caractéristiques physiques et fonctionnelles d'un individu.
Les chromosomes 16 sont l'une des 23 paires de chromosomes humains, ce qui signifie qu'il y a en tout 46 chromosomes dans chaque cellule du corps humain (à l'exception des spermatozoïdes et des ovules, qui n'en contiennent que 23). Les chromosomes 16 sont relativement grands et se situent au milieu de la gamme de taille des chromosomes humains.
Les gènes contenus dans les chromosomes 16 jouent un rôle important dans divers processus biologiques, notamment le développement du cerveau, le métabolisme des lipides et des glucides, la réponse immunitaire et la régulation de l'expression génétique. Les mutations dans ces gènes peuvent entraîner un certain nombre de maladies génétiques rares, telles que la neurofibromatose de type 1, le syndrome de Prader-Willi et le syndrome d'Angelman.
En résumé, les chromosomes humains de la paire 16 sont des structures en forme de bâtonnet qui se trouvent dans le noyau de chaque cellule du corps humain et contiennent des milliers de gènes responsables de nombreuses caractéristiques physiques et fonctionnelles d'un individu. Les mutations dans ces gènes peuvent entraîner un certain nombre de maladies génétiques rares.
Un déséquilibre allelique, également connu sous le nom de déséquilibre de liaison ou LD (Linkage Disequilibrium), est un phénomène dans lequel certaines combinaisons d'allèles à des loci génétiques adjacents se produisent plus fréquemment ou moins fréquemment que prévu par la fréquence d'allèle au hasard.
En d'autres termes, lorsque deux allèles de différents gènes sont hérités ensemble plus souvent qu'ils ne le seraient si les allèles étaient distribués indépendamment, on parle de déséquilibre allelique. Ce phénomène peut être utilisé pour détecter et localiser des gènes associés à des maladies ou des traits particuliers en analysant la fréquence relative des combinaisons d'allèles dans une population.
Le déséquilibre allelique est mesuré par le coefficient de corrélation de Lewontin, noté D', qui varie de -1 à 1. Une valeur positive de D' indique que les allèles sont en phase (c'est-à-dire qu'ils ont tendance à être hérités ensemble), tandis qu'une valeur négative indique qu'ils sont déséquilibrés dans la direction opposée. Une valeur de D' proche de zéro indique un équilibre allelique, ce qui signifie que les allèles sont distribués de manière aléatoire dans la population.
Les points de rupture des chromosomes (CBs) se réfèrent à des sites spécifiques sur les chromosomes où ils peuvent être fragiles et susceptibles de se casser pendant le processus de recombinaison génétique ou en raison d'une exposition à des agents mutagènes. Ces CBs sont souvent associés aux réarrangements chromosomiques structuraux, tels que les délétions, les duplications, les inversions et les translocations.
Les CBs peuvent être hérités ou acquérir de novo en raison d'erreurs pendant la méiose ou la mitose. Les mutations génétiques qui affectent la stabilité des chromosomes, telles que les mutations dans les gènes de réparation de l'ADN, peuvent augmenter la fréquence et la gravité des CBs.
Les CBs sont importants en médecine clinique car ils peuvent être associés à des maladies génétiques héréditaires ou sporadiques, telles que les troubles neurodéveloppementaux, les cancers et les maladies cardiovasculaires. Les techniques de cytogénétique moléculaire, telles que l'hybridation in situ fluorescente (FISH) et la séquençage de nouvelle génération (NGS), peuvent être utilisées pour identifier et caractériser les CBs à des fins diagnostiques et pronostiques.
L'analyse d'aggrégats est une méthode statistique utilisée en épidémiologie et en recherche médicale pour analyser des données regroupées ou agrégées, plutôt que des données individuelles. Cette méthode permet de protéger la confidentialité des données personnelles des patients, tout en fournissant des informations utiles sur les tendances et les schémas de santé dans une population donnée.
Dans l'analyse d'aggrégats, les données sont regroupées par catégories prédéfinies telles que l'âge, le sexe, la race/ethnicité, la région géographique, etc. Les statistiques telles que les taux de prévalence, d'incidence, de mortalité et de morbidité sont ensuite calculées pour chaque catégorie. Ces statistiques peuvent être comparées entre les catégories pour identifier les différences ou les similitudes dans les résultats de santé.
L'analyse d'aggrégats peut également être utilisée pour étudier l'association entre des facteurs de risque et des résultats de santé en examinant les taux de ces facteurs de risque et des résultats de santé dans différentes catégories. Cette méthode est particulièrement utile lorsque les données individuelles ne sont pas disponibles ou ne peuvent pas être partagées en raison de considérations de confidentialité.
Cependant, il est important de noter que l'analyse d'aggrégats a ses limites. Par exemple, elle peut ne pas tenir compte des facteurs de confusion potentiels qui peuvent affecter les résultats de santé. De plus, les catégories prédéfinies peuvent ne pas refléter la variabilité individuelle au sein des catégories, ce qui peut entraîner une perte d'information. Par conséquent, l'analyse d'aggrégats doit être utilisée en combinaison avec d'autres méthodes d'analyse pour fournir une image complète de l'association entre les facteurs de risque et les résultats de santé.
Les chromosomes humains de la paire 15, également connus sous le nom de chromosomes 15, sont des structures en forme de bâtonnet dans les cellules du corps humain qui contiennent des gènes et de l'ADN. Chaque personne a une paire de ces chromosomes, ce qui signifie qu'il y a deux chromosomes 15 dans chaque cellule.
Les chromosomes 15 sont responsables de la régulation de diverses fonctions corporelles et du développement de certaines caractéristiques physiques. Les gènes situés sur ces chromosomes jouent un rôle important dans le fonctionnement normal du cerveau, du système immunitaire, des hormones et d'autres systèmes corporels.
Les anomalies chromosomiques de la paire 15 peuvent entraîner des troubles génétiques tels que la syndrome de l'X fragile, le syndrome de Prader-Willi et le syndrome d'Angelman. Ces conditions sont caractérisées par une variété de symptômes, notamment des retards de développement, des problèmes d'apprentissage, des troubles du comportement et des anomalies physiques.
Il est important de noter que les tests génétiques peuvent être utilisés pour détecter les anomalies chromosomiques de la paire 15 et aider à poser un diagnostic pour les personnes atteintes de ces conditions.
Les malformations multiples, également connues sous le nom de malformations congénitales multiples, se réfèrent à la présence de deux ou plusieurs anomalies congénitales affectant différents organes ou systèmes du corps. Ces anomalies sont présentes dès la naissance et peuvent être causées par des facteurs génétiques, environnementaux ou une combinaison des deux.
Les malformations multiples peuvent affecter n'importe quelle partie du corps et peuvent varier en gravité, allant de légères à graves. Elles peuvent également affecter la fonctionnalité des organes touchés et dans les cas les plus sévères, peuvent être fatales.
Les exemples courants de malformations multiples comprennent le syndrome de Down (trisomie 21), qui est caractérisé par un retard mental, une apparence faciale distinctive et souvent d'autres anomalies telles que des problèmes cardiaques congénitaux ; le syndrome de Di George, qui affecte la croissance et le développement et peut causer des problèmes cardiaques, immunitaires et de développement du cerveau ; et le spina bifida, une anomalie de la colonne vertébrale qui peut causer des problèmes de mouvement et de sensation dans les jambes.
Le diagnostic et le traitement des malformations multiples dépendent du type et de la gravité des anomalies présentes. Les soins peuvent inclure une combinaison de chirurgie, de médicaments, de thérapies et de soutien de développement pour aider à gérer les symptômes et améliorer la qualité de vie de l'enfant affecté.
Les corps polaires sont de petites structures membranaires qui se forment pendant le processus de méiose dans les ovocytes (cellules reproductives femelles). Ils contiennent des matériaux génétiques et cytoplasmiques qui ne sont pas inclus dans l'ovule mature après la méiose. Les corps polaires résultent de la division inégale de la cellule mère ovocytaire, où un côté reçoit une quantité disproportionnée de cytoplasme et d'organites, tandis que l'autre côté devient le corps polar.
Habituellement, il y a deux corps polaires formés pendant la méiose : un après la première division méiotique (corps polar I) et un autre après la seconde division méiotique (corps polar II). Les corps polaires sont généralement éliminés du cytoplasme de l'ovule avant ou après la fécondation, ce qui permet d'éviter les anomalies chromosomiques dans le zygote en développement.
Cependant, il a été démontré que les corps polaires contiennent des informations génétiques et épigénétiques qui peuvent être utiles pour l'analyse du potentiel reproductif d'une femme, ainsi que pour le diagnostic de certaines maladies génétiques. Par conséquent, la recherche sur les corps polaires est un domaine en développement dans la médecine de la reproduction.
Les chromosomes humains de la paire 12, également connus sous le nom de chromosomes 12, sont des structures composées de ADN et des protéines appelées histones. Ils font partie d'un ensemble de 46 chromosomes dans chaque cellule humaine et se trouvent dans chaque noyau cellulaire.
Chaque personne a deux chromosomes 12, une héritée de chaque parent, ce qui signifie qu'il y a 23 paires de chromosomes au total dans le génome humain. Les chromosomes 12 sont subdivisés en plusieurs régions et bandes, chacune contenant des gènes spécifiques qui codent pour des protéines importantes pour le fonctionnement normal du corps.
Les mutations ou les variations dans les gènes situés sur les chromosomes 12 peuvent être associées à certaines maladies génétiques, telles que la neurofibromatose de type 1, le syndrome de Wilms et l'anémie de Fanconi. Des études continues sont en cours pour mieux comprendre les fonctions des gènes situés sur les chromosomes 12 et leur rôle dans le développement et la progression des maladies humaines.
La paraffine liquide, également connue sous le nom de paraffine liquide ou de vaseline liquide, est un liquide huileux inodore, insipide, incolore et visqueux dérivé du pétrole. Bien qu'il ne soit pas fréquemment utilisé dans le domaine médical, il peut être utilisé en tant que substance de remplissage pour les cavités corporelles lors d'interventions chirurgicales ou comme agent barrière pour protéger la peau.
Dans certains cas rares et spécifiques, la paraffine liquide peut être utilisée en médecine comme matériau d'inclusion dans le cadre de procédures diagnostiques particulières, telles que l'inclusion paraffinique. Ce processus consiste à injecter délicatement de petites quantités de paraffine liquide dans les tissus mous, généralement au niveau des seins ou des ganglions lymphatiques, afin de former une capsule protectrice autour d'une zone anormale ou suspecte. Cela permet aux médecins de prélever plus facilement et en toute sécurité un échantillon de tissu (biopsie) pour analyse ultérieure.
Il convient de noter que l'utilisation de la paraffine liquide à des fins médicales est limitée et doit être effectuée sous la supervision directe d'un professionnel de santé qualifié, en raison du risque potentiel d'effets indésirables tels que des réactions allergiques, une inflammation ou une infection.
Les chromosomes humains de la paire 22, également connus sous le nom de chromosomes 22, sont une partie importante du génome humain. Ils font partie des 23 paires de chromosomes trouvés dans chaque cellule humaine. Chaque chromosome 22 contient des milliers de gènes qui fournissent les instructions pour la production de protéines et d'autres produits fonctionnels nécessaires au bon fonctionnement de l'organisme.
Les chromosomes 22 sont parmi les plus petits des chromosomes humains, avec une longueur d'environ 50 millions de paires de bases. Ils représentent environ 1,5 à 3% du génome humain total. Les chromosomes 22 contiennent un certain nombre de gènes importants qui sont associés à diverses maladies et conditions médicales.
L'un des gènes les plus connus sur le chromosome 22 est le gène NF2, qui code pour une protéine appelée mercaptopurine nucléotide synthase (MER). Les mutations dans ce gène sont associées à la neurofibromatose de type 2, une maladie génétique caractérisée par la croissance de tumeurs bénignes le long des nerfs.
Les chromosomes 22 sont également importants dans l'étude de la schizophrénie, une maladie mentale grave qui affecte environ 1% de la population mondiale. Des études ont montré que certaines variations génétiques sur le chromosome 22 peuvent augmenter le risque de développer cette condition.
En plus des gènes, les chromosomes 22 contiennent également une grande quantité d'ADN non codant qui ne code pas directement pour des protéines. Cet ADN peut jouer un rôle important dans la régulation de l'expression génique et peut être associé à diverses maladies et conditions médicales.
En résumé, les chromosomes 22 sont importants dans l'étude de diverses maladies et conditions médicales, y compris la neurofibromatose de type 2 et la schizophrénie. Les mutations dans les gènes du chromosome 22 peuvent augmenter le risque de développer ces conditions, tandis que l'ADN non codant sur le chromosome 22 peut également jouer un rôle important dans la régulation de l'expression génique et être associé à diverses maladies.
Le phénotype est le résultat observable de l'expression des gènes en interaction avec l'environnement et d'autres facteurs. Il s'agit essentiellement des manifestations physiques, biochimiques ou développementales d'un génotype particulier.
Dans un contexte médical, le phénotype peut se rapporter à n'importe quelle caractéristique mesurable ou observable résultant de l'interaction entre les gènes et l'environnement, y compris la couleur des yeux, la taille, le poids, certaines maladies ou conditions médicales, voire même la réponse à un traitement spécifique.
Il est important de noter que deux individus ayant le même génotype (c'est-à-dire la même séquence d'ADN) ne seront pas nécessairement identiques dans leur phénotype, car des facteurs environnementaux peuvent influencer l'expression des gènes. De même, des individus avec des génotypes différents peuvent partager certains traits phénotypiques en raison de similitudes dans leurs environnements ou dans d'autres facteurs non génétiques.
La peinture chromosomique, également connue sous le nom de peinture génomique à l'aide de sonde complémentaire (Comprehensive Genomic Hybridization - CGH), est une technique de cytogénétique avancée utilisée pour détecter les anomalies chromosomiques invisibles aux méthodes conventionnelles. Elle permet l'identification des gains et pertes segmentaires d'ADN sur tous les chromosomes.
Dans cette technique, différentes sondes fluorescentes sont utilisées pour marquer chaque paire de chromosomes. Ces sondes se lient à des séquences spécifiques le long du génome et émettent une lumière fluorescente lorsqu'elles sont exposées à une source de lumière appropriée. La comparaison des profils de fluorescence entre les échantillons d'un patient et d'une référence normale permet de mettre en évidence toute différence dans la quantité relative d'ADN, indiquant ainsi des gains ou pertes chromosomiques.
La peinture chromosomique est particulièrement utile pour le diagnostic et le suivi des cancers, où elle peut révéler des changements complexes du génome qui ne seraient pas détectables par d'autres méthodes. Elle joue également un rôle important dans le diagnostic prénatal et postnatal de certaines maladies génétiques.
Une cassure chromosomique est un type d'anomalie chromosomique qui se produit lorsqu'un chromosome se brise en deux morceaux séparés. Cela peut être causé par des facteurs internes tels que des erreurs au cours de la réplication de l'ADN ou externe comme une exposition à des radiations ou certains produits chimiques.
Les cassures chromosomiques peuvent entraîner divers effets, selon leur localisation et leur gravité sur le chromosome. Parfois, les deux extrémités de la casse peuvent se rejoindre à nouveau de manière incorrecte, ce qui entraîne une recombinaison inégale des gènes et peut conduire à des mutations génétiques.
Dans certains cas, une cassure chromosomique peut également entraîner une délétion (perte d'une partie du chromosome) ou une insertion (ajout d'un morceau de chromosome dans un autre endroit). Si la cassure se produit pendant la division cellulaire, elle peut conduire à des cellules avec un nombre anormal de chromosomes, ce qui est appelé aneuploïdie.
Les cassures chromosomiques peuvent être responsables de certaines maladies génétiques héréditaires ou spontanées, ainsi que d'une susceptibilité accrue au cancer.
Les duplications segmentaires génomiques sont des événements de copie d'ADN qui se produisent lorsqu'un segment spécifique du génome est dupliqué, résultant en deux ou plusieurs copies de ce segment dans le même chromosome ou sur différents chromosomes. Ces duplications peuvent varier en taille, allant de quelques centaines de paires de bases à plusieurs méga-bases.
Les duplications segmentaires génomiques peuvent être classées en deux types principaux : les duplications intrachromosomiques et les duplications interchromosomiques. Les duplications intrachromosomiques se produisent lorsqu'un segment d'un chromosome est dupliqué sur le même chromosome, tandis que les duplications interchromosomiques se produisent lorsqu'un segment d'un chromosome est dupliqué sur un autre chromosome.
Les duplications segmentaires génomiques peuvent avoir des conséquences variées sur la fonction génétique et la santé humaine. Dans certains cas, ces duplications peuvent entraîner une augmentation de la production de protéines, ce qui peut être bénéfique ou préjudiciable, selon le gène concerné. Cependant, dans d'autres cas, les duplications segmentaires génomiques peuvent perturber l'expression génétique et entraîner des maladies génétiques ou des troubles du développement.
Les duplications segmentaires génomiques peuvent être héréditaires ou spontanées, et elles sont souvent associées à des variations du nombre de copies (CNV) dans le génome. Les CNV sont des variations de la quantité d'ADN qui peuvent entraîner des différences phénotypiques entre les individus, y compris la susceptibilité accrue à certaines maladies.
En résumé, les duplications segmentaires génomiques sont des événements de copie d'ADN qui se produisent lorsqu'un segment spécifique du génome est dupliqué, ce qui peut entraîner des variations du nombre de copies et des conséquences variées sur la fonction génétique et la santé humaine.
La régulation de l'expression génique tumorale dans un contexte médical se réfère aux mécanismes moléculaires et cellulaires qui contrôlent la manière dont les gènes s'expriment dans les cellules cancéreuses. Les changements dans l'expression des gènes peuvent entraîner une prolifération cellulaire accrue, une résistance à l'apoptose (mort cellulaire programmée), une angiogenèse (croissance de nouveaux vaisseaux sanguins) et une métastase, qui sont tous des processus clés dans le développement du cancer.
La régulation de l'expression génique tumorale peut être influencée par une variété de facteurs, y compris les mutations génétiques, les modifications épigénétiques (telles que la méthylation de l'ADN et l'acétylation des histones), les facteurs de transcription anormaux, les miARN (petits ARN non codants qui régulent l'expression des gènes) et les interactions entre les cellules tumorales et leur microenvironnement.
Comprendre la régulation de l'expression génique tumorale est crucial pour le développement de thérapies ciblées contre le cancer, car il permet d'identifier de nouvelles cibles thérapeutiques et de prédire la réponse des patients aux traitements existants. Des approches telles que l'édition du génome, la modulation épigénétique et l'interférence avec les miARN sont autant de stratégies prometteuses pour réguler l'expression des gènes dans le cancer et améliorer les résultats cliniques.
Les chromosomes humains de la paire 18, également connus sous le nom de chromosomes 18, sont des structures composées de ADN et protéines trouvés dans le noyau de chaque cellule du corps humain. Les humains ont normalement 23 paires de chromosomes, pour un total de 46 chromosomes, dans chaque cellule. Les chromosomes 18 sont la dix-huitième paire de ces chromosomes, et chaque personne hérite d'une copie de ce chromosome de chacun de ses parents.
Les chromosomes 18 sont des structures relativement grandes, représentant environ 2 à 2,5 % du total de l'ADN dans une cellule humaine. Ils contiennent des milliers de gènes qui fournissent des instructions pour la production de protéines et d'autres produits fonctionnels nécessaires au développement, à la fonction et à la survie de l'organisme.
Les anomalies chromosomiques des chromosomes 18 peuvent entraîner diverses conditions médicales. Par exemple, une trisomie 18, dans laquelle il y a trois copies du chromosome 18 au lieu des deux copies normales, est associée à une condition appelée syndrome d'Edwards. Cette condition est caractérisée par un ensemble de symptômes et de traits physiques distincts, notamment un retard de développement, des anomalies faciales, des problèmes cardiaques et d'autres anomalies congénitales.
D'autre part, une monosomie 18, dans laquelle il n'y a qu'une seule copie du chromosome 18 au lieu des deux copies normales, est associée à une condition appelée syndrome de Jacobsen. Cette condition est également caractérisée par un ensemble de symptômes et de traits physiques distincts, notamment un retard de développement, des anomalies faciales, des problèmes cardiaques et d'autres anomalies congénitales.
Dans l'ensemble, les chromosomes 18 jouent un rôle crucial dans le développement et la fonction normaux de l'organisme. Les anomalies chromosomiques de ces chromosomes peuvent entraîner divers problèmes de santé et des déficiences du développement.
L'instabilité chromosomique est un terme utilisé en génétique pour décrire une condition dans laquelle il y a une augmentation anormale du taux de changements structurels ou numériques des chromosomes. Cela peut inclure des choses comme des délétions, des duplications, des translocations, des inversions ou des gains ou pertes de chromosomes entiers. Ces changements peuvent se produire spontanément ou être hérités.
L'instabilité chromosomique peut entraîner une variété de problèmes de santé, en fonction de la région du génome touchée et de l'ampleur des changements. Par exemple, certaines formes d'instabilité chromosomique peuvent entraîner des maladies génétiques spécifiques, telles que le syndrome de Down (trisomie 21), le syndrome de Turner (monosomie X) ou le syndrome de Klinefelter (XXY). D'autres formes d'instabilité chromosomique peuvent prédisposer une personne à développer certains types de cancer.
Il existe deux principaux types d'instabilité chromosomique : l'instabilité numérique et l'instabilité structurelle. L'instabilité numérique fait référence à des anomalies dans le nombre de chromosomes, tandis que l'instabilité structurelle fait référence à des changements dans la structure des chromosomes.
L'instabilité chromosomique peut être causée par divers facteurs, notamment des mutations génétiques, une exposition à des agents environnementaux nocifs, certains médicaments ou une mauvaise réplication ou réparation de l'ADN. Dans certains cas, la cause est inconnue.
Un karyotype est un examen de laboratoire qui consiste à étudier l'ensemble des chromosomes d'une cellule humaine ou animale, généralement issus d'un prélèvement sanguin. Cette technique permet d'observer la forme, la taille et le nombre de chromosomes, ainsi que la présence de certaines anomalies structurales ou numériques.
Le karyotype est représenté sous la forme d'une image où les chromosomes sont disposés par paires, du plus grand au plus petit, et classés en fonction de leur morphologie (bras court et bras long). Les chromosomes sexuels (X et Y) sont également identifiables sur cette représentation.
L'analyse du karyotype est utilisée dans le diagnostic prénatal, le dépistage des maladies génétiques et la recherche de causes sous-jacentes à certaines affections médicales telles que les troubles de développement, les anomalies congénitales ou certains cancers.
Les ilôts génomiques, également connus sous le nom d'îlots CNV (Copies Number Variation), sont des régions particulières du génome qui présentent une variation dans le nombre de copies de certains gènes ou séquences d'ADN. Ces variations peuvent inclure des délétions, où une partie ou la totalité d'un ilôt est manquant, ou des duplications, où l'ilôt est présent en plus grand nombre de copies que la normale.
Les ilôts génomiques sont souvent localisés dans des régions du génome qui sont riches en répétitions en tandem d'ADN, ce qui les rend sujets à des erreurs de recombinaison pendant la méiose et la mitose. Ces erreurs peuvent entraîner des variations dans le nombre de copies de certains gènes, ce qui peut avoir un impact sur l'expression génique et la fonction des protéines.
Les ilôts génomiques sont souvent associés à des maladies génétiques et à des troubles du développement, tels que les troubles du spectre autistique, la schizophrénie, la déficience intellectuelle et certaines formes de retard mental. Ils peuvent également jouer un rôle dans la variabilité individuelle de la réponse aux médicaments et à l'environnement.
En médecine, la détection et l'analyse des ilôts génomiques peuvent être utiles pour le diagnostic et le suivi des maladies génétiques, ainsi que pour la recherche sur les causes sous-jacentes de certaines maladies complexes.
Les Techniques d'Amplification des Acides Nucléiques (NAAT, selon l'acronyme en anglais) sont un ensemble de méthodes moléculaires utilisées pour amplifier ou copier de manière exponentielle des séquences spécifiques d'ADN ou d'ARN. Ces techniques sont largement utilisées dans le domaine de la recherche et du diagnostic médical pour détecter, identifier et quantifier des agents pathogènes, des mutations génétiques ou des marqueurs biologiques.
Le procédé le plus connu et largement utilisé est la Réaction en Chaîne par Polymérase (PCR : Polymerase Chain Reaction), qui consiste à séparer les brins d'ADN, puis à synthétiser de nouveaux brins en utilisant des amorces spécifiques et une ADN polymérase thermostable. Ce processus est répété à plusieurs cycles, permettant ainsi l'amplification exponentielle de la séquence d'intérêt.
D'autres techniques d'amplification incluent la Transcription Inverse suivie de l'Amplification par PCR (RT-PCR : Reverse Transcription Polymerase Chain Reaction), qui permet d'amplifier des ARN en convertissant d'abord ces derniers en ADN complémentaire (ADNc) à l'aide d'une transcriptase inverse, suivie de la PCR. La LAMP (Loop-Mediated Isothermal Amplification) est une méthode isotherme d'amplification qui ne nécessite pas de thermocycleur et peut être réalisée à température constante, ce qui la rend plus accessible pour les tests sur le terrain ou dans des environnements à ressources limitées.
Les Techniques d'Amplification des Acides Nucléiques sont inestimables en médecine, notamment en microbiologie clinique, en génétique moléculaire et en oncologie, pour diagnostiquer rapidement et avec précision une grande variété de maladies infectieuses, de troubles héréditaires et de cancers.
Les gènes tumoraux, également connus sous le nom de gènes suppresseurs de tumeurs ou oncogènes, sont des gènes qui jouent un rôle crucial dans la régulation de la croissance et de la division cellulaires. Lorsqu'ils fonctionnent correctement, ces gènes aident à prévenir une croissance cellulaire excessive et cancéreuse en régulant l'activité des protéines qui favorisent ou inhibent la croissance cellulaire.
Cependant, lorsque ces gènes sont mutés ou endommagés, ils peuvent perdre leur capacité à fonctionner correctement, entraînant une division et une croissance cellulaires incontrôlées. Cela peut conduire au développement de tumeurs malignes ou cancéreuses.
Les mutations des gènes tumoraux peuvent être héréditaires ou acquises au cours de la vie d'un individu en raison de facteurs environnementaux, tels que l'exposition à des substances cancérigènes ou une mauvaise alimentation. Dans certains cas, les mutations peuvent également être causées par des erreurs aléatoires lors de la réplication de l'ADN.
L'identification et la compréhension des gènes tumoraux sont essentielles pour le diagnostic, le traitement et la prévention du cancer. Les tests génétiques peuvent être utilisés pour détecter les mutations des gènes tumoraux héréditaires, ce qui peut aider à identifier les personnes à risque de développer certains types de cancer. De plus, l'identification des mutations acquises des gènes tumoraux peut aider à guider le choix du traitement pour les patients atteints de cancer.
La délétion séquentielle est un terme utilisé en génétique et médecine moléculaire pour décrire la perte d'une séquence particulière de nucléotides dans une région spécifique du génome. Cela se produit lorsque des sections répétées de l'ADN, appelées répétitions en tandem, sont instables et ont tendance à se contractre, entraînant ainsi la suppression d'une partie du matériel génétique.
Dans une délétion séquentielle, cette perte de nucléotides se produit non pas une fois mais plusieurs fois de manière consécutive, ce qui entraîne l'effacement progressif d'une plus grande portion du gène ou de la région régulatrice. Cette répétition de délétions peut conduire à des mutations plus complexes et graves, augmentant ainsi le risque de développer certaines maladies génétiques.
Il est important de noter que les délétions séquentielles sont souvent associées aux expansions répétitives de nucléotides (ERN), qui sont des mutations génétiques caractérisées par la présence d'une section répétée anormalement longue d'un ou plusieurs nucléotides dans une région spécifique du génome. Les ERNs sont souvent liées à un large éventail de maladies neurodégénératives et neuromusculaires, telles que la maladie de Huntington, la sclérose latérale amyotrophique (SLA) et la myopathie facio-scapulo-humérale.
Les microsatellites, également connus sous le nom de "short tandem repeats" (STR), sont des séquences répétitives d'ADN qui se composent de motifs de deux à six paires de bases nucléiques répétées de manière consécutive. Ces séquences sont dispersées dans tout le génome et ont tendance à varier en longueur entre les individus, ce qui en fait des marqueurs utiles en médecine légale pour l'identification humaine et la paternité. En outre, certaines mutations de microsatellites sont associées à des maladies génétiques telles que la maladie de Huntington et la polyarthrite rhumatoïde. Les microsatellites peuvent également jouer un rôle dans la régulation de l'expression des gènes et la variabilité du transcriptome.
La translocation génétique est un type d'anomalie chromosomique où des segments entiers de deux chromosomes différents changent de place. Il existe deux types principaux de translocations génétiques : les translocations réciproques et les translocations Robertsoniennes.
Les translocations réciproques se produisent lorsque des segments de deux chromosomes différents sont échangés l'un avec l'autre. Ces translocations peuvent être équilibrées, ce qui signifie qu'aucun matériel génétique n'est ni gagné ni perdu dans le processus, ou déséquilibrée, ce qui entraîne une perte ou un gain de matériel génétique.
Les translocations Robertsoniennes, quant à elles, se produisent lorsque la partie distale (la partie la plus éloignée du centromère) de deux chromosomes acrocentriques (qui comprennent les chromosomes 13, 14, 15, 21 et 22) est interchangée, entraînant la fusion des deux chromosomes à leur centromère commun. Cela entraîne la formation d'un seul chromosome avec deux bras courts (p) et aucun bras long (q). Les translocations Robertsoniennes sont le plus souvent équilibrées, mais lorsqu'elles ne le sont pas, elles peuvent entraîner des anomalies génétiques et des troubles du développement.
Les translocations génétiques peuvent être héritées ou spontanées (de novo). Lorsqu'elles sont héritées, elles peuvent être asymptomatiques ou causer des problèmes de santé dépendamment de la façon dont les gènes affectés sont exprimés. Cependant, lorsqu'elles sont spontanées, elles peuvent entraîner des anomalies chromosomiques telles que le syndrome de Down (translocation entre les chromosomes 21 et un autre chromosome) ou le syndrome de Patau (translocation entre les chromosomes 13 et un autre chromosome).
En résumé, les translocations génétiques sont des réarrangements chromosomiques qui peuvent entraîner des problèmes de santé et des anomalies du développement. Elles peuvent être héritées ou spontanées et peuvent affecter n'importe quel chromosome. Les translocations Robertsoniennes sont un type spécifique de translocation qui implique la fusion de deux chromosomes à leur centromère commun, entraînant la formation d'un seul chromosome avec deux bras courts et aucun bras long.
Le Diagnostic Pré-Implantatoire (DPI) est une technique de diagnostic génétique utilisée en procréation médicalement assistée. Il consiste à analyser le matériel génétique des embryons conçus in vitro avant leur transfert dans l'utérus de la mère. Cette méthode permet d'identifier d'éventuelles anomalies chromosomiques ou génétiques chez les embryons, ce qui offre la possibilité aux médecins et aux couples de ne transférer que des embryons sains et exempts de maladies graves héréditaires. Le DPI est donc un outil précieux pour réduire le risque de transmission d'affections génétiques sévères à la descendance. Cependant, son utilisation fait l'objet de débats éthiques en raison des questions soulevées par la sélection embryonnaire et la manipulation génétique humaine.
La ploïdie est un terme utilisé en génétique pour décrire le nombre de jeu complet de chromosomes dans une cellule. Dans la plupart des espèces, y compris les humains, la ploïdie normale est diploïde, ce qui signifie qu'il y a deux jeux complets de chromosomes dans chaque cellule somatique (non reproductrice). Chez l'homme, cela se traduit par 46 chromosomes au total - 23 chromosomes proviennent du père et 23 proviennent de la mère.
Cependant, certaines cellules peuvent avoir des nombres anormaux de chromosomes, ce qui est appelé une anomalie de ploïdie. Par exemple, dans le syndrome de Down, il y a un chromosome supplémentaire 21, portant le total à 47 chromosomes. Cela peut entraîner des problèmes de développement et de santé.
La ploïdie est importante pour la stabilité génétique et la croissance et le développement normaux d'un organisme. Des changements dans le niveau de ploïdie peuvent entraîner des anomalies du développement, des maladies génétiques ou même la mort de l'organisme.
Les chromosomes humains de la paire 15, également connus sous le nom de chromosomes 15, sont des structures en forme de bâtonnet dans les cellules du corps humain qui contiennent des gènes et de l'ADN. Chaque personne a une paire de ces chromosomes, ce qui signifie qu'il y a deux chromosomes 15 dans chaque cellule.
Les chromosomes 15 sont responsables de la régulation de diverses fonctions corporelles et du développement de certaines caractéristiques physiques. Les gènes situés sur ces chromosomes jouent un rôle important dans le fonctionnement normal du cerveau, du système immunitaire, des hormones et d'autres systèmes corporels.
Les anomalies chromosomiques de la paire 15 peuvent entraîner des troubles génétiques tels que la syndrome de l'X fragile, le syndrome de Prader-Willi et le syndrome d'Angelman. Ces conditions sont caractérisées par une variété de symptômes, notamment des retards de développement, des problèmes d'apprentissage, des troubles du comportement et des anomalies physiques.
Il est important de noter que les tests génétiques peuvent être utilisés pour détecter les anomalies chromosomiques de la paire 15 et aider à poser un diagnostic pour les personnes atteintes de ces conditions.
En génétique, une mutation est une modification permanente et héréditaire de la séquence nucléotidique d'un gène ou d'une région chromosomique. Elle peut entraîner des changements dans la structure et la fonction des protéines codées par ce gène, conduisant ainsi à une variété de phénotypes, allant de neutres (sans effet apparent) à délétères (causant des maladies génétiques). Les mutations peuvent être causées par des erreurs spontanées lors de la réplication de l'ADN, l'exposition à des agents mutagènes tels que les radiations ou certains produits chimiques, ou encore par des mécanismes de recombinaison génétique.
Il existe différents types de mutations, telles que les substitutions (remplacement d'un nucléotide par un autre), les délétions (suppression d'une ou plusieurs paires de bases) et les insertions (ajout d'une ou plusieurs paires de bases). Les conséquences des mutations sur la santé humaine peuvent être très variables, allant de maladies rares à des affections courantes telles que le cancer.
L'ADN (acide désoxyribonucléique) est une molécule complexe qui contient les instructions génétiques utilisées dans le développement et la fonction de tous les organismes vivants connus et certains virus. L'ADN est un long polymère d'unités simples appelées nucléotides, avec des séquences de ces nucléotides qui forment des gènes. Ces gènes sont responsables de la synthèse des protéines et de la régulation des processus cellulaires.
L'ADN est organisé en une double hélice, où deux chaînes polynucléotidiques s'enroulent autour d'un axe commun. Les chaînes sont maintenues ensemble par des liaisons hydrogène entre les bases complémentaires : adénine (A) avec thymine (T), et guanine (G) avec cytosine (C).
L'ADN est présent dans le noyau de la cellule, ainsi que dans certaines mitochondries et chloroplastes. Il joue un rôle crucial dans l'hérédité, la variation génétique et l'évolution des espèces. Les mutations de l'ADN peuvent entraîner des changements dans les gènes qui peuvent avoir des conséquences sur le fonctionnement normal de la cellule et être associées à des maladies génétiques ou cancéreuses.
L'évolution moléculaire est un domaine de la biologie qui étudie les changements dans les séquences d'acides nucléiques et des protéines au fil du temps. Il s'appuie sur des disciplines telles que la génétique, la biochimie et la biophysique pour comprendre comment les organismes évoluent au niveau moléculaire.
L'évolution moléculaire examine les mutations, les réarrangements chromosomiques, les duplications de gènes, les transferts horizontaux de gènes et d'autres processus qui modifient la composition génétique des populations. Elle cherche à déterminer comment ces modifications influencent la forme, la fonction et le fitness des organismes.
Les analyses d'évolution moléculaire comprennent souvent des comparaisons de séquences entre différentes espèces ou populations, ainsi que l'inférence de phylogénies (arbres évolutifs) qui représentent les relations historiques entre ces groupes. Ces approches peuvent aider à répondre à des questions sur l'origine et la diversification des espèces, l'adaptation aux environnements changeants et l'évolution de la complexité biologique.
En résumé, l'évolution moléculaire est une branche importante de la biologie évolutive qui étudie les processus et les mécanismes sous-jacents à l'évolution des gènes et des protéines au fil du temps.
L'ADN bactérien fait référence à l'acide désoxyribonucléique présent dans les bactéries. Il s'agit du matériel génétique héréditaire des bactéries, qui contient toutes les informations nécessaires à leur croissance, leur développement et leur fonctionnement.
Contrairement à l'ADN des cellules humaines, qui est organisé en chromosomes situés dans le noyau de la cellule, l'ADN bactérien se présente sous forme d'une unique molécule circulaire située dans le cytoplasme de la cellule. Cette molécule d'ADN bactérien est également appelée chromosome bactérien.
L'ADN bactérien peut contenir des gènes codant pour des protéines, des ARN non codants et des éléments régulateurs qui contrôlent l'expression des gènes. Les bactéries peuvent également posséder de l'ADN extrachromosomique sous forme de plasmides, qui sont des petites molécules d'ADN circulaires contenant un ou plusieurs gènes.
L'étude de l'ADN bactérien est importante pour comprendre la physiologie et le métabolisme des bactéries, ainsi que pour développer des stratégies de lutte contre les infections bactériennes. Elle permet également d'identifier des marqueurs spécifiques qui peuvent être utilisés pour caractériser et classer différentes espèces bactériennes.
Je suis désolé, mais la phrase que vous avez demandée à définir ne semble pas être une expression ou un terme médical standard. "Spécificité Espèce" ne donne aucun résultat pertinent dans les contextes médicaux ou scientifiques.
Si vous cherchez des informations sur la spécificité en général dans le contexte médical, cela fait référence à la capacité d'un test diagnostique à correctement identifier les individus sans une certaine condition. En d'autres termes, la spécificité est le rapport entre le nombre de vrais négatifs et le total des personnes négatives (saines) dans une population donnée.
Si vous cherchiez des informations sur la taxonomie biologique ou l'identification des espèces, "spécificité d'espèce" pourrait faire référence à des caractéristiques uniques qui définissent et différencient une espèce donnée des autres.
Si vous pouviez me fournir plus de contexte ou clarifier votre question, je serais heureux de vous aider davantage.
Les chromosomes humains de la paire 14, également connus sous le nom de chromosomes 14, sont des structures composées de ADN et protéines qui contiennent des gènes et se trouvent dans le noyau de chaque cellule du corps. Les chromosomes 14 sont une paire de chromosomes homologues, ce qui signifie qu'ils ont la même taille, la même forme et contiennent des gènes similaires aux mêmes emplacements le long de la chromosome.
Chaque personne a 23 paires de chromosomes dans chaque cellule de leur corps, pour un total de 46 chromosomes. Les chromosomes 14 sont la 14ème paire de ces chromosomes, et ils sont numérotés de 14 à 14, ce qui signifie qu'ils sont présents deux fois dans chaque cellule du corps.
Les chromosomes 14 contiennent des centaines de gènes qui fournissent des instructions pour la production de protéines et d'autres produits génétiques importants pour le fonctionnement normal du corps. Les mutations dans ces gènes peuvent entraîner diverses maladies et conditions, telles que les troubles neurodégénératifs, les cancers et les maladies héréditaires.
En résumé, les chromosomes humains de la paire 14 sont des structures composées d'ADN et de protéines qui contiennent des gènes importants pour le fonctionnement normal du corps. Les mutations dans ces gènes peuvent entraîner diverses maladies et conditions.
Les tumeurs du sein sont des croissances anormales de cellules dans le tissu mammaire. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs bénignes ne se propagent pas au-delà du sein et ne mettent généralement pas la vie en danger, bien qu'elles puissent parfois causer des douleurs, des gonflements ou d'autres problèmes.
Les tumeurs malignes, en revanche, peuvent se propager (métastaser) à d'autres parties du corps et peuvent être mortelles. Le cancer du sein le plus courant est le carcinome canalaire infiltrant, qui commence dans les conduits qui transportent le lait vers l'extérieur du sein. Un autre type courant est le carcinome lobulaire infiltrant, qui se développe dans les glandes productrices de lait.
Les facteurs de risque de cancer du sein comprennent le sexe (être une femme), l'âge avancé, les antécédents familiaux de cancer du sein, les mutations génétiques héréditaires telles que BRCA1 et BRCA2, la densité mammaire élevée, les antécédents de radiothérapie dans la région du thorax, l'obésité, la consommation d'alcool, le début précoce des règles et la ménopause tardive.
Le dépistage régulier par mammographie est recommandé pour les femmes à risque élevé de cancer du sein. Le traitement peut inclure une combinaison de chirurgie, de radiothérapie, de chimiothérapie et d'hormonothérapie.
Le chromosome X humain est l'un des deux chromosomes sexuels chez les humains, l'autre étant le chromosome Y. Les humains ont normalement 46 chromosomes répartis en 23 paires, dont une paire de chromosomes sexuels. La plupart des femmes ont deux chromosomes X (XX), et la plupart des hommes ont un chromosome X et un chromosome Y (XY).
Le chromosome X est beaucoup plus grand que le chromosome Y et contient environ 155 millions de paires de bases, ce qui représente environ 5% du génome humain. Il code pour environ 1 098 protéines. Le chromosome X contient un certain nombre de gènes importants liés à la fonction cérébrale, à l'immunité et aux maladies génétiques.
Les femmes ont deux copies actives du chromosome X dans la plupart des cellules de leur corps, ce qui est appelé dosage génique. Cependant, les hommes n'ont qu'une seule copie active du chromosome X. Pour équilibrer le niveau d'expression des gènes sur le chromosome X chez les hommes, certains gènes sur le chromosome X sont soumis à une inactivation de l'X ou à la méthylation de l'ADN, ce qui entraîne leur silenciation.
Des mutations dans les gènes du chromosome X peuvent entraîner des maladies génétiques liées au sexe, telles que la dystrophie musculaire de Duchenne, le syndrome de l'X fragile et la maladie de Hunter. Les femmes qui sont porteuses d'une mutation sur un gène du chromosome X ont un risque accru de transmettre cette mutation à leurs enfants.
Un kératoacanthome est une tumeur cutanée benigne mais à croissance rapide qui se développe généralement sur les zones exposées au soleil telles que le visage, le dos des mains et les avant-bras. Elle a une apparence typique en forme de cratère avec un centre rempli de kératine, une protéine dure produite par la peau. Les bords de la tumeur sont souvent surélevés et ont une apparence en collerette. Bien que le kératoacanthome se résorbe généralement de lui-même en quelques mois, il peut néanmoins causer des cicatrices permanentes. Dans certains cas, il peut être confondu avec un carcinome épidermoïde malin, une forme agressive de cancer de la peau, ce qui nécessite une intervention chirurgicale pour un diagnostic et un traitement appropriés.
Un caryotype anormal, ou anomalie du caryotype, fait référence à une composition chromosomique anormale dans les cellules d'un individu. Cela peut inclure des changements dans le nombre de chromosomes (tels que la trisomie 21 ou syndrome de Down), des réarrangements structurels (tels que les délétions, duplications, inversions et translocations) ou d'autres anomalies chromosomiques. Ces variations peuvent entraîner une variété de problèmes de santé, selon la nature et l'ampleur de l'anomalie.
Les caryotypes anormaux sont généralement détectés par analyse du caryotype, qui implique l'observation des chromosomes d'un individu sous un microscope après avoir été colorés et disposés dans une configuration standardisée. Cette technique permet aux médecins et aux généticiens de visualiser la composition chromosomique d'un individu et de détecter toute anomalie qui pourrait être présente.
Les caryotypes anormaux peuvent être causés par des erreurs qui se produisent pendant la division cellulaire, telles que la non-disjonction, où les paires de chromosomes ne se séparent pas correctement pendant la méiose. Ils peuvent également résulter d'expositions environnementales nocives ou de mutations génétiques héréditaires. Dans certains cas, les caryotypes anormaux peuvent être associés à des problèmes de fertilité, de développement et de santé chez l'enfant.
Il est important de noter que tous les caryotypes anormaux ne sont pas nécessairement cliniquement significatifs et ne provoquent pas toujours des problèmes de santé évidents. Certains individus avec des caryotypes anormaux peuvent être asymptomatiques et mener une vie normale, tandis que d'autres peuvent présenter des anomalies congénitales graves ou un risque accru de maladies chroniques. Dans tous les cas, il est important de consulter un professionnel de la santé pour obtenir des conseils et une évaluation appropriés si vous soupçonnez que vous ou votre enfant avez un caryotype anormal.
Les sondes d'ADN sont des courtes séquences d'acides nucléiques (généralement d'ADN, mais parfois d'ARN) qui sont conçues pour rechercher et se lier spécifiquement à une séquence complémentaire particulière dans un échantillon d'ADN. Elles sont souvent utilisées en médecine et en biologie moléculaire pour identifier la présence de certains gènes ou mutations, détecter des agents pathogènes, ou analyser l'expression génétique.
Les sondes d'ADN peuvent être marquées avec des fluorophores ou d'autres étiquettes qui permettent de les détecter et de mesurer la force de leur liaison à la cible. Il existe différents types de sondes d'ADN, tels que les sondes linéaires, les sondes chevauchantes (overhang probes) et les sondes en grille (gridded probes), qui sont utilisées dans diverses techniques d'analyse, telles que la hybridation in situ, l'hybridation Southern, l'amplification en chaîne par polymérase (PCR) en temps réel et les microréseaux à ADN.
Une famille multigénique, dans le contexte de la génétique et de la médecine moléculaire, se réfère à un groupe de gènes apparentés qui ont évolué à partir d'un ancêtre commun par duplication génique et divergence subséquente. Ces gènes partagent souvent des séquences similaires et peuvent être impliqués dans des fonctions biologiques liées. Les membres de la famille multigénique peuvent être situés à proximité les uns des autres sur un chromosome, formant ainsi un cluster de gènes, ou ils peuvent être dispersés sur différents chromosomes. La compréhension des familles multigéniques est importante pour l'étude des mécanismes d'évolution génétique et de la fonction des gènes, ainsi que pour la recherche de variantes associées à des maladies héréditaires ou complexes.
En médecine, le terme "pronostic" se réfère à la prévision du résultat ou de l'issue attendue d'une maladie ou d'une blessure dans le corps humain. Il s'agit essentiellement d'une estimation de la probabilité du rétablissement complet, de l'amélioration continue, de l'évolution vers une invalidité permanente ou du décès d'un patient atteint d'une certaine maladie ou blessure.
Le pronostic est généralement fondé sur les antécédents médicaux du patient, les résultats des tests diagnostiques, l'étendue de la maladie ou de la lésion, la réponse au traitement et d'autres facteurs pertinents. Il peut être exprimé en termes généraux ou spécifiques, tels qu'un pronostic favorable, défavorable ou incertain.
Il est important de noter que le pronostic n'est pas une garantie et ne doit pas être considéré comme tel. Il s'agit simplement d'une estimation basée sur des données probantes et l'expérience clinique, qui peut varier d'un patient à l'autre. Les médecins doivent communiquer clairement le pronostic aux patients et à leur famille, en s'assurant qu'ils comprennent les risques, les avantages et les incertitudes associés au traitement et à la maladie sous-jacente.
Une lignée cellulaire tumorale, dans le contexte de la recherche en cancérologie, fait référence à une population homogène de cellules cancéreuses qui peuvent être cultivées et se diviser en laboratoire. Ces lignées cellulaires sont généralement dérivées de biopsies ou d'autres échantillons tumoraux prélevés sur des patients, et elles sont capables de se multiplier indéfiniment en culture.
Les lignées cellulaires tumorales sont souvent utilisées dans la recherche pour étudier les propriétés biologiques des cellules cancéreuses, tester l'efficacité des traitements anticancéreux et comprendre les mécanismes de progression du cancer. Cependant, il est important de noter que ces lignées cellulaires peuvent ne pas toujours se comporter ou réagir aux traitements de la même manière que les tumeurs d'origine dans le corps humain, ce qui peut limiter leur utilité en tant que modèles pour la recherche translationnelle.
Les chromosomes artificiels de bactériophage P1 sont des vecteurs d'ADN à base de bactériophage P1 utilisés dans la recherche en génétique et en biologie moléculaire. Les bactériophages sont des virus qui infectent les bactéries, et le bactériophage P1 est un type spécifique de bactériophage qui se lie et infecte la bactérie Escherichia coli (E. coli).
Les chromosomes artificiels de bactériophage P1 sont créés en insérant des fragments d'ADN étranger dans le génome du bactériophage P1. Ces fragments d'ADN peuvent provenir d'une variété de sources, y compris d'autres bactéries, de virus, de cellules animales ou humaines, et peuvent être utilisés pour étudier la fonction et l'expression des gènes, la régulation génétique, et d'autres processus biologiques.
Les chromosomes artificiels de bactériophage P1 sont particulièrement utiles en raison de leur capacité à accueillir de longs fragments d'ADN (jusqu'à 100 kilobases) et de leur stabilité relative une fois insérés dans le génome du bactériophage. De plus, les bactériophages P1 peuvent être facilement manipulés et amplifiés en laboratoire, ce qui permet aux chercheurs de produire de grandes quantités de chromosomes artificiels pour une utilisation dans des expériences à grande échelle.
En résumé, les chromosomes artificiels de bactériophage P1 sont des vecteurs d'ADN artificiels créés en insérant des fragments d'ADN étranger dans le génome du bactériophage P1. Ils sont utilisés dans la recherche en génétique et en biologie moléculaire pour étudier divers processus biologiques, tels que la fonction et la régulation des gènes.
Les chromosomes en anneau sont un type rare d'anomalie chromosomique structurale qui se produit lorsqu'il y a une perte des extrémités (télomères) de deux bras d'un chromosome, ce qui entraîne la jonction des deux extrémités restantes pour former un anneau. Cette anomalie peut être causée par des recombinaisons inégales pendant la méiose ou par des réarrangements chromosomiques au cours du développement de l'embryon.
Les chromosomes en anneau peuvent entraîner une variété de symptômes, selon la taille de l'anneau et la quantité de matériel génétique perdu. Les conséquences les plus courantes sont des retards de développement, des anomalies physiques, des problèmes d'apprentissage et des troubles du comportement. Dans certains cas, les chromosomes en anneau peuvent également augmenter le risque de développer certaines maladies ou conditions, telles que des cancers.
Le diagnostic des chromosomes en anneau peut être posé par analyse cytogénétique, qui consiste à examiner les chromosomes sous un microscope après avoir cultivé des cellules du patient. Cette technique permet de visualiser l'anneau et d'identifier le chromosome concerné. Des techniques plus récentes, telles que la puce à ADN et le séquençage de nouvelle génération, peuvent également être utilisées pour déterminer avec précision les régions du chromosome qui sont touchées.
Le traitement des chromosomes en anneau dépend de la gravité des symptômes et peut inclure une prise en charge médicale, une thérapie physique ou occupante, une éducation spécialisée et un soutien psychologique. Dans certains cas, une intervention chirurgicale peut être nécessaire pour corriger les anomalies physiques associées à l'anomalie chromosomique.
Je suis désolé, mais il y a une certaine confusion dans votre question. Un "algorithme" n'est pas un terme médical, c'est un terme technique utilisé en informatique et en mathématiques pour décrire un ensemble fini d'instructions bien définies pour résoudre un problème particulier. Il peut être utilisé dans le développement de systèmes et d'outils médicaux, mais ce n'est pas une définition médicale en soi.
Les chromosomes sont des structures situées dans le noyau de chaque cellule de notre corps qui contiennent la majeure partie de l'ADN humain. Ils sont constitués de molécules d'ADN enroulées autour de protéines histones, formant une structure compacte et stable.
Chaque chromosome est une longue molécule d'ADN qui contient des milliers de gènes responsables de la détermination des caractéristiques héréditaires telles que la couleur des yeux, la taille et le groupe sanguin. Les humains ont 23 paires de chromosomes, ce qui signifie qu'il y a un total de 46 chromosomes dans chaque cellule du corps à l'exception des cellules reproductrices (spermatozoïdes et ovules) qui n'en contiennent que 23.
Les chromosomes sont numérotés de 1 à 22, selon leur taille décroissante, et la 23ème paire est composée d'un chromosome X et d'un chromosome Y chez les hommes, tandis que chez les femmes, il y a deux chromosomes X. Les anomalies dans le nombre ou la structure des chromosomes peuvent entraîner des maladies génétiques telles que la trisomie 21 (syndrome de Down) ou la mucoviscidose.
En résumé, les chromosomes sont des structures cruciales dans notre corps qui contiennent l'ADN et les gènes responsables de notre hérédité et de notre développement. Les anomalies chromosomiques peuvent entraîner des maladies génétiques graves.
Un modèle génétique est une représentation théorique ou mathématique d'un trait, d'une maladie ou d'une caractéristique héréditaire donnée, qui tente de décrire et d'expliquer la manière dont les gènes et l'environnement interagissent pour influencer ce trait. Il s'appuie sur des études épidémiologiques, statistiques et moléculaires pour comprendre la transmission héréditaire d'un trait particulier au sein d'une population. Les modèles génétiques peuvent aider à prédire le risque de développer une maladie, à identifier les gènes associés à un trait et à élucider les mécanismes sous-jacents des maladies complexes.
Les modèles génétiques peuvent être simples ou complexes, selon la nature du trait étudié. Dans le cas d'un trait monogénique, où une seule mutation dans un gène spécifique est suffisante pour provoquer la maladie, le modèle peut être relativement simple et basé sur les lois de Mendel. Cependant, pour les traits complexes ou quantitatifs, qui sont influencés par plusieurs gènes et l'environnement, les modèles génétiques peuvent être plus sophistiqués et prendre en compte des facteurs tels que la pénétrance incomplète, l'effet de dosage, l'épistasie et l'interaction entre gènes et environnement.
Les modèles génétiques sont largement utilisés dans la recherche médicale et la médecine prédictive pour comprendre les causes sous-jacentes des maladies et améliorer le diagnostic, le pronostic et le traitement des patients.
'Brucella ovis' est une espèce de bactérie qui appartient au genre 'Brucella'. Cette bactérie est connue pour causer une maladie chez les moutons et les chèvres, appelée brucellose ovine. La brucellose est une maladie infectieuse qui peut entraîner des avortements, une réduction de la production laitière et une baisse de la fertilité chez les animaux infectés.
Les symptômes de la brucellose ovine peuvent inclure des avortements spontanés, des naissances prématurées ou des mort-nés, ainsi qu'une diminution de la production laitière et une perte de poids chez les animaux infectés. La maladie peut également se propager aux humains qui entrent en contact avec des animaux infectés ou avec des produits contaminés, tels que le lait non pasteurisé.
La bactérie 'Brucella ovis' est généralement transmise entre les animaux par contact direct avec des sécrétions infectées, telles que l'avorton ou les liquides génitaux. Les humains peuvent être infectés en inhalant des particules contaminées ou en entrant en contact direct avec des animaux infectés ou des produits contaminés.
Le diagnostic de la brucellose ovine repose généralement sur des tests sérologiques et des cultures de sang ou de tissus. Le traitement peut inclure des antibiotiques, mais il est souvent difficile d'éradiquer complètement l'infection. Les mesures préventives comprennent la vaccination des animaux et la mise en place de contrôles stricts pour empêcher la propagation de la maladie.
La microdissection est une technique chirurgicale mini-invasive utilisée en médecine. Elle consiste à isoler et à séparer des structures ou des tissus anatomiques très petits, généralement mesurant moins d'un millimètre, à l'aide de microinstruments spéciaux et sous un fort grossissement offert par un microscope. Cette technique permet aux chirurgiens d'opérer avec une grande précision, réduisant ainsi les risques de dommages aux tissus environnants. La microdissection est souvent utilisée dans le domaine de la neurochirurgie, de l'ophtalmologie et de l'otologie pour traiter des affections telles que les tumeurs cérébrales, les décollements de rétine ou les cholestéatomes de l'oreille moyenne.
La "hybridation génétique" est un terme utilisé en génétique et en biologie pour décrire le processus de croisement entre deux individus d'espèces, de sous-espèces ou de variétés différentes, mais qui sont suffisamment étroitement liées pour être capable de se reproduire et de produire des descendants fertiles. Les descendants de cette hybridation sont appelés hybrides et héritent généralement des caractéristiques génétiques des deux parents, ce qui peut entraîner une combinaison unique de traits.
L'hybridation génétique peut se produire naturellement dans la nature lorsque différentes populations d'une même espèce se rencontrent et se croisent, ou elle peut être provoquée expérimentalement en laboratoire par des scientifiques pour étudier les effets de la combinaison de gènes de différentes souches.
L'hybridation génétique peut avoir des conséquences importantes sur l'évolution des espèces, car elle peut entraîner l'introduction de nouveaux gènes dans une population, ce qui peut conduire à une augmentation de la variabilité génétique et à une adaptation accrue aux changements environnementaux. Cependant, l'hybridation peut également entraîner une perte de diversité génétique et de biodiversité si elle conduit à la disparition de sous-espèces ou de variétés uniques.
ARN messager (ARNm) est une molécule d'acide ribonucléique simple brin qui transporte l'information génétique codée dans l'ADN vers les ribosomes, où elle dirige la synthèse des protéines. Après la transcription de l'ADN en ARNm dans le noyau cellulaire, ce dernier est transloqué dans le cytoplasme et fixé aux ribosomes. Les codons (séquences de trois nucléotides) de l'ARNm sont alors traduits en acides aminés spécifiques qui forment des chaînes polypeptidiques, qui à leur tour se replient pour former des protéines fonctionnelles. Les ARNm peuvent être régulés au niveau de la transcription, du traitement post-transcriptionnel et de la dégradation, ce qui permet une régulation fine de l'expression génique.
Dans le contexte actuel, les vaccins à ARNm contre la COVID-19 ont été développés en utilisant des morceaux d'ARNm synthétiques qui codent pour une protéine spécifique du virus SARS-CoV-2. Lorsque ces vaccins sont administrés, les cellules humaines produisent cette protéine virale étrangère, ce qui déclenche une réponse immunitaire protectrice contre l'infection par le vrai virus.
Le Southern Blot est une méthode de laboratoire utilisée en biologie moléculaire pour détecter et identifier des séquences d'ADN spécifiques dans un échantillon d'acide désoxyribonucléique (ADN). Cette technique a été nommée d'après son inventeur, le scientifique britannique Edwin Southern.
Le processus implique plusieurs étapes :
1. L'échantillon d'ADN est d'abord coupé en fragments de taille égale à l'aide d'une enzyme de restriction.
2. Ces fragments sont ensuite séparés par électrophorèse sur gel d'agarose, une méthode qui permet de les organiser selon leur longueur.
3. Le gel est ensuite transféré sur une membrane de nitrocellulose ou de nylon, créant ainsi un "blot" du patron de bandes des fragments d'ADN.
4. La membrane est alors exposée à une sonde d'ADN marquée, qui se lie spécifiquement aux séquences d'intérêt.
5. Enfin, l'emplacement des bandes sur la membrane est détecté par autoradiographie ou par d'autres méthodes de visualisation, révélant ainsi la présence et la quantité relative des séquences d'ADN cibles dans l'échantillon.
Le Southern Blot est une technique sensible et spécifique qui permet non seulement de détecter des séquences d'ADN particulières, mais aussi de distinguer des variantes subtiles telles que les mutations ponctuelles ou les polymorphismes. Il s'agit d'une méthode fondamentale en biologie moléculaire et en génétique, largement utilisée dans la recherche et le diagnostic de maladies génétiques, ainsi que dans l'analyse des gènes et des génomes.
Le formaldéhyde est un gaz incolore, à odeur forte et irritante. À température ambiante, il se présente généralement sous forme de solution aqueuse, appelée formol, qui contient environ 37% de formaldéhyde. Le formaldéhyde est un composé organique volatil (COV) qui est largement utilisé dans l'industrie pour produire des résines et des matériaux de construction, tels que les mousses isolantes et les contreplaqués. Il est également utilisé comme conservateur dans certains produits de soins personnels et comme désinfectant dans les solutions d'embaumement utilisées dans les établissements médicaux et funéraires.
Dans le corps humain, le formaldéhyde peut être produit naturellement par des processus métaboliques, tels que la décomposition des acides aminés. Cependant, une exposition excessive à cette substance peut être nocive pour la santé. L'inhalation de formaldéhyde peut provoquer des irritations des yeux, du nez et de la gorge, ainsi que des symptômes respiratoires tels que la toux et l'essoufflement. Une exposition prolongée ou à fortes doses peut entraîner des problèmes de santé plus graves, notamment un risque accru de cancer du nasopharynx et de leucémie. Par conséquent, les réglementations gouvernementales limitent généralement l'exposition professionnelle au formaldéhyde à des niveaux inférieurs à 0,75 partie par million (ppm) dans l'air.
Le test génétique est un type d'examen diagnostique qui consiste à analyser les gènes d'un individu dans le but d'identifier des modifications ou des variations dans l'ADN qui peuvent être associées à un risque accru de développer une maladie héréditaire ou d'autres conditions médicales. Les tests génétiques peuvent également être utilisés pour déterminer la susceptibilité d'un individu à répondre à certains traitements médicaux ou pour établir des relations familiales.
Les tests génétiques peuvent impliquer l'analyse de l'ADN, de l'ARN ou des protéines, et peuvent être effectués sur des échantillons de sang, de salive, de cheveux ou d'autres tissus. Les résultats du test génétique peuvent aider les médecins à poser un diagnostic, à prévoir le risque de développer une maladie à l'avenir, à déterminer le meilleur traitement pour une maladie existante ou à fournir des conseils en matière de planification familiale.
Il est important de noter que les tests génétiques peuvent avoir des implications émotionnelles et sociales importantes pour les individus et leur famille, et qu'il est donc essentiel de recevoir un counseling génétique avant et après le test pour comprendre pleinement les résultats et les conséquences potentielles.
En médecine, un fixateur est un appareil utilisé pour maintenir un os ou un tissu dans une position spécifique pendant la guérison. Il aide à stabiliser les fractures complexes, les lésions des tissus mous et les déformations osseuses sévères. Les fixateurs peuvent être externes, où des broches sont insérées dans l'os et reliées à une structure externe, ou internes, où des plaques et des vis sont utilisées pour maintenir l'os en place à l'intérieur du corps. Ils permettent aux os de cicatriser correctement tout en permettant aux médecins d'ajuster la position des fragments osseux si nécessaire.
Un marqueur génétique est un segment spécifique de l'ADN qui est variable d'une personne à l'autre. Il peut être utilisé pour identifier des individus ou des groupes d'individus partageant des caractéristiques particulières, comme une prédisposition à certaines maladies. Les marqueurs génétiques ne causent pas directement la maladie, mais ils peuvent indiquer une région du génome où un gène associé à la maladie peut être situé.
Les marqueurs génétiques sont souvent utilisés dans la recherche médicale et la médecine prédictive pour aider à diagnostiquer des conditions héréditaires, prédire le risque de développer certaines maladies, suivre l'évolution d'une maladie ou déterminer la réponse à un traitement spécifique. Ils peuvent également être utiles dans les enquêtes médico-légales pour identifier des victimes ou des auteurs de crimes.
Les marqueurs génétiques peuvent prendre différentes formes, telles que des variations dans la longueur des séquences répétitives d'ADN (VNTR), des polymorphismes nucléotidiques simples (SNP) ou des insertions/délétions de quelques paires de bases.
L'analyse des mutations de l'ADN est une méthode d'examen génétique qui consiste à rechercher des modifications (mutations) dans la séquence de l'acide désoxyribonucléique (ADN). L'ADN est le matériel génétique présent dans les cellules de tous les organismes vivants et contient les instructions pour le développement, la fonction et la reproduction des organismes.
Les mutations peuvent survenir spontanément ou être héritées des parents d'un individu. Elles peuvent entraîner des changements dans la structure et la fonction des protéines, ce qui peut à son tour entraîner une variété de conséquences, allant de mineures à graves.
L'analyse des mutations de l'ADN est utilisée dans un large éventail d'applications, y compris le diagnostic et le suivi des maladies génétiques, la détermination de la susceptibilité à certaines maladies, l'identification des auteurs de crimes, la recherche sur les maladies et le développement de médicaments.
Il existe différentes méthodes pour analyser les mutations de l'ADN, notamment la séquençage de nouvelle génération (NGS), la PCR en temps réel, la PCR quantitative et la Southern blotting. Le choix de la méthode dépend du type de mutation recherchée, de la complexité du test et des besoins du patient ou du chercheur.
La réaction de polymérisation en chaîne par transcriptase inverse (RT-PCR en anglais) est une méthode de laboratoire utilisée pour amplifier des fragments d'ARN spécifiques. Cette technique combine deux processus distincts : la transcription inverse, qui convertit l'ARN en ADN complémentaire (ADNc), et la polymérisation en chaîne, qui permet de copier rapidement et de manière exponentielle des millions de copies d'un fragment d'ADN spécifique.
La réaction commence par la transcription inverse, où une enzyme appelée transcriptase inverse utilise un brin d'ARN comme matrice pour synthétiser un brin complémentaire d'ADNc. Ce processus est suivi de la polymérisation en chaîne, où une autre enzyme, la Taq polymérase, copie le brin d'ADNc pour produire des millions de copies du fragment d'ADN souhaité.
La RT-PCR est largement utilisée dans la recherche médicale et clinique pour détecter et quantifier l'expression génétique, diagnostiquer les maladies infectieuses, détecter les mutations génétiques et effectuer des analyses de génome. Elle est également utilisée dans les tests de diagnostic COVID-19 pour détecter le virus SARS-CoV-2.
La reproductibilité des résultats, également connue sous le nom de réplicabilité, est un principe fondamental en recherche médicale qui décrit la capacité d'un résultat expérimental ou d'une observation à être reproduit ou répliqué lorsqu'un même protocole ou une méthodologie similaire est utilisée par différents chercheurs ou dans différents échantillons.
En d'autres termes, la reproductibilité des résultats signifie que si une étude est menée à plusieurs reprises en suivant les mêmes procédures et méthodes, on devrait obtenir des résultats similaires ou identiques. Cette capacité à reproduire des résultats est importante pour établir la validité et la fiabilité d'une recherche médicale, car elle aide à éliminer les biais potentiels, les erreurs aléatoires et les facteurs de confusion qui peuvent affecter les résultats.
La reproductibilité des résultats est particulièrement importante en médecine, où les décisions de traitement peuvent avoir un impact important sur la santé et le bien-être des patients. Les études médicales doivent être conçues et menées de manière à minimiser les sources potentielles d'erreur et à maximiser la reproductibilité des résultats, ce qui peut inclure l'utilisation de protocoles standardisés, la randomisation des participants, le double aveugle et l'analyse statistique appropriée.
Cependant, il est important de noter que même avec les meilleures pratiques de recherche, certains résultats peuvent ne pas être reproductibles en raison de facteurs imprévus ou inconnus. Dans ces cas, les chercheurs doivent travailler ensemble pour comprendre les raisons de l'absence de reproductibilité et pour trouver des moyens d'améliorer la conception et la méthodologie des études futures.
La bioinformatique est une discipline interdisciplinaire qui combine les méthodes et les théories des informatiques, des mathématiques et des sciences de la vie pour analyser et interpréter les données biologiques, en particulier les données génomiques. Elle implique l'utilisation d'algorithmes, de modèles statistiques et d'outils logiciels pour comprendre et organiser les informations biologiques à grande échelle. Les domaines d'application comprennent la découverte de gènes, la génomique comparative, l'analyse des réseaux de régulation génique, la protéomique, la modélisation structurale et fonctionnelle des protéines, et la médecine personnalisée.
La bioinformatique est un domaine en pleine croissance qui aide à accélérer les découvertes scientifiques dans le domaine de la biologie moléculaire et cellulaire, ainsi qu'à améliorer la compréhension des maladies humaines et le développement de thérapies ciblées. Elle est également utilisée pour l'analyse des données de séquençage à haut débit, telles que les données du génome entier, de l'exome et de la transcriptomique, ainsi que pour l'intégration et l'interprétation des données issues de différentes sources expérimentales.
Le terme "léiomyosarcome" est utilisé en médecine pour désigner un type rare de cancer qui se développe dans les muscles lisses, qui sont les muscles involontaires trouvés dans les parois des vaisseaux sanguins, du tube digestif, de l'utérus et d'autres organes.
Les leiomyosarcomes peuvent survenir n'importe où dans le corps, mais ils sont le plus souvent trouvés dans le muscle lisse de l'abdomen ou du bassin. Ces tumeurs peuvent être très agressives et ont tendance à se développer rapidement.
Les symptômes du leiomyosarcome dépendent de la localisation de la tumeur, mais peuvent inclure des douleurs abdominales ou pelviennes, une sensation de masse dans l'abdomen, des saignements anormaux et une perte de poids inexpliquée.
Le diagnostic d'un leiomyosarcome est généralement posé après une biopsie de la tumeur suspecte, suivie d'une analyse histopathologique pour confirmer le type de cancer. Le traitement dépend du stade et de l'emplacement de la tumeur, mais peut inclure une chirurgie pour enlever la tumeur, une radiothérapie ou une chimiothérapie pour aider à détruire les cellules cancéreuses restantes.
L'immunohistochimie est une technique de laboratoire utilisée en anatomopathologie pour localiser les protéines spécifiques dans des tissus prélevés sur un patient. Elle combine l'utilisation d'anticorps marqués, généralement avec un marqueur fluorescent ou chromogène, et de techniques histologiques standard.
Cette méthode permet non seulement de déterminer la présence ou l'absence d'une protéine donnée dans une cellule spécifique, mais aussi de déterminer sa localisation précise à l'intérieur de cette cellule (noyau, cytoplasme, membrane). Elle est particulièrement utile dans le diagnostic et la caractérisation des tumeurs cancéreuses, en permettant d'identifier certaines protéines qui peuvent indiquer le type de cancer, son stade, ou sa réponse à un traitement spécifique.
Par exemple, l'immunohistochimie peut être utilisée pour distinguer entre différents types de cancers du sein en recherchant des marqueurs spécifiques tels que les récepteurs d'œstrogènes (ER), de progestérone (PR) et HER2/neu.
La trisomie est un trouble génétique qui se produit lorsqu'un individu a trois instances d'un chromosome particulier au lieu des deux normales. Cela se produit généralement en raison d'une erreur de division cellulaire pendant la formation des ovules ou des spermatozoïdes, ou très tôt dans le développement du fœtus.
La forme la plus courante de trisomie est la trisomie 21, également connue sous le nom de syndrome de Down. Les personnes atteintes de trisomie 21 ont généralement un retard mental et physique, des caractéristiques faciales distinctives et une augmentation du risque de certains problèmes de santé.
D'autres types courants de trisomie comprennent la trisomie 18 (syndrome d'Edwards) et la trisomie 13 (syndrome de Patau), qui sont associées à des anomalies graves et souvent fatales.
La trisomie peut également être causée par une translocation, où une partie du chromosome se détache pendant la division cellulaire et s'attache à un autre chromosome, entraînant une copie supplémentaire de ce matériel génétique.
Dans l'ensemble, la trisomie est un trouble génétique complexe qui peut entraîner une variété de symptômes et de complications de santé, selon le chromosome impliqué et le degré de surcharge génétique.
Un réarrangement de gène, également connu sous le nom de translocation chromosomique, est un type d'anomalie génétique qui se produit lorsqu'un segment d'un chromosome se brise et se rejoint à un autre chromosome, entraînant un réarrangement des gènes. Ce phénomène peut entraîner une activation ou une inactivation anormale de certains gènes, ce qui peut conduire au développement de certaines maladies génétiques telles que la leucémie et d'autres cancers. Les réarrangements de gènes peuvent être héréditaires ou acquises au cours de la vie en raison de l'exposition à des agents mutagènes tels que les radiations et certains produits chimiques. Ils peuvent également se produire lors de la division cellulaire normale, en particulier pendant la méiose, qui est le processus de formation des gamètes (spermatozoïdes et ovules). Les réarrangements de gènes peuvent être détectés par diverses techniques de laboratoire telles que la cytogénétique et les tests moléculaires.
Les chromosomes humains de la paire 19, également connus sous le nom de chromosomes 19, sont l'une des 23 paires de chromosomes présentes dans les cellules humaines. Chaque personne hérite d'une copie de chaque chromosome de chaque parent, ce qui signifie que nous avons deux copies du chromosome 19 en tout.
Le chromosome 19 est l'un des plus grands chromosomes humains et contient un grand nombre de gènes, estimés à environ 1 400. Il code pour une variété de protéines et de molécules impliquées dans divers processus biologiques, tels que le métabolisme, la réponse immunitaire, la fonction nerveuse et la croissance cellulaire.
Des variations dans les gènes situés sur le chromosome 19 ont été associées à un certain nombre de maladies génétiques, notamment la maladie d'Alzheimer, la thrombose veineuse profonde, la sclérose latérale amyotrophique et la surdité neurosensorielle. Les chercheurs continuent d'étudier le chromosome 19 pour comprendre son rôle dans la santé humaine et les maladies.
La maladie de Hodgkin à grandes cellules B, diffuse (LBDC) est un type de lymphome, qui est un cancer du système lymphatique. Le lymphome se développe lorsque les lymphocytes, un type de globule blanc, se divisent et se multiplient de manière incontrôlable. Dans le cas du LBDC, il s'agit spécifiquement de cellules B anormales qui se multiplient de manière agressive dans les ganglions lymphatiques, la moelle osseuse, le sang et d'autres tissus corporels. Le terme "diffus" décrit la façon dont ces cellules cancéreuses se propagent dans les tissus affectés.
Le LBDC est généralement diagnostiqué par une biopsie d'un ganglion lymphatique enflé ou d'autres tissus suspects, suivie d'examens complémentaires pour évaluer l'étendue de la maladie. Le traitement dépend du stade et de l'emplacement de la maladie, ainsi que de l'âge et de l'état général de santé du patient. Les options de traitement peuvent inclure une chimiothérapie, une radiothérapie, une greffe de cellules souches ou une combinaison de ces thérapies.
Il est important de noter que le LBDC peut être une maladie grave et agressive, mais avec un diagnostic et un traitement précoces, les taux de réussite sont généralement bons. Les patients doivent travailler en étroite collaboration avec leur équipe de soins de santé pour élaborer un plan de traitement personnalisé et obtenir des soins de suivi réguliers pour surveiller la maladie et gérer les effets secondaires du traitement.
Le terme 'faciès' est utilisé en médecine pour décrire l'apparence générale ou les caractéristiques distinctives du visage d'une personne. Il s'agit essentiellement d'un terme descriptif qui permet de reconnaître certaines conditions médicales ou syndromes en observant les traits faciaux spécifiques. Par exemple, le faciès acromégale décrit un visage élargi et bouffi associé à une production excessive d'hormone de croissance, tandis que le faciès de Down réfère aux caractéristiques faciales distinctives observées chez les personnes atteintes du syndrome de Down. Il est important de noter que l'utilisation du terme 'faciès' ne doit pas être stigmatisante ou péjorative, mais plutôt servir comme un outil descriptif et diagnostic dans le domaine médical.
Un gène suppresseur de tumeur, également connu sous le nom de gène oncosuppresseur, est un type de gène qui code des protéines responsables de la régulation négative de la croissance cellulaire et de la division. Ces gènes jouent un rôle crucial dans la prévention de la transformation maligne des cellules et aident à maintenir l'homéostasie cellulaire en réprimant la prolifération cellulaire excessive et en favorisant l'apoptose (mort cellulaire programmée) lorsque les cellules présentent des anomalies ou des dommages.
Les mutations ou altérations de ces gènes suppresseurs de tumeur peuvent entraîner une diminution ou une perte de leur activité, ce qui peut conduire à une augmentation de la division cellulaire incontrôlée et, finalement, à la formation de tumeurs malignes. Les exemples bien connus de gènes suppresseurs de tumeur comprennent le gène TP53 (alias p53), qui est le plus souvent muté dans les cancers humains, ainsi que d'autres gènes tels que RB1, BRCA1 et BRCA2.
Il est important de noter que la plupart des cancers sont causés par l'accumulation de plusieurs altérations génétiques, y compris des mutations dans les gènes suppresseurs de tumeur, des mutations activatrices dans les gènes proto-oncogènes et des modifications épigénétiques.
Le diagnostic prénatal (DPN) est un ensemble de techniques médicales utilisées pour déterminer la présence ou l'absence de maladies, d'anomalies congénitales ou de conditions génétiques dans le fœtus avant sa naissance. Il s'agit d'un processus invasif ou non invasif qui permet aux médecins d'identifier précocement les problèmes de santé potentiels du fœtus, offrant ainsi la possibilité aux parents et aux prestataires de soins de prendre des décisions éclairées sur la poursuite ou non de la grossesse, la planification des soins prénatals et postnatals, et la préparation à l'accueil d'un enfant malade.
Les méthodes de diagnostic prénatal comprennent les échographies, les analyses sanguines maternelles, les tests invasifs tels que l'amniocentèse et la biopsie des villosités choriales, ainsi que les examens génétiques préimplantatoires dans le cadre d'une fécondation in vitro. Ces procédures permettent de rechercher des anomalies chromosomiques, des mutations génétiques spécifiques et d'autres problèmes structurels ou fonctionnels du fœtus.
Il est important de noter que le diagnostic prénatal soulève des questions éthiques complexes et controversées concernant l'eugénisme, la sélection des embryons et les droits des personnes handicapées. Les professionnels de santé doivent donc veiller à fournir des informations complètes, impartiales et sensibles sur les avantages, les risques et les implications potentielles des tests de diagnostic prénatal, afin que les parents puissent prendre une décision éclairée et en accord avec leurs valeurs personnelles et leur contexte culturel.
Le génotype, dans le contexte de la génétique et de la médecine, se réfère à l'ensemble complet des gènes héréditaires d'un individu, y compris toutes les variations alléliques (formes alternatives d'un gène) qu'il a héritées de ses parents. Il s'agit essentiellement de la constitution génétique innée d'un organisme, qui détermine en grande partie ses caractéristiques et prédispositions biologiques.
Les différences génotypiques peuvent expliquer pourquoi certaines personnes sont plus susceptibles à certaines maladies ou répondent différemment aux traitements médicaux. Par exemple, dans le cas de la mucoviscidose, une maladie génétique potentiellement mortelle, les patients ont généralement un génotype particulier : deux copies du gène CFTR muté.
Il est important de noter que le génotype ne définit pas entièrement les caractéristiques d'un individu ; l'expression des gènes peut être influencée par divers facteurs environnementaux et épigénétiques, ce qui donne lieu à une grande variabilité phénotypique (manifestations observables des traits) même entre les personnes partageant le même génotype.
Le histiocytose fibreuse bénigne (HFB) est une affection cutanée rare et généralement bénigne caractérisée par la prolifération anormale de histiocytes, qui sont des cellules du système immunitaire qui aident à combattre l'infection. Ces histiocytes produisent une accumulation anormale de tissu cicatriciel dans la peau, formant une masse ou une papule ferme et souvent indolore.
Les lésions d'HFB sont généralement solitaires, mais peuvent parfois être multiples. Ils se produisent le plus souvent sur les membres inférieurs, en particulier autour des chevilles et des pieds, bien qu'ils puissent apparaître n'importe où sur la peau. Les lésions ont tendance à se développer lentement au fil du temps et peuvent atteindre jusqu'à plusieurs centimètres de diamètre.
Bien que l'HFB soit généralement bénin, il peut parfois être difficile de le distinguer d'autres affections cutanées plus graves, telles que les sarcomes ou les carcinomes. Par conséquent, une biopsie et un examen histopathologique sont souvent nécessaires pour confirmer le diagnostic.
Le traitement de l'HFB dépend généralement de la taille et de la localisation des lésions. Dans de nombreux cas, aucun traitement n'est nécessaire car les lésions peuvent rester stables ou même régresser spontanément avec le temps. Cependant, si les lésions sont grandes, ennuyeuses ou situées dans une zone esthétiquement sensible, des options de traitement telles que l'excision chirurgicale, la cryochirurgie ou le laser peuvent être envisagées.
La cartographie physique des chromosomes est une méthode de détermination de l'emplacement et de l'ordre relatif des gènes et des séquences d'ADN spécifiques sur un chromosome. Cette technique utilise des sondes d'ADN marquées pour identifier et localiser des séquences spécifiques sur un chromosome.
Le processus implique généralement la fragmentation de l'ADN chromosomique en morceaux plus petits, qui sont ensuite triés par taille et hybridés avec des sondes d'ADN marquées. Les modèles de signal de fluorescence résultants sont utilisés pour construire une carte physique du chromosome, indiquant l'emplacement relatif des différentes séquences d'ADN.
La cartographie physique est un outil important en génétique et en biologie moléculaire, car elle permet de comprendre la structure et l'organisation des gènes sur un chromosome, ce qui peut aider à identifier les mutations et les variations associées aux maladies. Elle est également utilisée dans la recherche sur le génome humain et dans la médecine génomique pour étudier les variations du génome humain et leur impact sur la santé.
L'alignement des séquences en génétique et en bioinformatique est un processus permettant d'identifier et d'afficher les similitudes entre deux ou plusieurs séquences biologiques, telles que l'ADN, l'ARN ou les protéines. Cette méthode consiste à aligner les séquences de nucléotides ou d'acides aminés de manière à mettre en évidence les régions similaires et les correspondances entre elles.
L'alignement des séquences peut être utilisé pour diverses applications, telles que l'identification des gènes et des fonctions protéiques, la détection de mutations et de variations génétiques, la phylogénie moléculaire et l'analyse évolutive.
Il existe deux types d'alignement de séquences : l'alignement global et l'alignement local. L'alignement global compare l'intégralité des séquences et est utilisé pour aligner des séquences complètes, tandis que l'alignement local ne compare qu'une partie des séquences et est utilisé pour identifier les régions similaires entre des séquences partiellement homologues.
Les algorithmes d'alignement de séquences utilisent des matrices de score pour évaluer la similarité entre les nucléotides ou les acides aminés correspondants, en attribuant des scores plus élevés aux paires de résidus similaires et des scores plus faibles ou négatifs aux paires dissemblables. Les algorithmes peuvent également inclure des pénalités pour les écarts entre les séquences, tels que les insertions et les délétions.
Les méthodes d'alignement de séquences comprennent la méthode de Needleman-Wunsch pour l'alignement global et la méthode de Smith-Waterman pour l'alignement local, ainsi que des algorithmes plus rapides tels que BLAST (Basic Local Alignment Search Tool) et FASTA.
Le carcinome in situ est une forme précoce et localisée de cancer. Il s'agit d'une prolifération anormale et non invasive de cellules cancéreuses dans un tissu ou une glande, généralement la peau ou les muqueuses. Ces cellules cancéreuses se multiplient et peuvent se développer en amas (appelés « nids ») dans le tissu affecté, mais elles ne traversent pas la membrane basale qui sépare ce tissu des structures sous-jacentes saines.
En d'autres termes, les cellules cancéreuses restent confinées à l'endroit où elles se sont développées et n'ont pas encore acquis la capacité à envahir les tissus voisins ou à se propager (métastaser) vers d'autres parties du corps.
Le carcinome in situ est souvent classé comme un stade précoce de cancer, bien qu'il puisse évoluer en une forme invasive et plus agressive s'il n'est pas traité. Il est important de diagnostiquer et de traiter le carcinome in situ à un stade précoce pour empêcher sa progression vers des stades plus avancés et réduire le risque de récidive après le traitement.
Les types courants de carcinomes in situ comprennent le carcinome basocellulaire superficiel, le carcinome squameux in situ (également connu sous le nom d'érythroplasie de Queyrat ou de kératose actinique in situ), et le carcinome du sein in situ (DCIS).
Un oncogène est un gène qui, lorsqu'il est muté ou surexprimé, peut contribuer au développement du cancer. Dans des conditions normales, ces gènes jouent des rôles importants dans la régulation de la croissance cellulaire, la différenciation et l'apoptose (mort cellulaire programmée).
Cependant, lorsqu'ils sont altérés, ils peuvent entraîner une prolifération cellulaire incontrôlable et d'autres caractéristiques typiques des cellules cancéreuses. Les oncogènes peuvent provenir de mutations spontanées, être hérités ou être causés par des facteurs environnementaux tels que l'exposition aux radiations ou aux produits chimiques toxiques.
Certains oncogènes sont associés à des types spécifiques de cancer, tandis que d'autres peuvent être liés à plusieurs types de cancer. L'étude des oncogènes et de leur rôle dans la cancérogenèse aide au développement de thérapies ciblées pour le traitement du cancer.
Les Troubles du Développement (TTD) sont des perturbations dans l'acquisition ou l'utilisation des compétences psychologiques, physiques, et/ou langagières qui se manifestent pendant la période de développement de l'enfant, allant de la naissance à l'âge adulte jeune. Ces troubles peuvent affecter plusieurs domaines du développement, tels que la communication, les interactions sociales, le raisonnement, le comportement, l'apprentissage, et l'adaptation à l'environnement. Les TTD comprennent un large éventail de conditions, y compris les troubles du spectre autistique, les troubles déficitaires de l'attention/hyperactivité (TDAH), les troubles spécifiques des apprentissages, et les troubles moteurs ou langagiers. Les causes sous-jacentes peuvent être génétiques, environnementales, ou liées à des facteurs combinés. Le diagnostic et la prise en charge précoces sont cruciaux pour améliorer le pronostic et favoriser une meilleure adaptation de l'enfant dans son milieu familial, scolaire et social.
Un syndrome, dans le contexte médical, est un ensemble de symptômes ou de signes cliniques qui, considérés dans leur globalité, suggèrent l'existence d'une pathologie spécifique ou d'un état anormal dans le fonctionnement de l'organisme. Il s'agit essentiellement d'un ensemble de manifestations cliniques qui sont associées à une cause sous-jacente commune, qu'elle soit connue ou inconnue.
Un syndrome n'est pas une maladie en soi, mais plutôt un regroupement de signes et symptômes qui peuvent être liés à différentes affections médicales. Par exemple, le syndrome métabolique est un ensemble de facteurs de risque qui augmentent la probabilité de développer des maladies cardiovasculaires et du diabète de type 2. Ces facteurs comprennent l'obésité abdominale, l'hypertension artérielle, l'hyperglycémie à jeun et les taux élevés de triglycérides et de faibles taux de HDL-cholestérol.
La définition d'un syndrome peut évoluer avec le temps, alors que la compréhension des mécanismes sous-jacents s'améliore grâce aux recherches médicales et scientifiques. Certains syndromes peuvent être nommés d'après les professionnels de la santé qui ont contribué à leur identification ou à leur description, comme le syndrome de Down (trisomie 21) ou le syndrome de Klinefelter (XXY).
Il est important de noter que la présence d'un syndrome ne permet pas toujours d'établir un diagnostic définitif, car plusieurs affections médicales peuvent partager des symptômes similaires. Cependant, l'identification d'un syndrome peut aider les professionnels de la santé à orienter le diagnostic et le traitement vers des causes probables ou à fournir des informations sur le pronostic et la prise en charge globale du patient.
Un ependymome est un type rare de tumeur cérébrale qui se développe à partir des cellules ependymaires tapissant les ventricules cérébraux et le canal central de la moelle épinière. Ces tumeurs peuvent survenir à tout âge, mais elles sont plus fréquentes chez les enfants et les jeunes adultes. Les symptômes dépendent de la localisation de la tumeur et peuvent inclure des maux de tête, des nausées, des vomissements, des troubles de l'équilibre et de la coordination, une faiblesse musculaire, des changements de vision et des crises d'épilepsie. Le traitement dépend du grade et de la localisation de la tumeur et peut inclure une chirurgie pour enlever autant de la tumeur que possible, suivie de radiothérapie ou de chimiothérapie.
Je suis désolé, mais il semble y avoir une certaine confusion dans votre question. Un "logiciel" ne fait pas partie des définitions médicales traditionnelles. Le terme "logiciel" est d'origine informatique et désigne les programmes, instructions et données qui sont exécutées par un ordinateur pour effectuer une tâche spécifique. Cependant, dans le contexte de la médecine numérique ou de la santé connectée, le logiciel peut faire référence aux applications ou systèmes informatiques utilisés dans les soins de santé, tels que les dossiers médicaux électroniques, les systèmes de gestion des rendez-vous et les outils d'analyse des données.
Les tumeurs de l'uvée se réfèrent à des croissances anormales dans la région de l'œil appelée uvée, qui comprend l'iris, le corps ciliaire et la choroïde. Ces tumeurs peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs bénignes courantes de l'uvée comprennent les néviomes et les adénomes, tandis que les tumeurs malignes comprennent le mélanome uvéal et la métastase oculaire.
Les symptômes des tumeurs de l'uvée peuvent inclure une vision floue ou déformée, des taches sombres dans le champ visuel, des changements de couleur de l'iris, une douleur oculaire ou un gonflement de l'œil. Le diagnostic est généralement posé par un examen ophtalmologique complet comprenant une dilatation pupillaire et éventuellement des tests d'imagerie tels que l'échographie ou l'angiographie.
Le traitement dépend du type, de la taille et de l'emplacement de la tumeur. Les petites tumeurs bénignes peuvent ne nécessiter aucun traitement, tandis que les tumeurs plus grandes ou malignes peuvent être traitées par une chirurgie, une radiothérapie, une thérapie photodynamique ou une ablation au laser. Il est important de noter que les tumeurs malignes de l'uvée peuvent se propager à d'autres parties du corps et nécessitent donc un traitement agressif pour prévenir la propagation et améliorer les chances de guérison.
Les gènes bactériens sont des segments d'ADN dans le génome d'une bactérie qui portent l'information génétique nécessaire à la synthèse des protéines et à d'autres fonctions cellulaires essentielles. Ils contrôlent des caractéristiques spécifiques telles que la croissance, la reproduction, la résistance aux antibiotiques et la production de toxines. Chaque gène a un code spécifique qui détermine la séquence d'acides aminés dans une protéine particulière. Les gènes bactériens peuvent être étudiés pour comprendre les mécanismes de la maladie, développer des thérapies et des vaccins, et améliorer les processus industriels tels que la production de médicaments et d'aliments.
Les sondes oligonucléotides sont des courtes séquences d'acides nucléiques simples ou modifiés, généralement constituées de 15 à 30 nucléotides, qui sont utilisées pour détecter ou cibler spécifiquement des séquences complémentaires particulières dans l'ADN ou l'ARN. Elles sont souvent utilisées en biologie moléculaire et en génie génétique pour diverses applications, telles que la détection de gènes spécifiques, l'hybridation in situ, l'amplification génique (comme dans la réaction en chaîne par polymérase ou PCR), la transcription inverse et l'édition de gènes. Les sondes oligonucléotides peuvent être marquées avec des fluorophores, des biotines ou d'autres étiquettes pour faciliter leur détection et leur quantification.
Un adénocarcinome est un type de cancer qui se développe dans les cellules glandulaires. Ces cellules sont présentes dans de nombreux tissus et organes du corps, et elles produisent des substances telles que des mucus ou des hormones.
Les adénocarcinomes peuvent survenir dans divers endroits, notamment les poumons, le sein, le côlon, le rectum, l'estomac, la prostate et le pancréas. Ils se développent à partir d'une tumeur bénigne appelée adénome, qui peut devenir cancéreuse au fil du temps.
Les symptômes de l'adénocarcinome dépendent de son emplacement dans le corps. Par exemple, un adénocarcinome du sein peut provoquer une masse ou une grosseur palpable, tandis qu'un adénocarcinome du poumon peut causer une toux persistante, des douleurs thoraciques et des expectorations sanglantes.
Le traitement de l'adénocarcinome dépend également de son emplacement et de son stade. Les options de traitement peuvent inclure la chirurgie, la radiothérapie, la chimiothérapie ou une combinaison de ces traitements. Le pronostic varie en fonction du type et du stade du cancer, ainsi que de facteurs tels que l'âge et l'état de santé général du patient.
Les bases de données génétiques sont des systèmes de gestion et d'organisation de l'information génétique et moléculaire sur les gènes, les mutations, les variations génétiques, les maladies héréditaires, les populations et les familles. Elles peuvent contenir des informations telles que des séquences d'ADN, des cartes génétiques, des données cliniques, des antécédents familiaux, des résultats de tests génétiques et des renseignements sur la recherche en génétique.
Les bases de données génétiques peuvent être utilisées à des fins diverses, telles que la recherche scientifique, le diagnostic clinique, la médecine personnalisée, la médecine prédictive et la médecine préventive. Elles sont également utiles pour l'identification de gènes associés à des maladies héréditaires, la compréhension des mécanismes moléculaires sous-jacents aux maladies et le développement de thérapies ciblées.
Les bases de données génétiques peuvent être publiques ou privées, accessibles à tous ou restreintes à un usage spécifique. Il est important de noter que l'utilisation de ces bases de données doit respecter les lois et règlements en vigueur en matière de confidentialité et d'éthique, ainsi que les normes et directives édictées par les organismes professionnels et les autorités réglementaires.
Les chromosomes humains de la paire 10, également connus sous le nom de chromosomes 10, sont une partie importante du matériel génétique d'un être humain. Chaque personne a deux chromosomes 10, une héritée de chaque parent, ce qui signifie qu'il y a 46 chromosomes au total dans le noyau de chaque cellule humaine (à l'exception des cellules reproductives, qui ne contiennent que 23 chromosomes).
Les chromosomes 10 sont des structures en forme de bâtonnet composées de molécules d'ADN et de protéines appelées histones. Ils portent des milliers de gènes, qui sont des segments d'ADN qui contiennent les instructions pour la production de protéines spécifiques.
Les chromosomes 10 jouent un rôle crucial dans le développement et le fonctionnement normaux du corps humain. Des mutations dans les gènes situés sur ces chromosomes peuvent entraîner des maladies génétiques telles que la sclérose tubéreuse de Bourneville, une maladie neurologique rare caractérisée par l'apparition de tumeurs bénignes dans le cerveau et d'autres organes.
Les chercheurs continuent à étudier les chromosomes 10 et les gènes qui y sont contenus pour en apprendre davantage sur leur rôle dans la santé humaine et les maladies, ainsi que sur les moyens de prévenir ou de traiter ces maladies.
Une séquence d'acides aminés est une liste ordonnée d'acides aminés qui forment une chaîne polypeptidique dans une protéine. Chaque protéine a sa propre séquence unique d'acides aminés, qui est déterminée par la séquence de nucléotides dans l'ADN qui code pour cette protéine. La séquence des acides aminés est cruciale pour la structure et la fonction d'une protéine. Les différences dans les séquences d'acides aminés peuvent entraîner des différences importantes dans les propriétés de deux protéines, telles que leur activité enzymatique, leur stabilité thermique ou leur interaction avec d'autres molécules. La détermination de la séquence d'acides aminés d'une protéine est une étape clé dans l'étude de sa structure et de sa fonction.
Les tumeurs de la peau sont des croissances anormales qui se forment dans les tissus cutanés. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs bénignes ne mettent généralement pas la vie en danger et ne se propagent pas à d'autres parties du corps, mais elles peuvent parfois causer des problèmes esthétiques ou fonctionnels. Les tumeurs malignes, en revanche, peuvent envahir les tissus environnants et se propager (métastases) à d'autres parties du corps.
Les types courants de tumeurs bénignes de la peau comprennent les naevus (grains de beauté), les kystes epidermiques, les lipomes, les fibromes, et les papillomes. Les naevus melanocytaires sont les grains de beauté les plus courants et sont généralement inoffensifs, bien que certains puissent évoluer en mélanomes malins.
Les types courants de tumeurs malignes de la peau comprennent le carcinome basocellulaire, le carcinome spinocellulaire (ou carcinome épidermoïde), et le mélanome malin. Le carcinome basocellulaire est le type le plus courant de cancer de la peau et se développe généralement à partir des cellules basales de la peau. Il se propage rarement aux autres parties du corps, mais peut détruire les tissus environnants s'il n'est pas traité. Le carcinome spinocellulaire est moins courant que le carcinome basocellulaire, mais il a un potentiel de métastase plus élevé. Le mélanome malin est le type de cancer de la peau le plus agressif et peut se propager rapidement aux autres parties du corps s'il n'est pas traité à temps.
Les facteurs de risque pour les cancers de la peau comprennent une exposition excessive au soleil, des antécédents personnels ou familiaux de cancer de la peau, une peau claire, des grains de beauté anormaux, un système immunitaire affaibli, et l'utilisation de certains médicaments. Il est important de se protéger du soleil en portant des vêtements protecteurs, en utilisant un écran solaire avec un FPS d'au moins 30, en évitant les heures les plus chaudes de la journée, et en effectuant des auto-examens réguliers de la peau pour détecter tout changement anormal.
Un astrocytome est un type de tumeur cérébrale qui se développe à partir des cellules gliales appelées astrocytes, qui soutiennent et protègent les neurones dans le cerveau. Les astrocytomes peuvent être classés en fonction de leur grade, qui indique leur niveau de malignité ou d'agressivité.
Les grades vont de I à IV, où les astrocytomies de grade I sont les moins agressifs et ont tendance à se développer lentement, tandis que les astrocytomes de grade IV sont les plus agressifs et peuvent se propager rapidement dans le cerveau. Les symptômes d'un astrocytome dépendent de la localisation et de la taille de la tumeur, mais peuvent inclure des maux de tête, des convulsions, des nausées, des vomissements, une vision floue, des changements de personnalité ou de comportement, et des problèmes de coordination ou d'équilibre.
Le traitement dépend du grade et de la localisation de la tumeur, mais peut inclure une chirurgie pour enlever autant de la tumeur que possible, une radiothérapie pour aider à détruire les cellules cancéreuses restantes, et une chimiothérapie pour aider à ralentir ou arrêter la croissance de la tumeur. Dans certains cas, une thérapie ciblée ou une immunothérapie peuvent également être utilisées pour traiter l'astrocytome.
La fusion de gènes est un événement moléculaire dans lequel deux ou plusieurs gènes se combinent pour former un nouveau gène unique avec une séquence d'acides nucléiques et une fonction protéique altérées. Cela peut se produire à la suite de divers mécanismes, tels que des translocations chromosomiques, des inversions ou des insertions. Dans certaines leucémies et lymphomes, par exemple, des fusions de gènes peuvent entraîner une production accrue de protéines anormales qui favorisent la cancérogenèse et la progression tumorale. La détection de fusions de gènes spécifiques peut être utile pour le diagnostic, la stratification des risques et la planification du traitement des maladies malignes hématologiques et d'autres affections.
La variation structurale du génome (GSV) fait référence à des changements dans la structure et l'organisation des gènes ou des séquences d'ADN à grande échelle, qui peuvent affecter un seul gène ou s'étendre sur de vastes régions du génome. Ces variations comprennent des événements tels que les délétions, les duplications, les inversions et les translocations.
Les délétions sont des pertes de segments d'ADN allant de quelques centaines à plusieurs millions de paires de bases. Les duplications sont des copies supplémentaires de segments d'ADN qui peuvent varier en taille, allant de quelques centaines à des millions de paires de bases.
Les inversions sont des événements où un segment d'ADN est retourné dans le sens inverse par rapport à son orientation d'origine sur le chromosome. Les translocations se produisent lorsqu'un segment d'ADN est déplacé d'un chromosome vers un autre chromosome, entraînant une recombinaison entre les deux chromosomes.
Ces variations structurelles peuvent avoir des effets importants sur la fonction génétique et sont associées à un large éventail de phénotypes, y compris des maladies génétiques complexes telles que les troubles du développement, les maladies neurodégénératives et le cancer.
Un simple nucléotide polymorphisme (SNP) est un type courant de variation génétique chez les êtres humains. Il s'agit d'une substitution d'une seule paire de bases dans le DNA qui se produit lorsque une paire de bases du DNA est remplacée par une autre. Par exemple, une paire A-T peut être remplacée par une paire G-C. Ces variations se produisent environ une fois sur 300 paires de bases dans le génome humain et chaque personne a environ 4 à 5 millions de SNPs dans son génome.
Les SNPs peuvent se trouver dans les régions codantes (qui codent pour des protéines) ou non codantes du génome. Lorsqu'ils se produisent dans les régions codantes, ils peuvent entraîner des changements dans l'aminoacide qui est codé par ce segment de DNA, ce qui peut affecter la fonction de la protéine. Cependant, la plupart des SNPs n'ont pas d'effet sur la fonction des protéines et sont considérés comme neutres.
Les SNPs peuvent être utiles dans la recherche médicale pour identifier des susceptibilités génétiques à certaines maladies, suivre la propagation de maladies infectieuses, déterminer les réponses aux traitements médicamenteux et établir des relations entre les individus.
Les cellules cancéreuses en culture sont des cellules cancéreuses prélevées sur un être humain ou un animal, qui sont ensuite cultivées et multipliées dans un laboratoire. Ce processus est souvent utilisé pour la recherche médicale et biologique, y compris l'étude de la croissance et du comportement des cellules cancéreuses, la découverte de nouveaux traitements contre le cancer, et les tests de sécurité et d'efficacité des médicaments et des thérapies expérimentales.
Les cellules cancéreuses en culture sont généralement prélevées lors d'une biopsie ou d'une intervention chirurgicale, puis transportées dans un milieu de culture spécial qui contient les nutriments et les facteurs de croissance nécessaires à la survie et à la reproduction des cellules. Les cellules sont maintenues dans des conditions stériles et sous observation constante pour assurer leur santé et leur pureté.
Les cultures de cellules cancéreuses peuvent être utilisées seules ou en combinaison avec d'autres méthodes de recherche, telles que l'imagerie cellulaire, la génomique, la protéomique et la biologie des systèmes. Ces approches permettent aux chercheurs d'étudier les mécanismes moléculaires du cancer à un niveau granulaire, ce qui peut conduire à une meilleure compréhension de la maladie et au développement de nouveaux traitements plus efficaces.
Les isochromosomes sont des anomalies chromosomiques caractérisées par la présence d'un chromosome avec deux bras identiques, contrairement à la structure normale où chaque chromosome a un bras court (p) et un bras long (q). Cela se produit lorsque le centromère, qui est la région où les deux bras du chromosome sont reliés, se divise de manière inégale pendant la mitose ou la méiose. En conséquence, l'un des bras se perd et l'autre se duplique, entraînant un déséquilibre dans la quantité et le type de matériel génétique présent sur ce chromosome.
Les isochromosomes peuvent être formés à partir de n'importe quel chromosome, mais ils sont plus fréquemment observés sur les chromosomes 8, 12, 15 et 18. Selon le chromosome impliqué, la présence d'isochromosomes peut entraîner diverses conséquences cliniques, y compris des anomalies congénitales, des retards de développement, des malformations et une prédisposition à certains types de cancer.
Par exemple, l'isochromosome i(12p) est souvent associé au syndrome de Pallister-Killian, qui se caractérise par une dysmorphie faciale, des anomalies du système nerveux central et des membres, ainsi qu'un retard mental. De même, l'isochromosome i(17q) a été impliqué dans la pathogenèse de certaines leucémies aiguës myéloblastiques (LAM).
En résumé, les isochromosomes sont des anomalies chromosomiques qui se caractérisent par un chromosome avec deux bras identiques. Elles peuvent entraîner divers problèmes de santé en fonction du chromosome impliqué et ont été associées à certaines maladies génétiques et cancers.
En biologie cellulaire et en particulier dans la mitose ou la méiose, la métaphase est une phase du cycle cellulaire où les chromosomes, qui ont déjà répliqué leur matériel génétique et se sont condensés, sont correctement alignés au milieu de la cellule. Cela se produit après que les centrosomes aient migré vers les pôles opposés de la cellule et formé des faisceaux mitotiques (ou spindle fibers en anglais), qui attachent les chromosomes aux pôles par leurs kinétochores. Durant cette phase, il y a égal segregation des chromatides sorores vers les pôles opposés de la cellule, préparant ainsi à l'anaphase où chaque pôle recevra un ensemble complet de chromosomes.
Le clonage moléculaire est une technique de laboratoire qui permet de créer plusieurs copies identiques d'un fragment d'ADN spécifique. Cette méthode implique l'utilisation de divers outils et processus moléculaires, tels que des enzymes de restriction, des ligases, des vecteurs d'ADN (comme des plasmides ou des phages) et des hôtes cellulaires appropriés.
Le fragment d'ADN à cloner est d'abord coupé de sa source originale en utilisant des enzymes de restriction, qui reconnaissent et coupent l'ADN à des séquences spécifiques. Le vecteur d'ADN est également coupé en utilisant les mêmes enzymes de restriction pour créer des extrémités compatibles avec le fragment d'ADN cible. Les deux sont ensuite mélangés dans une réaction de ligation, où une ligase (une enzyme qui joint les extrémités de l'ADN) est utilisée pour fusionner le fragment d'ADN et le vecteur ensemble.
Le produit final de cette réaction est un nouvel ADN hybride, composé du vecteur et du fragment d'ADN cloné. Ce nouvel ADN est ensuite introduit dans un hôte cellulaire approprié (comme une bactérie ou une levure), où il peut se répliquer et produire de nombreuses copies identiques du fragment d'ADN original.
Le clonage moléculaire est largement utilisé en recherche biologique pour étudier la fonction des gènes, produire des protéines recombinantes à grande échelle, et développer des tests diagnostiques et thérapeutiques.
Les protéines tumorales, également connues sous le nom de marqueurs tumoraux, sont des substances (généralement des protéines) que l'on peut trouver en quantités anormalement élevées dans le sang, l'urine ou d'autres tissus du corps lorsqu'une personne a un cancer. Il est important de noter que ces protéines peuvent également être présentes en petites quantités chez les personnes sans cancer.
Il existe différents types de protéines tumorales, chacune étant associée à un type spécifique de cancer ou à certains stades de développement du cancer. Par exemple, la protéine tumorale PSA (antigène prostatique spécifique) est souvent liée au cancer de la prostate, tandis que l'ACE (antigène carcinoembryonnaire) peut être associé au cancer colorectal.
L'utilisation des protéines tumorales dans le diagnostic et le suivi du cancer est un domaine en évolution constante de la recherche médicale. Elles peuvent aider au dépistage précoce, à l'établissement d'un diagnostic, à la planification du traitement, à la surveillance de la réponse au traitement et à la détection des récidives. Cependant, leur utilisation doit être soigneusement évaluée en raison de leur faible spécificité et sensibilité, ce qui signifie qu'elles peuvent parfois donner des résultats faussement positifs ou négatifs. Par conséquent, les protéines tumorales sont généralement utilisées en combinaison avec d'autres tests diagnostiques et cliniques pour obtenir une image plus complète de la santé du patient.
Le carcinome canalaire du sein est une forme courante de cancer du sein qui commence dans les cellules des conduits ou canaux qui transportent le lait du sein vers l'extérieur. Dans la plupart des cas, ce type de cancer se développe d'abord dans les cellules à l'intérieur des parois des canaux milkaires. Au fur et à mesure que ces cellules cancéreuses se multiplient, elles peuvent traverser la paroi du canal et envahir le tissu mammaire environnant.
Les symptômes courants du carcinome canalaire du sein comprennent une grosseur ou un durcissement dans le sein, un écoulement anormal du mamelon (qui peut être teinté de sang), un changement de la forme ou de la taille du sein, des rougeurs ou des démangeaisons de la peau du sein, et une rétraction ou un renfoncement du mamelon.
Le traitement du carcinome canalaire du sein dépend du stade et du grade du cancer, ainsi que de facteurs tels que l'âge et l'état de santé général du patient. Les options de traitement peuvent inclure une chirurgie pour enlever la tumeur, la radiothérapie, la chimiothérapie et/ou le traitement hormonal. Dans certains cas, une thérapie ciblée ou une immunothérapie peut également être recommandée.
Il est important de noter que le dépistage précoce du cancer du sein par la mammographie et l'examen clinique réguliers peuvent aider à détecter ce type de cancer à un stade précoce, lors il est plus facile à traiter. Les femmes présentant des facteurs de risque élevés pour le cancer du sein, telles que les antécédents familiaux de la maladie ou les mutations génétiques connues, peuvent être recommandées pour un dépistage plus fréquent et/ou plus précoce.
Le terme "stade cancer" fait référence à un système de classification qui évalue le degré d'avancement d'une tumeur maligne dans l'organisme. Il est généralement déterminé par la taille de la tumeur primitive, l'envahissement des ganglions lymphatiques environnants et la présence ou non de métastases à distance. Le système de stadification le plus couramment utilisé est le système TNM (Tumor, Node, Metastasis), qui est basé sur les caractéristiques tumorales, les ganglions lymphatiques et les métastases.
Le stade I correspond à une tumeur localisée et de petite taille sans envahissement des ganglions lymphatiques ni métastases. Les stades II et III décrivent des tumeurs plus larges ou qui ont commencé à se propager aux ganglions lymphatiques voisins. Le stade IV, également connu sous le nom de cancer avancé ou métastatique, indique que la tumeur s'est propagée à d'autres parties du corps, telles que les poumons, le foie ou les os.
La détermination du stade du cancer est importante pour planifier le traitement approprié et prévoir le pronostic du patient.
L'ordre des gènes, également connu sous le nom d'orientation génomique, se réfère à l'organisation linéaire des gènes sur un chromosome ou un segment d'ADN. Il décrit la séquence dans laquelle les gènes sont disposés les uns par rapport aux autres sur une molécule d'ADN.
Dans le génome humain, il y a environ 20 000 à 25 000 gènes répartis sur 23 paires de chromosomes. Chaque chromosome contient des centaines ou des milliers de gènes, qui sont disposés dans un ordre spécifique. Les gènes peuvent être orientés dans les deux sens, c'est-à-dire qu'ils peuvent être lus soit de gauche à droite (sens 5' vers 3'), soit de droite à gauche (sens 3' vers 5') sur l'ADN.
L'ordre des gènes est important car il peut influencer la régulation de l'expression génique et la manière dont les gènes interagissent entre eux. Des mutations dans l'ordre des gènes peuvent entraîner des maladies génétiques ou des troubles du développement. Par conséquent, la compréhension de l'ordre des gènes est essentielle pour comprendre les mécanismes moléculaires qui sous-tendent le fonctionnement normal et anormal du génome humain.
La progression d'une maladie, également appelée évolution de la maladie, se réfère à la manifestation temporelle des stades ou étapes d'une maladie chez un patient. Il s'agit essentiellement de la détérioration continue ou de l'aggravation d'un trouble médical au fil du temps, qui peut entraîner une augmentation de la gravité des symptômes, une déficience accrue, une invalidité et, éventuellement, la mort. La progression de la maladie est généralement mesurée en termes de déclin fonctionnel ou de dommages aux organes affectés. Elle peut être influencée par divers facteurs, notamment l'âge du patient, la durée de la maladie, le traitement et les comorbidités sous-jacentes. Le suivi de la progression de la maladie est crucial pour évaluer l'efficacité des interventions thérapeutiques et pour la planification des soins futurs.
Le médulloblastome est un type de tumeur cérébrale maligne (cancéreuse) qui se développe dans le cervelet, une partie du cerveau située à l'arrière de la tête. Il s'agit d'une tumeur primitive neuroectodermique, ce qui signifie qu'elle se forme à partir des cellules qui sont normalement utilisées pour former les tissus nerveux et le cerveau pendant le développement embryonnaire.
Les médulloblastomes sont plus fréquents chez les enfants que chez les adultes, avec environ 50% des cas diagnostiqués avant l'âge de 10 ans. Les symptômes peuvent inclure des maux de tête, des nausées et des vomissements, une perte d'équilibre et de coordination, une faiblesse musculaire, une vision double ou floue, et des changements de comportement ou de personnalité.
Le traitement du médulloblastome implique généralement une combinaison de chirurgie pour enlever autant de la tumeur que possible, de radiothérapie pour détruire les cellules cancéreuses restantes, et de chimiothérapie pour aider à prévenir la récidive. Le pronostic dépend du stade et du grade de la tumeur, ainsi que de l'âge et de la santé générale du patient. Les médulloblastomes ont un taux de survie global à cinq ans d'environ 60 à 70%, mais cela peut varier en fonction des facteurs mentionnés ci-dessus.
Les tumeurs cérébrales sont des croissances anormales de cellules à l'intérieur ou autour du cerveau. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs cérébrales bénignes ont tendance à se développer plus lentement et sont moins susceptibles de se propager hors du cerveau. Cependant, elles peuvent encore être dangereuses si elles pressent sur des parties vitales du cerveau. Les tumeurs cérébrales malignes se développent rapidement, ont tendance à envahir les tissus cérébraux voisins et peuvent se propager à d'autres parties du corps.
Les symptômes des tumeurs cérébrales dépendent de leur taille, de leur emplacement et de la vitesse à laquelle elles se développent. Ils peuvent inclure des maux de tête sévères et persistants, des nausées ou des vomissements, une vision floue ou double, des problèmes d'équilibre ou de coordination, des changements de personnalité ou de comportement, des convulsions, des problèmes de mémoire ou de concentration, et dans certains cas, une faiblesse ou un engourdissement d'un côté du corps.
Le traitement dépend du type, de la taille et de l'emplacement de la tumeur, de son caractère bénin ou malin, de l'âge et de l'état général du patient. Il peut inclure une chirurgie pour enlever la tumeur, une radiothérapie pour détruire les cellules cancéreuses, une chimiothérapie pour tuer les cellules cancéreuses, ou une thérapie ciblée qui utilise des médicaments pour cibler spécifiquement les changements dans les cellules cancéreuses.
Les amorces d'ADN sont de courtes séquences de nucléotides, généralement entre 15 et 30 bases, qui sont utilisées en biologie moléculaire pour initier la réplication ou l'amplification d'une région spécifique d'une molécule d'ADN. Elles sont conçues pour être complémentaires à la séquence d'ADN cible et se lier spécifiquement à celle-ci grâce aux interactions entre les bases azotées complémentaires (A-T et C-G).
Les amorces d'ADN sont couramment utilisées dans des techniques telles que la réaction en chaîne par polymérase (PCR) ou la séquençage de l'ADN. Dans ces méthodes, les amorces d'ADN se lient aux extrémités des brins d'ADN cibles et servent de point de départ pour la synthèse de nouveaux brins d'ADN par une ADN polymérase.
Les amorces d'ADN sont généralement synthétisées chimiquement en laboratoire et peuvent être modifiées chimiquement pour inclure des marqueurs fluorescents ou des groupes chimiques qui permettent de les détecter ou de les séparer par électrophorèse sur gel.
Le dermatofibrosarcome est un type rare de cancer qui se développe dans les tissus conjonctifs profonds sous la peau. Il commence généralement par former une petite masse ou une plaque ferme et souvent indolore sur la peau. Ces lésions peuvent ressembler à des cicatrices, à des plaques cutanées épaissies ou à des taches rouges, brunes ou bleuâtres.
Ce type de sarcome a tendance à se développer lentement et peut rester localisé pendant plusieurs années avant de s'étendre davantage dans les tissus sous-jacents ou de se métastaser vers d'autres parties du corps, comme les poumons. Le dermatofibrosarcome est souvent résistant aux traitements traditionnels tels que la chimiothérapie et la radiothérapie, ce qui rend sa prise en charge particulièrement difficile.
Les facteurs de risque associés au développement du dermatofibrosarcome sont mal compris, mais il semble qu'il y ait une prédisposition génétique dans certains cas. Les personnes ayant un système immunitaire affaibli peuvent également être plus à risque de développer cette forme de cancer. Le diagnostic repose sur l'examen histopathologique d'une biopsie cutanée, qui permettra de confirmer la nature maligne des cellules et d'identifier le sous-type spécifique du dermatofibrosarcome.
Le traitement dépend du stade et de l'emplacement de la tumeur. Dans les cas précoces, une excision chirurgicale large peut être suffisante pour éliminer complètement la tumeur. Cependant, en raison de sa nature infiltrative, il est fréquent que des récidives locales se produisent après l'intervention initiale. Par conséquent, une surveillance étroite et des interventions supplémentaires peuvent être nécessaires pour prévenir la progression de la maladie. Dans les cas avancés ou récurrents, d'autres options thérapeutiques telles que la radiothérapie, la chimiothérapie ou l'immunothérapie peuvent être envisagées en fonction des caractéristiques moléculaires de la tumeur et de l'état général du patient.
Un marqueur biologique tumoral, également connu sous le nom de biomarqueur tumoral, est une substance ou un signe que l'on peut détecter dans le sang, d'autres fluides corporels, ou des tissus qui peuvent indiquer la présence d'une tumeur cancéreuse ou d'un processus pathologique spécifique. Ces marqueurs peuvent être des protéines, des gènes, des hormones ou d'autres molécules produites par les cellules cancéreuses ou par l'organisme en réponse à la présence de la tumeur.
Les marqueurs biologiques tumoraux sont souvent utilisés pour aider au diagnostic, au staging (détermination du degré d'avancement) et au suivi du traitement du cancer. Cependant, il est important de noter que ces marqueurs ne sont pas spécifiques à un seul type de cancer et peuvent être présents dans d'autres conditions médicales. Par conséquent, ils doivent être utilisés en combinaison avec d'autres tests diagnostiques pour confirmer le diagnostic de cancer.
Exemples courants de marqueurs biologiques tumoraux comprennent l'antigène prostatique spécifique (PSA) pour le cancer de la prostate, l'alpha-fœtoprotéine (AFP) pour le cancer du foie, et l'antigène carcinoembryonnaire (CEA) pour le cancer colorectal.
L'expression génétique est un processus biologique fondamental dans lequel l'information génétique contenue dans l'ADN est transcritte en ARN, puis traduite en protéines. Ce processus permet aux cellules de produire les protéines spécifiques nécessaires à leur fonctionnement, à leur croissance et à leur reproduction.
L'expression génétique peut être régulée à différents niveaux, y compris la transcription de l'ADN en ARNm, la maturation de l'ARNm, la traduction de l'ARNm en protéines et la modification post-traductionnelle des protéines. Ces mécanismes de régulation permettent aux cellules de répondre aux signaux internes et externes en ajustant la production de protéines en conséquence.
Des anomalies dans l'expression génétique peuvent entraîner des maladies génétiques ou contribuer au développement de maladies complexes telles que le cancer. L'étude de l'expression génétique est donc essentielle pour comprendre les mécanismes moléculaires de la maladie et développer de nouvelles stratégies thérapeutiques.
La définition médicale de « Traitement Image Assisté Ordinateur » (TIAO) est le processus qui consiste à utiliser un ordinateur et des logiciels spécialisés pour traiter, analyser et afficher des images médicales. Les images peuvent être acquises à partir de divers modèles d'imagerie tels que la radiographie, l'échographie, la tomodensitométrie (TDM), l'imagerie par résonance magnétique (IRM) et la médecine nucléaire.
Le TIAO permet aux professionnels de la santé d'améliorer la qualité des images, d'effectuer des mesures précises, de détecter les changements anormaux dans le corps, de simuler des procédures et des interventions chirurgicales, et de partager des images avec d'autres médecins. Il peut également aider au diagnostic, à la planification du traitement, à la thérapie guidée par l'image et au suivi des patients.
Les outils de TIAO comprennent des algorithmes d'amélioration de l'image tels que la correction de l'intensité, la suppression du bruit, la segmentation des structures anatomiques, l'enregistrement d'images et la reconstruction 3D. Ces outils peuvent être utilisés pour détecter et diagnostiquer les maladies telles que le cancer, les maladies cardiovasculaires, les troubles neurologiques et les traumatismes.
En résumé, le Traitement Image Assisté Ordinateur est une technologie médicale importante qui permet d'améliorer la qualité des images médicales, de faciliter le diagnostic et la planification du traitement, et d'améliorer les résultats pour les patients.
En termes médicaux, la généalogie est l'étude systématique des antécédents familiaux et médicaux d'une personne ou d'une famille sur plusieurs générations. Elle vise à identifier les modèles de maladies héréditaires ou génétiques dans une famille, ce qui peut aider à évaluer le risque de développer certaines affections et à mettre en œuvre des stratégies de prévention et de dépistage appropriées.
Les professionnels de la santé utilisent souvent des arbres généalogiques pour représenter visuellement les relations familiales et les antécédents médicaux. Ces outils peuvent être particulièrement utiles dans la pratique clinique, en particulier lorsqu'il s'agit de maladies rares ou complexes qui ont tendance à se produire dans certaines familles en raison de facteurs génétiques sous-jacents.
En plus d'être un outil important pour la médecine préventive, la généalogie peut également fournir des informations précieuses sur l'histoire naturelle de diverses maladies et conditions, ce qui contribue à faire progresser notre compréhension globale de la pathogenèse et de la physiopathologie. Par conséquent, elle joue un rôle crucial dans la recherche médicale et les soins cliniques.
La monosomie est un type d'anomalie chromosomique caractérisée par la présence d'un seul chromosome au lieu de deux dans un ensemble de chromosomes. Cela signifie qu'un individu atteint de monosomie ne possède qu'une copie d'un chromosome particulier au lieu des deux copies normales. Cette condition génétique est généralement associée à diverses maladies congénitales et handicaps mentaux, selon le chromosome concerné. Un exemple bien connu de monosomie est la trisomie 21 (syndrome de Down), où il y a une copie supplémentaire du chromosome 21. Cependant, dans le cas de la monosomie, un chromosome entier est manquant au lieu d'être présent en triple exemplaire. La monosomie peut survenir soit en raison d'un événement aléatoire pendant la division cellulaire, soit par héritage d'un parent atteint d'une translocation équilibrée.
Les néoplasmes des cellules plasmatiques, également connus sous le nom de gammapathies monoclonales, sont des affections caractérisées par la prolifération anormale et excessive de cellules plasmatiques dans la moelle osseuse et/ou les tissus extra-médullaires. Les cellules plasmatiques sont un type de globule blanc qui produit des anticorps ou des immunoglobulines pour aider à combattre les infections.
Dans les néoplasmes des cellules plasmatiques, ces cellules se multiplient de manière incontrôlable et peuvent produire un seul type d'immunoglobuline anormale, appelée paraprotéine. Cette prolifération anormale peut entraîner une variété de symptômes, tels qu'une fatigue excessive, des douleurs osseuses, des infections fréquentes, une insuffisance rénale et des saignements anormaux.
Le type le plus courant de néoplasme des cellules plasmatiques est le myélome multiple, qui se caractérise par la présence de lésions osseuses multiples et la production de grandes quantités de paraprotéines. D'autres types de néoplasmes des cellules plasmatiques comprennent le plasma cellulaires solitaires (ou monoclonaux), qui se produisent généralement dans les tissus extra-médullaires et sont souvent asymptomatiques, et l'amylose à chaînes légères, qui se caractérise par l'accumulation de chaînes légères d'immunoglobulines dans divers organes et tissus.
Le diagnostic des néoplasmes des cellules plasmatiques repose sur une combinaison de tests de laboratoire, d'imagerie et de biopsie de la moelle osseuse. Le traitement dépend du type et de la gravité de la maladie et peut inclure une chimiothérapie, une radiothérapie, une greffe de cellules souches et des thérapies ciblées.
Les tumeurs du foie sont des growths anormales qui se produisent dans cet organe. Ils peuvent être bénins (non cancéreux) ou malins (cancéreux).
Les tumeurs bénignes du foie comprennent les hémangiomes, les adénomes et les hyperplasies nodulaires focales. Ces types de tumeurs ne se propagent pas à d'autres parties du corps et peuvent souvent être surveillés sans traitement. Cependant, dans certains cas, ils peuvent causer des problèmes s'ils deviennent grands ou si leur croissance comprime les structures voisines.
Les tumeurs malignes du foie comprennent le carcinome hépatocellulaire (CHC) et les métastases hépatiques. Le CHC est une forme de cancer qui commence dans les cellules du foie, tandis que les métastases hépatiques sont des cancers qui se sont propagés au foie à partir d'autres parties du corps. Les deux types peuvent causer des dommages importants aux fonctions hépatiques et nécessitent un traitement agressif.
Les facteurs de risque pour le développement des tumeurs malignes du foie comprennent l'infection par le virus de l'hépatite B ou C, la consommation excessive d'alcool, l'obésité, le diabète et l'exposition à certains produits chimiques. Les symptômes des tumeurs du foie peuvent inclure une douleur ou une sensation de plénitude dans le haut de l'abdomen, une perte de poids inexpliquée, une jaunisse (jaunissement de la peau et du blanc des yeux), des nausées et des vomissements. Le diagnostic est généralement posé par imagerie médicale telle qu'une échographie ou une tomodensitométrie, suivie d'une biopsie pour confirmer le type de tumeur.
En génétique, un exon est une séquence d'ADN qui code pour une partie spécifique d'une protéine. Après la transcription de l'ADN en ARN messager (ARNm), les exons sont conservés et assemblés dans le processus de maturation de l'ARNm, tandis que les introns, qui sont les séquences non codantes, sont éliminés. Les exons forment ainsi la section codante finale de l'ARNm mature, qui est ensuite traduite en une chaîne polypeptidique lors de la synthèse des protéines.
En bref, un exon est une région d'un gène qui contribue à la séquence d'acides aminés d'une protéine après le traitement et l'assemblage de l'ARNm mature. Les mutations dans les exons peuvent entraîner des modifications dans la structure des protéines, ce qui peut conduire à des maladies génétiques ou à des changements phénotypiques.
Un oligodendrogliome est un type rare de tumeur cérébrale qui se développe à partir des cellules gliales du cerveau appelées oligodendrocytes. Ces cellules sont responsables de la production et du maintien de la gaine de myéline, une substance grasse qui entoure et protège les fibres nerveuses dans le cerveau et la moelle épinière.
Les oligodendrogliomes se développent généralement dans les lobes frontaux ou temporaux du cerveau et peuvent être à croissance lente ou rapide. Ils ont tendance à affecter des personnes d'âge moyen, bien que certains cas puissent survenir chez les enfants.
Les symptômes de l'oligodendrogliome dépendent de la taille et de la localisation de la tumeur. Ils peuvent inclure des maux de tête, des convulsions, des nausées, des vomissements, une faiblesse musculaire, des changements de comportement ou de personnalité, des problèmes de vision, de l'ouïe ou du langage.
Le diagnostic d'un oligodendrogliome est généralement posé après un examen d'imagerie cérébrale, tel qu'une tomodensitométrie (TDM) ou une imagerie par résonance magnétique (IRM). Une biopsie ou une résection chirurgicale de la tumeur peut être nécessaire pour confirmer le diagnostic et déterminer le grade de la tumeur.
Le traitement de l'oligodendrogliome dépend du grade de la tumeur, de sa taille et de sa localisation. Les options de traitement peuvent inclure une chirurgie pour enlever autant de la tumeur que possible, une radiothérapie et/ou une chimiothérapie. Dans certains cas, un traitement d'attente peut être recommandé si la tumeur est à croissance lente et ne cause pas de symptômes importants.
L'ADN complémentaire (cADN) est une copie d'ADN synthétisée à partir d'ARN messager (ARNm) à l'aide d'une enzyme appelée transcriptase inverse. Ce processus est souvent utilisé dans la recherche scientifique pour étudier et analyser les gènes spécifiques. Le cADN est complémentaire à l'original ARNm, ce qui signifie qu'il contient une séquence nucléotidique qui est complémentaire à la séquence de l'ARNm. Cette technique permet de créer une copie permanente et stable d'un gène spécifique à partir de l'ARN transitoire et instable, ce qui facilite son analyse et sa manipulation en laboratoire.
En médecine, une tumeur est une augmentation anormale et localisée de la taille d'un tissu corporel due à une croissance cellulaire accrue. Les tumeurs peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses).
Les tumeurs bénignes sont généralement des masses arrondies, bien circonscrites et ne se propagent pas aux tissus environnants. Elles peuvent cependant causer des problèmes si elles compriment ou déplacent des organes vitaux.
Les tumeurs malignes, en revanche, ont tendance à envahir les tissus voisins et peuvent se propager (métastaser) vers d'autres parties du corps via le système sanguin ou lymphatique. Elles sont souvent désignées sous le terme de «cancer».
Il est important de noter que toutes les augmentations anormales de la taille d'un tissu ne sont pas nécessairement des tumeurs. Par exemple, un œdème (gonflement) ou une inflammation peuvent également entraîner une augmentation temporaire de la taille d'une zone spécifique du corps.
Une hernie diaphragmatique est un problème médical congénital dans lequel le contenu de l'abdomen, généralement l'estomac et l'intestin grêle, passe à travers une ouverture dans le diaphragme vers le thorax. Le diaphragme est un muscle qui sépare la cavité thoracique de la cavité abdominale et joue un rôle crucial dans la respiration.
Dans une hernie diaphragmatique, cette ouverture peut être causée par un développement anormal pendant la grossesse, où certaines parties du diaphragme ne se forment pas correctement, ce qui entraîne une faiblesse dans la paroi musculaire. Cela permet aux organes abdominaux de migrer vers le thorax, comprimant ainsi les poumons et affectant la capacité respiratoire du nouveau-né.
Les hernies diaphragmatiques peuvent être classées en deux types principaux : hernie diaphragmatique postérieure (hernie de Bochdalek) et hernie diaphragmatique antérieure (hernie de Morgagni ou hernie de Larrey). Les hernies diaphragmatiques peuvent également être associées à d'autres anomalies congénitales, telles que des malformations cardiaques et des troubles du tube digestif.
Le traitement d'une hernie diaphragmatique nécessite généralement une intervention chirurgicale pour réparer l'ouverture dans le diaphragme et remettre les organes abdominaux à leur place respective. Le pronostic dépend de la taille de l'hernie, de la présence d'autres anomalies congénitales et du moment où le diagnostic est posé et le traitement est initié.
Les malformations crâniofaciales sont des anomalies congénitales qui affectent la formation et le développement appropriés du crâne et du visage. Elles peuvent varier d'une apparence mineure à une déformation sévère qui peut causer des problèmes fonctionnels importants.
Ces malformations résultent généralement de perturbations survenant pendant la période de développement fœtal, entre le 20ème et le 50ème jour après la conception. Les facteurs causaux peuvent être génétiques, environnementaux ou une combinaison des deux.
Les exemples courants de malformations crâniofaciales comprennent :
1. Le bec-de-lièvre : une fente dans la lèvre supérieure et parfois le palais.
2. La fente labiale palatine : une fente plus large affectant à la fois la lèvre et le palais.
3. L'anencéphalie : absence de certaines parties du cerveau et du crâne.
4. Le cyclopisme : fusion des yeux en un seul œil central.
5. La microtie : sous-développement des oreilles.
6. L'holoprosencéphalie : anomalie du cerveau antérieur et du visage.
Le traitement dépendra de la gravité et du type spécifique de malformation. Il peut inclure une intervention chirurgicale, une thérapie orthodontique, une réadaptation auditive ou vocale, et des soins médicaux continus.
Le mélanome est un type de cancer qui se développe à partir des cellules pigmentées de la peau connues sous le nom de mélanocytes. Il se caractérise généralement par la croissance anormale et incontrôlée de ces cellules, aboutissant souvent à la formation de taches ou de bosses sur la peau qui peuvent être plates ou surélevées, lisses ou rugueuses, et qui peuvent présenter une variété de couleurs, allant du brun foncé au noir.
Les mélanomes peuvent se produire n'importe où sur le corps, mais ils sont les plus fréquents dans les zones exposées au soleil, telles que le dos, les bras et les jambes. Ils peuvent également se développer dans des parties du corps qui ne sont pas exposées au soleil, comme les muqueuses, les yeux et les ongles.
Le mélanome est considéré comme l'un des cancers de la peau les plus agressifs et les plus dangereux en raison de sa capacité à se propager rapidement à d'autres parties du corps s'il n'est pas détecté et traité à un stade précoce. Les facteurs de risque comprennent une exposition excessive au soleil, des antécédents personnels ou familiaux de mélanome, des grains de beauté atypiques et une peau claire qui se brûle facilement au soleil.
Le traitement du mélanome dépend du stade et de l'étendue de la maladie. Les options de traitement peuvent inclure la chirurgie, la radiothérapie, la chimiothérapie, l'immunothérapie et la thérapie ciblée. La prévention consiste à se protéger du soleil en utilisant un écran solaire, en portant des vêtements protecteurs et en évitant les heures de fort ensoleillement. Il est également important de subir des examens réguliers de la peau pour détecter tout changement ou toute nouvelle croissance suspecte.
Le Trouble Autistique, également connu sous le nom de Autisme Classique ou Autisme de l'Enfant, est un trouble du développement neurologique caractérisé par trois principaux symptômes :
1. Déficits persistants en matière de communication et d'interaction sociales dans divers contextes, comme des difficultés à établir des relations sociales appropriées à l'âge, des problèmes de réciprocité socio-émotionnelle, ou une absence de partage des intérêts, des émotions ou du plaisir avec d'autres personnes.
2. Des patterns restreints et répétitifs de comportements, d'intérêts ou d'activités, comme des mouvements stéréotypés ou répétitifs, une insistance sur la sameness, des routines inflexibles, des intérêts très restreints et fixes, ou une hyper- ou hypo-réactivité sensorielle ou intérêt inhabituel pour des aspects sensoriels de l'environnement.
3. Les symptômes doivent être présents dans le courant de la petite enfance, généralement avant l'âge de trois ans.
4. Ces symptômes causent une altération cliniquement significative du fonctionnement social, scolaire, professionnel ou autre domaine important de fonctionnement.
5. Ces troubles ne sont pas mieux expliqués par un retard global du développement ou par un trouble spécifique du développement de la communication comme le Rett ou le syndrome d'Asperger.
Les études d'association génétique sont un type courant de recherche en génétique visant à identifier les relations entre des variations spécifiques du matériel génétique, telles que les polymorphismes nucléotidiques simples (SNP), et la présence ou l'absence d'une maladie ou d'un trait particulier. Ces études comparent généralement des groupes de personnes atteintes d'une maladie avec des groupes de personnes sans la maladie, en examinant les fréquences des variations génétiques entre les deux groupes.
L'objectif d'une étude d'association génétique est de déterminer si une certaine variante génétique est plus susceptible d'être trouvée chez les personnes atteintes d'une maladie que chez celles qui ne le sont pas. Si une telle association est constatée, cela peut fournir des indices importants sur les gènes et les voies biologiques qui peuvent contribuer au développement ou à la progression de la maladie.
Cependant, il est important de noter que même si une association génétique est constatée, cela ne prouve pas nécessairement que la variante génétique en question est la cause directe de la maladie. D'autres facteurs, tels que des variations dans d'autres gènes ou des facteurs environnementaux, peuvent également jouer un rôle important dans le développement de la maladie.
Les études d'association génétique sont largement utilisées dans la recherche sur les maladies complexes, telles que le cancer, les maladies cardiovasculaires et les troubles neurologiques, qui sont susceptibles d'être influencés par de multiples facteurs génétiques et environnementaux.
L'hétérogénéité génétique est un terme utilisé en génétique pour décrire la situation où différents gènes ou différentes mutations dans le même gène peuvent conduire à des phénotypes similaires ou identiques. Cela signifie qu'il existe plusieurs façons dont une maladie particulière peut se manifester, en fonction de la manière dont les gènes sont hérités ou altérés.
L'hétérogénéité génétique peut être classée en deux types : l'hétérogénéité génétique d'un gène (ou hétérogénéité algique) et l'hétérogénéité génétique intergénique (ou hétérogénéité non algique).
L'hétérogénéité génétique d'un gène se produit lorsque différentes mutations dans le même gène entraînent la même maladie. Par exemple, il existe de nombreuses mutations différentes dans le gène CFTR qui peuvent causer la mucoviscidose, une maladie génétique grave qui affecte principalement les poumons et le système digestif.
D'autre part, l'hétérogénéité génétique intergénique se produit lorsque des mutations dans différents gènes entraînent la même maladie. Par exemple, il existe plusieurs types de cancer du sein qui peuvent être causés par des mutations dans différents gènes, tels que BRCA1, BRCA2, P53 et autres.
L'hétérogénéité génétique peut compliquer le diagnostic et le traitement des maladies génétiques, car il peut être difficile de déterminer quelle mutation ou quel gène est à l'origine de la maladie chez un patient donné. Cependant, la compréhension de l'hétérogénéité génétique peut également fournir des informations importantes sur les mécanismes sous-jacents des maladies et aider au développement de nouveaux traitements personnalisés pour les patients atteints de maladies rares ou complexes.
Dans le domaine de la génétique, un individu homozygote est une personne qui hérite de deux allèles identiques pour un trait spécifique, ayant reçu ce même allèle de chacun de ses deux parents. Cela peut se traduire par l'expression d'un caractère particulier ou l'apparition d'une maladie génétique dans le cas où ces allèles sont défectueux.
On distingue deux types d'homozygotes : les homozygotes dominants et les homozygotes récessifs. Les homozygotes dominants présentent le phénotype lié au gène dominant, tandis que les homozygotes récessifs expriment le phénotype associé au gène récessif.
Par exemple, dans le cas de la drépanocytose, une maladie génétique héréditaire affectant l'hémoglobine des globules rouges, un individu homozygote pour cette affection possède deux allèles mutés du gène de l'hémoglobine et exprimera donc les symptômes de la maladie.
Le lymphome B est un type de cancer qui affecte les lymphocytes B, un type de globules blancs qui jouent un rôle crucial dans le système immunitaire. Les lymphocytes B aident à combattre les infections en produisant des anticorps pour neutraliser ou éliminer les agents pathogènes.
Dans le cas d'un lymphome B, les lymphocytes B subissent une transformation maligne et se multiplient de manière incontrôlable, formant une masse tumorale dans les ganglions lymphatiques ou d'autres tissus lymphoïdes. Ces tumeurs peuvent se propager à d'autres parties du corps, affectant ainsi plusieurs organes et systèmes.
Les lymphomes B peuvent être classés en deux catégories principales : les lymphomes hodgkiniens et les lymphomes non hodgkiniens. Les lymphomes hodgkiniens sont caractérisés par la présence de cellules de Reed-Sternberg, qui sont des cellules malignes spécifiques à ce type de cancer. Les lymphomes non hodgkiniens, en revanche, n'incluent pas ces cellules et peuvent être subdivisés en plusieurs sous-types en fonction de leur apparence au microscope, de leur comportement clinique et de leur réponse au traitement.
Les facteurs de risque du lymphome B comprennent l'âge avancé, une immunodéficience due à une maladie sous-jacente ou à des médicaments immunosuppresseurs, une infection par le virus d'Epstein-Barr, une exposition à certains produits chimiques et une prédisposition génétique. Les symptômes courants du lymphome B peuvent inclure des ganglions lymphatiques hypertrophiés, de la fièvre, des sueurs nocturnes, une perte de poids inexpliquée, de la fatigue et des douleurs articulaires. Le diagnostic repose généralement sur une biopsie d'un ganglion lymphatique ou d'un autre tissu affecté, suivie d'examens complémentaires pour évaluer l'étendue de la maladie et planifier le traitement.
La synténie est un terme utilisé en génétique et en génomique pour décrire la présence des gènes homologues sur les mêmes chromosomes entre différentes espèces. Il s'agit essentiellement de la conservation du même arrangement relatif des gènes sur un chromosome donné au cours de l'évolution.
Dans un contexte médical, la synténie peut être particulièrement utile dans l'étude des maladies génétiques évolutives et de la fonction des gènes. Par exemple, en identifiant les segments de synténie entre les humains et les modèles animaux, les chercheurs peuvent déterminer quels gènes spécifiques sont associés à certaines maladies et étudier ces gènes dans des systèmes modèles pour mieux comprendre leur fonction et développer de nouvelles stratégies thérapeutiques.
Cependant, il est important de noter que la synténie n'est pas toujours parfaite entre les espèces, car des réarrangements chromosomiques tels que les inversions, les translocations et les duplications peuvent se produire au cours de l'évolution. Ces événements peuvent entraîner une perte ou un gain de gènes dans certains segments synténiques, ce qui peut compliquer l'analyse comparative entre espèces.
Une banque de gènes est une collection organisée et stockée de matériel génétique, y compris l'ADN, les ARN et les cellules, prélevés sur des humains, des animaux ou d'autres organismes. Ces échantillons sont généralement prélevés dans le but de les utiliser à des fins de recherche scientifique, de diagnostic médical ou de thérapie génique. Les banques de gènes peuvent être spécialisées dans un type particulier d'échantillon, comme les échantillons tumoraux, ou contenir une variété d'échantillons provenant de diverses sources.
Les banques de gènes sont importantes pour la recherche biomédicale car elles fournissent des matériaux standardisés et bien caractérisés qui peuvent être utilisés dans différentes études. Elles permettent également le partage des ressources entre les chercheurs, ce qui peut accélérer les progrès de la recherche et réduire les coûts.
Dans le domaine de la médecine personnalisée, les banques de gènes peuvent être utilisées pour stocker des échantillons d'ADN de patients atteints de maladies spécifiques, ce qui permet aux chercheurs d'étudier les variations génétiques associées à ces maladies. Ces informations peuvent ensuite être utilisées pour développer des traitements personnalisés pour chaque patient.
Cependant, l'utilisation de banques de gènes soulève également des questions éthiques et juridiques importantes concernant la confidentialité, le consentement et la propriété des échantillons et des informations génétiques qu'ils contiennent. Il est donc essentiel de mettre en place des politiques et des procédures claires pour garantir que les banques de gènes soient utilisées de manière responsable et éthique.
En médecine, la fixation tissulaire est un processus qui consiste à attacher, à maintenir ou à stabiliser un tissu, un organe ou une structure corporelle à sa place anatomique correcte en utilisant diverses méthodes et matériaux. Cela peut être nécessaire pour faciliter la guérison après une intervention chirurgicale, une blessure ou une maladie.
Les méthodes de fixation tissulaire comprennent l'utilisation de sutures, d'agrafes, de clips, de colle biologique et de plaques et vis métalliques. Le choix de la méthode dépend du type de tissu ou de structure à fixer, de la localisation anatomique, de la force nécessaire pour maintenir la réparation et des préférences personnelles du chirurgien.
Par exemple, dans la chirurgie orthopédique, les plaques et les vis sont souvent utilisées pour fixer les os fracturés, tandis que dans la chirurgie générale, les sutures sont couramment utilisées pour fermer les incisions cutanées. Dans la chirurgie oculaire, une colle biologique peut être utilisée pour fixer les tissus délicats de l'œil.
Il est important de noter que la fixation tissulaire doit être effectuée avec soin et précision pour éviter toute complication postopératoire, telles que des infections, des saignements ou une mauvaise guérison.
Un adénome est un type de tumeur non cancéreuse (bénigne) qui se développe dans les glandes. Il peut se former dans divers endroits du corps où il y a des glandes, mais ils sont le plus souvent trouvés dans la prostate, les glandes surrénales et les glandes hypophysaires. Les adénomes sont généralement lents à se développer et ne se propagent pas à d'autres parties du corps. Cependant, selon leur taille et leur emplacement, ils peuvent causer des problèmes de santé en comprimant les tissus voisins ou en interférant avec leur fonction normale.
Les adénomes peuvent ne pas provoquer de symptômes, surtout s'ils sont petits. Cependant, selon l'emplacement et la taille de la tumeur, des symptômes peuvent apparaître. Par exemple, un adénome de la prostate peut causer des problèmes de miction, tandis qu'un adénome de la glande pituitaire peut entraîner une production excessive d'hormones ou une vision floue.
Le traitement dépend de la taille et de l'emplacement de la tumeur, ainsi que des symptômes qu'elle provoque. Dans certains cas, aucun traitement n'est nécessaire et la tumeur est simplement surveillée. Dans d'autres cas, une intervention chirurgicale ou une radiothérapie peut être recommandée pour enlever la tumeur ou réduire sa taille.
Les tumeurs sus-tentoriellees sont des tumeurs cérébrales benignes ou malignes qui se développent au-dessus du tentorium cerebelli, une membrane qui sépare les hémisphères cérébraux du cervelet dans le crâne. Ces tumeurs peuvent inclure des méningiomes, des gliomes, des schwannomes, et d'autres types de tumeurs. Elles peuvent causer une variété de symptômes, en fonction de leur taille et de leur emplacement, y compris des maux de tête, des convulsions, des nausées, des vomissements, des troubles de la vision, des problèmes d'équilibre et de coordination, et des changements cognitifs ou comportementaux. Le traitement dépend du type et de l'emplacement de la tumeur, mais peut inclure une chirurgie, une radiothérapie ou une chimiothérapie.
Une apraxie est un trouble du mouvement caractérisé par l'incapacité d'effectuer des mouvements volontaires ou des séquences de mouvements complexes, malgré un contrôle musculaire et cognitif préservé. Ce trouble est généralement causé par une lésion cérébrale, le plus souvent dans les zones du cerveau responsables du traitement et de la planification des mouvements volontaires, telles que les lobes frontaux ou pariétaux.
Les apraxies peuvent affecter différents types de mouvements, tels que la coordination des membres (appelée apraxie motrice), l'utilisation d'outils et d'objets (apraxie idéomotrice), la parole (apraxie verbale) ou les expressions faciales et les gestes sociaux (apraxie bucco-faciale). Les symptômes peuvent inclure des mouvements maladroits, lents, imprécis ou inappropriés, ainsi qu'une difficulté à initier, arrêter ou modifier des mouvements volontaires.
Le diagnostic d'apraxie repose sur une évaluation neurologique approfondie, comprenant un examen physique et neuropsychologique détaillé. Le traitement peut inclure la réadaptation et la thérapie des mouvements, ainsi que l'utilisation d'aides techniques pour faciliter les activités quotidiennes. Dans certains cas, des médicaments peuvent être prescrits pour aider à gérer les symptômes moteurs associés à l'apraxie.
Les tumeurs pulmonaires sont des croissances anormales dans les tissus du poumon. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses).
Les tumeurs pulmonaires bénignes ne se propagent pas au-delà du poumon et sont généralement traitées par une intervention chirurgicale pour enlever la tumeur. Cependant, même si elles sont bénignes, certaines d'entre elles peuvent continuer à se développer et provoquer des problèmes respiratoires en raison de l'occupation d'espace dans le poumon.
Les tumeurs pulmonaires malignes, également appelées cancer du poumon, sont beaucoup plus graves. Elles peuvent se propager à d'autres parties du corps par le système sanguin ou lymphatique. Il existe deux principaux types de cancer du poumon : le carcinome pulmonaire à petites cellules et le carcinome pulmonaire non à petites cellules. Le premier type se développe plus rapidement et a tendance à se propager plus tôt que le second.
Le tabagisme est la cause la plus fréquente de cancer du poumon. L'exposition à certains produits chimiques, la pollution atmosphérique ou l'hérédité peuvent également contribuer au développement de ces tumeurs. Les symptômes courants incluent une toux persistante, des douleurs thoraciques, un essoufflement, des expectorations sanglantes et une perte de poids inexpliquée. Le traitement dépend du type et du stade de la tumeur, mais peut inclure une combinaison de chirurgie, de radiothérapie et de chimiothérapie.
Un carcinome est un type de cancer qui commence dans les cellules épithéliales, qui sont les cellules qui tapissent la surface des organes et des glandes. Ces cellules ont une forme aplatie et une fonction de protection ou de sécrétion. Les carcinomes peuvent se développer à partir de divers types d'épithélium dans tout le corps, y compris la peau, les poumons, le sein, le côlon, la prostate et le rein.
Les carcinomes peuvent être classés en plusieurs sous-types en fonction de leur apparence au microscope et de leurs caractéristiques moléculaires. Certains des sous-types courants comprennent les carcinomes squameux, les adénocarcinomes, les carcinomes à cellules basales et les carcinomes à cellules rénales.
Les facteurs de risque pour le développement d'un carcinome peuvent inclure l'exposition aux rayonnements, au tabagisme, à certaines substances chimiques, à une infection virale ou bactérienne, à des antécédents familiaux de cancer et au vieillissement.
Le traitement d'un carcinome dépend du type et du stade du cancer, ainsi que de la santé générale du patient. Les options de traitement peuvent inclure une chirurgie pour enlever la tumeur, une radiothérapie pour détruire les cellules cancéreuses à l'aide de rayonnements, une chimiothérapie pour tuer les cellules cancéreuses avec des médicaments, ou une thérapie ciblée pour attaquer des caractéristiques spécifiques des cellules cancéreuses.
'Clostridium botulinum Type B' fait référence à un type spécifique de bactérie anaérobie gram-positive, qui produit une toxine neurotoxique appelée toxine botulique de type B. Cette bactérie est souvent trouvée dans le sol et l'eau et peut se développer dans des environnements sans oxygène. La toxine botulique de type B est responsable de la maladie du botulisme, qui est une affection neuroparalytique grave caractérisée par une faiblesse musculaire, une vision double, une difficulté à avaler et à respirer.
Le botulisme de type B est relativement moins fréquent que les autres types de botulisme, mais il peut être tout aussi dangereux. Il peut se produire lorsque des aliments contaminés par la bactérie sont consommés ou lorsque la toxine elle-même pénètre dans le corps par d'autres moyens, tels que des plaies cutanées.
Bien que le botulisme de type B soit une maladie grave, il peut être traité avec succès s'il est diagnostiqué et traité rapidement. Le traitement standard consiste en l'administration d'antitoxines pour neutraliser la toxine botulique et des soins de soutien pour gérer les symptômes. La prévention du botulisme implique des pratiques adéquates de manipulation et de conservation des aliments, ainsi que la vaccination contre le botulisme dans certains cas.
Les "Urinary Bladder Neoplasms" sont des tumeurs anormales et anarchiques qui se développent dans la paroi de la vessie. Elles peuvent être bénignes ou malignes (cancéreuses). Les cancers de la vessie, qui représentent la majorité des urinary bladder neoplasms, sont classés en fonction de leur aspect cellulaire et de leur comportement invasif.
Les cancers de la vessie les plus fréquents sont les carcinomes urothéliaux, qui prennent naissance dans les cellules qui tapissent l'intérieur de la vessie. Ils peuvent être superficiels, n'envahissant que la muqueuse ou profonds, s'étendant jusqu'aux couches musculaires plus profondes de la paroi vésicale.
Les facteurs de risque incluent le tabagisme, une exposition professionnelle à certains produits chimiques, des antécédents familiaux de cancer de la vessie et certaines maladies préexistantes telles que des infections urinaires récurrentes ou des calculs vésicaux.
Le diagnostic est généralement posé par cystoscopie (examen endoscopique de la vessie) associée à une biopsie pour analyse histopathologique. Le traitement dépend du type et du stade de la tumeur, allant d'une résection endoscopique pour les tumeurs superficielles à une chirurgie radicale, une chimiothérapie ou une radiothérapie pour les tumeurs plus avancées.
Le gène P53, également connu sous le nom de TP53 (tumor protein p53), est un gène suppresseur de tumeurs humain crucial qui code pour la protéine p53. Cette protéine joue un rôle essentiel dans la régulation du cycle cellulaire, de l'apoptose (mort cellulaire programmée) et de la réparation de l'ADN. Lorsque le gène P53 fonctionne correctement, il aide à prévenir la transformation des cellules normales en cellules cancéreuses en arrêtant la prolifération des cellules présentant des dommages à l'ADN ou en déclenchant leur apoptose.
Des mutations du gène P53 peuvent entraîner une production de protéines p53 anormales ou non fonctionnelles, ce qui peut perturber la régulation du cycle cellulaire et favoriser la croissance des tumeurs. Environ 50 % des cancers humains présentent des mutations du gène P53, ce qui en fait l'un des gènes les plus fréquemment altérés dans les cancers. Par conséquent, le gène P53 est considéré comme un important marqueur moléculaire et un facteur pronostique pour divers types de cancer.
La transcription génétique est un processus biologique essentiel à la biologie cellulaire, impliqué dans la production d'une copie d'un brin d'ARN (acide ribonucléique) à partir d'un brin complémentaire d'ADN (acide désoxyribonucléique). Ce processus est catalysé par une enzyme appelée ARN polymérase, qui lit la séquence de nucléotides sur l'ADN et synthétise un brin complémentaire d'ARN en utilisant des nucléotides libres dans le cytoplasme.
L'ARN produit pendant ce processus est appelé ARN pré-messager (pré-mRNA), qui subit ensuite plusieurs étapes de traitement, y compris l'épissage des introns et la polyadénylation, pour former un ARN messager mature (mRNA). Ce mRNA sert ensuite de modèle pour la traduction en une protéine spécifique dans le processus de biosynthèse des protéines.
La transcription génétique est donc un processus crucial qui permet aux informations génétiques codées dans l'ADN de s'exprimer sous forme de protéines fonctionnelles, nécessaires au maintien de la structure et de la fonction cellulaires, ainsi qu'à la régulation des processus métaboliques et de développement.
Un paragangliome est un type rare et souvent bénin de tumeur qui se développe à partir des cellules du système nerveux sympathique ou parasympathique. Ces tumeurs peuvent être trouvées dans divers endroits du corps, y compris la tête, le cou, l'abdomen, la poitrine et la région pelvienne. Les paragangliomes qui se développent dans la région de la tête et du cou sont souvent appelés phéochromocytomes lorsqu'ils surviennent dans les glandes surrénales.
Les paragangliomes peuvent produire des hormones telles que l'adrénaline et la noradrénaline, ce qui peut entraîner une hypertension artérielle, des palpitations cardiaques, des sueurs, des tremblements, des maux de tête et d'autres symptômes associés à une activation excessive du système nerveux sympathique. Cependant, certaines tumeurs peuvent être non fonctionnelles et ne pas produire de symptômes hormonaux.
Le traitement des paragangliomes dépend généralement de leur localisation et de leur taille. Les options thérapeutiques comprennent la chirurgie, la radiothérapie externe, la radioembolisation et la thérapie médicamenteuse. Dans certains cas, une surveillance attentive peut être recommandée si la tumeur est petite et ne provoque pas de symptômes.
Le gène Myc, également connu sous le nom de gène c-myc, est un gène qui code pour la protéine Myc, qui joue un rôle crucial dans la régulation de la croissance cellulaire, du métabolisme et de l'apoptose (mort cellulaire programmée). Cette protéine forme un complexe avec d'autres protéines pour activer ou réprimer la transcription de divers gènes.
L'activation anormale ou la surexpression du gène Myc a été associée à plusieurs types de cancer, tels que le cancer du sein, des ovaires, des poumons et du côlon. Elle peut entraîner une prolifération cellulaire incontrôlée, une résistance à l'apoptose et une angiogenèse (formation de nouveaux vaisseaux sanguins), ce qui favorise la croissance tumorale.
Par conséquent, le gène Myc est considéré comme un oncogène, c'est-à-dire un gène capable de provoquer une transformation maligne des cellules lorsqu'il est activé ou surexprimé de manière anormale. Il constitue donc une cible thérapeutique potentielle pour le traitement du cancer.
La méthylation de l'ADN est un processus épigénétique impliquant l'ajout d'un groupe méthyle (-CH3) à l'une des bases azotées de l'ADN, généralement à la cytosine. Cette modification se produit principalement dans les régions promotrices des gènes et joue un rôle crucial dans la régulation de l'expression génétique.
La méthylation de l'ADN est catalysée par une enzyme appelée ADN méthyltransférase, qui transfère le groupe méthyle du donneur, généralement la S-adénosylméthionine (SAM), vers l'ADN cible.
La méthylation de l'ADN peut entraîner une répression de l'expression génique en empêchant la liaison des facteurs de transcription aux séquences d'ADN promotrices méthylées. Cela peut conduire à un large éventail de conséquences physiologiques, y compris le développement et la progression de diverses maladies, telles que les cancers.
Par conséquent, la méthylation de l'ADN est un processus dynamique et réversible qui joue un rôle essentiel dans la régulation des fonctions cellulaires normales et anormales.
La grossesse, également connue sous le nom de gestation, est un état physiologique dans lequel un ovule fécondé, ou zygote, s'implante dans l'utérus et se développe pendant environ 40 semaines, aboutissant à la naissance d'un bébé. Ce processus complexe implique des changements significatifs dans le corps de la femme, affectant presque tous les systèmes organiques.
Au cours des premières semaines de grossesse, l'embryon se développe rapidement, formant des structures vitales telles que le cœur, le cerveau et le tube neural. Après environ huit semaines, l'embryon est appelé fœtus et poursuit son développement, y compris la croissance des membres, des organes sensoriels et du système nerveux.
La grossesse est généralement divisée en trois trimestres, chacun marqué par des stades spécifiques de développement fœtal:
1. Premier trimestre (jusqu'à 12 semaines): Pendant cette période, l'embryon subit une croissance et un développement rapides. Les structures vitales telles que le cœur, le cerveau, les yeux et les membres se forment. C'est également lorsque le risque d'anomalies congénitales est le plus élevé.
2. Deuxième trimestre (13 à 26 semaines): Durant ce stade, le fœtus continue de croître et se développer. Les organes commencent à fonctionner de manière autonome, et le fœtus peut entendre et répondre aux stimuli externes. Le risque d'anomalies congénitales est considérablement réduit par rapport au premier trimestre.
3. Troisième trimestre (27 semaines jusqu'à la naissance): Au cours de ces dernières semaines, le fœtus prend du poids et se prépare à la vie en dehors de l'utérus. Les poumons mûrissent, et le cerveau continue de se développer rapidement.
Tout au long de la grossesse, il est crucial que les femmes enceintes maintiennent un mode de vie sain, comprenant une alimentation équilibrée, l'exercice régulier et l'évitement des substances nocives telles que l'alcool, le tabac et les drogues illicites. De plus, il est essentiel de suivre les soins prénataux recommandés pour assurer la santé et le bien-être de la mère et du fœtus.
Un locus génétique (pluriel : loci) est un emplacement spécifique sur un chromosome où se trouve un gène donné. Il s'agit essentiellement d'une position fixe et définie sur un chromosome où réside une séquence d'ADN particulière, qui code généralement pour un trait ou une caractéristique spécifique.
Les chercheurs utilisent souvent l'expression "locus génétique" dans le contexte de la cartographie génétique et de l'analyse des maladies héréditaires. Par exemple, dans l'étude des maladies monogéniques (causées par une seule mutation génétique), les scientifiques peuvent rechercher un locus spécifique sur un chromosome qui est associé à la maladie en question.
Il convient de noter que plusieurs gènes ou marqueurs moléculaires peuvent être localisés au même locus génétique, ce qui peut compliquer l'analyse et l'interprétation des résultats. De plus, les variations dans ces régions peuvent entraîner des différences phénotypiques entre les individus, telles que la couleur des yeux, la taille ou le risque de développer certaines maladies.
Les tumeurs ovariennes sont des croissances anormales qui se forment dans les ovaires. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs ovariennes peuvent être classées en fonction de leur type cellulaire, y compris les tumeurs épithéliales, les tumeurs stromales et les tumeurs germinales.
Les tumeurs épithéliales sont le type le plus courant de tumeur ovarienne et se développent à partir des cellules qui recouvrent la surface de l'ovaire. Les tumeurs épithéliales peuvent être bénignes ou malignes, et les formes malignes peuvent être classées en fonction de leur agressivité et de leur potentiel de propagation.
Les tumeurs stromales sont des tumeurs qui se développent à partir des cellules du tissu conjonctif de l'ovaire, y compris les cellules qui produisent les hormones sexuelles. La plupart des tumeurs stromales sont bénignes, mais certaines peuvent être malignes et se propager à d'autres parties du corps.
Les tumeurs germinales sont des tumeurs qui se développent à partir des cellules qui produisent les ovules. Elles sont généralement rares et ont tendance à se produire chez les femmes plus jeunes. Les tumeurs germinales peuvent être bénignes ou malignes, et certaines formes peuvent produire des quantités excessives d'hormones sexuelles.
Les symptômes des tumeurs ovariennes peuvent inclure des douleurs abdominales ou pelviennes, des ballonnements, une sensation de satiété rapide, des changements dans les habitudes intestinales ou urinaires, et des saignements vaginaux anormaux. Cependant, de nombreuses tumeurs ovariennes ne présentent aucun symptôme précoce et sont découvertes lors d'examens de routine ou lorsqu'elles ont déjà atteint une taille importante.
Le traitement des tumeurs ovariennes dépend du type, de la taille et de l'étendue de la tumeur, ainsi que de l'âge et de l'état de santé général de la patiente. Les options de traitement peuvent inclure une chirurgie pour enlever la tumeur, une chimiothérapie ou une radiothérapie pour détruire les cellules cancéreuses, et des médicaments hormonaux pour contrôler la production d'hormones sexuelles. Dans certains cas, une surveillance attentive peut être recommandée pour surveiller l'évolution de la tumeur.
Un gène dupliqué, dans le contexte de la génétique, fait référence à une situation où un segment d'ADN contenant un gène spécifique est copié une ou plusieurs fois, ce qui entraîne la présence de deux ou plusieurs copies du même gène sur un seul chromosome ou sur différents chromosomes. Cette duplication peut être le résultat de divers mécanismes, tels que les translocations chromosomiques, les inversions chromosomiques ou les réplications d'ADN défectueuses.
Les gènes dupliqués peuvent avoir différents effets sur l'expression des gènes et la fonction génétique. Dans certains cas, les copies supplémentaires de gènes peuvent être silencieuses ou non fonctionnelles, ce qui n'a pas d'impact significatif sur l'organisme. Cependant, dans d'autres situations, les gènes dupliqués peuvent entraîner une augmentation de la production de protéines, ce qui peut conduire à des avantages évolutifs ou, inversement, à des maladies génétiques.
Les gènes dupliqués sont considérés comme un moteur important de l'évolution moléculaire et de la diversification des génomes. Ils offrent une source de matériel génétique supplémentaire qui peut être soumis à des mutations aléatoires, conduisant éventuellement à l'apparition de nouvelles fonctions ou à des adaptations utiles pour l'organisme. Toutefois, les gènes dupliqués peuvent également être associés à des maladies génétiques, telles que les troubles du développement, les maladies neurodégénératives et certains types de cancer, lorsque la régulation de l'expression des gènes est altérée ou lorsque les copies supplémentaires entraînent une production excessive de protéines toxiques.
La diploidie est un terme utilisé en génétique et en cytologie pour décrire la condition dans laquelle une cellule contient deux jeux complets de chromosomes. Dans la plupart des espèces, les cellules somatiques (cellules du corps qui ne sont pas des cellules reproductives) sont diploïdes et possèdent deux copies de chaque chromosome, une copie héritée de chaque parent.
Chez l'homme, par exemple, les cellules somatiques normales contiennent 46 chromosomes au total, organisés en 23 paires. Les cellules reproductives (ovules et spermatozoïdes) sont des exceptions à cette règle, car elles ne possèdent qu'un seul jeu de chromosomes, ce qui les rend haploïdes.
La diploidie est un état normal pour la plupart des cellules somatiques et est essentielle au bon fonctionnement de l'organisme. Cependant, certaines conditions médicales peuvent entraîner une altération du nombre de chromosomes dans les cellules, ce qui peut entraîner des problèmes de santé graves. Par exemple, la trisomie 21 (syndrome de Down) est une condition dans laquelle une personne a trois copies du chromosome 21 au lieu des deux normales, ce qui entraîne un ensemble de caractéristiques physiques et développementales uniques.
Les maladies génétiques congénitales sont des affections médicales présentes dès la naissance, causées par des anomalies dans les gènes ou le matériel chromosomique. Ces anomalies peuvent être héritées des parents (maladies génétiques héréditaires) ou résulter de mutations spontanées qui se produisent pendant la formation des ovules, des spermatozoïdes ou très tôt dans le développement embryonnaire.
Les maladies génétiques congénitales peuvent affecter n'importe quelle partie du corps et entraîner une grande variété de symptômes, allant d'une légère incapacité à une invalidité sévère ou même mettre la vie en danger. Certaines maladies génétiques congénitales sont apparentes à la naissance, tandis que d'autres ne deviennent évidentes qu'à mesure que l'enfant se développe et grandit.
Les exemples de maladies génétiques congénitales comprennent la fibrose kystique, la mucoviscidose, la drépanocytose, la phénylcétonurie (PKU), la trisomie 21 (syndrome de Down), le nanisme et certaines formes d'infirmité motrice cérébrale. Le traitement de ces maladies dépend du type spécifique de l'affection et peut inclure des médicaments, une thérapie, une intervention chirurgicale ou un régime alimentaire spécial. Dans certains cas, il n'existe pas encore de traitement curatif pour ces maladies, mais la recherche se poursuit activement dans le but d'améliorer les soins et les résultats pour les personnes atteintes de ces affections.
Les techniques génétiques sont des procédures et méthodes scientifiques spécialisées utilisées dans le domaine de la génétique pour étudier, manipuler et comprendre l'information génétique. Elles comprennent une variété de processus allant de l'analyse de l'ADN et de l'ARN à la modification directe du matériel génétique.
Certaines de ces techniques incluent :
1. La séquençage de l'ADN : Il s'agit d'une méthode permettant de déterminer l'ordre des nucléotides dans une molécule d'ADN. Cela révèle la séquence exacte du code génétique, offrant ainsi des informations précieuses sur les gènes et les mutations associées à diverses maladies.
2. La PCR (Polymerase Chain Reaction) : Cette technique permet d'amplifier rapidement et spécifiquement une petite quantité d'ADN en plusieurs millions de copies. Elle est largement utilisée dans la recherche, le diagnostic et les applications médico-légales.
3. L'analyse des ARN : Elle consiste à étudier l'ARN (acide ribonucléique), une molécule essentielle au processus de traduction du code génétique en protéines. Cette analyse peut fournir des informations sur les niveaux d'expression des gènes et aider à identifier les voies moléculaires impliquées dans divers processus biologiques et maladies.
4. L'édition de gènes : Il s'agit d'une technique permettant de modifier directement le matériel génétique en insérant, supprimant ou remplaçant des séquences spécifiques d'ADN dans le génome. Les outils couramment utilisés pour l'édition de gènes comprennent CRISPR-Cas9 et TALENs (Transcription Activator-Like Effector Nucleases).
5. La thérapie génique : Elle consiste à introduire des gènes fonctionnels dans les cellules d'un patient pour traiter ou prévenir une maladie. Cette approche peut être utilisée pour remplacer un gène défectueux, augmenter la production d'une protéine manquante ou inhiber l'expression d'une protéine nocive.
6. La biobanque : Elle désigne une collection de matériel biologique (tissus, sang, ADN) et de données associées qui sont stockés et utilisés à des fins de recherche. Les biobanques permettent aux chercheurs d'accéder à de vastes ensembles de données et de matériel pour étudier les maladies et développer de nouvelles thérapies.
7. La génomique des populations : Elle consiste à analyser le génome complet (génome entier) d'un grand nombre d'individus au sein d'une population donnée. Cette approche permet d'identifier les variations génétiques courantes et rares, ainsi que leur distribution dans la population.
8. La pharmacogénomique : Elle désigne l'utilisation de l'information génétique pour prédire la réponse d'un patient à un médicament donné. Cette approche permet de personnaliser le traitement en fonction du profil génétique du patient, améliorant ainsi son efficacité et réduisant les risques d'effets indésirables.
9. La médecine de précision : Elle consiste à utiliser l'information génétique, moléculaire et clinique pour personnaliser le diagnostic, le traitement et le suivi des patients atteints de maladies complexes. Cette approche vise à améliorer les résultats thérapeutiques en prenant en compte les caractéristiques uniques de chaque patient.
10. La génétique comportementale : Elle désigne l'étude des liens entre les variations génétiques et le comportement humain, y compris la cognition, l'humeur, la personnalité et les troubles psychiatriques. Cette approche vise à identifier les facteurs génétiques qui contribuent au risque de développer ces conditions et à élaborer des stratégies de prévention et de traitement plus efficaces.
Les tumeurs du cervelet sont des growths anormaux qui se développent dans ou sur le cervelet, qui est la partie du cerveau située à la base du crâne et responsable du contrôle de la coordination musculaire, du maintien de l'équilibre et du traitement des informations sensorielles. Ces tumeurs peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses) et peuvent entraîner une variété de symptômes, en fonction de leur taille, de leur emplacement et de leur croissance.
Les types courants de tumeurs du cervelet comprennent les médulloblastomes, les astrocytomes, les épendymomes et les hémangioblastomes. Les symptômes peuvent inclure des maux de tête, des nausées, des vomissements, une perte d'équilibre, une faiblesse musculaire, des troubles de la parole et de la déglutition, ainsi que des changements cognitifs ou émotionnels.
Le traitement dépend du type, de la taille et de l'emplacement de la tumeur, ainsi que de l'âge et de l'état général du patient. Les options de traitement peuvent inclure une chirurgie pour enlever la tumeur, une radiothérapie pour détruire les cellules cancéreuses, et/ou une chimiothérapie pour ralentir ou arrêter la croissance de la tumeur. La réadaptation peut également être nécessaire pour aider le patient à retrouver des fonctions normales après le traitement.
Le mésothéliome est un type rare et agressif de cancer qui se développe dans la membrane protectrice (la mésothélium) qui recouvre les poumons, le cœur, l'estomac et d'autres organes internes. La majorité des cas de mésothéliome sont liés à une exposition antérieure à l'amiante.
Il existe plusieurs types de mésothéliomes, mais le plus courant est le mésothéliome pleural, qui affecte les membranes entourant les poumons. Les symptômes peuvent inclure une douleur thoracique, une toux persistante, un essoufflement, une perte de poids et une accumulation de liquide dans la cavité thoracique (dite épanchement pleural).
Le diagnostic de mésothéliome est souvent difficile car les symptômes peuvent être similaires à ceux d'autres maladies pulmonaires. Des examens d'imagerie, tels que des radiographies ou des tomodensitométries, ainsi qu'une biopsie sont généralement nécessaires pour confirmer le diagnostic.
Le traitement du mésothéliome peut inclure une chirurgie, une chimiothérapie et/ou une radiothérapie, en fonction du stade et de l'emplacement de la tumeur. Malheureusement, le pronostic pour les personnes atteintes de mésothéliome est généralement mauvais, avec un taux de survie à cinq ans inférieur à 10%.
Il est important de noter que l'exposition à l'amiante est la principale cause de mésothéliome et qu'il existe des lois et réglementations pour protéger les travailleurs contre l'exposition à ce matériau dangereux. Si vous pensez avoir été exposé à l'amiante, il est important de consulter un médecin pour un dépistage et une surveillance régulière de votre santé pulmonaire.
Un méningiome est un type de tumeur non cancéreuse (bénigne) qui se développe à partir des membranes qui entourent le cerveau et la moelle épinière (les méninges). Ces tumeurs grossissent généralement lentement et peuvent devenir assez grandes avant de présenter des symptômes. Les symptômes dépendent de la localisation et de la taille de la tumeur mais peuvent inclure des maux de tête, des convulsions, des problèmes de vision, des troubles auditifs, des difficultés à avaler ou des faiblesses musculaires. Dans certains cas, les méningiomes ne causent aucun symptôme et sont découverts par hasard lors d'examens d'imagerie pour d'autres problèmes de santé. Le traitement dépend de la taille, de la localisation et des symptômes de la tumeur. Il peut inclure une surveillance attentive, une radiothérapie ou une chirurgie pour enlever la tumeur.
La séquentielle acide nucléique homologie (SANH) est un concept dans la biologie moléculaire qui décrit la similarité ou la ressemblance dans la séquence de nucléotides entre deux ou plusieurs brins d'acide nucléique (ADN ou ARN). Cette similitude peut être mesurée et exprimée en pourcentage, représentant le nombre de nucléotides correspondants sur une certaine longueur de la séquence.
La SANH est souvent utilisée dans l'étude de l'évolution moléculaire, où elle peut indiquer une relation évolutive entre deux organismes ou gènes. Plus la similarité de la séquence est élevée, plus les deux séquences sont susceptibles d'avoir un ancêtre commun récent.
Dans le contexte médical, la SANH peut être utilisée pour diagnostiquer des maladies génétiques ou infectieuses. Par exemple, l'analyse de la SANH entre un échantillon inconnu et une base de données de séquences connues peut aider à identifier le pathogène responsable d'une infection. De même, la comparaison de la séquence d'un gène suspect dans un patient avec des séquences normales peut aider à détecter les mutations associées à une maladie génétique particulière.
Cependant, il est important de noter que la SANH seule ne suffit pas pour établir une relation évolutive ou diagnostiquer une maladie. D'autres facteurs tels que la longueur de la séquence comparée, le contexte biologique et les preuves expérimentales doivent également être pris en compte.
Les tumeurs des gaines nerveuses, également connues sous le nom de tumeurs des gaines de Schwann ou neurinomes, sont des types de tumeurs benignes qui se développent à partir de la gaine de myéline entourant les nerfs périphériques. Elles peuvent se produire n'importe où dans le corps où il y a des nerfs périphériques, mais elles sont le plus souvent trouvées sur le nerf vestibulocochléaire dans l'oreille interne (schwannome vestibulaire) ou sur les membres.
Les symptômes dépendent de la localisation et de la taille de la tumeur, mais peuvent inclure des douleurs, des engourdissements, des faiblesses musculaires, des picotements ou un manque de coordination dans la zone innervée par le nerf affecté. Dans certains cas, les tumeurs peuvent devenir assez grandes pour exercer une pression sur les structures voisines, entraînant des symptômes supplémentaires.
Le traitement dépend de la taille et de l'emplacement de la tumeur, ainsi que des symptômes associés. Dans certains cas, une simple observation peut être recommandée si la tumeur est petite et ne cause pas de symptômes. Cependant, si la tumeur continue à se développer ou provoque des symptômes invalidants, une intervention chirurgicale peut être nécessaire pour enlever la tumeur. La radiothérapie peut également être utilisée dans certains cas pour aider à réduire la taille de la tumeur.
Le gène P16, également connu sous le nom de CDKN2A ou INK4a, est un gène suppresseur de tumeurs situé sur le chromosome 9 humain. Il code pour une protéine qui joue un rôle crucial dans la régulation du cycle cellulaire en inhibant l'activité des kinases cyclines dépendantes, ce qui entraîne l'arrêt de la phase G1 du cycle cellulaire et empêche ainsi la division cellulaire désordonnée et non contrôlée.
Des mutations ou une hyperméthylation du promoteur de ce gène peuvent entraîner une inactivation du gène P16, ce qui peut contribuer au développement de divers types de cancer, tels que le cancer du poumon, de la tête et du cou, de l'ovaire et de la peau. Par conséquent, la détection des altérations du gène P16 est importante dans le diagnostic, le pronostic et le traitement du cancer.
Le neuroblastome est une tumeur maligne rare qui se développe à partir de cellules nerveuses immatures (cellules neuroblastiques) trouvées dans les ganglions sympathiques, qui sont des groupes de cellules nerveuses situés le long de la colonne vertébrale. Les ganglions sympathiques font partie du système nerveux sympathique, qui est responsable de notre réaction "combat ou fuite" face au danger.
Le neuroblastome se produit généralement dans les glandes surrénales, deux petites glandes situées juste au-dessus des reins, mais il peut également se développer dans d'autres parties du système nerveux sympathique le long de la colonne vertébrale.
Les neuroblastomes peuvent se propager (métastaser) à d'autres parties du corps, y compris les os, la peau, le foie et les ganglions lymphatiques. Les symptômes dépendent de l'emplacement et de la taille de la tumeur ainsi que de la propagation du cancer.
Les neuroblastomes sont généralement diagnostiqués chez les enfants de moins de 5 ans, bien qu'ils puissent se produire à tout âge. Le traitement dépend du stade et de la gravité du cancer, ainsi que de l'âge et de l'état de santé général de l'enfant. Les options de traitement peuvent inclure une chirurgie pour enlever la tumeur, une chimiothérapie, une radiothérapie et des thérapies ciblées qui attaquent spécifiquement les cellules cancéreuses.
Les tumeurs rénales sont des croissances anormales dans ou sur les reins. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs rénales bénignes ne se propagent pas généralement à d'autres parties du corps et peuvent ne pas nécessiter de traitement, selon leur taille et leur localisation. Cependant, certaines tumeurs rénales bénignes peuvent causer des problèmes si elles pressent ou endommagent les tissus environnants.
Les tumeurs rénales malignes, également connues sous le nom de cancer du rein, se développent dans les cellules du rein et peuvent se propager à d'autres parties du corps. Le type le plus courant de cancer du rein est le carcinome à cellules rénales, qui représente environ 80 à 85% des cas. D'autres types comprennent le sarcome du rein, le lymphome du rein et le cancer des cellules transitionnelles du haut appareil urinaire.
Les facteurs de risque de développer un cancer du rein comprennent le tabagisme, l'obésité, l'hypertension artérielle, l'exposition à certaines substances chimiques et les antécédents familiaux de cancer du rein. Les symptômes peuvent inclure du sang dans les urines, des douleurs au dos ou aux flancs, une perte de poids inexpliquée, une fièvre persistante et une fatigue extrême. Le traitement dépend du stade et du grade de la tumeur, ainsi que de la santé globale du patient. Les options de traitement peuvent inclure la chirurgie, la radiothérapie, la chimiothérapie et l'immunothérapie.
Le mosaïcisme est un terme utilisé en génétique pour décrire la présence de deux ou plusieurs populations de cellules avec des génotypes différents dans un même individu. Cela se produit lorsqu'un événement mutationnel, comme une mutation génétique ou un réarrangement chromosomique, se produit après la fécondation et affecte seulement certaines cellules de l'organisme en développement.
Par exemple, dans le cas d'une mutation génétique, certaines cellules peuvent contenir la version mutée du gène, tandis que d'autres cellules peuvent contenir la version non-mutée du gène. Le mosaïcisme peut affecter n'importe quelle partie de l'organisme, y compris les tissus somatiques (corps) et germinaux (gamètes).
Le mosaïcisme peut être à l'origine de variations subtiles dans les caractéristiques physiques ou des différences plus importantes dans la fonction et la structure des organes. Dans certains cas, le mosaïcisme peut entraîner des troubles génétiques ou prédisposer une personne à certaines maladies. Cependant, dans d'autres cas, le mosaïcisme peut ne pas avoir de conséquences cliniquement significatives.
Le carcinome hépatocellulaire (CHC) est le type le plus commun de cancer primitif du foie, ce qui signifie qu'il se développe à partir des cellules hépatiques (hépatocytes). Cette tumeur maligne se forme généralement dans un foie déjà endommagé par une maladie chronique comme l'hépatite B ou C, la cirrhose alcoolique ou la stéatohépatite non alcoolique.
Le CHC se caractérise par la prolifération anarchique de cellules hépatiques qui forment une masse tumorale. Ces cellules peuvent envahir les tissus avoisinants et se propager à d'autres parties du corps via la circulation sanguine ou lymphatique, ce qui complique le traitement et réduit les chances de guérison.
Les symptômes du carcinome hépatocellulaire peuvent inclure une perte de poids inexpliquée, une fatigue excessive, une perte d'appétit, des douleurs abdominales, une sensation de plénitude dans le quadrant supérieur droit de l'abdomen, des nausées et des vomissements, une jaunisse (ictère), une ascite (accumulation de liquide dans l'abdomen) et des troubles de la coagulation sanguine.
Le diagnostic du CHC repose sur des examens d'imagerie médicale tels que l'échographie, la tomographie computérisée (CT scan) ou l'imagerie par résonance magnétique (IRM). Dans certains cas, une biopsie peut être nécessaire pour confirmer le diagnostic et déterminer le type de cellules cancéreuses.
Le traitement du carcinome hépatocellulaire dépend de plusieurs facteurs, tels que l'étendue de la maladie, la fonction hépatique, l'état général du patient et les comorbidités existantes. Les options thérapeutiques comprennent la chirurgie (résection hépatique ou transplantation hépatique), la radiothérapie, la chimiothérapie, l'ablation par radiofréquence, la cryoablation et les thérapies ciblées. Dans certains cas, une combinaison de plusieurs traitements peut être proposée pour améliorer les chances de guérison ou de contrôle de la maladie.
Les tumeurs des méninges sont des growths anormaux qui se développent dans les membranes protectrices (les méninges) qui enveloppent le cerveau et la moelle épinière. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les symptômes dépendent de la taille et de l'emplacement de la tumeur, mais peuvent inclure des maux de tête, des nausées, des vomissements, des convulsions, des changements de comportement ou de personnalité, des problèmes d'équilibre et de coordination, une faiblesse musculaire ou une paralysie. Le traitement dépend du type et de l'emplacement de la tumeur et peut inclure une chirurgie, une radiothérapie et/ou une chimiothérapie.
L'analyse de survie est une méthode statistique utilisée dans l'évaluation de la durée de temps jusqu'à un événement particulier, tel que la récidive d'une maladie ou le décès. Elle permet de déterminer et de comparer la probabilité de survie entre différents groupes de patients, tels que ceux traités par différents protocoles thérapeutiques.
L'analyse de survie prend en compte les données censurées, c'est-à-dire les cas où l'événement n'a pas été observé pendant la durée de l'étude. Cette méthode permet d'utiliser des courbes de survie pour visualiser et comparer les résultats entre différents groupes.
Les courbes de survie peuvent être présentées sous forme de graphiques Kaplan-Meier, qui montrent la probabilité cumulative de survie au fil du temps. Les tests statistiques tels que le test log-rank peuvent être utilisés pour comparer les différences entre les courbes de survie de différents groupes.
L'analyse de survie est largement utilisée dans la recherche médicale, en particulier dans l'évaluation des traitements du cancer et d'autres maladies graves. Elle permet aux chercheurs de déterminer l'efficacité relative des différents traitements et de comparer les risques et les avantages associés à chacun.
Les tumeurs de l'estomac, également connues sous le nom de tumeurs gastriques, se réfèrent à des growths anormaux dans la muqueuse de l'estomac. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses).
Les tumeurs bénignes comprennent les polypes gastriques, les leiomyomes et les lipomes. Les polypes gastriques sont des growths sur la muqueuse de l'estomac qui peuvent devenir cancéreux s'ils ne sont pas enlevés. Les leiomyomes sont des tumeurs des muscles lisses de l'estomac, et les lipomes sont des tumeurs graisseuses.
Les tumeurs malignes, ou cancers gastriques, peuvent se propager à d'autres parties du corps et sont souvent fatales. Les adénocarcinomes sont le type le plus courant de cancer de l'estomac et se développent à partir des cellules glandulaires de la muqueuse de l'estomac. D'autres types de tumeurs malignes comprennent les lymphomes, les sarcomes, et les carcinoïdes.
Les facteurs de risque de développement d'un cancer de l'estomac incluent l'infection à Helicobacter pylori, le tabagisme, une alimentation riche en aliments salés ou fumés, un faible apport en fruits et légumes, une histoire de maladie de reflux gastro-oesophagien (RGO), et des antécédents familiaux de cancer gastrique. Les symptômes peuvent inclure des douleurs abdominales, des nausées et des vomissements, une perte d'appétit, une perte de poids involontaire, et une sensation de satiété après avoir mangé seulement de petites quantités.
Les protéines oncogènes sont des protéines qui, lorsqu'elles sont surexprimées ou mutées, peuvent contribuer au développement du cancer. Elles jouent un rôle crucial dans la régulation de la croissance et de la division cellulaires. Cependant, certaines modifications de ces gènes peuvent entraîner une production excessive de protéines oncogènes ou des protéines qui fonctionnent de manière anormale. Cela peut conduire à une multiplication et une division cellulaires incontrôlées, ce qui est caractéristique d'un cancer. Les protéines oncogènes peuvent provenir de gènes normaux (proto-oncogènes) qui deviennent anormaux en raison de mutations, ou elles peuvent être issues de virus cancérigènes.
Une métastase tumorale se réfère à la propagation de cancer à partir d'un site primaire (où le cancer a commencé) vers un autre organe ou tissu distant dans le corps. Cela se produit généralement lorsque les cellules cancéreuses se détachent du site tumoral initial, pénètrent dans la circulation sanguine ou lymphatique, et migrent ensuite pour former une nouvelle tumeur dans un autre endroit.
Les métastases peuvent affecter pratiquement n'importe quel organe du corps, mais elles sont le plus souvent trouvées dans les poumons, le foie, les os et le cerveau. Les symptômes de métastase dépendent de l'emplacement et de la taille de la tumeur secondaire. Par exemple, une métastase au cerveau peut causer des maux de tête, des convulsions ou des problèmes de vision, tandis qu'une métastase osseuse peut entraîner des douleurs osseuses et une augmentation du risque de fractures.
Le diagnostic d'une métastase tumorale implique généralement des examens d'imagerie tels que la tomodensitométrie (TDM), l'imagerie par résonance magnétique (IRM) ou la scintigraphie osseuse, ainsi qu'éventuellement une biopsie pour confirmer le diagnostic et déterminer le type de cancer. Le traitement des métastases peut inclure une chimiothérapie, une radiothérapie, une thérapie ciblée ou une intervention chirurgicale, en fonction du type de cancer, de l'emplacement et de l'étendue des métastases.
ERBB-2, également connu sous le nom de HER2/neu, est un gène qui produit une protéine appelée récepteur 2 du facteur de croissance épidermique humain (HER2/neu). Cette protéine se trouve à la surface des cellules et joue un rôle important dans la croissance et la division des cellules.
Dans certains types de cancer, en particulier le cancer du sein, ce gène peut être surexprimé ou amplifié, entraînant une production excessive de la protéine HER2/neu. Cette surexpression est associée à un cancer plus agressif et à un pronostic moins favorable.
Le test ERBB-2 est utilisé pour déterminer si un cancer du sein est positif au gène ERBB-2 ou non. Les cancers du sein qui sont positifs pour le gène ERBB-2 peuvent être traités avec des thérapies ciblées, telles que le trastuzumab (Herceptin), qui se lie spécifiquement à la protéine HER2/neu et empêche la croissance et la division des cellules cancéreuses.
La microcéphalie est une condition médicale dans laquelle le périmètre crânien d'un individu est inférieur à deux écarts-types de la moyenne pour l'âge, le sexe et la race. Cela se traduit généralement par une tête anormalement petite et souvent accompagnée d'un retard mental variable, allant de léger à sévère. La microcéphalie peut être causée par des facteurs génétiques ou environnementaux, tels que des infections maternelles pendant la grossesse, une exposition à des toxines ou un manque d'oxygène pendant le développement du fœtus. Dans certains cas, la cause de la microcéphalie est inconnue. Il s'agit d'une affection rare, et son incidence varie selon les populations et les critères diagnostiques utilisés.
Un glioblastome est un type agressif et malin de tumeur cérébrale qui se développe à partir des cellules gliales, qui soutiennent et protègent les neurones dans le cerveau. Les glioblastomes sont classés comme grade IV selon la classification de l'Organisation mondiale de la santé (OMS) pour les tumeurs du système nerveux central.
Ces tumeurs se caractérisent par une croissance rapide, une vascularisation abondante et une infiltration profonde dans les tissus cérébraux environnants. Elles sont composées de plusieurs types cellulaires différents, dont des astrocytes anormaux et des vaisseaux sanguins anarchiques.
Les glioblastomes peuvent survenir à tout âge mais sont plus fréquents chez les adultes entre 45 et 70 ans. Les symptômes courants incluent des maux de tête sévères, des nausées, des vomissements, des convulsions, une perte d'équilibre, une faiblesse musculaire, une confusion mentale et des changements de personnalité ou de comportement.
Le traitement standard consiste généralement en une chirurgie suivie de radiothérapie et/ou de chimiothérapie. Cependant, le pronostic est souvent défavorable, avec une survie médiane d'environ 15 mois après le diagnostic. La récidive est fréquente malgré le traitement initial en raison de la nature infiltrante de ces tumeurs.
Je suis désolé, mais la phrase "Cadre Lecture Ouvert" ne correspond pas à une définition médicale établie. Le terme "cadre de lecture ouvert" peut être utilisé dans un contexte littéraire ou universitaire pour décrire un cadre théorique ou conceptuel qui est large et inclusif, permettant différentes interprétations et perspectives.
Cependant, si vous cherchez des informations médicales sur un sujet spécifique, n'hésitez pas à me fournir plus de détails et je ferai de mon mieux pour vous aider.
En biologie et en médecine, l'interphase est la phase du cycle cellulaire durant laquelle la cellule prépare la division cellulaire suivante (mitose ou méiose) en répliquant son matériel génétique et en assurant sa croissance. Cette phase représente la période la plus longue du cycle cellulaire, occupant environ 90% du temps total.
Au cours de l'interphase, le chromosome se décondense et devient invisible sous un microscope optique. Le matériel génétique, constitué d'ADN et de protéines histones, forme une structure appelée chromatine qui occupe la majeure partie du noyau cellulaire.
L'interphase est divisée en trois sous-phases :
1. Phase G1 (Gap 1) : C'est la première phase de l'interphase, où la cellule se prépare à la réplication de son ADN. Elle connaît une croissance active et synthétise des protéines et d'autres molécules nécessaires à sa survie et à sa division.
2. Phase S (Synthesis) : Durant cette phase, le matériel génétique est répliqué de manière exacte pour assurer la transmission fidèle de l'information génétique aux cellules filles. Les chromosomes sont dupliqués et deviennent des structures diploïdes composées de deux chromatides sœurs identiques liées par le centromère.
3. Phase G2 (Gap 2) : La dernière phase de l'interphase est consacrée à la préparation de la division cellulaire suivante. Les cellules synthétisent des protéines et des organites supplémentaires, réparent d'éventuels dommages à l'ADN et s'assurent que tous les systèmes sont en place pour assurer une division cellulaire réussie.
Après la phase G2, la cellule entre dans la prophase, marquant le début de la mitose ou de la méiose, selon le type de division cellulaire.
La chaîne de Markov est un concept utilisé en probabilité et statistique, qui est parfois appliqué dans le domaine médical pour modéliser des processus complexes et aléatoires. Un processus de Markov est un type de processus stochastique qui évolue au fil du temps en passant d'un état à un autre selon une certaine probabilité.
Dans le contexte médical, la chaîne de Markov peut être utilisée pour décrire l'évolution d'une maladie ou d'un état de santé au fil du temps, en prenant en compte les différents facteurs qui peuvent influencer cette évolution. Par exemple, un modèle de chaîne de Markov peut être utilisé pour prédire l'évolution de la maladie d'Alzheimer chez une personne âgée, en tenant compte de son état cognitif actuel, de son âge, de ses antécédents médicaux et d'autres facteurs pertinents.
Dans un modèle de chaîne de Markov, chaque état de santé est représenté par un nœud dans le graphe, et les probabilités de transition entre ces états sont représentées par des arcs reliant les nœuds. Les probabilités de transition peuvent être estimées à partir de données empiriques ou théoriques, en fonction de la complexité du modèle et de la disponibilité des données.
Il est important de noter que la chaîne de Markov repose sur l'hypothèse de Markov forte, qui stipule que la probabilité de transition entre deux états ne dépend que de l'état actuel et non des états précédents. Cette hypothèse peut être restrictive dans certains contextes médicaux, où les antécédents peuvent avoir une influence importante sur l'évolution future de la maladie.
En résumé, la chaîne de Markov est un outil probabiliste utilisé en médecine pour modéliser et prédire l'évolution des maladies en fonction de différents facteurs pertinents. Bien que cette méthode repose sur certaines hypothèses simplificatrices, elle peut être utile dans de nombreux contextes où les données sont limitées ou incertaines.
Un allèle est une forme alternative d'un gène donné qui est localisé à la même position (locus) sur un chromosome homologue. Les allèles peuvent produire des protéines ou des ARNm avec des séquences différentes, entraînant ainsi des différences phénotypiques entre les individus.
Les gènes sont des segments d'ADN qui contiennent les instructions pour la production de protéines spécifiques ou pour la régulation de l'expression génique. Chaque personne hérite de deux copies de chaque gène, une copie provenant de chaque parent. Ces deux copies peuvent être identiques (homozygotes) ou différentes (hétérozygotes).
Les allèles différents peuvent entraîner des variations subtiles dans la fonction protéique, ce qui peut se traduire par des différences phénotypiques entre les individus. Certaines de ces variations peuvent être bénéfiques, neutres ou préjudiciables à la santé et à la survie d'un organisme.
Les allèles sont importants en génétique car ils permettent de comprendre comment des caractères héréditaires sont transmis d'une génération à l'autre, ainsi que les mécanismes sous-jacents aux maladies génétiques et aux traits complexes.
Le chondrosarcome est un type rare de cancer qui se développe dans le cartilage, un tissu conjonctif flexible qui recouvre et protège la plupart des extrémités osseuses. Ce cancer se produit généralement dans les os pelviens, la base du crâne, les côtes ou les membres longs.
Les chondrosarcomes sont classés en fonction de leur apparence au microscope et comprennent plusieurs sous-types, notamment:
1. Chondrosarcome conventionnel : C'est le type le plus courant et il se développe généralement dans les os des membres longs ou du bassin. Il est divisé en trois grades (I, II, III) basés sur son agressivité et sa vitesse de croissance.
2. Chondrosarcome dédifférencié : Ce sous-type se produit lorsqu'un chondrosarcome conventionnel devient plus agressif et envahit les tissus environnants. Il est considéré comme un cancer de haut grade et a un pronostic moins favorable.
3. Chondrosarcome mésenchymateux : C'est un type rare qui se produit principalement chez les enfants et les jeunes adultes. Il peut se développer dans n'importe quel os, mais il est plus fréquent dans la mâchoire inférieure (mandibule) et le bassin.
4. Chondrosarcome à cellules claires : Ce sous-type est également rare et a tendance à se produire chez les adultes plus âgés. Il peut affecter n'importe quel os, mais il est plus fréquent dans la colonne vertébrale et le bassin.
Les symptômes du chondrosarcome peuvent inclure une masse ou un gonflement douloureux dans la zone touchée, des douleurs articulaires, des fractures osseuses spontanées et une limitation de la mobilité. Le diagnostic est généralement posé après avoir effectué une biopsie pour examiner les tissus affectés. Le traitement dépend du stade et du type de cancer, mais il peut inclure une chirurgie pour enlever la tumeur, une radiothérapie ou une chimiothérapie.
L'exome est la partie de l'ADN humain qui code les protéines, représentant environ 1% du génome entier. Il se compose d'environ 22 000 gènes et contient environ 85% des mutations associées aux maladies héréditaires. L'exome est souvent séquencé dans le cadre de l'analyse génétique pour identifier les variantes qui peuvent être à l'origine de certaines maladies ou conditions médicales.
Les tumeurs des tissus mous sont des growths anormales qui se développent dans les muscles, les tendons, les ligaments, les nerfs, les graisses et les fibres conjonctives qui forment la majorité du corps humain. Ces tumeurs peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs bénignes ne se propagent généralement pas à d'autres parties du corps et sont moins susceptibles de réapparaître après le traitement. D'autre part, les tumeurs malignes peuvent envahir les tissus voisins et se propager à d'autres parties du corps, ce qui rend le traitement plus difficile.
Les symptômes des tumeurs des tissus mous dépendent de la taille et de l'emplacement de la tumeur. Certaines personnes peuvent ne présenter aucun symptôme, tandis que d'autres peuvent ressentir une masse ou un gonflement dans la région affectée, des douleurs, des picotements, des faiblesses ou une limitation de la mobilité.
Le traitement dépend du type et du stade de la tumeur. Les options de traitement peuvent inclure la chirurgie, la radiothérapie, la chimiothérapie ou une combinaison de ces méthodes. Dans certains cas, une surveillance attentive peut être recommandée si la tumeur est bénigne et ne provoque aucun symptôme.
Il est important de noter que les tumeurs des tissus mous peuvent se développer n'importe où dans le corps, y compris dans les zones difficiles à atteindre ou à traiter. Par conséquent, il est crucial de consulter un médecin dès que possible si vous remarquez des symptômes suspects, tels qu'une masse ou une grosseur inhabituelle, des douleurs ou une limitation de la mobilité.
Les facteurs de transcription sont des protéines qui régulent l'expression des gènes en se liant aux séquences d'ADN spécifiques, appelées éléments de réponse, dans les régions promotrices ou enhancers des gènes. Ces facteurs peuvent activer ou réprimer la transcription des gènes en recrutant ou en éloignant d'autres protéines impliquées dans le processus de transcription, y compris l'ARN polymérase II, qui synthétise l'ARN messager (ARNm). Les facteurs de transcription peuvent être régulés au niveau de leur activation, de leur localisation cellulaire et de leur dégradation, ce qui permet une régulation complexe et dynamique de l'expression des gènes en réponse à différents signaux et stimuli cellulaires. Les dysfonctionnements des facteurs de transcription ont été associés à diverses maladies, y compris le cancer et les maladies neurodégénératives.
Les protéines fixant l'ADN, également connues sous le nom de protéines liant l'ADN ou protéines nucléaires, sont des protéines qui se lient spécifiquement à l'acide désoxyribonucléique (ADN). Elles jouent un rôle crucial dans la régulation de la transcription et de la réplication de l'ADN, ainsi que dans la maintenance de l'intégrité du génome.
Les protéines fixant l'ADN se lient à des séquences d'ADN spécifiques grâce à des domaines de liaison à l'ADN qui reconnaissent et se lient à des motifs particuliers dans la structure de l'ADN. Ces protéines peuvent agir comme facteurs de transcription, aidant à activer ou à réprimer la transcription des gènes en régulant l'accès des polymérases à l'ADN. Elles peuvent également jouer un rôle dans la réparation de l'ADN, en facilitant la reconnaissance et la réparation des dommages à l'ADN.
Les protéines fixant l'ADN sont souvent régulées elles-mêmes par des mécanismes post-traductionnels tels que la phosphorylation, la méthylation ou l'acétylation, ce qui permet de moduler leur activité en fonction des besoins cellulaires. Des anomalies dans les protéines fixant l'ADN peuvent entraîner diverses maladies génétiques et sont souvent associées au cancer.
Le chromosome Y humain est un des deux chromosomes sexuels chez les êtres humains (l'autre étant le chromosome X). Il est présent en double exemplaire chez les hommes, associé à un chromosome X, formant ainsi les chromosomes sexuels XY. Chez la femme, il n'y a qu'un seul chromosome X (XX).
Le chromosome Y humain est beaucoup plus petit que le chromosome X et contient environ 50 à 60 millions de paires de bases. Il code pour moins de 100 gènes, comparativement aux milliers de gènes présents sur le chromosome X.
Le chromosome Y humain est important pour la détermination du sexe masculin et contient des gènes qui sont essentiels au développement des organes reproducteurs mâles, tels que le gène SRY (Sex-determining Region Y). Il joue également un rôle dans la spermatogenèse et la différenciation sexuelle.
Des mutations ou des anomalies sur le chromosome Y peuvent entraîner des troubles de la fertilité, des maladies génétiques rares et d'autres problèmes de santé. Par exemple, une délétion du bras court du chromosome Y peut entraîner un syndrome de dysérérosine, qui se caractérise par une petite taille, une faible masse musculaire, des anomalies génitales et une intelligence inférieure à la moyenne.
Les techniques de typage bactérien sont des méthodes utilisées en microbiologie pour identifier et clasifier les bactéries au-delà du niveau de genre et d'espèce. Elles permettent de distinguer des souches bactériennes similaires mais pas identiques, ce qui est crucial dans la surveillance des maladies infectieuses, l'épidémiologie, le contrôle de la contamination et la recherche.
Plusieurs techniques sont couramment utilisées pour le typage bactérien, y compris :
1. **Sérotypage** : Cette méthode consiste à classer les bactéries en fonction des antigènes présents à leur surface. Les antigènes sont des molécules reconnues par le système immunitaire et qui peuvent déclencher une réponse immune spécifique. Dans le cadre du sérotypage, on utilise des sérums contenant des anticorps spécifiques pour identifier les différents types d'antigènes présents à la surface des bactéries.
2. **Phagotypage** : Cette technique est semblable au sérotypage, mais elle utilise des phages (des virus qui infectent les bactéries) au lieu d'anticorps pour identifier les souches bactériennes. Les phages se lient aux récepteurs spécifiques situés à la surface des bactéries, ce qui permet de distinguer différents types de bactéries.
3. **Bactériophagage** : Cette méthode consiste à utiliser des bactériophages pour infecter et tuer des bactéries spécifiques. Elle est souvent utilisée dans le contrôle de la contamination, en particulier dans l'industrie alimentaire.
4. **Profilage biochimique** : Cette technique consiste à analyser les profils métaboliques des bactéries pour les distinguer. Les bactéries sont incubées dans des milieux contenant différents nutriments et substrats, et on observe leur capacité à dégrader ou à utiliser ces substances pour produire de l'énergie.
5. **Analyse génétique** : Cette méthode consiste à analyser l'ADN des bactéries pour identifier les différences entre les souches. Les techniques d'analyse génétique comprennent la PCR (réaction en chaîne par polymérase), le séquençage de l'ADN et l'hybridation de l'ADN.
6. **Protéomique** : Cette technique consiste à analyser les protéines produites par les bactéries pour identifier les différences entre les souches. Les techniques de protéomique comprennent la spectrométrie de masse et l'analyse des profils d'expression des gènes.
En combinant ces différentes méthodes, il est possible de distinguer et d'identifier avec précision les différents types de bactéries, ce qui est important pour la recherche médicale, la sécurité alimentaire et la lutte contre les maladies infectieuses.
En médecine et en biologie, la virulence d'un agent pathogène (comme une bactérie ou un virus) se réfère à sa capacité à provoquer des maladies chez un hôte. Plus précisément, elle correspond à la quantité de toxines sécrétées par l'agent pathogène ou au degré d'invasivité de celui-ci dans les tissus de l'hôte. Une souche virulente est donc capable d'entraîner des symptômes graves, voire fatals, contrairement à une souche moins virulente qui peut ne provoquer qu'une infection bénigne ou asymptomatique.
Il est important de noter que la virulence n'est pas un attribut fixe et immuable d'un agent pathogène ; elle peut varier en fonction de divers facteurs, tels que les caractéristiques propres de l'hôte (son âge, son état immunitaire, etc.) et les conditions environnementales dans lesquelles se déroule l'infection. Par ailleurs, la virulence est un concept distinct de la contagiosité, qui renvoie à la facilité avec laquelle un agent pathogène se transmet d'un hôte à un autre.
Une empreinte génétique, également appelée profilage ADN ou analyse de l'ADN, est une méthode d'identification individuelle basée sur l'analyse des séquences répétitives uniques et variables dans l'ADN. Cela implique l'examen des régions non codantes du génome, appelées marqueurs génétiques, qui se trouvent dans presque tous les noyaux cellulaires. Les modèles de ces marqueurs varient d'une personne à l'autre (sauf dans le cas de jumeaux identiques), ce qui permet une identification précise et fiable d'un individu.
L'empreinte génétique est largement utilisée en médecine légale pour aider à identifier des suspects ou des victimes dans les affaires criminelles, ainsi que dans la recherche médicale et biologique pour étudier l'hérédité, la parenté et d'autres caractéristiques génétiques. Il est important de noter que le processus d'obtention d'une empreinte génétique nécessite généralement un échantillon de cellules, comme du sang, de la salive ou des cheveux, et doit être effectué dans des laboratoires spécialisés avec un équipement approprié et des techniciens qualifiés.
Je suis désolé, mais il semble y avoir une certaine confusion dans votre question. Le terme "Interface Utilisateur" est généralement associé au domaine de l'informatique et de la conception de logiciels, plutôt qu'à la médecine.
Une interface utilisateur (IU) est la zone où un humain interagit avec une machine ou un appareil, en l'occurrence, un logiciel. Elle comprend typically the layout of the software's buttons, menus, and other visual elements—like text, images, and sounds—that allow people to interact with electronic devices. Une interface utilisateur bien conçue devrait être intuitive, facile à utiliser et adaptée aux besoins de l'utilisateur.
Si vous cherchez des termes similaires dans le domaine médical, vous pourriez peut-être rechercher des termes tels que "dispositifs médicaux interactifs" ou "systèmes de santé numériques", qui décrivent les technologies conçues spécifiquement pour être utilisées dans un contexte médical et qui comprennent une interface avec laquelle l'utilisateur peut interagir.
Les aberrations des chromosomes sexuels sont des anomalies chromosomiques qui affectent les chromosomes sexuels, X et Y. Les variations les plus courantes concernent le nombre ou la structure de ces chromosomes.
Voici quelques exemples d'aberrations des chromosomes sexuels :
1. Syndrome de Klinefelter : Il s'agit d'une anomalie chromosomique où un individu a au moins une copie supplémentaire du chromosome X, ce qui donne lieu à la formule chromosomique XXY. Les personnes atteintes de cette condition sont généralement masculines, mais présentent des caractéristiques sexuelles secondaires anormales, telles qu'une augmentation de la taille des seins (gynécomastie), une stérilité et un développement musculaire insuffisant.
2. Syndrome de Turner : Dans cette condition, une personne n'a qu'un seul chromosome X, ce qui donne lieu à la formule chromosomique 45,X0. Les personnes atteintes de cette condition sont généralement féminines, mais présentent des caractéristiques sexuelles secondaires anormales, telles qu'un nanisme, un cou court et large, des oreilles bas implantées et une stérilité.
3. Syndrome de Jacob : Il s'agit d'une anomalie chromosomique où un individu a au moins trois chromosomes X, ce qui donne lieu à la formule chromosomique XXX. Les personnes atteintes de cette condition sont généralement féminines, mais présentent des caractéristiques sexuelles secondaires anormales, telles qu'une stérilité et un risque accru de développer des troubles mentaux.
4. Syndrome de superfemme : Dans cette condition, une personne a plus d'un chromosome Y en plus du chromosome X, ce qui donne lieu à des formules chromosomiques telles que XXXY ou XXXXY. Les personnes atteintes de cette condition sont généralement féminines, mais présentent des caractéristiques sexuelles secondaires anormales, telles qu'un nanisme et une stérilité.
5. Mosaïcisme : Dans ce cas, certaines cellules d'une personne ont un nombre anormal de chromosomes X ou Y, tandis que d'autres cellules ont un nombre normal. Les personnes atteintes de cette condition peuvent présenter des caractéristiques sexuelles secondaires anormales et des troubles mentaux.
Les anomalies chromosomiques peuvent entraîner des problèmes de développement, des malformations congénitales et des handicaps intellectuels. Les personnes atteintes d'anomalies chromosomiques peuvent également présenter un risque accru de développer certains types de cancer.
Les Tumeurs Neuroectodermiques Primaires (TNEP) sont un groupe hétérogène de tumeurs malignes cérébrales qui se développent à partir des cellules neuroectodermiques du système nerveux central. Elles sont caractérisées par une croissance rapide, une forte tendance à l'invasion locale et une propension à la métastase vers d'autres parties du cerveau ou de la moelle épinière, ainsi que dans des cas plus rares, vers d'autres organes.
Les TNEP comprennent plusieurs sous-types histologiques, dont les plus courants sont le médulloblastome, l'astrocytome primitif à grande cellule, l'ectoméningeome et le pineoblastome. Chaque sous-type a ses propres caractéristiques cliniques, radiologiques et moléculaires.
Le traitement des TNEP implique généralement une combinaison de chirurgie, de radiothérapie et de chimiothérapie. Le pronostic dépend du sous-type de tumeur, de l'âge du patient, de l'étendue de la maladie au moment du diagnostic et de la réponse au traitement. Les TNEP sont généralement des tumeurs agressives avec un mauvais pronostic, en particulier pour les patients atteints de médulloblastomes de haut grade ou d'astrocytomes primitifs à grande cellule. Cependant, certains sous-types, comme les ectoméningeomes, peuvent avoir un meilleur pronostic avec un traitement approprié.
Le polymorphisme génétique fait référence à la présence de plus d'un allèle pour un gène donné dans une population, ce qui entraîne une variabilité génétique. Il s'agit d'une variation normale et courante du matériel génétique chez les êtres humains et d'autres organismes. Ce polymorphisme peut se produire en raison de divers types de mutations, telles que des substitutions de base, des insertions ou des délétions d'une ou plusieurs paires de bases dans le gène.
Les polymorphismes génétiques peuvent avoir différents effets sur la fonction du gène et de son produit protéique associé. Dans certains cas, ils peuvent ne pas affecter la fonction du tout, tandis que dans d'autres, ils peuvent entraîner des changements mineurs ou même majeurs dans la structure et la fonction de la protéine. Ces variations peuvent contribuer à la diversité phénotypique observée au sein d'une population, y compris la susceptibilité aux maladies et les réponses aux traitements médicaux.
Les polymorphismes génétiques sont souvent utilisés en médecine et en recherche biomédicale pour identifier des marqueurs génétiques associés à des maladies ou à des traits spécifiques. Ils peuvent également être utiles dans l'identification individuelle, la parenté et les études d'ascendance.
Les conditions précancéreuses, également appelées lésions précancéreuses ou états précancéreux, sont des changements anormaux dans les cellules qui peuvent évoluer vers un cancer si elles ne sont pas détectées et traitées à temps. Ces conditions ne sont pas encore cancéreuses, mais elles présentent un risque accru de développer un cancer. Elles peuvent apparaître sous forme de croissance ou de lésion anormale dans un tissu ou un organe spécifique.
Les exemples courants de conditions précancéreuses comprennent les suivantes :
1. Dysplasie : Il s'agit d'une anomalie cellulaire qui se produit lorsque les cellules présentent des caractéristiques atypiques, telles qu'un noyau plus grand ou une division cellulaire anormale. La dysplasie peut survenir dans divers tissus et organes, y compris la bouche, le col de l'utérus, le poumon, le sein et l'intestin.
2. Leucoplasie : Il s'agit d'une lésion blanche et squameuse qui se forme sur la muqueuse, généralement dans la bouche ou le vagin. Bien que la plupart des leucoplasies soient bénignes, certaines peuvent évoluer vers un cancer si elles ne sont pas traitées.
3. Érythroplasie : Il s'agit d'une lésion rouge et veloutée qui se forme sur la muqueuse, généralement dans la bouche ou le vagin. Comme la leucoplasie, l'érythroplasie peut être bénigne, mais elle présente un risque accru de développer un cancer.
4. Kératose actinique : Il s'agit d'une croissance squameuse et rugueuse qui se forme sur la peau, généralement exposée au soleil. Bien que la kératose actinique ne soit pas cancéreuse en soi, elle peut évoluer vers un carcinome spinocellulaire, une forme de cancer de la peau.
5. Dysplasie : Il s'agit d'une anomalie cellulaire anormale qui peut se produire dans n'importe quelle partie du corps. La dysplasie peut être bénigne ou pré-cancéreuse, selon la gravité de l'anomalie cellulaire.
Il est important de noter que toutes ces conditions ne sont pas nécessairement cancéreuses ou pré-cancéreuses, mais elles présentent un risque accru de développer un cancer si elles ne sont pas traitées. Si vous présentez l'un de ces symptômes, il est important de consulter un médecin pour un examen et un diagnostic appropriés.
Une tumeur trophoblastique placentaire est un type rare de tumeur qui se développe dans le tissu qui forme le placenta pendant la grossesse. Ces tumeurs peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs trophoblastiques bénignes sont appelées hydatiformes molaires, tandis que les tumeurs trophoblastiques malignes sont appelées choriocarcinomes.
Les hydatiformes molaires se produisent lorsque l'ovule est fécondé par un spermatozoïde, mais il n'y a pas de développement embryonnaire normal. Au lieu de cela, le tissu placentaire se multiplie rapidement et forme une masse de vésicules remplies de liquide. Les hydatiformes molaires sont généralement traitées par l'ablation chirurgicale du tissu affecté, suivie d'un traitement supplémentaire avec des médicaments anticancéreux si nécessaire.
Les choriocarcinomes sont des tumeurs trophoblastiques malignes qui se développent à partir du tissu placentaire et peuvent se propager rapidement dans tout le corps. Ils peuvent survenir après une grossesse normale, une fausse couche ou un avortement, bien que cela soit très rare. Le traitement des choriocarcinomes implique généralement une combinaison de chimiothérapie et de chirurgie pour éliminer la tumeur et prévenir sa propagation.
Il est important de noter que les tumeurs trophoblastiques placentaires sont très rares, mais elles peuvent être traitées avec succès si elles sont détectées et traitées rapidement. Les femmes qui ont eu une grossesse compliquée ou qui présentent des symptômes inhabituels après une grossesse devraient consulter un médecin pour un examen approfondi.
Le chromosome X est l'un des deux chromosomes sexuels, l'autre étant le chromosome Y. Les humains ont généralement 46 chromosomes répartis en 23 paires, dont une paire de chromosomes sexuels. La plupart des femmes ont deux chromosomes X (XX), tandis que la plupart des hommes ont un chromosome X et un chromosome Y (XY).
Le chromosome X est beaucoup plus grand que le chromosome Y et contient environ 1 500 gènes, ce qui représente environ 7 % du nombre total de gènes dans une cellule humaine. Il code des protéines importantes pour le développement et le fonctionnement du corps, y compris certaines qui sont essentielles au cerveau et aux systèmes nerveux.
Des anomalies chromosomiques sur le chromosome X peuvent entraîner divers troubles génétiques, tels que la syndromes de l'X fragile, le syndrome de Turner (monosomie X) et le syndrome de Klinefelter (XXY). Ces conditions peuvent affecter le développement physique, intellectuel et neurologique.
La liaison génétique est un phénomène dans lequel des gènes ou des marqueurs génétiques spécifiques situés à proximité les uns des autres sur un chromosome ont tendance à être hérités ensemble au cours de la méiose, car il est moins probable qu'ils soient séparés par recombinaison génétique. Plus la distance entre deux gènes ou marqueurs est petite, plus ils sont susceptibles d'être co-transmis et donc considérés comme étant liés. La mesure de cette liaison est exprimée en unités de carte génétique, telles que les centimorgans (cM), qui représentent environ un échange réciproque par recombinaison sur 100 meioses.
Ce concept est fondamental dans la cartographie génétique et l'analyse de l'expression des gènes, ainsi que dans l'identification des mutations causales de maladies monogéniques et complexes. Il permet également d'identifier des susceptibilités génétiques à certaines conditions médicales ou traits, ce qui peut conduire à un counseling génétique plus précis et à une médecine personnalisée.
Le dépistage néonatal est un processus systématique de détection précoce, à grande échelle et généralisée, de certaines conditions médicales congénitales ou acquises à la naissance chez les nouveau-nés. Il est réalisé en prenant des échantillons de sang, d'urine ou d'autres tissus peu après la naissance, puis en analysant ces échantillons à l'aide de divers tests de laboratoire.
Le dépistage néonatal vise à identifier rapidement les nouveau-nés qui présentent un risque accru de développer des problèmes de santé graves et potentiellement évitables, tels que les troubles métaboliques héréditaires, les maladies du sang, les déficits hormonaux et d'autres affections congénitales. Une détection précoce permet une intervention thérapeutique rapide, ce qui peut améliorer considérablement les résultats pour la santé des nourrissons concernés, réduire la morbidité et la mortalité, et améliorer leur qualité de vie globale.
Les programmes de dépistage néonatal sont généralement mis en œuvre par les autorités sanitaires publiques ou les établissements de santé, et ils sont recommandés dans de nombreux pays développés pour tous les nouveau-nés à moins que des contre-indications médicales ne soient présentes. Les conditions ciblées par le dépistage néonatal peuvent varier selon les pays et les régions en fonction des ressources disponibles, des priorités de santé publique et des prévalences locales des différentes affections.
L'informatique graphique, également connue sous le nom de computer graphics ou computational visualization, est une branche de l'informatique qui se concentre sur la création et l'affichage d'images numériques et d'animations. Elle combine des concepts issus de plusieurs domaines, tels que les mathématiques, la physique, la psychologie et le design, pour produire des représentations visuelles réalistes ou stylisées sur un écran d'ordinateur.
L'informatique graphique implique généralement trois étapes principales :
1. Modélisation : Cette étape consiste à définir les objets 3D et leurs propriétés, telles que la forme, la taille, la couleur et la texture. Les modèles géométriques peuvent être créés manuellement à l'aide de logiciels spécialisés ou générés automatiquement à partir de données réelles (par exemple, des scanners 3D).
2. Rendu : Le processus de rendu convertit les données géométriques en une image numérique en calculant la manière dont la lumière interagit avec chaque objet et sa surface. Cela inclut le calcul des ombres, des réflexions, des refractions et d'autres phénomènes optiques pour produire une représentation visuelle réaliste.
3. Animation : Dans cette étape, on définit les mouvements et les transformations temporelles des objets dans la scène. Cela peut inclure des déplacements, des rotations, des changements de forme ou d'autres modifications apportées aux propriétés des objets au fil du temps.
L'informatique graphique a de nombreuses applications dans divers domaines, notamment les jeux vidéo, le cinéma, l'architecture, l'ingénierie, la médecine et la recherche scientifique. Dans le contexte médical, elle est utilisée pour la visualisation de structures anatomiques, la planification chirurgicale, l'enseignement et la formation, ainsi que dans le développement d'outils d'imagerie diagnostique tels que les tomodensitomètres et les IRM.
Les protéines gènes suppresseurs de tumeurs sont des protéines qui jouent un rôle crucial dans la régulation du cycle cellulaire et la prévention de la croissance cellulaire anormale. Elles aident à prévenir la transformation des cellules normales en cellules cancéreuses en supprimant l'activation des gènes responsables de la division et de la prolifération cellulaires.
Les gènes qui codent pour ces protéines suppressives de tumeurs sont souvent appelés "gènes suppresseurs de tumeurs". Lorsque ces gènes sont mutés ou endommagés, ils peuvent perdre leur capacité à produire des protéines fonctionnelles, ce qui peut entraîner une augmentation de la division cellulaire et de la croissance tumorale.
Les exemples bien connus de gènes suppresseurs de tumeurs comprennent le gène TP53, qui code pour la protéine p53, et le gène RB1, qui code pour la protéine Rb. Les mutations de ces gènes sont associées à un risque accru de développer certains types de cancer.
En résumé, les protéines gènes suppresseurs de tumeurs sont des protéines qui aident à réguler la croissance cellulaire et à prévenir la formation de tumeurs en supprimant l'activation des gènes responsables de la division et de la prolifération cellulaires.
Une métastase lymphatique est le processus par lequel le cancer se propage d'un organe primaire vers les ganglions lymphatiques voisins ou éloignés. Cela se produit lorsque les cellules cancéreuses se détachent de la tumeur primitive, pénètrent dans les vaisseaux lymphatiques et sont transportées jusqu'aux ganglions lymphatiques où elles peuvent former une nouvelle tumeur. Ce phénomène est souvent associé à un pronostic plus défavorable car il indique que le cancer a déjà commencé à se propager dans le corps.
Les tumeurs de l'intestin sont des croissances anormales qui se forment dans l'un des composants du système digestif communément appelé l'intestin. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses).
Les tumeurs bénignes incluent les adénomes, les lipomes, les leiomyomes et les hémangiomes. Elles sont généralement traitées par résection chirurgicale si elles causent des symptômes, tels que saignements, obstruction ou perforation.
Les tumeurs malignes, également connues sous le nom de cancers de l'intestin, comprennent les carcinomes adénocarcinomes, les lymphomes et les sarcomes. Le traitement dépend du type, de la taille, de l'emplacement et de l'étendue de la tumeur, mais il peut inclure une combinaison de chirurgie, de radiothérapie et de chimiothérapie.
Les facteurs de risque de développer un cancer colorectal, qui est le type le plus courant de cancer de l'intestin, comprennent l'âge avancé, les antécédents familiaux de cancer colorectal, certaines conditions médicales telles que la maladie de Crohn et la colite ulcéreuse, un régime alimentaire riche en viandes rouges et transformées, le tabagisme et l'obésité.
Blastomères sont les cellules précoces qui résultent du processus de division cellulaire très précoce dans l'embryogenèse. Ces cellules sont formées après la fécondation, lorsque le zygote (la cellule œuf fécondée) subit plusieurs divisions mitotiques pour former une petite boule de cellules, appelée morula.
Au fur et à mesure que les blastomères se divisent et se multiplient, ils forment une cavité au centre de la morula, ce qui entraîne la formation d'un stade suivant de développement embryonnaire connu sous le nom de blastocyste. Les cellules du blastocyste sont différenciées en deux types: les cellules extérieures, appelées trophoblastes, et un amas de cellules intérieures, appelé masse cellulaire interne.
Les blastomères jouent donc un rôle crucial dans le développement précoce de l'embryon et sont essentiels à la formation des structures qui deviendront plus tard le placenta et les tissus de l'embryon lui-même. Les anomalies dans le nombre, la forme ou la division des blastomères peuvent entraîner des problèmes de développement et des malformations congénitales.
Les facteurs de virulence sont des propriétés ou caractéristiques que possèdent certains micro-organismes (comme les bactéries, les champignons et les virus) qui leur permettent de causer des infections et des maladies chez l'hôte. Ces facteurs peuvent être des molécules ou des structures situées à la surface de l'agent pathogène ou produites par celui-ci. Ils contribuent à différentes étapes du processus infectieux, comme l'adhésion aux cellules de l'hôte, l'entrée et la multiplication dans les tissus, l'évasion du système immunitaire et les dommages causés aux tissus.
Les facteurs de virulence peuvent être classés en plusieurs catégories, telles que :
1. Adhésines : protéines ou polysaccharides qui favorisent l'adhérence des micro-organismes aux cellules de l'hôte, facilitant ainsi l'établissement de l'infection.
2. Invasines : molécules qui permettent aux micro-organismes d'envahir et de se multiplier dans les tissus de l'hôte.
3. Exotoxines : protéines sécrétées par certaines bactéries qui ont des effets délétères sur les cellules de l'hôte, comme l'entrée cellulaire, la modification du métabolisme ou la lyse cellulaire.
4. Endotoxines : composants de la membrane externe de certaines bactéries gram-négatives qui déclenchent une réponse inflammatoire lorsqu'ils sont libérés, par exemple, après la mort de la bactérie.
5. Systèmes de sécrétion : complexes protéiques permettant aux bactéries d'injecter des protéines effectrices dans les cellules de l'hôte pour manipuler leur fonction et favoriser la survie et la multiplication de la bactérie.
6. Capsules et autres structures de protection : polysaccharides ou protéines qui recouvrent certaines bactéries, les protégeant des défenses de l'hôte et facilitant leur persistance dans l'organisme.
7. Facteurs de résistance à l'immunité : molécules produites par les micro-organismes pour échapper aux mécanismes de défense de l'hôte, comme le système du complément ou les cellules immunitaires.
La compréhension des facteurs virulents et des stratégies d'évasion immunitaire utilisées par les micro-organismes permet de développer des approches thérapeutiques et préventives visant à contrer ces mécanismes et à améliorer la prise en charge des infections.
BRCA1 (Breast Cancer Gene 1) est un gène suprimeur de tumeurs qui code une protéine impliquée dans la réparation des dommages à l'ADN, en particulier ceux causés par les cassures double brin. Les mutations du gène BRCA1 augmentent considérablement le risque de développer un cancer du sein et de l'ovaire héréditaire. Ce gène est localisé sur le chromosome 17q21.
Les femmes porteuses d'une mutation du gène BRCA1 ont un risque estimé à environ 55-65% de développer un cancer du sein avant l'âge de 70 ans, et jusqu'à 45% peuvent développer un cancer de l'ovaire. Les hommes porteurs d'une mutation BRCA1 ont également un risque accru de cancers du sein, de la prostate et des testicules.
Il est important de noter que tous les individus héritant une mutation BRCA1 ne développeront pas nécessairement un cancer. Des facteurs supplémentaires tels que l'âge, l'environnement, le mode de vie et d'autres facteurs génétiques peuvent influencer le risque global de développer un cancer. La détection précoce et la surveillance rapprochée sont cruciales pour la prise en charge des personnes atteintes de mutations BRCA1.
Le génome des plantes se réfère à l'ensemble complet de gènes et d'autres séquences d'ADN présent dans le noyau cellulaire des cellules végétales. Il comprend tous les gènes héréditaires qui déterminent les caractéristiques et les fonctions des plantes, y compris leur développement, la croissance, la reproduction et la réponse aux stimuli environnementaux. Le génome des plantes est contenu dans plusieurs chromosomes et peut varier considérablement en taille et en complexité entre différentes espèces végétales. Récemment, les progrès de la technologie de séquençage de l'ADN ont permis le séquençage complet du génome de plusieurs plantes, ce qui a conduit à une meilleure compréhension de leur évolution, de leur diversité et de leur physiologie.
Une séquence conservée, dans le contexte de la biologie moléculaire et de la génétique, se réfère à une section spécifique d'une séquence d'ADN ou d'ARN qui reste essentiellement inchangée au fil de l'évolution chez différentes espèces. Ces séquences sont souvent impliquées dans des fonctions biologiques cruciales, telles que la régulation de l'expression des gènes ou la structure des protéines. Parce qu'elles jouent un rôle important dans la fonction cellulaire, les mutations dans ces régions sont généralement désavantageuses et donc sélectionnées contre au cours de l'évolution.
La conservation des séquences peut être utilisée pour identifier des gènes ou des fonctions similaires entre différentes espèces, ce qui est utile dans les études comparatives et évolutives. Plus une séquence est conservée à travers divers organismes, plus il est probable qu'elle ait une fonction importante et similaire chez ces organismes.
Le lymphome du manteau est un type rare de lymphome non hodgkinien, qui se développe dans les cellules B du système immunitaire. Il tire son nom des "cellules du manteau" qui entourent et protègent les follicules des ganglions lymphatiques. Ce cancer affecte généralement les personnes âgées de plus de 60 ans et est plus fréquent chez les hommes que chez les femmes.
Les symptômes du lymphome du manteau peuvent inclure des gonflements des ganglions lymphatiques, une fatigue persistante, des sueurs nocturnes, une perte de poids inexpliquée, des douleurs articulaires ou musculaires, et des démangeaisons cutanées. Le diagnostic est généralement posé à partir d'une biopsie d'un ganglion lymphatique enflé, qui permet d'identifier les cellules cancéreuses caractéristiques du lymphome du manteau.
Le traitement dépend du stade et de la gravité de la maladie, ainsi que de l'âge et de l'état général de santé du patient. Les options de traitement peuvent inclure une chimiothérapie, une radiothérapie, une greffe de cellules souches ou une thérapie ciblée avec des médicaments qui attaquent spécifiquement les cellules cancéreuses. Malheureusement, le lymphome du manteau a tendance à être agressif et difficile à traiter, bien que certains patients puissent bénéficier d'une rémission prolongée ou même d'une guérison complète.
Les tumeurs colorectales sont des croissances anormales dans le côlon ou le rectum, qui peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Elles se développent à partir de cellules qui tapissent la paroi interne du côlon ou du rectum et se multiplient de manière incontrôlable.
Les adénomes sont les types de tumeurs colorectales bénignes les plus courants. Bien qu'ils ne soient pas cancéreux, certains d'entre eux peuvent évoluer vers un cancer s'ils ne sont pas enlevés. Les carcinomes sont des tumeurs colorectales malignes qui se développent à partir des cellules glandulaires de la muqueuse intestinale.
Les facteurs de risque de développement des tumeurs colorectales comprennent l'âge avancé, un régime alimentaire riche en graisses et pauvre en fibres, le tabagisme, une consommation excessive d'alcool, l'obésité, une histoire personnelle ou familiale de polypes intestinaux ou de cancer colorectal, certaines maladies inflammatoires de l'intestin telles que la maladie de Crohn et la colite ulcéreuse, ainsi qu'une prédisposition génétique héréditaire.
Les symptômes des tumeurs colorectales peuvent inclure des saignements rectaux, des modifications de l'habitude intestinale telles que la constipation ou la diarrhée persistante, une sensation de satiété après avoir mangé seulement une petite quantité de nourriture, des douleurs abdominales, une fatigue excessive et une perte de poids inexpliquée.
Le diagnostic des tumeurs colorectales peut être posé grâce à des tests d'imagerie tels qu'une colonoscopie ou une sigmoïdoscopie, qui permettent de visualiser l'intérieur du côlon et de prélever des échantillons de tissus pour analyse. Le traitement dépend du stade et de la localisation de la tumeur, mais peut inclure une chirurgie pour enlever la tumeur, une radiothérapie ou une chimiothérapie pour détruire les cellules cancéreuses restantes.
Une mutation de lignée germinale fait référence à une modification permanente et héréditaire du matériel génétique qui se produit dans les cellules reproductrices (gamètes) d'un individu, c'est-à-dire dans les spermatozoïdes chez l'homme ou dans les ovocytes chez la femme. Ces mutations sont transmises à la descendance et peuvent entraîner des changements héréditaires dans les caractéristiques génétiques, y compris les prédispositions aux maladies génétiques. Contrairement aux mutations somatiques, qui ne concernent que les cellules non reproductrices et n'affectent donc pas la lignée germinale, les mutations de lignée germinale ont un impact sur la constitution génétique des générations futures.
Il est important de noter qu'une mutation dans une lignée germinale ne signifie pas nécessairement que la maladie se développera chez l'individu qui en hérite, car certains gènes peuvent tolérer des modifications sans provoquer de problèmes de santé. Cependant, dans d'autres cas, ces mutations peuvent entraîner des maladies graves ou même mettre la vie en danger, selon le type et l'emplacement de la mutation.
Les mutations de lignée germinale peuvent être détectées par des tests génétiques spécifiques, tels que l'analyse du séquençage de l'exome ou du génome entier, qui permettent d'identifier les variations dans le matériel génétique hérité. Ces informations peuvent être utiles pour la planification familiale et la gestion des risques de maladies héréditaires.
La leucémie lymphoïde aiguë précurseur des lymphocytes T (Precursor T-Cell Lymphoblastic Leukemia-Lymphoma, ou PTLL) est un type rare et agressif de cancer qui affecte les cellules souches immatures du système immunitaire, appelées lymphoblastes T. Ce cancer peut se développer à la fois dans la moelle osseuse (leucémie) et dans les ganglions lymphatiques (lymphome).
Dans le PTLL, les lymphoblastes T anormaux se multiplient de manière incontrôlable et s'accumulent dans la moelle osseuse, empêchant ainsi la production de cellules sanguines normales. Ces cellules cancéreuses peuvent également se propager à d'autres parties du corps, telles que les ganglions lymphatiques, le foie, la rate et le cerveau.
Les symptômes courants du PTLL comprennent la fatigue, des ecchymoses ou des saignements faciles, des infections fréquentes, des douleurs osseuses ou articulaires, des sueurs nocturnes, une perte de poids et des ganglions lymphatiques enflés. Le diagnostic repose généralement sur une biopsie de la moelle osseuse et/ou des ganglions lymphatiques, ainsi que sur d'autres tests de laboratoire et d'imagerie.
Le traitement du PTLL implique généralement une chimiothérapie intense, suivie souvent d'une greffe de moelle osseuse. Le pronostic dépend de plusieurs facteurs, tels que l'âge du patient, le stade et l'extension de la maladie au moment du diagnostic, ainsi que la réponse au traitement.
L'ARN ribosomique 16S est une molécule d'acide ribonucléique (ARN) qui fait partie du petit ribosome dans les cellules vivantes. Les ribosomes sont des complexes protéiques et ARN qui jouent un rôle central dans la synthèse des protéines.
L'ARN ribosomique 16S est spécifiquement utilisé en biologie moléculaire pour identifier et classer les bactéries. Il s'agit d'un ARN conservé qui contient des séquences régionales variables qui peuvent être utilisées pour distinguer différentes espèces bactériennes.
En particulier, la région hypervariable de l'ARN ribosomique 16S est souvent ciblée pour l'amplification par PCR et la séquençage dans les études de microbiologie moléculaire. Ces techniques permettent aux chercheurs d'identifier et de caractériser rapidement et précisément les espèces bactériennes présentes dans un échantillon, ce qui est particulièrement utile dans des domaines tels que la médecine, l'agriculture et l'environnement.
L'ostéosarcome est un type agressif et rare de cancer des os. Il se développe généralement à partir des cellules qui forment l'os (les ostéoblastes), entraînant ainsi une production anormale d'os ou d'une masse tissulaire osseuse dans la moelle osseuse. Bien que ce cancer puisse se développer dans n'importe quel os, il est le plus souvent localisé près des articulations longues telles que les genoux et les hanches.
Les symptômes de l'ostéosarcome peuvent inclure une douleur osseuse ou articulaire, un gonflement autour de la zone touchée, des difficultés à bouger la partie affectée du corps, une fatigue générale et une perte de poids involontaire. Les ostéosarcomes sont généralement traités par une combinaison de chirurgie, de chimiothérapie et de radiothérapie, en fonction de l'étendue et de la localisation du cancer.
Le pronostic pour les personnes atteintes d'ostéosarcome dépend de plusieurs facteurs, tels que l'âge du patient, la taille et la localisation de la tumeur, le stade du cancer au moment du diagnostic et la réponse au traitement. Les taux de survie à cinq ans pour les patients atteints d'ostéosarcome peuvent varier considérablement, allant de 20% à 70%, selon ces facteurs.
La "transformation cellulaire néoplasique" est un processus biologique dans lequel une cellule normale et saine se transforme en une cellule cancéreuse anormale et autonome. Ce processus est caractérisé par des changements irréversibles dans la régulation de la croissance et de la division cellulaire, entraînant la formation d'une tumeur maligne ou d'un néoplasme.
Les facteurs qui peuvent contribuer à la transformation cellulaire néoplasique comprennent des mutations génétiques aléatoires, l'exposition à des agents cancérigènes environnementaux tels que les radiations et les produits chimiques, ainsi que certains virus oncogènes.
Les changements cellulaires qui se produisent pendant la transformation néoplasique comprennent des anomalies dans les voies de signalisation cellulaire, une régulation altérée de l'apoptose (mort cellulaire programmée), une activation anormale des enzymes impliquées dans la réplication de l'ADN et une augmentation de l'angiogenèse (croissance de nouveaux vaisseaux sanguins pour fournir de l'oxygène et des nutriments à la tumeur).
La transformation cellulaire néoplasique est un processus complexe qui peut prendre plusieurs années, voire plusieurs décennies, avant qu'une tumeur maligne ne se développe. Cependant, une fois que cela se produit, les cellules cancéreuses peuvent envahir les tissus environnants et se propager à d'autres parties du corps, ce qui peut entraîner des complications graves et même la mort.
Un carcinome lobulaire est un type spécifique de cancer du sein qui commence dans les lobules, qui sont les glandes situées dans le sein qui produisent du lait. Dans ce type de cancer, les cellules cancéreuses se développent et forment des amas ou des masses dans les lobules.
Les carcinomes lobulaires peuvent être invasifs ou non invasifs (in situ). Les carcinomes lobulaires in situ (CLIS) sont confinés aux lobules et ne se sont pas propagés aux tissus environnants. Bien que le CLIS ne soit pas considéré comme une forme de cancer à part entière, il peut augmenter le risque de développer un cancer du sein invasif à l'avenir.
Les carcinomes lobulaires invasifs (CLI), en revanche, se sont propagés au-delà des lobules et peuvent se propager aux tissus environnants, y compris les vaisseaux sanguins et lymphatiques. Les CLI peuvent se propager à d'autres parties du corps, ce qui peut entraîner des complications graves et mettre la vie en danger.
Les symptômes du carcinome lobulaire peuvent inclure une masse ou un gonflement dans le sein, une modification de la forme ou de la taille du sein, une douleur ou une sensation de tiraillement dans le sein, une rougeur ou une éruption cutanée sur le sein, et un écoulement du mamelon. Cependant, ces symptômes peuvent également être causés par d'autres affections moins graves, il est donc important de consulter un médecin si vous remarquez des changements dans vos seins.
Le traitement du carcinome lobulaire dépend du stade et du grade du cancer, ainsi que de facteurs tels que l'âge et l'état de santé général du patient. Les options de traitement peuvent inclure la chirurgie, la radiothérapie, la chimiothérapie et/ou le traitement hormonal.
Une tumeur carcinoïde est un type rare et spécifique de tumeur neuroendocrine qui se développe à partir des cellules du système neuroendocrinien, qui régule les hormones et d'autres substances chimiques dans le corps. Ces tumeurs peuvent se former dans divers endroits, y compris l'intestin grêle, l'appendice, le poumon, le pancréas, le foie et d'autres organes. Les tumeurs carcinoïdes sont généralement petites et à croissance lente, mais elles peuvent devenir plus agressives et se propager (métastaser) vers d'autres parties du corps au fil du temps.
Les symptômes des tumeurs carcinoïdes dépendent de leur emplacement et de la sécrétion hormonale associée. Les symptômes courants comprennent des diarrhées, des rougeurs cutanées (flush), une respiration sifflante, des douleurs abdominales, des battements de cœur rapides ou irréguliers et une fatigue extrême. Le diagnostic est généralement posé par l'intermédiaire d'une biopsie et confirmé par des tests d'imagerie tels qu'un scanner ou une IRM.
Le traitement dépend de la taille, de la localisation et de l'étendue de la tumeur. Les options thérapeutiques peuvent inclure la chirurgie pour enlever la tumeur, la radiothérapie, la chimiothérapie, la thérapie ciblée et les médicaments pour contrôler les symptômes associés à la sécrétion hormonale excessive. Le pronostic varie considérablement en fonction du stade de la maladie au moment du diagnostic et de la réponse au traitement.
La tumeur de Wilms, également connue sous le nom de néphroblastome, est un type rare de cancer qui se développe dans les reins. Elle est généralement diagnostiquée chez les enfants âgés de 3 à 4 ans, bien qu'elle puisse affecter des enfants de tous âges. La tumeur de Wilms prend naissance dans les cellules du rein appelées blastèmes néphrogéniques qui sont responsables de la formation du rein pendant le développement fœtal.
Les symptômes de la tumeur de Wilms peuvent inclure une masse abdominale douloureuse ou indolore, des douleurs abdominales, des nausées et des vomissements, une hypertension artérielle et une hématurie (présence de sang dans les urines). Le diagnostic est généralement posé par imagerie médicale, telle qu'une échographie ou une tomographie computérisée (CT scan), suivie d'une biopsie pour confirmer le type de tumeur.
Le traitement de la tumeur de Wilms dépend du stade et du grade de la tumeur, ainsi que de l'âge et de l'état de santé général de l'enfant. Les options de traitement peuvent inclure la chirurgie pour enlever la tumeur, la radiothérapie et la chimiothérapie. Dans les cas avancés ou avec une récidive de la maladie, une greffe de moelle osseuse peut être envisagée.
Le pronostic de la tumeur de Wilms est généralement bon, avec un taux de survie à cinq ans supérieur à 90% pour les enfants atteints d'une tumeur localisée et traités de manière agressive. Cependant, le pronostic dépend du stade et du grade de la tumeur, ainsi que de l'âge et de l'état de santé général de l'enfant. Les enfants atteints d'une tumeur avancée ou récidivante peuvent avoir un pronostic plus défavorable.
Les tumeurs de la bouche sont des croissances anormales qui peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses) et qui se forment dans les tissus de la bouche, y compris les lèvres, le palais, la langue, les gencives, les joues, le plancher de la bouche et les amygdales.
Les tumeurs bénignes sont généralement des croissances non cancéreuses qui se développent lentement et ne se propagent pas à d'autres parties du corps. Elles peuvent inclure des granulomes, des kystes, des papillomes et des fibromes. Bien que ces tumeurs soient généralement sans danger, elles peuvent causer des problèmes si elles continuent de croître et compriment les structures voisines.
Les tumeurs malignes, en revanche, sont des cancers qui se développent dans les cellules des tissus de la bouche. Les carcinomes épidermoïdes représentent environ 90 % des cancers de la bouche et se forment généralement sur les lèvres, le plancher de la bouche, les côtés ou le dessous de la langue et les gencives. Les sarcomes, les mélanomes et les lymphomes peuvent également affecter la bouche, mais ils sont beaucoup plus rares.
Les facteurs de risque des tumeurs de la bouche comprennent le tabagisme, la consommation excessive d'alcool, une mauvaise hygiène bucco-dentaire, une exposition prolongée au soleil sur les lèvres, une mauvaise alimentation et certaines infections virales telles que le papillomavirus humain (VPH).
Les symptômes des tumeurs de la bouche peuvent inclure des bosses ou des ulcères qui ne guérissent pas, des douleurs ou des saignements dans la bouche, des difficultés à mâcher, à avaler ou à parler, un engourdissement ou une faiblesse de la langue ou des joues et des ganglions lymphatiques enflés dans le cou.
Le diagnostic des tumeurs de la bouche implique généralement une biopsie pour confirmer le type et l'étendue de la tumeur. Le traitement dépend du type et du stade de la tumeur, mais peut inclure une chirurgie, une radiothérapie, une chimiothérapie ou une thérapie ciblée. La prévention des tumeurs de la bouche implique l'adoption d'un mode de vie sain, y compris l'arrêt du tabac et de la consommation excessive d'alcool, une bonne hygiène bucco-dentaire et une alimentation équilibrée.
Un inhibiteur de kinase cycline-dépendante P16, également connu sous le nom d'inhibiteur de CDK4/6, est un type de médicament utilisé en oncologie pour traiter certains types de cancer. Ces médicaments agissent en inhibant l'activité des kinases cycline-dépendantes CDK4 et CDK6, qui sont des protéines clés dans le cycle cellulaire et la régulation de la croissance cellulaire.
Plus précisément, les kinases CDK4/6 jouent un rôle important dans la phase G1 du cycle cellulaire, où elles phosphorylent la protéine Rb (rétinoblastome), ce qui permet la transcription des gènes nécessaires à la progression vers la phase S du cycle cellulaire. En inhibant l'activité de ces kinases, les inhibiteurs de CDK4/6 empêchent la phosphorylation de Rb et donc la progression du cycle cellulaire, entraînant ainsi une arrestation de la croissance des cellules cancéreuses.
Le P16 est un inhibiteur naturel de CDK4/6 qui régule négativement leur activité. Cependant, dans certains types de cancer, comme le cancer du sein, de l'ovaire ou du poumon, il peut y avoir une inactivation du gène P16 ou une surexpression des kinases CDK4/6, ce qui entraîne une prolifération cellulaire incontrôlée. Les inhibiteurs de CDK4/6 sont donc utilisés pour restaurer l'équilibre et ralentir la croissance des cellules cancéreuses.
Les exemples d'inhibiteurs de kinase cycline-dépendante P16 comprennent le palbociclib, le ribociclib et l'abémaciclib, qui sont approuvés par la Food and Drug Administration (FDA) pour le traitement du cancer du sein avancé ou métastatique.
Un gliome est un type de tumeur cérébrale qui prend origine dans les cellules gliales du cerveau. Les cellules gliales sont des cellules de soutien et de nutrition pour les neurones (cellules nerveuses) du système nerveux central. Il existe plusieurs types de gliomes, selon le type de cellule gliale à partir duquel ils se développent. Certains gliomes peuvent être bénins et croître lentement, tandis que d'autres sont malins et se développent rapidement, envahissant et détruisant les tissus cérébraux sains avoisinants. Les symptômes dépendent de la localisation et de la taille de la tumeur, mais peuvent inclure des maux de tête, des convulsions, des nausées, des vomissements, une vision floue, des problèmes d'équilibre et de coordination, ainsi que des changements cognitifs ou de la personnalité. Le traitement dépend du type et de la gravité de la tumeur et peut inclure une chirurgie, une radiothérapie et/ou une chimiothérapie.
Les protéines bactériennes se réfèrent aux différentes protéines produites et présentes dans les bactéries. Elles jouent un rôle crucial dans divers processus métaboliques, structurels et fonctionnels des bactéries. Les protéines bactériennes peuvent être classées en plusieurs catégories, notamment :
1. Protéines structurales : Ces protéines sont impliquées dans la formation de la paroi cellulaire, du cytosquelette et d'autres structures cellulaires importantes.
2. Protéines enzymatiques : Ces protéines agissent comme des catalyseurs pour accélérer les réactions chimiques nécessaires au métabolisme bactérien.
3. Protéines de transport : Elles facilitent le mouvement des nutriments, des ions et des molécules à travers la membrane cellulaire.
4. Protéines de régulation : Ces protéines contrôlent l'expression génétique et la transduction du signal dans les bactéries.
5. Protéines de virulence : Certaines protéines bactériennes contribuent à la pathogénicité des bactéries, en facilitant l'adhésion aux surfaces cellulaires, l'invasion tissulaire et l'évasion du système immunitaire de l'hôte.
L'étude des protéines bactériennes est importante dans la compréhension de la physiologie bactérienne, le développement de vaccins et de thérapies antimicrobiennes, ainsi que dans l'élucidation des mécanismes moléculaires de maladies infectieuses.
Les tumeurs de l'œsophage se réfèrent à des croissances anormales dans la muqueuse de l'œsophage, qui est le tube musculaire reliant la gorge à l'estomac. Ces tumeurs peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses).
Les tumeurs bénignes de l'œsophage comprennent les leiomyomes, les lipomes, et les fibromes. Elles sont généralement traitées par résection si elles causent des symptômes comme la dysphagie (difficulté à avaler) ou la douleur.
Les tumeurs malignes de l'œsophage, quant à elles, sont principalement des carcinomes épidermoïdes et des adénocarcinomes. Le tabagisme, la consommation excessive d'alcool, le reflux gastro-oesophagien (RGO) sévère et persistant, l'obésité et certains types de troubles précancéreux de l'œsophage peuvent augmenter le risque de développer ces tumeurs malignes.
Le traitement des tumeurs malignes de l'œsophage dépend du stade de la maladie, de la localisation de la tumeur et de l'état général de santé du patient. Les options thérapeutiques peuvent inclure la chirurgie, la radiothérapie, la chimiothérapie ou une combinaison de ces traitements.
Les tumeurs de la plèvre, également connues sous le nom de mésothéliomes pleuraux, sont des cancers agressifs qui se développent dans les fines membranes (la plèvre) qui entourent les poumons. La plupart des cas de mésothéliomes pleuraux sont liés à une exposition antérieure à l'amiante.
Il existe deux types principaux de tumeurs de la plèvre : le mésothéliome épithélial et le mésothéliome sarcomatoïde. Le mésothéliome épithélial est le type le plus courant et a généralement un meilleur pronostic que le mésothéliome sarcomatoïde.
Les symptômes des tumeurs de la plèvre peuvent inclure une douleur thoracique, une toux persistante, une difficulté à respirer, une perte d'appétit et une perte de poids involontaire. Le diagnostic est souvent difficile car les symptômes sont similaires à ceux d'autres maladies pulmonaires courantes.
Le traitement dépend du stade et de l'emplacement de la tumeur, ainsi que de l'état général de santé du patient. Les options de traitement peuvent inclure une chirurgie pour enlever la tumeur, une radiothérapie pour détruire les cellules cancéreuses, et/ou une chimiothérapie pour ralentir la croissance de la tumeur. Malheureusement, le pronostic est généralement mauvais pour les patients atteints de tumeurs de la plèvre, avec un taux de survie à cinq ans inférieur à 10%.
Les modes d'hérédité sont des schémas ou des modèles selon lesquels un trait ou une maladie génétique est transmis des parents aux enfants par le biais des gènes. Il existe plusieurs modes d'hérédité, dont les plus courants sont la dominance mendélienne, la récessivité mendélienne, le lien avec le sexe (ou hérédité liée au chromosome X), l'hérédité mitochondriale et les interactions gène-environnement.
1. Dominance mendélienne : Lorsqu'un seul gène altéré est suffisant pour provoquer un trait ou une maladie, on parle de dominance mendélienne. Dans ce cas, un individu ne nécessite qu'une copie du gène anormal (allèle) d'un parent pour exprimer le trait ou la maladie. Les personnes hétérozygotes (portant une copie normale et une copie altérée du gène) présentent généralement les mêmes caractéristiques que celles des homozygotes (portant deux copies altérées du gène). Un exemple classique de dominance mendélienne est la maladie de Huntington.
2. Récessivité mendélienne : Lorsque les deux allèles d'un gène doivent être altérés pour exprimer un trait ou une maladie, on parle de récessivité mendélienne. Dans ce cas, un individu doit hériter d'une copie altérée du gène de chaque parent pour développer le trait ou la maladie. Les personnes hétérozygotes (portant une copie normale et une copie altérée du gène) ne présentent généralement pas les caractéristiques associées au trait récessif. La fibrose kystique est un exemple de maladie causée par la récessivité mendélienne.
3. Lien avec le sexe (ou hérédité liée au chromosome X) : Les gènes sont situés sur les chromosomes sexuels, X et Y. La plupart des personnes ont deux copies du chromosome X (XX chez les femmes et XY chez les hommes). Lorsqu'un gène associé à une maladie est situé sur le chromosome X, il peut être transmis de manière récessive ou dominante. Cependant, comme les hommes n'ont qu'une seule copie du chromosome X, ils sont plus susceptibles d'exprimer des gènes liés au chromosome X que les femmes. La dystrophie musculaire de Duchenne est un exemple de maladie causée par une mutation dans un gène lié au chromosome X.
4. Hérédité mitochondriale : Les mitochondries sont des organites présents dans les cellules qui produisent de l'énergie. Elles contiennent leur propre ADN, distinct de l'ADN nucléaire présent dans le noyau de la cellule. Lorsqu'un gène associé à une maladie est situé sur l'ADN mitochondrial, il peut être transmis par les mères aux enfants des deux sexes. L'acidose lactique et la neuropathie sont des exemples de maladies causées par des mutations dans l'ADN mitochondrial.
5. Hérédité multifactorielle : De nombreuses maladies sont dues à une combinaison de facteurs génétiques et environnementaux. Par exemple, le cancer du sein peut être causé par des mutations dans certains gènes, mais également par des facteurs environnementaux tels que l'exposition aux radiations ou au tabac.
6. Hérédité complexe : Certaines maladies sont dues à une combinaison de plusieurs gènes et facteurs environnementaux. Par exemple, la schizophrénie peut être causée par des mutations dans plusieurs gènes différents, ainsi que par des facteurs environnementaux tels que le stress ou l'utilisation de drogues.
7. Hérédité mitochondriale : Certaines maladies sont dues à des mutations dans l'ADN mitochondrial, qui est transmis par les mères aux enfants des deux sexes. Par exemple, certaines formes de diabète et de surdité peuvent être causées par des mutations dans l'ADN mitochondrial.
8. Hérédité autosomique récessive : Certaines maladies sont dues à des mutations dans les gènes situés sur les chromosomes non sexuels (autosomes). Ces maladies ne se manifestent que si une personne hérite de deux copies du gène muté, une de chaque parent. Par exemple, la fibrose kystique et l'anémie falciforme sont des exemples de maladies autosomiques récessives.
9. Hérédité autosomique dominante : Certaines maladies sont dues à des mutations dans les gènes situés sur les chromosomes non sexuels (autosomes). Ces maladies se manifestent même si une personne n'hérite que d'une seule copie du gène muté. Par exemple, la maladie de Huntington et le syndrome de Marfan sont des exemples de maladies autosomiques dominantes.
10. Hérédité liée au chromosome X : Certaines maladies sont dues à des mutations dans les gènes situés sur le chromosome X. Ces maladies affectent principalement les hommes, car ils n'ont qu'une seule copie du chromosome X. Les femmes ont deux copies du chromosome X et sont donc moins susceptibles d'être touchées par ces maladies. Par exemple, la dystrophie musculaire de Duchenne et l'hémophilie sont des exemples de maladies liées au chromosome X.
Une banque de gènes est une installation qui collecte, stocke et distribue des échantillons de matériel génétique, tels que l'ADN, les cellules souches ou les tissus. Ces échantillons peuvent provenir de diverses sources, y compris des donneurs humains sains, des patients atteints de certaines maladies et des espèces animales ou végétales menacées.
Les banques de gènes ont plusieurs objectifs importants. L'un d'eux est de préserver la diversité génétique pour les générations futures, en particulier dans le cas de plantes et d'animaux en voie de disparition. Les échantillons stockés peuvent également être utilisés à des fins de recherche scientifique, y compris l'étude des maladies héréditaires et la découverte de nouveaux traitements médicaux.
Dans le domaine de la médecine, les banques de gènes peuvent fournir des échantillons de tissus sains qui peuvent être utilisés pour la recherche sur les maladies génétiques et le développement de thérapies géniques. Les cellules souches stockées dans les banques de gènes peuvent également être utilisées pour le traitement de certaines maladies, telles que le cancer et les maladies du sang.
Il est important de noter que les banques de gènes sont soumises à des réglementations strictes en matière de confidentialité et d'éthique, afin de protéger les droits des donneurs et de garantir que les échantillons soient utilisés de manière responsable.
Une biopsie est un procédé médical dans lequel un échantillon de tissu corporel est prélevé et examiné de près, généralement au microscope, pour déterminer la présence ou l'absence d'une maladie ou d'une condition spécifique. Ce processus aide les médecins à poser un diagnostic précis et à planifier le traitement approprié.
Il existe plusieurs types de biopsies, en fonction de la partie du corps concernée et de la méthode utilisée pour prélever l'échantillon :
1. Biopsie par incision/excision : Dans ce type de biopsie, une incision est pratiquée dans la peau pour accéder au tissu cible. L'échantillon peut être retiré en entier (biopsie excisionnelle) ou seulement une partie du tissu peut être prélevée (biopsie incisionnelle). Ce type de biopsie est couramment utilisé pour les grains de beauté suspects, les lésions cutanées et les nodules.
2. Biopsie par aspiration : Cette procédure implique l'utilisation d'une aiguille fine pour prélever un échantillon de tissu ou de liquide dans une zone spécifique du corps. La biopsie à l'aiguille fine est souvent utilisée pour les ganglions lymphatiques enflés, les glandes thyroïdiennes et d'autres organes superficiels.
3. Biopsie au trocart : Dans ce type de biopsie, un trocart (une aiguille creuse) est inséré dans le corps pour prélever un échantillon de tissu. Ce type de biopsie est souvent utilisé pour les organes profonds comme le foie, les poumons et les os.
4. Biopsie chirurgicale : Lors d'une biopsie chirurgicale, une incision est pratiquée dans la peau pour accéder à l'organe ou au tissu cible. Une partie de l'organe ou du tissu est ensuite retirée pour analyse. Ce type de biopsie est souvent utilisé pour les tumeurs malignes et les lésions suspectes dans des organes comme le sein, le poumon et le foie.
5. Biopsie liquide : Cette procédure consiste à analyser un échantillon de sang ou d'autres fluides corporels pour détecter la présence de cellules tumorales ou d'ADN circulant provenant d'une tumeur cancéreuse. La biopsie liquide est une méthode non invasive qui peut être utilisée pour le suivi du traitement et la détection précoce des récidives.
Les résultats de la biopsie sont généralement examinés par un pathologiste, qui fournit un rapport décrivant les caractéristiques du tissu ou des cellules prélevés. Ce rapport peut aider à déterminer le diagnostic et le traitement appropriés pour une maladie spécifique.
La récidive tumorale locale est un terme médical utilisé pour décrire la réapparition d'une tumeur maligne (cancer) dans la même région où elle s'est développée initialement, après un traitement initial qui avait apparemment réussi à éliminer la tumeur. Cela signifie que des cellules cancéreuses sont restées dans le corps, inaperçues par les médecins, et ont recommencé à se multiplier pour former une nouvelle tumeur au même endroit.
Une récidive tumorale locale peut survenir des mois ou même des années après le traitement initial. Elle est souvent associée à un pronostic moins favorable que lors du diagnostic initial, car les cellules cancéreuses peuvent avoir développé une résistance aux thérapies précédemment utilisées. Toutefois, la prise en charge d'une récidive dépend du type de cancer, de son stade au moment du diagnostic de la récidive et des antécédents thérapeutiques du patient.
Les Expressed Sequence Tags (EST) sont des courts fragments d'ADN qui représentent une partie spécifique d'un gène exprimé. Ils sont produits par le séquençage de l'extrémité 5' ou 3' des ARNm complémentaires (cDNAs) obtenus à partir d'une source d'ARNm spécifique, comme un tissu ou une cellule. Les EST sont utiles pour la découverte et la cartographie de gènes, car ils fournissent des informations sur l'expression génique dans différents tissus et sous différentes conditions physiologiques ou pathologiques. Cependant, il est important de noter qu'un seul EST ne représente généralement qu'une partie d'un gène et peut contenir des erreurs de séquençage, ce qui nécessite la confirmation par d'autres méthodes pour une utilisation fonctionnelle.
Une lignée cellulaire est un groupe homogène de cellules dérivées d'un seul type de cellule d'origine, qui se divisent et se reproduisent de manière continue dans des conditions de culture en laboratoire. Ces cellules sont capables de maintenir certaines caractéristiques spécifiques à leur type cellulaire d'origine, telles que la forme, les fonctions et les marqueurs moléculaires, même après plusieurs générations.
Les lignées cellulaires sont largement utilisées dans la recherche biomédicale pour étudier divers processus cellulaires et moléculaires, tester de nouveaux médicaments, développer des thérapies et comprendre les mécanismes sous-jacents aux maladies humaines. Il est important de noter que certaines lignées cellulaires peuvent présenter des anomalies chromosomiques ou génétiques dues à leur manipulation en laboratoire, ce qui peut limiter leur utilisation dans certains contextes expérimentaux ou cliniques.
Les tumeurs du thymus sont des growchs anormaux qui se forment dans le thymus, une glande située dans la partie supérieure de la poitrine derrière le sternum. Le thymus fait partie du système immunitaire et est important pour le développement et la maturation des lymphocytes T, un type de globules blancs qui aident à protéger le corps contre les infections et les maladies.
Les tumeurs du thymus peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs bénignes sont appelées thymomes et les tumeurs malignes sont appelées carcinomes du thymus.
Les symptômes des tumeurs du thymus peuvent varier en fonction de la taille et de l'emplacement de la tumeur, ainsi que de son type. Les symptômes courants comprennent une douleur thoracique, une toux sèche, une respiration difficile et une fatigue. Dans certains cas, les tumeurs du thymus peuvent produire des hormones ou d'autres substances qui peuvent affecter d'autres parties du corps, entraînant ainsi des symptômes supplémentaires.
Le traitement des tumeurs du thymus dépend du type et de l'étendue de la tumeur. Les options de traitement peuvent inclure une chirurgie pour enlever la tumeur, une radiothérapie pour détruire les cellules cancéreuses, et une chimiothérapie pour tuer les cellules cancéreuses. Dans certains cas, une greffe de moelle osseuse peut être recommandée pour reconstituer le système immunitaire après un traitement agressif.
Il est important de noter que les tumeurs du thymus sont relativement rares et que la plupart d'entre elles peuvent être traitées avec succès si elles sont détectées et traitées à un stade précoce. Si vous présentez des symptômes qui pourraient indiquer une tumeur du thymus, il est important de consulter un médecin dès que possible pour obtenir un diagnostic et un plan de traitement appropriés.
La séquence d'acides aminés homologue se réfère à la similarité dans l'ordre des acides aminés dans les protéines ou les gènes de différentes espèces. Cette similitude est due au fait que ces protéines ou gènes partagent un ancêtre commun et ont évolué à partir d'une séquence originale par une série de mutations.
Dans le contexte des acides aminés, l'homologie signifie que les deux séquences partagent une similitude dans la position et le type d'acides aminés qui se produisent à ces positions. Plus la similarité est grande entre les deux séquences, plus il est probable qu'elles soient étroitement liées sur le plan évolutif.
L'homologie de la séquence d'acides aminés est souvent utilisée dans l'étude de l'évolution des protéines et des gènes, ainsi que dans la recherche de fonctions pour les nouvelles protéines ou gènes. Elle peut également être utilisée dans le développement de médicaments et de thérapies, en identifiant des cibles potentielles pour les traitements et en comprenant comment ces cibles interagissent avec d'autres molécules dans le corps.
Les tumeurs osseuses sont des croissances anormales qui se forment dans les os. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs bénignes ne se propagent pas à d'autres parties du corps et ont tendance à croître lentement. Dans de nombreux cas, elles ne causent aucun symptôme et peuvent être découvertes par hasard lors d'examens médicaux ou radiologiques effectués pour d'autres raisons. Cependant, certaines tumeurs bénignes peuvent devenir assez grandes et affaiblir l'os, ce qui peut entraîner des fractures.
Les tumeurs malignes, en revanche, ont le potentiel de se propager à d'autres parties du corps. Elles sont souvent plus agressives que les tumeurs bénignes et peuvent croître rapidement. Les symptômes associés aux tumeurs osseuses malignes dépendent de la taille et de l'emplacement de la tumeur, mais peuvent inclure des douleurs osseuses, des gonflements, des fractures osseuses spontanées, une fatigue excessive et une perte de poids involontaire.
Le traitement des tumeurs osseuses dépend du type, de la taille, de l'emplacement et du stade de la tumeur. Les options de traitement peuvent inclure la surveillance attentive, la chirurgie, la radiothérapie, la chimiothérapie ou une combinaison de ces approches. Dans certains cas, des thérapies ciblées ou des immunothérapies peuvent également être utilisées pour traiter les tumeurs osseuses malignes.
Un modèle statistique est une représentation mathématique d'un ensemble de données ou d'un phénomène, conçue pour expliquer les relations entre différentes variables et prévoir les résultats futurs. Il s'agit essentiellement d'une équation ou d'un ensemble d'équations qui décrivent la relation entre des variables dépendantes (variables d'intérêt ou de réponse) et des variables indépendantes (variables explicatives ou prédictives). Les modèles statistiques peuvent être paramétriques, semi-paramétriques ou non paramétriques et peuvent inclure des termes d'interaction et des effets aléatoires pour capturer la complexité de la relation entre les variables.
Les modèles statistiques sont largement utilisés dans le domaine médical pour analyser les données de recherche, tester les hypothèses, évaluer l'association et la causalité, et concevoir des études expérimentales. Par exemple, les modèles de régression linéaire, logistique et de Cox sont fréquemment utilisés pour analyser les données cliniques et épidémiologiques. Les modèles statistiques permettent aux chercheurs d'estimer les effets des facteurs de risque sur les issues de santé, d'ajuster les biais de confusion et d'évaluer l'incertitude dans les estimations grâce à des intervalles de confiance et des tests d'hypothèses.
Cependant, il est important de noter que la qualité et l'utilité d'un modèle statistique dépendent de plusieurs facteurs, notamment la validité des hypothèses sous-jacentes, la pertinence des variables incluses, la taille de l'échantillon et la qualité des données. Par conséquent, les chercheurs doivent être conscients des limites et des hypothèses de leurs modèles statistiques et interpréter les résultats avec prudence.
Un inhibiteur P15 de kinase cycline-dépendante, également connu sous le nom d'inhibiteur CDK4/6, est une classe de médicaments qui ciblent et bloquent l'activité des kinases cycline-dépendantes CDK4 et CDK6. Ces enzymes jouent un rôle crucial dans la régulation du cycle cellulaire, plus précisément au cours de la phase G1, où elles phosphorylent les protéines de la famille des rétinoblastomes (pRb), entraînant ainsi leur inactivation. L'inactivation de pRb permet la progression du cycle cellulaire et l'entrée dans la phase S, où l'ADN est répliqué.
L'inhibition des kinases CDK4/6 empêche la phosphorylation et l'inactivation de pRb, ce qui entraîne une accumulation de protéines hypophosphorylées pRb et un blocage subséquent du cycle cellulaire en phase G1. Ce mécanisme d'action est particulièrement important dans le traitement des tumeurs malignes, où la régulation du cycle cellulaire est souvent altérée, entraînant une prolifération incontrôlée des cellules cancéreuses.
Les inhibiteurs P15 de kinase cycline-dépendante sont couramment utilisés dans le traitement de divers types de cancer, tels que les cancers du sein et les lymphomes, en monothérapie ou en combinaison avec d'autres agents thérapeutiques. Les représentants de cette classe de médicaments comprennent le palbociclib, le ribociclib et l'abémaciclib.
Un avortement spontané, également connu sous le nom de fausse couche, est la perte naturelle d'une grossesse avant la vingtième semaine. Cela se produit généralement au cours du premier trimestre de la grossesse. Les causes peuvent être liées à des problèmes chromosomiques, à des anomalies structurelles de l'utérus ou des trompes de Fallope, à des maladies maternelles sous-jacentes, à des traumatismes ou à des facteurs environnementaux.
Les symptômes d'un avortement spontané peuvent inclure des saignements vaginaux, des crampes abdominales et la perte de tissus provenant de la muqueuse utérine. Dans certains cas, l'avortement spontané peut ne pas être accompagné de symptômes évidents, ce qui est appelé un avortement septique ou manqué.
Le diagnostic d'un avortement spontané est généralement posé sur la base des antécédents médicaux et d'un examen physique, ainsi que d'une échographie pour confirmer l'absence de battements cardiaques fœtaux. Le traitement dépend de la gravité de la perte et peut inclure une surveillance attentive, des médicaments pour aider à expulser les tissus restants ou une intervention chirurgicale mineure.
Il est important de noter que l'avortement spontané est relativement courant et ne doit pas nécessairement être un signe de problèmes de fertilité futurs. Cependant, il est recommandé de consulter un médecin après une fausse couche pour déterminer la cause sous-jacente et discuter des options de planification familiale à long terme.
Je suis désolé, mais votre question est un peu mal formulée. Le terme "Internet" n'est pas un concept médical. Il s'agit d'un réseau informatique mondial qui relie des millions d'ordinateurs et de réseaux dans le monde entier. Cependant, je peux vous fournir des informations sur la façon dont l'Internet est utilisé dans un contexte médical.
Dans le domaine médical, Internet est souvent utilisé comme un outil pour faciliter la communication et le partage d'informations entre les professionnels de la santé, les chercheurs, les établissements de santé et les patients. Il existe de nombreuses façons dont Internet est utilisé en médecine, notamment :
1. La télémédecine : Il s'agit de l'utilisation de technologies de communication à distance pour fournir des soins médicaux aux patients qui se trouvent dans des endroits éloignés ou qui ont des difficultés à se déplacer. Les médecins peuvent utiliser Internet pour consulter des patients, examiner des images médicales et discuter de traitements.
2. La recherche médicale : Les chercheurs en médecine utilisent Internet pour accéder à des bases de données d'articles de recherche, partager des données et collaborer sur des projets de recherche.
3. L'éducation médicale : Les étudiants en médecine et les professionnels de la santé utilisent Internet pour accéder à des ressources d'apprentissage en ligne, assister à des conférences et des séminaires en direct et collaborer avec d'autres professionnels de la santé dans le monde entier.
4. La gestion des dossiers médicaux électroniques : De nombreux établissements de santé utilisent des dossiers médicaux électroniques pour stocker et gérer les informations sur les patients. Internet permet aux professionnels de la santé d'accéder à ces dossiers à partir de n'importe où, ce qui facilite la coordination des soins et la prise de décisions éclairées.
5. La télémédecine : Les médecins peuvent utiliser Internet pour fournir des consultations médicales à distance, surveiller les patients à domicile et offrir des soins de suivi après une hospitalisation.
En génétique, un hétérozygote est un individu qui possède deux allèles différents d'un même gène sur les deux chromosomes homologues. Cela signifie que l'individu a hérité d'un allèle particulier du gène en question de chacun de ses parents, et ces deux allèles peuvent être différents l'un de l'autre.
Dans le contexte de la génétique mendélienne classique, un hétérozygote est représenté par une notation avec une lettre majuscule suivie d'un signe plus (+) pour indiquer que cet individu est hétérozygote pour ce gène spécifique. Par exemple, dans le cas d'un gène avec deux allèles A et a, un hétérozygote serait noté Aa.
La présence d'hétérozygotie peut entraîner des phénotypes variés, en fonction du type de gène concerné et de la nature des allèles en présence. Dans certains cas, l'allèle dominant (généralement représenté par une lettre majuscule) détermine le phénotype, tandis que dans d'autres cas, les deux allèles peuvent contribuer au phénotype de manière égale ou interactive.
Il est important de noter qu'être hétérozygote pour certains gènes peut conférer des avantages ou des inconvénients en termes de santé, de résistance aux maladies et d'autres caractéristiques. Par exemple, l'hétérozygotie pour certaines mutations associées à la mucoviscidose (fibrose kystique) peut offrir une protection contre certaines bactéries nocives de l'appareil respiratoire.
Les tumeurs du pancréas sont des croissances anormales qui se forment dans le tissu du pancréas. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs bénignes ne se propagent pas à d'autres parties du corps et peuvent souvent être traitées par une intervention chirurgicale. Cependant, certaines tumeurs bénignes peuvent devenir cancéreuses avec le temps.
Les tumeurs malignes du pancréas sont des cancers qui se forment dans les cellules du pancréas et peuvent se propager à d'autres parties du corps. Les adénocarcinomes sont les types de cancer du pancréas les plus courants et représentent environ 90% de tous les cas. Ils se développent dans les cellules qui tapissent les conduits du pancréas qui produisent des enzymes digestives.
Les autres types de tumeurs malignes du pancréas comprennent les tumeurs neuroendocrines, qui se forment dans les cellules hormonales du pancréas, et les sarcomes, qui se développent dans le tissu conjonctif du pancréas.
Les symptômes des tumeurs du pancréas peuvent inclure une douleur abdominale supérieure, une perte de poids inexpliquée, une jaunisse (jaunissement de la peau et du blanc des yeux), des nausées et des vomissements. Le traitement dépend du type et de l'étendue de la tumeur, mais peut inclure une intervention chirurgicale, une chimiothérapie ou une radiothérapie.
Les protéines nucléaires sont des protéines qui se trouvent dans le noyau des cellules et jouent un rôle crucial dans la régulation de l'expression des gènes, la réplication de l'ADN, la réparation de l'ADN, la transcription de l'ARN et d'autres processus essentiels à la survie et à la reproduction des cellules.
Il existe plusieurs types de protéines nucléaires, y compris les histones, qui sont des protéines structurelles qui aident à compacter l'ADN en chromosomes, et les facteurs de transcription, qui se lient à l'ADN pour réguler l'expression des gènes. Les protéines nucléaires peuvent également inclure des enzymes qui sont impliquées dans la réplication et la réparation de l'ADN, ainsi que des protéines qui aident à maintenir l'intégrité structurelle du noyau.
Les protéines nucléaires peuvent être régulées au niveau de leur expression, de leur localisation dans la cellule et de leur activité enzymatique. Des anomalies dans les protéines nucléaires peuvent entraîner des maladies génétiques et contribuer au développement du cancer. Par conséquent, l'étude des protéines nucléaires est importante pour comprendre les mécanismes moléculaires qui sous-tendent la régulation de l'expression des gènes et d'autres processus cellulaires essentiels.
L'ADN ribosomal (rDNA) est un type spécifique d'acide désoxyribonucléique qui code pour les ARN ribosomaux, qui sont des composants structurels et fonctionnels essentiels des ribosomes. Les ribosomes sont des complexes macromoléculaires trouvés dans les cellules de tous les organismes vivants et jouent un rôle crucial dans la synthèse des protéines en facilitant le processus de traduction de l'ARN messager (ARNm) en chaînes polypeptidiques.
Les gènes rDNA sont généralement organisés en plusieurs centaines à quelques milliers de copies dans le génome d'un organisme donné, ce qui permet une expression abondante et régulée des ARN ribosomaux nécessaires pour soutenir la synthèse constante des protéines. Les séquences rDNA sont souvent utilisées comme marqueurs dans l'étude de l'évolution moléculaire, de la systématique et de la biodiversité en raison de leur conservation relative entre les espèces et de leur variabilité au sein des populations.
Les ARN ribosomaux sont classés en deux catégories principales : les ARN ribosomaux 18S, 5,8S et 28S (eucaryotes) ou 16S et 23S (procaryotes), qui composent le noyau des ribosomes et sont directement impliqués dans la catalyse de la formation des liaisons peptidiques pendant la traduction, et les ARN ribosomaux 5S, qui sont associés aux sous-unités ribosomales mineures.
En résumé, l'ADN ribosomal est un type d'acide désoxyribonucléique qui code pour les ARN ribosomaux essentiels à la synthèse des protéines dans les cellules de tous les organismes vivants. Les gènes rDNA sont souvent utilisés comme marqueurs dans l'étude de l'évolution moléculaire, de la systématique et de la biodiversité en raison de leur conservation relative entre les espèces et de leur variabilité au sein des populations.
Les techniques de diagnostic moléculaire sont des procédures de laboratoire qui identifient et analysent les changements dans la séquence d'acide désoxyribonucléique (ADN), d'acide ribonucléique (ARN) ou de protéines pour diagnostiquer des maladies spécifiques, telles que les infections, les maladies génétiques et le cancer.
Ces techniques comprennent la réaction en chaîne par polymérase (PCR), la séquençage de l'ADN, la hybridation in situ en fluorescence (FISH), la microréseau d'expression génique (GEP) et les tests immunologiques moléculaires.
Elles sont devenues des outils essentiels pour le diagnostic et la surveillance des maladies, offrant une précision et une sensibilité accrues par rapport aux méthodes de diagnostic traditionnelles. Les techniques de diagnostic moléculaire peuvent également être utilisées pour déterminer la susceptibilité d'un patient à un traitement spécifique, ce qui permet une médecine personnalisée et une prise en charge plus efficace des patients.
La composition en bases nucléiques fait référence à la proportion ou au pourcentage des quatre différentes bases nucléotidiques dans une molécule d'acide nucléique donnée, comme l'ADN ou l'ARN. Les quatre bases nucléotidiques sont l'adénine (A), la thymine (T) ou l'uracile (U) et la guanine (G) pour l'ADN et l'ARN respectivement, et la cytosine (C).
Le calcul de la composition en bases nucléiques peut être utile dans divers contextes, tels que l'analyse des séquences génomiques ou d'autres acides nucléiques. Par exemple, une composition en bases nucléiques déséquilibrée peut indiquer la présence de régions répétitives, de gènes viraux intégrés ou d'autres caractéristiques structurelles et fonctionnelles importantes dans l'acide nucléique.
Il est important de noter que la composition en bases nucléiques peut varier considérablement entre différents organismes, tissus et types d'acides nucléiques. Par exemple, les gènes codants pour des protéines ont tendance à avoir une composition en bases nucléiques différente de celle des régions non codantes de l'ADN. De même, l'ARN messager a généralement une composition en bases nucléiques différente de celle de l'ADN génomique.
Dans l'ensemble, la composition en bases nucléiques est un aspect important de l'analyse des acides nucléiques et peut fournir des informations précieuses sur leur structure, leur fonction et leur évolution.
L'ARN tumoral, ou ARN non codant des tumeurs, fait référence aux types d'acide ribonucléique (ARN) présents dans les cellules cancéreuses qui ne sont pas traduits en protéines. Contrairement à l'ARN messager (ARNm), qui sert de modèle pour la synthèse des protéines, l'ARN tumoral est principalement impliqué dans la régulation de l'expression génétique et d'autres processus cellulaires.
Les chercheurs ont identifié plusieurs types d'ARN tumoraux qui peuvent contribuer au développement et à la progression du cancer, notamment :
1. ARN non codants longs (lncRNA) : Ces molécules d'ARN de plus de 200 nucléotides sont souvent exprimées de manière anormale dans les tumeurs et peuvent réguler l'expression des gènes en interagissant avec l'ADN, l'ARN ou les protéines.
2. microARN (miRNA) : Ces petites molécules d'ARN de 18 à 25 nucléotides peuvent réguler l'expression des gènes en se liant aux ARNm et en favorisant leur dégradation ou en inhibant leur traduction en protéines.
3. Petits ARN interférents (siRNA) : Similaires aux miARN, ces petites molécules d'ARN de 20 à 25 nucléotides peuvent réguler l'expression des gènes en ciblant et en dégradant les ARNm complémentaires.
4. ARN circulaires (circRNA) : Ces molécules d'ARN fermées en boucle peuvent agir comme éponges pour les miARN, régulant ainsi l'expression des gènes cibles.
L'étude de l'ARN tumoral peut fournir des informations importantes sur la biologie du cancer et offrir de nouvelles cibles thérapeutiques potentielles. Les techniques de séquençage à haut débit, telles que le séquençage de l'ARN à grande échelle (RNA-seq), ont permis d'identifier et de caractériser les profils d'expression des ARN non codants dans divers types de cancer. Ces connaissances peuvent contribuer au développement de biomarqueurs diagnostiques et pronostiques, ainsi qu'à la conception de thérapies ciblées pour traiter le cancer.
Le récepteur ErbB-2, également connu sous le nom de HER2/neu ou c-erbB-2, est un membre de la famille des récepteurs du facteur de croissance épidermique (EGFR). Il s'agit d'une protéine transmembranaire qui joue un rôle crucial dans la signalisation cellulaire et la régulation de la croissance et de la division cellulaires.
Dans des conditions normales, le récepteur ErbB-2 est exprimé à faible niveau à la surface des cellules épithéliales. Cependant, dans certaines formes de cancer du sein, une amplification génétique ou une surexpression du gène ERBB2 entraîne une production excessive de la protéine ErbB-2. Cette surexpression favorise la croissance et la division cellulaires incontrôlées, contribuant ainsi à la progression du cancer.
Le récepteur ErbB-2 est donc un marqueur important dans le diagnostic et le traitement du cancer du sein. Des thérapies ciblées, telles que le trastuzumab (Herceptin), ont été développées pour cibler spécifiquement cette protéine et sont utilisées en combinaison avec d'autres traitements dans le traitement des cancers du sein HER2-positifs.
L'interprétation statistique des données est le processus d'analyse, d'évaluation et d'explication des résultats numériques obtenus à partir de diverses méthodes statistiques. Dans un contexte médical, cela peut inclure l'analyse de données recueillies lors d'essais cliniques, d'enquêtes épidémiologiques ou d'autres études de recherche.
Les statisticiens médicaux utilisent une variété de tests et d'outils statistiques pour déterminer la signification des résultats bruts. Cela peut inclure le calcul de moyennes, de médianes, d'écarts types et d'autres mesures de tendance centrale et de dispersion. Ils peuvent également effectuer des tests d'hypothèse pour déterminer si les différences observées entre les groupes sont statistiquement significatives.
L'interprétation des données statistiques dans un contexte médical nécessite une compréhension approfondie de la maladie ou du phénomène étudié, ainsi que des méthodes statistiques utilisées. Les résultats doivent être présentés clairement et de manière accessible, en expliquant ce qu'ils signifient dans le contexte plus large de la recherche médicale.
Il est important de noter que l'interprétation statistique des données ne doit jamais être faite de manière isolée, mais doit toujours être considérée en conjonction avec d'autres preuves telles que les résultats de la recherche qualitative, les connaissances cliniques et le jugement professionnel.
Les tumeurs primitives multiples (TPM) est un terme utilisé en oncologie pour décrire une situation où plusieurs types différents de tumeurs malignes se développent simultanément ou séquentiellement chez un même patient, sans preuve de métastases à partir d'une tumeur primitive initiale. Chaque tumeur est considérée comme indépendante et primaire, d'où le nom de "tumeurs primitives multiples".
Cette condition est relativement rare et peut être causée par des facteurs génétiques ou environnementaux. Dans certains cas, les TPM peuvent être associées à des syndromes héréditaires tels que le syndrome de Li-Fraumeni, le syndrome de Von Hippel-Lindau, et d'autres prédispositions génétiques au cancer.
Le diagnostic et la prise en charge des TPM peuvent être complexes, nécessitant une évaluation approfondie par une équipe multidisciplinaire de spécialistes pour déterminer le type et l'origine de chaque tumeur, ainsi que les options de traitement appropriées pour chacune d'entre elles.
Le génome viral se réfère à l'ensemble complet de gènes ou matériel génétique qu'un virus contient. Il peut être composé d'ADN (acide désoxyribonucléique) ou d'ARN (acide ribonucléique), et peut être soit à double brin, soit à simple brin. La taille du génome viral varie considérablement selon les différents types de virus, allant de quelques kilobases à plusieurs centaines de kilobases. Le génome viral contient toutes les informations nécessaires à la réplication et à la propagation du virus dans l'hôte infecté.
Le génome fongique se réfère à l'ensemble complet de gènes et d'autres matériels génétiques trouvés dans un champignon. Il est contenu dans le noyau cellulaire, ainsi que dans certaines mitochondries et chloroplastes, sous la forme d'ADN circulaire. Le génome fongique peut varier considérablement en taille et en complexité d'une espèce à l'autre.
Les champignons ont des génomes uniques par rapport aux autres organismes, tels que les plantes et les animaux. Par exemple, leur code génétique contient moins de redondance, ce qui signifie qu'un seul nucléotide peut souvent faire la différence entre deux acides aminés différents. De plus, les champignons ont tendance à avoir des introns plus longs et plus nombreux, ce qui sont des séquences d'ADN non codantes qui sont retirées après la transcription de l'ARNm.
L'étude du génome fongique peut fournir des informations importantes sur la biologie et l'évolution des champignons, ainsi que sur leur potentiel pour causer des maladies humaines, animales et végétales. Elle peut également aider à identifier de nouvelles cibles thérapeutiques pour le développement de médicaments antifongiques.
Les tumeurs surrénaliennes sont des growths anormaux qui se développent dans les glandes surrénales. Les glandes surrénales sont des petites glandes situées au-dessus des reins qui produisent plusieurs hormones importantes telles que l'adrénaline, le cortisol et les androgènes.
Les tumeurs surrénaliennes peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses). Les tumeurs bénignes sont appelées adénomes surrénaliens et sont relativement courantes, affectant environ 3 à 10 personnes sur 100 000. La plupart des adénomes surrénaliens ne causent pas de symptômes et ne nécessitent aucun traitement.
Cependant, certaines tumeurs surrénales peuvent produire des niveaux excessifs d'hormones, ce qui peut entraîner une variété de symptômes. Par exemple, les tumeurs surrénales qui produisent de l'adrénaline peuvent causer des palpitations cardiaques, de l'anxiété, de la transpiration et des tremblements. Les tumeurs surrénales qui produisent du cortisol peuvent entraîner une prise de poids, un visage bouffi, une pression artérielle élevée, une faiblesse musculaire et une fragilité osseuse.
Les tumeurs surrénales malignes sont appelées phéochromocytomes ou corticosurrénalomes, selon l'hormone qu'elles produisent. Ces tumeurs sont rares mais peuvent être très dangereuses car elles peuvent entraîner une crise cardiaque, un accident vasculaire cérébral ou même la mort si elles ne sont pas traitées rapidement.
Le diagnostic des tumeurs surrénales implique généralement une combinaison de tests d'imagerie et de tests sanguins pour déterminer la taille, l'emplacement et le type de tumeur. Le traitement dépend du type et de la gravité de la tumeur mais peut inclure une chirurgie, une radiothérapie ou une chimiothérapie.
Le potentiel invasif des tumeurs est un terme utilisé en oncologie pour décrire la capacité d'une tumeur à envahir les tissus adjacents et à se propager (métastaser) dans d'autres parties du corps. Cela dépend de plusieurs facteurs, y compris le type et le grade de la tumeur, ainsi que la présence ou l'absence de certaines protéines qui favorisent la croissance des vaisseaux sanguins (angiogenèse) et la migration cellulaire.
Les tumeurs avec un haut potentiel invasif sont plus agressives et ont tendance à se développer et à se propager rapidement, ce qui peut rendre le traitement plus difficile. Le potentiel invasif d'une tumeur est généralement évalué par l'analyse de biopsies ou d'échantillons chirurgicaux de la tumeur, et il est souvent pris en compte lors du choix du traitement et de la planification du suivi.
Les tumeurs de la prostate se réfèrent à toute croissance anormale des cellules dans la glande prostates. Elles peuvent être bénignes (non cancéreuses) ou malignes (cancéreuses).
1. Tumeurs Prostatiques Bénignes: Les tumeurs bénignes de la prostate sont courantes, surtout chez les hommes âgés. Le type le plus commun est l'hyperplasie bénigne de la prostate (HBP), également appelée adénome de la prostate. Cette condition se caractérise par une augmentation du volume de la glande prostates due à la croissance des cellules, ce qui peut entraîner des symptômes urinaires tels que difficulté à uriner, miction fréquente, besoin urgent d'uriner, ou sensation de vidange incomplète de la vessie.
2. Tumeurs Prostatiques Malignes: Les cancers de la prostate sont des tumeurs malignes qui se développent dans les cellules de la glande prostates. Le cancer de la prostate se développe généralement lentement et peut ne pas provoquer de symptômes pendant des années. Cependant, certains types peuvent être agressifs et se propager rapidement à d'autres parties du corps. Les facteurs de risque comprennent l'âge avancé, les antécédents familiaux de cancer de la prostate et certaines mutations génétiques.
Les tumeurs de la prostate sont généralement détectées par un toucher rectal ou un test sanguin appelé dosage du PSA (antigène spécifique de la prostate). Des examens d'imagerie, tels que l'échographie ou l'IRM, peuvent également être utilisés pour aider au diagnostic et au staging. Le traitement dépend du type et du stade de la tumeur, ainsi que de l'âge et de l'état de santé général du patient. Il peut inclure une surveillance active, une chirurgie, une radiothérapie ou une thérapie hormonale.
La protéine suppresseur de tumeur P53, également connue sous le nom de protéine tumorale suppressrice p53, est un type de protéine qui joue un rôle crucial dans la prévention de la croissance et de la division cellulaires anormales. Elle est codée par le gène TP53, qui est l'un des gènes les plus fréquemment mutés dans les cancers humains.
La protéine P53 est souvent appelée "gardienne du génome" car elle régule la réponse cellulaire aux dommages de l'ADN en arrêtant le cycle cellulaire, ce qui permet à la cellule de réparer les dommages avant que la division ne se produise. Si les dommages sont trop graves et ne peuvent être réparés, la protéine P53 déclenche l'apoptose, ou mort cellulaire programmée, pour éliminer la cellule anormale et prévenir la formation de tumeurs.
Les mutations du gène TP53 peuvent entraîner une protéine P53 non fonctionnelle ou dysfonctionnelle, ce qui peut entraîner une accumulation de cellules anormales et augmenter le risque de développement de tumeurs malignes. En fait, des mutations du gène TP53 ont été identifiées dans environ la moitié de tous les cancers humains. Par conséquent, la protéine P53 est considérée comme un important biomarqueur tumoral et une cible thérapeutique prometteuse pour le traitement du cancer.
Les régions promotrices génétiques sont des séquences d'ADN situées en amont du gène, qui servent à initier et à réguler la transcription de l'ARN messager (ARNm) à partir de l'ADN. Ces régions contiennent généralement des séquences spécifiques appelées "sites d'initiation de la transcription" où se lie l'ARN polymérase, l'enzyme responsable de la synthèse de l'ARNm.
Les régions promotrices peuvent être courtes ou longues et peuvent contenir des éléments de régulation supplémentaires tels que des sites d'activation ou de répression de la transcription, qui sont reconnus par des facteurs de transcription spécifiques. Ces facteurs de transcription peuvent activer ou réprimer la transcription du gène en fonction des signaux cellulaires et des conditions environnementales.
Les mutations dans les régions promotrices peuvent entraîner une altération de l'expression génique, ce qui peut conduire à des maladies génétiques ou à une susceptibilité accrue aux maladies complexes telles que le cancer. Par conséquent, la compréhension des mécanismes régissant les régions promotrices est essentielle pour comprendre la régulation de l'expression génique et son rôle dans la santé et la maladie.
La leucémie lymphoïde chronique à cellules B (LLC-B) est un type de cancer qui affecte les globules blancs appelés lymphocytes B. Cette forme de leucémie se caractérise par la présence de lymphocytes anormaux dans la moelle osseuse et le sang, ce qui entraîne une accumulation progressive de ces cellules anormales dans le corps.
Les cellules cancéreuses dans la LLC-B sont des lymphocytes B immatures ou immaturés qui ne fonctionnent pas correctement et peuvent s'accumuler dans la moelle osseuse, le sang, les ganglions lymphatiques, la rate et d'autres organes. Cela peut entraîner une augmentation du nombre de globules blancs dans le sang, ce qui peut interférer avec la fonction normale des autres cellules sanguines telles que les globules rouges et les plaquettes.
Les symptômes de la LLC-B peuvent inclure fatigue, essoufflement, sensation de faiblesse, perte de poids, sueurs nocturnes, frissons, fièvre, infections fréquentes, gonflement des ganglions lymphatiques et une sensation d'inconfort ou de plénitude dans l'abdomen due à un foie ou une rate hypertrophiés.
Le diagnostic de la LLC-B est généralement posé sur la base d'un examen physique, d'analyses sanguines et d'autres tests tels qu'une biopsie de moelle osseuse ou un scanner. Le traitement dépend du stade de la maladie et peut inclure une surveillance attentive, une chimiothérapie, une immunothérapie, une thérapie ciblée ou une greffe de cellules souches.
La cycline D1 est une protéine qui joue un rôle crucial dans le cycle cellulaire, plus précisément au stade G1. Elle se lie à des kinases dépendantes des cyclines et forme le complexe CDK4/6-cycline D, qui phosphoryle les protéines de la famille des rb (comme RB1), entraînant ainsi la libération du facteur de transcription E2F et l'activation de la transcription des gènes nécessaires à l'entrée dans la phase S. La cycline D1 est régulée par divers signaux, dont les voies de signalisation mitogène et celles impliquant les récepteurs d'hormones stéroïdiennes, comme les récepteurs des androgènes et des œstrogènes. Des niveaux accrus ou une activation anormale de la cycline D1 peuvent conduire à un déséquilibre du contrôle du cycle cellulaire, ce qui peut favoriser la tumorigenèse.
Une étude d'association à l'échelle du génome (GWAS) est une méthode d'analyse statistique utilisée en génétique pour identifier des variations dans l'ADN, appelées polymorphismes nucléotidiques simples (SNP), qui sont associés à un trait particulier ou à une maladie. Dans une GWAS, les chercheurs examinent simultanément plusieurs centaines de milliers, voire des millions de SNP répartis sur l'ensemble du génome humain, dans des groupes de cas (personnes atteintes d'une maladie) et de témoins (personnes sans la maladie).
L'objectif est d'identifier les variations génétiques qui sont statistiquement plus fréquentes chez les personnes atteintes de la maladie que chez les personnes sans la maladie. Ces SNP identifiés peuvent ensuite être utilisés pour localiser des gènes ou des régions du génome associés à la maladie, ce qui peut contribuer à une meilleure compréhension des mécanismes sous-jacents de la maladie et éventuellement conduire au développement de nouveaux traitements.
Il est important de noter que les résultats d'une GWAS ne peuvent pas prouver qu'un SNP particulier cause une maladie, mais seulement qu'il existe une association entre le SNP et la maladie. D'autres études sont souvent nécessaires pour confirmer ces associations et comprendre les mécanismes biologiques sous-jacents.
Un néphroblastome, également connu sous le nom de tumeur de Wilms, est un type rare et agressif de cancer qui se développe dans les reins. Il s'agit d'une forme de tumeur rénale maligne qui affecte généralement les enfants, bien que des cas chez les adultes aient également été signalés.
Le néphroblastome se développe à partir de cellules souches embryonnaires résiduelles dans le rein, appelées néphroblastes, qui ne se sont pas développées correctement pendant la période prénatale. Ces tumeurs peuvent être unilatérales ou bilatérales et peuvent se propager à d'autres parties du corps par le biais de la circulation sanguine ou lymphatique.
Les symptômes courants du néphroblastome comprennent une masse abdominale indolore, des douleurs abdominales, des nausées et des vomissements, une hypertension artérielle et une hématurie (présence de sang dans les urines). Le diagnostic est généralement posé par imagerie médicale, telle qu'une échographie ou une tomographie computérisée (CT scan), suivie d'une biopsie pour confirmer le type de tumeur.
Le traitement du néphroblastome implique généralement une combinaison de chirurgie, de radiothérapie et de chimiothérapie. Le pronostic dépend du stade et de l'extension de la maladie au moment du diagnostic, ainsi que de l'âge et de l'état de santé général du patient. Les taux de survie à cinq ans sont généralement élevés pour les patients atteints de néphroblastome, en particulier lorsqu'il est diagnostiqué et traité à un stade précoce.
Les sondes d'ARN, également connues sous le nom de sondes à capture d'ARN ou de sondes à capture de molécules d'intérêt, sont des outils de diagnostic et de recherche utilisés pour détecter et identifier spécifiquement des séquences d'ARN particulières dans un échantillon.
Les sondes d'ARN sont généralement constituées d'une chaîne d'oligonucléotides synthétiques, souvent de 15 à 30 nucléotides de longueur, qui sont complémentaires à une séquence cible spécifique de l'ARN. Ces sondes peuvent être marquées avec des fluorophores ou d'autres étiquettes pour permettre la détection et la quantification de l'ARN cible.
Les sondes d'ARN sont souvent utilisées dans les techniques de hybridation in situ, où elles se lient spécifiquement à leur ARN cible dans un échantillon tissulaire ou cellulaire, permettant ainsi la visualisation et l'analyse de l'expression génique au niveau cellulaire. Elles sont également utilisées dans les techniques de PCR en temps réel pour détecter et quantifier des ARN spécifiques dans un échantillon.
Les sondes d'ARN peuvent être conçues pour cibler des séquences spécifiques d'ARN messager (ARNm), d'ARN non codant ou de virus, ce qui en fait des outils précieux pour la recherche et le diagnostic de maladies telles que les infections virales, les cancers et les troubles génétiques.
Je suis désolé, "facteur temps" n'a pas de définition spécifique dans le domaine médical. Le terme "facteur temps" est plutôt utilisé en général pour décrire la durée pendant laquelle quelque chose se produit ou évolue, ou il peut également faire référence à l'importance de considérer le moment et la planification dans un contexte médical. Par exemple, l'administration d'un médicament à un moment précis ("facteur temps critique") ou la progression d'une maladie au fil du temps ("évolution temporelle de la maladie") peuvent être décrites en utilisant le terme "facteur temps". Cependant, il n'y a pas de définition médicale universellement acceptée pour ce terme.
La simulation informatique en médecine est l'utilisation de modèles et d'algorithmes informatiques pour imiter ou reproduire des processus, des conditions ou des situations médicales. Elle permet aux professionnels de la santé et aux apprenants de pratiquer, d'analyser et d'évaluer divers scénarios médicaux dans un environnement virtuel et contrôlé, sans mettre en danger les patients réels.
Les simulations informatiques peuvent varier en complexité, allant des modèles simples de physiologie humaine aux représentations détaillées de systèmes organiques complets. Elles sont utilisées dans divers domaines médicaux, tels que l'anesthésie, la chirurgie, la médecine d'urgence, la radiologie et la formation des infirmières, pour améliorer les compétences cliniques, la prise de décision et la gestion des situations critiques.
Les avantages de la simulation informatique en médecine incluent :
1. Apprentissage sans risque : Les apprenants peuvent s'entraîner dans un environnement virtuel sans mettre en danger les patients réels.
2. Répétition et pratique : Les utilisateurs peuvent répéter des scénarios autant de fois que nécessaire pour maîtriser une compétence ou une procédure.
3. Évaluation objective : La simulation informatique permet d'enregistrer et d'analyser les performances, offrant ainsi un retour d'information objectif sur les forces et les faiblesses des apprenants.
4. Personnalisation de l'apprentissage : Les simulations peuvent être adaptées aux besoins spécifiques des apprenants en fonction de leur niveau d'expérience, de leurs connaissances et de leurs objectifs d'apprentissage.
5. Accessibilité : La simulation informatique est accessible à distance, ce qui permet aux professionnels de la santé de se former et de maintenir leurs compétences quel que soit leur emplacement géographique.
Les protéines proto-oncogènes C-MYC sont des facteurs de transcription qui jouent un rôle crucial dans la régulation de l'expression des gènes impliqués dans la croissance cellulaire, la différentiation, la prolifération et l'apoptose (mort cellulaire programmée). Ces proto-oncogènes sont souvent surexprimés ou mutés dans divers types de cancer, ce qui entraîne une activation ou une dysrégulation anormales des voies de signalisation cellulaires.
La protéine C-MYC forme un complexe avec d'autres facteurs de transcription et se lie à des séquences spécifiques d'ADN appelées E-boxes, situées dans les promoteurs ou les enhancers de gènes cibles. Cette liaison permet la transcription de ces gènes et l'activation de processus cellulaires tels que la synthèse des protéines, la progression du cycle cellulaire et la métabolisme énergétique.
Dans un contexte cancéreux, une activation ou une amplification anormale de C-MYC peut entraîner une prolifération cellulaire incontrôlée, une résistance à l'apoptose et une évasion des mécanismes de contrôle cellulaires. Ces altérations contribuent au développement et à la progression de divers types de tumeurs solides et de leucémies.
En bref, les protéines proto-oncogènes C-MYC sont des régulateurs clés de l'expression génique et des processus cellulaires, et leur dysrégulation est associée à la pathogenèse du cancer.
 Cytogénétique
Cytogénétique Technologies de diagnostic génétique - Sujets spéciaux - Édition professionnelle du Manuel MSD
Technologies de diagnostic génétique - Sujets spéciaux - Édition professionnelle du Manuel MSD Alteration of cohesin genes in myeloid diseases.
Alteration of cohesin genes in myeloid diseases. Permalien vers Distinct chromosomal abnormality pattern in primary liver cancer of non-B, non-C patients
Permalien vers Distinct chromosomal abnormality pattern in primary liver cancer of non-B, non-C patients Membres
Membres Coordination nationale</span>
Coordination nationale</span> Amniocentèse ou ponction de la cavité amniotique
Amniocentèse ou ponction de la cavité amniotique Nouvelles données en génétique chromosomique | médecine/sciences
Nouvelles données en génétique chromosomique | médecine/sciences DeCS
DeCS