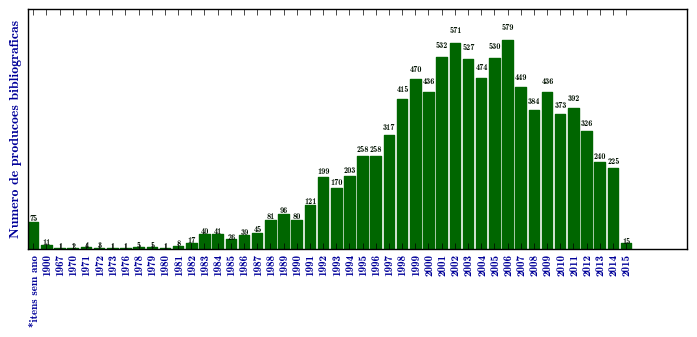
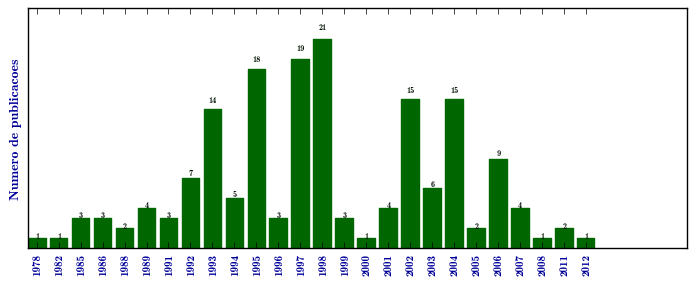
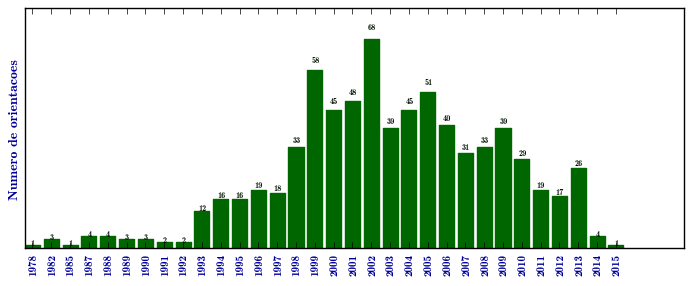
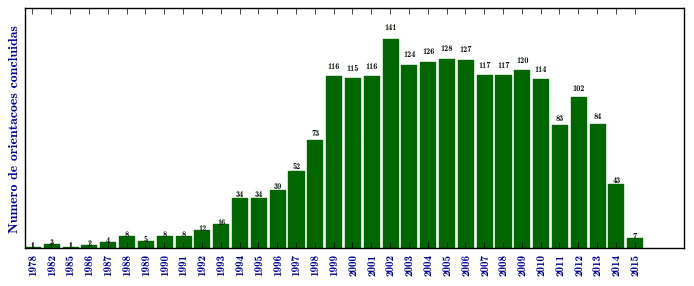
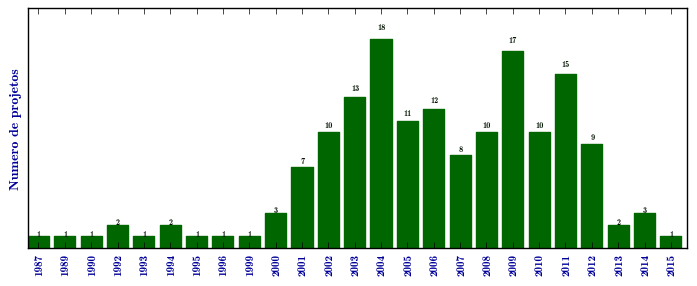
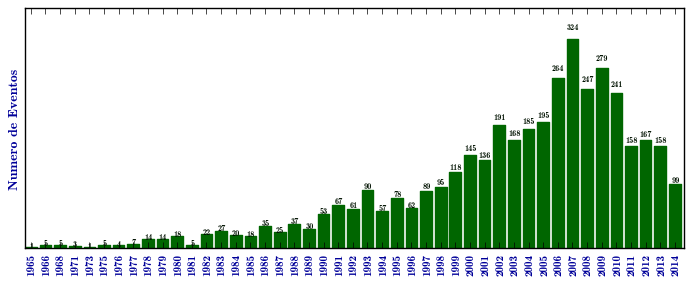
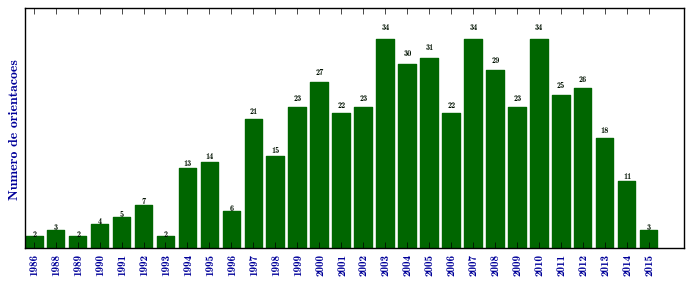
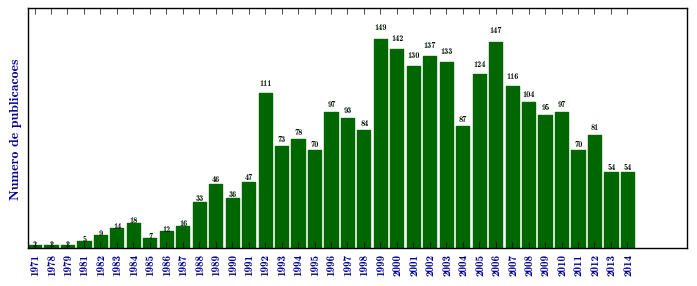
Mucopolissacaridose I
Iduronidase
Mucopolissacaridose VI
Mucopolissacaridose VII
Mucopolissacaridose III
Terapia de Reposição de Enzimas
Mucopolissacaridose II
Mucopolissacaridoses
Glicosaminoglicanas
Mucopolissacaridose IV
Ácido Idurônico
N-Acetilgalactosamina-4-Sulfatase
Cães
Condro-4-Sulfatase
Iduronato Sulfatase
Condroitina Sulfatases
Glucuronidase
Mucopolissacaridose Tipo I (MPS I) é um distúrbio genético raro que afeta o metabolismo dos mucopolissacarídeos, um tipo de carboidrato complexo encontrado em todos os tecidos corporais. Existem três subtipos da doença: Hurler, Hurler-Scheie e Scheie, que variam em gravidade.
MPS I é causada por uma deficiência na enzima alpha-L-iduronidase, que é necessária para descompor os mucopolissacarídeos. Quando essa enzima falta ou está presente em níveis reduzidos, os mucopolissacarídeos não podem ser adequadamente descompostos e acumulam-se dentro das células, causando danos a vários órgãos e sistemas corporais.
Os sinais e sintomas da doença geralmente começam a aparecer durante os primeiros seis meses de vida e podem incluir: desenvolvimento anormal ou atrasado, baixa estatura, freqüentes infecções respiratórias, problemas cardíacos, anormalidades esqueléticas, danos ao sistema nervoso central, problemas de visão e audição, e fígado e baço ingrossados.
O tratamento para MPS I geralmente inclui terapia de reposição enzimática (ERT), que pode ajudar a reduzir a acumulação de mucopolissacarídeos no corpo e prevenir ou retardar alguns dos sintomas da doença. Também podem ser necessários outros tratamentos, como cirurgia e fisioterapia, dependendo da gravidade dos sintomas e das complicações associadas à doença.
Iduronidase é uma enzima lisossomal importante que desempenha um papel crucial no metabolismo de certos glicosaminoglicanos (GAG), também conhecidos como mucopolissacarídeos. A iduronidase catalisa a remoção de resíduos de iduronida de GAGs, como dermatana sulfato e heparana sulfato, durante o processo de degradação lisossomal.
Em indivíduos sadios, a iduronidase é produzida pelas células e funciona normalmente para manter a homeostase dos GAGs. No entanto, em algumas condições genéticas raras, como a doença de Hurler, a doença de Scheie e a síndrome de Hunter (também conhecidas como mucopolissacaridose tipo I e II, respectivamente), há deficiências ou mutações nesta enzima, o que leva à acumulação de GAGs nos tecidos corporais. Isso pode resultar em sintomas graves, como atraso no desenvolvimento, anormalidades ósseas e faciais, problemas cardiovasculares e neurológicos, entre outros.
O tratamento para essas condições geralmente inclui terapia de reposição enzimática, na qual a iduronidase é administrada por via intravenosa para ajudar a restaurar os níveis normais da enzima e prevenir a acumulação de GAGs.
Mucopolissacaridose Tipo VI, também conhecida como Síndrome de Maroteaux-Lamy, é uma doença genética rara que pertence ao grupo das Mucopolissacaridoses (MPS). É causada por deficiência da enzima N-acetilgalactosamina 4-sulfatase, resultando em acúmulo de certos tipos de glicosaminoglicanas (GAGs) ou mucopolissacarídeos nos lisossomos das células.
Este acúmulo leva a diversos sintomas clínicos, como alterações esqueléticas, problemas cardiovasculares, danos aos órgãos internos e deficiências cognitivas. Os sinais e sintomas geralmente começam a aparecer entre os 2 e 6 anos de idade e podem incluir:
1. Crescimento lento ou atrasado
2. Baixa estatura
3. Cara alongada com características distintas (como focinho alongado, olhos grandes e pálpebras inferiores voltadas para baixo)
4. Problemas ósseos e articulares, como rigidez articular, luxação da articulação do quadril e curvatura anormal da coluna (escoliose)
5. Doença cardiovascular, incluindo valvopatia e hipertrofia do ventrículo esquerdo
6. Problemas respiratórios, como obstrução das vias aéreas superiores e apneia do sono
7. Inchaço dos tecidos moles (edema) em diferentes partes do corpo
8. Perda auditiva progressiva
9. Problemas de visão, incluindo opacidade da córnea e glaucoma
10. Hérnias abdominais
O tratamento para a Mucopolissacaridose Tipo VI geralmente inclui terapia de reposição enzimática (ERT) com alglucosidase alfa, fisioterapia, cuidados odontológicos regulares e cirurgias corretivas para problemas ósseos e articulares. O manejo dos sintomas e complicações associadas à doença é essencial para melhorar a qualidade de vida dos pacientes.
Mucopolissacaridose VII, também conhecida como Síndrome de Sly, é uma doença genética rara e séria que afeta o metabolismo dos mucopolissacarídeos (GAGs), um tipo de carboidrato complexo encontrado em todos os tecidos corporais.
Esta condição é causada por deficiência da enzima beta-glucuronidase, que é necessária para a decomposição e reciclagem adequados dos GAGs. Como resultado, os GAGs se acumulam em células e tecidos corporais, causando danos progressivos à saúde do indivíduo.
Os sintomas da mucopolissacaridose VII geralmente começam a aparecer nos primeiros meses de vida e podem incluir:
* Desenvolvimento lento ou atrasado
* Baixa estatura
* Fonte de voz grossa
* Problemas respiratórios e infeções frequentes
* Doença hepática e esplênica
* Problemas cardíacos
* Anormalidades ósseas, incluindo a displasia esquelética e a formação anormal dos ossos do crânio
* Problemas de visão e audição
A mucopolissacaridose VII é uma condição hereditária e autossômica recessiva, o que significa que os indivíduos afetados recebem uma cópia defeituosa do gene responsável pela doença de cada pai.
O tratamento para a mucopolissacaridose VII geralmente se concentra em aliviar os sintomas e melhorar a qualidade de vida do paciente, pois não existe cura conhecida para a doença. Os cuidados de suporte podem incluir fisioterapia, terapia da fala, tratamento ortopédico, medicação para controlar as infecções e outras complicações, e possivelmente um transplante de células-tronco hematopoiéticas.
Mucopolissacaridose Tipo III, também conhecida como Síndrome de Sanfilippo, é um distúrbio metabólico hereditário que pertence ao grupo das Mucopolissacaridoses (MPS). É causado por deficiências em uma das quatro enzimas (heparan N-sulfatase, alpha-N-acetilglucosaminidase, acetil-CoAlpha-glucosaminide aciltransferase ou N-acetilglucosamina 6-sulphatase) necessárias para quebrar down os mucopolissacarídeos (GAGs), especificamente heparan sulfato.
A acumulação de GAGs nos lisossomos leva a danos em vários órgãos e sistemas corporais, incluindo o cérebro. Os sintomas geralmente começam entre os 2 e 6 anos de idade e podem incluir problemas de comportamento e aprendizagem, perda auditiva, atraso no crescimento, dismorfia facial, hepatomegalia (fígado aumentado), esplenomegalia (baço aumentado) e problemas cardiovasculares. A doença progressivamente piora ao longo do tempo, levando a complicações neurológicas graves e morte prematura, geralmente na adolescência ou na idade adulta jovem.
A Mucopolissacaridose Tipo III é herdada como um traço autossômico recessivo, o que significa que os indivíduos afetados recebem uma cópia defeituosa do gene de cada pai.
Terapia de reposição de enzimas (ERT, do inglés Enzyme Replacement Therapy) refere-se a um tratamento médico que consiste na administração de uma enzima deficiente ou ausente no organismo de um indivíduo, geralmente devido a uma doença genética. Essa terapia tem como objetivo substituir a enzima deficiente e prevenir ou minimizar os sintomas da doença.
A ERT é frequentemente utilizada no tratamento de doenças lisossomais, um grupo de condições genéticas que afetam as lisossomos, compartimentos celulares responsáveis pelo processamento e reciclagem de materiais dentro da célula. Em muitas dessas doenças, a falta ou deficiência de uma enzima específica leva à acumulação de substâncias tóxicas dentro das células, resultando em danos teciduais e diversos sintomas clínicos.
A terapia de reposição de enzimas envolve a produção da enzima deficiente em laboratório, geralmente por meio de tecnologias de biotecnologia avançadas, como a engenharia genética. A enzima é então purificada e administrada ao paciente por via intravenosa (injetada diretamente na corrente sanguínea) em doses regulares, geralmente a cada uma ou duas semanas.
A ERT pode ajudar a melhorar os sintomas da doença, reduzir o progresso da doença e aumentar a esperança de vida dos pacientes. No entanto, a terapia não consegue corrigir as causas subjacentes da doença e pode apresentar limitações e efeitos colaterais. Além disso, a ERT pode ser extremamente cara, o que limita seu acesso a um grande número de pacientes em todo o mundo.
Mucopolissacaridose II, também conhecida como MPS II ou Doença de Hunter, é uma doença genética rara e progressiva que afeta o metabolismo dos mucopolissacarídeos (GAGs), um tipo de carboidrato complexo encontrado nas células. Essa doença é causada por uma deficiência da enzima iduronato-2-sulfatase, que é necessária para descompor e reciclar os GAGs. Como resultado, os GAGs se acumulam dentro dos lisossomos das células, levando a danos em vários órgãos e sistemas corporais.
A Doença de Hunter é herdada como um traço recessivo ligado ao cromossomo X, o que significa que os meninos são mais frequentemente afetados do que as meninas. Os sinais e sintomas da doença geralmente começam a aparecer entre os 2 e 4 anos de idade e podem incluir:
* Baixo crescimento
* Desenvolvimento mental lento ou estagnado
* Problemas respiratórios
* Doença cardiovascular
* Inchaço dos ganchos do nariz, orelhas e pálpebras
* Hérnias abdominais
Diagnóstico: O diagnóstico da MPS II geralmente é confirmado por meio de um exame de sangue que mede os níveis de enzimas. Também podem ser usadas outras técnicas, como análises genéticas e biópsias de tecidos.
Tratamento: Atualmente, não existe cura para a MPS II. O tratamento geralmente se concentra em gerenciar os sintomas e prevenir complicações. A terapia de reposição enzimática (ERT) pode ser usada para substituir a enzima deficiente, mas seu efeito sobre a progressão da doença é limitado. Outros tratamentos podem incluir fisioterapia, cirurgias e medicamentos para controlar os sintomas.
Mucopolissacaridoses (MPS) são um grupo de doenças genéticas raras e progressivas causadas por deficiências em enzimas específicas que resultam na acumulação de glicosaminoglicanos (GAGs), também conhecidos como mucopolissacarídeos, nas células. Essa acumulação leva a danos em tecidos e órgãos, causando uma variedade de sintomas que podem incluir problemas ósseos e articulares, fígado e baço agrandados, anormalidades faciais, problemas cardiovasculares, comprometimento cognitivo e neurológico, entre outros.
Existem diferentes tipos de MPS, cada um deles causado por uma deficiência específica em determinada enzima. Alguns dos tipos mais comuns incluem a doença de Hurler (MPS IH), a doença de Scheie (MPS IS), a síndrome de Hunter (MPS II), a síndrome de Sanfilippo (MPS III) e a síndrome de Morquio (MPS IV). Cada tipo tem sua própria progressão clínica, sintomas e expectativa de vida.
O tratamento para MPS geralmente inclui terapia de reposição enzimática, cuidados de suporte e, em alguns casos, transplante de medula óssea. O diagnóstico precoce é importante para iniciar o tratamento o mais cedo possível e ajudar a melhorar os resultados clínicos e a qualidade de vida dos pacientes.
Glicosaminoglicanos (GAGs) são longas cadeias polissacarídeas compostas por repetições de disacáridos, que consistem em um hexoseamina e um urônico ou hexurônico ácido. Eles são frequentemente encontrados na matriz extracelular e ligados à proteínas formando proteoglicanos.
Existem diferentes tipos de GAGs, incluindo condroitin sulfato, dermatan sulfato, heparan sulfato, heparina e queratân sulfato. Cada tipo tem uma composição específica de disacáridos e é encontrado em tecidos diferentes do corpo.
As funções dos GAGs incluem fornecer estrutura mecânica aos tecidos, regulando a atividade de fatores de crescimento e citocinas, e participando na interação entre células e matriz extracelular. Alterações nos níveis ou estruturas dos GAGs têm sido associadas a diversas doenças, incluindo oenartrose, distúrbios da hemorragia e câncer.
Mucopolissacaridose IV, também conhecida como Síndrome de Morquio, é uma doença genética rara e progressiva que afeta o metabolismo dos mucopolissacarídeos (GAGs), um tipo de carboidrato complexo encontrado em tecidos conjuntivos, cartilagem e órgãos internos.
A doença é causada por deficiência nas enzimas N-acetilgalactosamina-6-sulfatase (GALNS) ou beta-galactosidase, que são necessárias para a degradação dos GAGs. Como resultado, os GAGs se acumulam nos tecidos e órgãos do corpo, causando danos progressivos e sintomas clínicos.
Existem duas formas da doença: a forma A, causada pela deficiência de GALNS, e a forma B, causada pela deficiência de beta-galactosidase. Ambas as formas afetam principalmente o crescimento ósseo e a estrutura dos tecidos conjuntivos, levando a anormalidades esqueléticas, problemas respiratórios, cardiovasculares e auditivos, entre outros sintomas.
A mucopolissacaridose IV é herdada de forma autossômica recessiva, o que significa que os indivíduos afetados recebem uma cópia do gene defeituoso de cada pai. A doença geralmente se manifesta durante a infância e sua gravidade varia consideravelmente entre os pacientes. Atualmente, não existe cura para a mucopolissacaridose IV, mas o tratamento pode incluir fisioterapia, terapia de reabilitação, medicação para aliviar os sintomas e, em alguns casos, terapia enzimática substitutiva.
O Ácido Idurónico é um glicosaminoglicano, um tipo de carboidrato complexo encontrado em tecidos conectivos e mucopolissacarídeos do corpo humano. Ele é um componente importante da proteoglicana, uma macromolécula formada pela união de glicosaminoglicanos com uma proteína central.
O Ácido Idurónico é um açúcar hexurônico que pode ser encontrado em duas formas isoméricas: iduronato e iduronato-6-sulfato. Ele desempenha um papel importante na estrutura e função dos tecidos conectivos, como o cartilagem, os vasos sanguíneos e a pele.
Além disso, o Ácido Idurónico é também um marcador de doenças hereditárias raras, como a síndrome de Hurler e a síndrome de Hunter, que são doenças metabólicas causadas por deficiências nos enzimas responsáveis pela degradação dos glicosaminoglicanos. Isso pode resultar em acúmulo de glicosaminoglicanos e outros materiais na célula, levando a sintomas como anormalidades esqueléticas, problemas cardiovasculares e neurológicos.
N-Acetilgalactosamina-4-Sulfatase é uma enzima (proteína) que desempenha um papel importante no metabolismo dos mucopolissacarídeos, um tipo de carboidrato complexo presente em nosso corpo. Essa enzima é especificamente responsável por catalisar a remoção do grupo sulfato da posição 4 da N-acetilgalactosamina, um açúcar encontrado nesses mucopolissacarídeos.
A deficiência dessa enzima causa uma doença genética rara conhecida como Mucopolissacaridose tipo VI ou Maroteaux-Lamy, que é caracterizada por um acúmulo progressivo de mucopolissacarídeos sulfatados nos tecidos e órgãos do corpo. Isso pode levar a diversos sintomas clínicos, como problemas ósseos e articulares, anormalidades faciais, baixa estatura, hepatomegalia (fígado aumentado de tamanho), entre outros. O tratamento geralmente inclui terapias de substituição enzimática e cuidados de suporte para gerenciar os sintomas e prevenir complicações.
A definição médica de "cães" se refere à classificação taxonômica do gênero Canis, que inclui várias espécies diferentes de canídeos, sendo a mais conhecida delas o cão doméstico (Canis lupus familiaris). Além do cão doméstico, o gênero Canis também inclui lobos, coiotes, chacais e outras espécies de canídeos selvagens.
Os cães são mamíferos carnívoros da família Canidae, que se distinguem por sua habilidade de correr rápido e perseguir presas, bem como por seus dentes afiados e poderosas mandíbulas. Eles têm um sistema sensorial aguçado, com visão, audição e olfato altamente desenvolvidos, o que lhes permite detectar e rastrear presas a longa distância.
No contexto médico, os cães podem ser estudados em vários campos, como a genética, a fisiologia, a comportamento e a saúde pública. Eles são frequentemente usados como modelos animais em pesquisas biomédicas, devido à sua proximidade genética com os humanos e à sua resposta semelhante a doenças humanas. Além disso, os cães têm sido utilizados com sucesso em terapias assistidas e como animais de serviço para pessoas com deficiências físicas ou mentais.
A condro-4-sulfatase é uma enzima (proteína que catalisa reações químicas) envolvida no metabolismo dos glicosaminoglicanos, um tipo de carboidrato complexo presente na matriz extracelular dos tecidos conjuntivos e ósseos.
Especificamente, a condro-4-sulfatase é responsável por remover o grupo sulfato do carbono 4 da galactoseamine nos glicosaminoglicanos chamados condroitin sulfatos. Essa modificação é importante para regular a interação entre as moléculas de proteoglicano e outras moléculas na matriz extracelular, como fatores de crescimento e citocinas.
Deficiências nesta enzima podem resultar em doenças genéticas raras, como a síndrome de Morquio A (Mucopolissacaridose IVA), que é caracterizada por anormalidades esqueléticas, cardiovasculares e respiratórias, entre outros sintomas.
Iduronato sulfatase é uma enzima (proteína que causa reações químicas no corpo) que desempenha um papel importante na decomposição e reciclagem de certos tipos de moléculas complexas chamadas glicosaminoglicanos (GAGs). Esses GAGs são encontrados nas membranas celulares e no tecido conjuntivo, onde desempenham um papel importante na estrutura e função dos tecidos.
A iduronato sulfatase é especificamente responsável por quebrar a ligação entre dois açúcares específicos (iduronato e N-acetilgalactosamina) em um tipo de GAG chamado dermatan sulfato. A falta ou deficiência dessa enzima leva à acumulação de dermatan sulfato no corpo, o que pode causar vários sintomas graves e progressivos, incluindo problemas ósseos e articulares, anormalidades faciais, problemas cardíacos e respiratórios, e deficiência intelectual. Essa condição é conhecida como mucopolissacaridose do tipo II (MPS II) ou síndrome de Hunter.
As condroitinase sulfatases são um grupo de enzimas que desempenham um papel importante no metabolismo dos glicosaminoglicanos (GAGs), particularmente da condroitina sulfato e dermatan sulfato, que são componentes importantes da matriz extracelular em tecidos conjuntivos, cartilaginosos e ósseos.
Essas enzimas catalisam a remoção de grupos sulfato específicos dos resíduos de condroitina sulfato e dermatan sulfato, o que é essencial para a manutenção do equilíbrio normal entre a síntese e a degradação desses GAGs. A deficiência ou disfunção das condroitinase sulfatases pode resultar em várias doenças genéticas raras, como a mucopolissacaridose tipo IV (MPS IV), também conhecida como síndrome de Morquio.
Existem quatro tipos principais de condroitinase sulfatases: sulfatases A, B, C e D, cada uma das quais remove um tipo específico de grupo sulfato dos resíduos de GAGs. As mutações em qualquer um desses genes podem resultar em diferentes formas clínicas da doença, dependendo do grau e do local da deficiência enzimática.
Glucuronidase é uma enzima que catalisa a reação de hidrólise da glucurónide, que é um éter formado durante o processo de glucuronidação. A glucuronidação é uma importante via metabólica no organismo dos mamíferos, na qual substratos endógenos e exógenos são convertidos em glucurónides mais solúveis em água, facilitando assim a sua excreção.
A enzima Glucuronidase é encontrada em vários tecidos e fluidos corporais, incluindo o fígado, rins, intestino, plasma sanguíneo e leite materno. A atividade da glucuronidase pode ser medida em diferentes fontes biológicas como um indicador de função celular ou patologia. Por exemplo, a atividade da glucuronidase no líquido sinovial pode estar aumentada em pacientes com artrite reumatoide.
A Glucuronidase é clinicamente relevante porque pode desempenhar um papel na ativação de fármacos inativos e também na inativação de fármacos ativos, dependendo do substrato específico. Além disso, a medição da atividade da glucuronidase pode ser útil no diagnóstico e monitorização de algumas condições clínicas.