Granulomatosis de Wegener
Mieloblastina
Anticuerpos Anticitoplasma de Neutrófilos
Granulomatosis Linfomatoide
Vasculitis
Duramadre
Poliangeítis Microscópica
Granulomatosis Orofacial
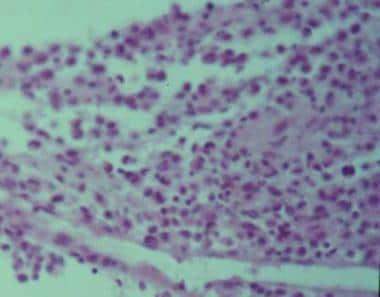
La granulomatosis de Wegener, también conocida como granulomatosis with polyangiitis (GPA), es una enfermedad inflamatoria grave y rara que afecta varios sistemas corporales, especialmente los vasos sanguíneos pequeños en los pulmones, los riñones y las vías respiratorias superiores. Se caracteriza por la inflamación y formación de granulomas (pequeñas masas de tejido anormal) en los vasos sanguíneos y los órganos afectados.
La enfermedad se manifiesta clínicamente con una variedad de síntomas, que pueden incluir:
1. Síntomas respiratorios: tos crónica, dolor torácico, hemoptisis (toser sangre), neumonía y disnea (dificultad para respirar).
2. Síntomas renales: hematuria (sangre en la orina), proteinuria (proteínas en la orina) e insuficiencia renal aguda o crónica.
3. Síntomas de las vías respiratorias superiores: rinitis, sinusitis, dolor facial y pérdida del sentido del olfato.
4. Síntomas sistémicos: fiebre, fatiga, pérdida de apetito y pérdida de peso.
5. Otras manifestaciones clínicas: artralgias (dolores articulares), erupciones cutáneas, úlceras orales y oculares, y neuropatías periféricas (daño a los nervios).
El diagnóstico de la granulomatosis de Wegener se basa en una combinación de hallazgos clínicos, radiológicos e histopatológicos. El análisis de laboratorio puede mostrar niveles elevados de proteína C-reactiva (PCR) y velocidad de sedimentación globular (VSG), así como la presencia de anticuerpos citoplasmáticos anti-neutrófilos (ANCA) específicos, especialmente los dirigidos contra la proteinasa 3 (PR3). Sin embargo, estos últimos no son siempre detectables y su ausencia no excluye el diagnóstico.
El tratamiento de la granulomatosis de Wegener requiere una combinación de inmunosupresión y terapia antiinflamatoria. La terapia de primera línea implica el uso de corticosteroides en combinación con ciclofosfamida o rituximab, seguido de un mantenimiento con azatioprina o metotrexato. En casos graves o refractarios, se pueden considerar otras opciones terapéuticas, como plasmaféresis y terapia dirigida contra las interleucinas proinflamatorias.
La supervivencia a largo plazo de los pacientes con granulomatosis de Wegener ha mejorado significativamente en las últimas décadas gracias al desarrollo de nuevos tratamientos y a un mejor manejo clínico. Sin embargo, la enfermedad sigue siendo una afección grave que puede causar complicaciones importantes y reducir la calidad de vida de los pacientes. Por lo tanto, es fundamental un seguimiento estrecho y una monitorización regular de la respuesta al tratamiento y de los efectos secundarios de la terapia inmunosupresora.
La mieloblastina es un tipo de proteína que se encuentra en la sangre y se asocia con la producción de glóbulos blancos inmaduros, también conocidos como blastos. Es específicamente una proteinasa serina, una enzima que ayuda a descomponer otras proteínas. La mieloblastina se produce principalmente en la médula ósea y se libera al torrente sanguíneo durante la fase de diferenciación de los glóbulos blancos.
Es importante destacar que los niveles elevados de mieloblastina en la sangre pueden ser un indicador de una afección médica subyacente, particularmente ciertos tipos de leucemia, como la leucemia mieloide aguda. Sin embargo, un diagnóstico definitivo requeriría una evaluación más detallada y pruebas adicionales.
Por lo tanto, mientras que la mieloblastina es interesante desde un punto de vista bioquímico, su presencia o niveles elevados en la sangre tienen implicaciones clínicas significativas y pueden ser indicativos de condiciones médicas graves.
Los anticuerpos anticitoplasma de neutrófilos (ANCA) son un tipo de anticuerpo que se encuentra en el torrente sanguíneo y están dirigidos contra los componentes del citoplasma de los neutrófilos, un tipo de glóbulo blanco importante en la respuesta inmunitaria. Los ANCA se asocian con varias enfermedades autoinmunitarias, como la granulomatosis de Wegener, la poliangitis microscópica y la colitis ulcerosa.
Existen dos tipos principales de ANCA: los ANCA perinucleares (pANCA) y los ANCA citoplasmáticos (cANCA). Los pANCA se dirigen contra los antígenos presentes en el núcleo de los neutrófilos, mientras que los cANCA se dirigen contra un antígeno específico llamado proteinasa 3 (PR3) en el citoplasma de estas células.
La presencia de ANCA en sangre puede ayudar a diagnosticar y monitorizar el tratamiento de las enfermedades autoinmunitarias asociadas con estos anticuerpos. Sin embargo, también pueden encontrarse ANCA en personas sin ninguna enfermedad relacionada, por lo que su detección debe interpretarse junto con otros hallazgos clínicos y de laboratorio.
La granulomatosis linfomatoide es un trastorno inflamtorio crónico y raro que afecta predominantemente los tejidos pulmonares y la piel, aunque también puede involucrar otros órganos. Se caracteriza por la presencia de granulomas no caseificantes, que son agregados de células inflamatorias, en particular linfocitos T activados y células gigantes multinucleadas.
Existen dos tipos principales de granulomatosis linfomatoide: la forma clásica y la forma pulmonar. La forma clásica se presenta más comúnmente en adultos jóvenes y afecta principalmente los ganglios linfáticos, la piel y el sistema nervioso central. Por otro lado, la forma pulmonar es más frecuente en fumadores y se limita al tejido pulmonar.
Ambas formas de granulomatosis linfomatoide pueden causar síntomas variables dependiendo del órgano afectado. Los síntomas más comunes incluyen tos crónica, dificultad para respirar, fatiga, fiebre, sudoración nocturna y pérdida de peso involuntaria. En la forma clásica, también pueden presentarse lesiones cutáneas, dolores de cabeza y problemas neurológicos.
Aunque la causa exacta de la granulomatosis linfomatoide sigue siendo desconocida, se cree que está relacionada con una respuesta inmunológica anormal a antígenos desconocidos en individuos genéticamente predispuestos. El diagnóstico de granulomatosis linfomatoide generalmente requiere una biopsia tisular y un examen histopatológico para confirmar la presencia de granulomas característicos.
El tratamiento de la granulomatosis linfomatoide puede incluir corticosteroides, inmunosupresores y agentes biológicos dirigidos contra citoquinas específicas involucradas en la patogénesis de la enfermedad. La respuesta al tratamiento varía entre los pacientes, y algunos pueden requerir terapia de mantenimiento a largo plazo para controlar los síntomas y prevenir recaídas.
La vasculitis es un término médico que se refiere a la inflamación de los vasos sanguíneos. Puede afectar a vasos de diferentes tamaños, desde pequeñas capilares hasta grandes arterias. La inflamación puede causar estrechamiento, debilitamiento o bloqueo de los vasos sanguíneos, lo que puede impedir el flujo sanguíneo adecuado a los tejidos y órganos del cuerpo.
Los síntomas de la vasculitis pueden variar ampliamente dependiendo de la gravedad de la inflamación y la ubicación de los vasos sanguíneos afectados. Algunas personas con vasculitis pueden experimentar fiebre, fatiga, pérdida de apetito y dolores articulares. Otros síntomas más específicos pueden incluir erupciones cutáneas, debilidad muscular, entumecimiento o dolor en los brazos o las piernas, y problemas respiratorios o renales.
La causa de la vasculitis no siempre está clara, pero se cree que puede estar relacionada con respuestas anormales del sistema inmunológico a infecciones, medicamentos, toxinas u otras sustancias extrañas en el cuerpo. En algunos casos, la vasculitis puede ser una complicación de otras enfermedades autoinmunitarias como lupus eritematoso sistémico o artritis reumatoide.
El tratamiento de la vasculitis depende del tipo y gravedad de la enfermedad, pero generalmente implica el uso de medicamentos para reducir la inflamación y suprimir el sistema inmunológico. Los corticosteroides como la prednisona son comúnmente utilizados, al igual que los fármacos inmunosupresores como la ciclofosfamida o el metotrexato. En casos graves, puede ser necesario un tratamiento adicional con plasmaféresis o terapia de reemplazo renal.
La duramadre, también conocida como dura mater en terminología latina, es la capa más externa y resistente de las meninges, las membranas que recubren el sistema nervioso central. Está compuesta principalmente por tejido conectivo denso y rico en colágeno, proporcionando una protección mecánica importante al cerebro y la médula espinal.
La duramadre se adhiere firmemente a las estructuras esqueléticas que rodean el cerebro y la médula espinal, como el cráneo y la columna vertebral. A diferencia de las otras meninges (la aracnoides y la piamadre), la duramadre no tiene un revestimiento celular interno, lo que la hace menos propensa a sufrir lesiones o inflamaciones.
Entre la duramadre y la aracnoides existe un espacio potencial llamado espacio epidural, donde se pueden acumular líquidos o sangre en caso de traumatismos o patologías específicas, como hemorragias o infecciones. La correcta integridad estructural y funcional de la duramadre es crucial para mantener la homeostasis del sistema nervioso central y preservar su integridad fisiológica.
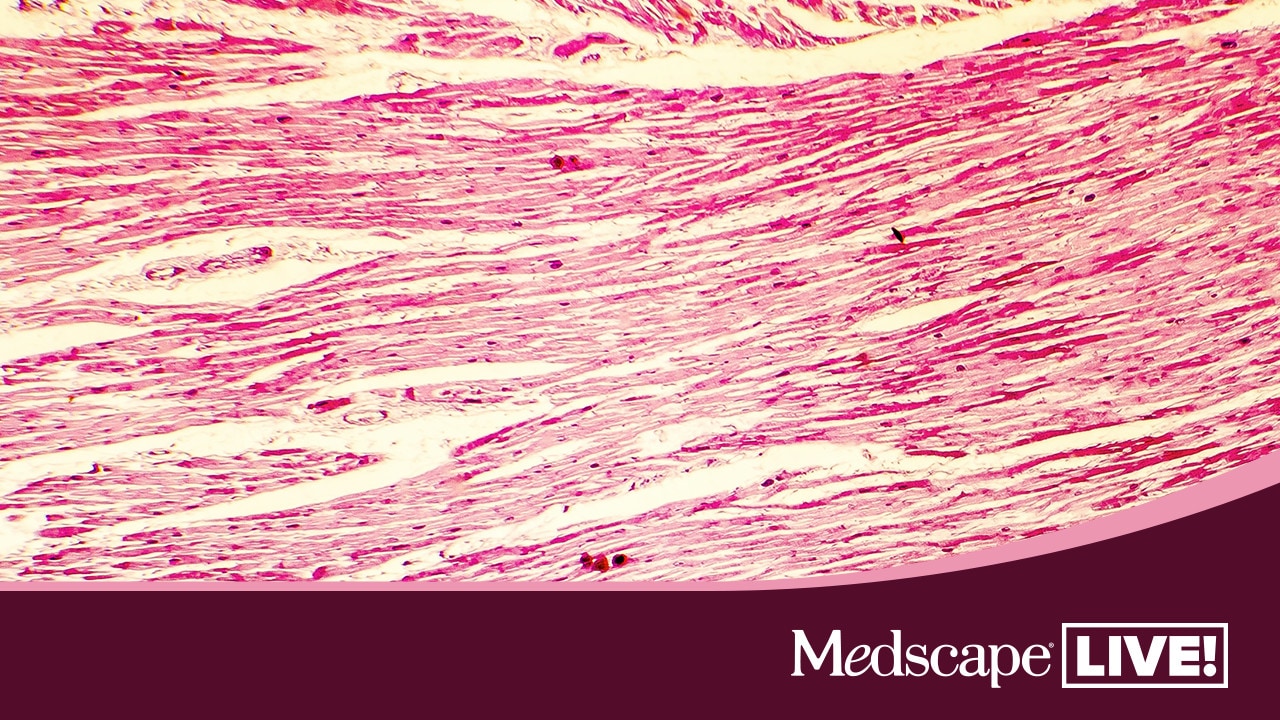
La Poliangeítis Microscópica es una enfermedad inflamatoria de los vasos sanguíneos pequeños (vasculitis) que afecta predominantemente a los glomérulos renales y a las arteriolas pulmonares. Se caracteriza por una necrosis fibrinoide segmentaria y una infiltración inflamatoria de la pared vascular, con predominio de neutrófilos.
Esta enfermedad puede presentarse de forma aislada o asociada a otras enfermedades autoinmunes, como el lupus eritematoso sistémico o la granulomatosis de Wegener. Los síntomas más comunes incluyen hematuria (sangre en la orina), proteinuria (proteínas en la orina), y disfunción renal, aunque también pueden presentarse síntomas pulmonares, cutáneos o neurológicos.
El diagnóstico se realiza mediante biopsia renal o pulmonar, que muestra los cambios característicos en la pared vascular. El tratamiento suele incluir corticosteroides y otros fármacos inmunosupresores, como ciclofosfamida o micofenolato mofetil, con el objetivo de controlar la inflamación y prevenir daños permanentes en los órganos afectados.
La granulomatosis orofacial es una enfermedad crónica que afecta principalmente a los tejidos blandos de la cara y la boca. Aunque no se conoce completamente su causa, se cree que está relacionada con un trastorno del sistema inmunológico. La característica distintiva de esta afección son los granulomas, pequeñas masas de tejido formadas por la acumulación de células inflamatorias.
Los síntomas más comunes incluyen hinchazón y tumefacción en los labios, mejillas u otras áreas de la cara, úlceras orales recurrentes, especialmente en el interior de los labios, y dolor o sensibilidad en las zonas afectadas. Algunos pacientes también pueden experimentar sequedad bucal, dificultad para masticar o tragar, y cambios en la voz.
El diagnóstico de granulomatosis orofacial se realiza generalmente mediante una biopsia, donde se toma una pequeña muestra de tejido para su examen al microscopio. El tratamiento puede incluir medicamentos que reducen la inflamación, como corticosteroides o agentes inmunomoduladores, y en casos graves o refractarios a dicho tratamiento, se podría considerar la cirugía para extirpar los granulomas.
Es importante distinguir la granulomatosis orofacial de otras condiciones que pueden presentarse con síntomas similares, como el cáncer bucal o enfermedades infecciosas, por lo que es crucial buscar atención médica especializada en caso de presentar estos síntomas.
El síndrome de Churg-Strauss, también conocido como granulomatosis eosinofílica con poliangeítis, es una forma rara de vasculitis sistémica, una inflamación de los vasos sanguíneos que puede afectar a varios órganos y sistemas corporales. Se caracteriza por la inflamación de los vasos sanguíneos pequeños y medianos, lo que lleva a una serie de complicaciones clínicas.
Este síndrome se asocia con frecuencia con antecedentes de asma alérgica y sinusalitis crónica. Los síntomas pueden variar ampliamente, dependiendo de los órganos afectados, e incluyen: dolor articular, erupciones cutáneas, neumonía eosinofílica, insuficiencia cardíaca congestiva, y daño renal.
El diagnóstico se realiza mediante una combinación de hallazgos clínicos, análisis de sangre que muestran eosinofilia (un aumento en el número de glóbulos blancos llamados eosinófilos), y pruebas de imagen o biopsias que revelan la inflamación de los vasos sanguíneos. El tratamiento generalmente implica el uso de corticosteroides y otros fármacos inmunosupresores para controlar la inflamación y prevenir daños adicionales a los órganos.
 Vasculitis
Vasculitis Granulomatosis de Wegener | Poliangitis granulomatosa | MedlinePlus en español
Granulomatosis de Wegener | Poliangitis granulomatosa | MedlinePlus en español Actualización en el estudio de Granulomatosis con poliangeitis (Granulomatosis de Wegener)
Actualización en el estudio de Granulomatosis con poliangeitis (Granulomatosis de Wegener) Etiopatogenia, compromiso ocular. Granulomatosis de Wegener - Conde Médicos
Etiopatogenia, compromiso ocular. Granulomatosis de Wegener - Conde Médicos Guía clínica de Vasculitis leucocitoclástica - Fisterra
Guía clínica de Vasculitis leucocitoclástica - Fisterra CRITERIOS DIAGNÓSTICOSL: ENFERMEDAD DE CHURG-STRAUSS
CRITERIOS DIAGNÓSTICOSL: ENFERMEDAD DE CHURG-STRAUSS Granulomatosis con poliangeítis - Trastornos de los tejidos musculoesquelético y conectivo - Manual MSD versión para...
Granulomatosis con poliangeítis - Trastornos de los tejidos musculoesquelético y conectivo - Manual MSD versión para... Comunicación Inflamación/Enfermedades autoinmunes (Pósters) | Revista Española de Cardiología
Comunicación Inflamación/Enfermedades autoinmunes (Pósters) | Revista Española de Cardiología Vasculitis y Riñón | PPT
Vasculitis y Riñón | PPT cap06
cap06 Listado de enfermedades reumáticas | Clínica Reumatológica Dr. Ponce
Listado de enfermedades reumáticas | Clínica Reumatológica Dr. Ponce Causas del mareo - Vestibular Disorders Association
Causas del mareo - Vestibular Disorders Association Liquen plano hipertrófico. A propósito de un caso - Revista Argentina de Dermatología
Liquen plano hipertrófico. A propósito de un caso - Revista Argentina de Dermatología Fascitis eosinofilica.A proposito de un caso clinico | Soanre
Fascitis eosinofilica.A proposito de un caso clinico | Soanre Búsqueda | BVS CLAP/SMR-OPS/OMS
Búsqueda | BVS CLAP/SMR-OPS/OMS DeCS 2015 - Términos alterados
DeCS 2015 - Términos alterados EL RINCÓN DE LA MEDICINA INTERNA.
EL RINCÓN DE LA MEDICINA INTERNA.  Colesteatoma: Qué es, síntomas, tratamiento, diagnóstico y más
Colesteatoma: Qué es, síntomas, tratamiento, diagnóstico y más Vasculitis retiniana - Copro, la enciclopedia libre
Vasculitis retiniana - Copro, la enciclopedia libre