Neurofibromatosis 1
Neurofibromatosis 2
Genes de Neurofibromatosis 1
Genes de la Neurofibromatosis 2
Neurofibromatosis
Neurofibromina 1
Neurofibromina 2
Neurofibroma Plexiforme
Neurofibroma
Manchas Café con Leche
Glioma del Nervio Óptico
Neoplasias de la Vaina del Nervio
Neurilemoma
Neoplasias del Nervio Óptico
Neuroma Acústico
Neoplasias del Sistema Nervioso Periférico
Neurofibrosarcoma
Neoplasias de los Nervios Craneales
Neoplasias del Sistema Nervioso
Meningioma
Implantes Auditivos de Tronco Encefálico
Meningocele
Hamartoma
Seudoartrosis
Células de Schwann
Trastornos del Aprendizaje
Síndrome de Noonan
Mosaicismo
Enfermedades del Desarrollo Óseo
Cromosomas Humanos Par 22
Neoplasias Primarias Múltiples
Piebaldismo
Feocromocitoma
Imagen por Resonancia Magnética
Neoplasias Meníngeas
Somatostatinoma
Descalcificación Patológica
Mutación
Trastornos de la Pigmentación
Tumor Glómico
Quiasma Óptico
Neoplasias Torácicas
Linaje
Astrocitoma
Síndrome de Proteo
Leucemia Mielomonocítica Juvenil
Tomografía Computarizada por Rayos X
Neoplasias del Iris
Angiomatosis
Aracnoides
La neurofibromatosis 1 (NF1), también conocida como enfermedad de Von Recklinghausen, es un trastorno genético que afecta al crecimiento y desarrollo del sistema nervioso. Se caracteriza por la aparición de tumores benignos llamados neurofibromas que se desarrollan a lo largo de los nervios, así como por la presencia de manchas café con leche en la piel y lunares anormales.
Los síntomas suelen aparecer durante la infancia o la adolescencia y pueden variar mucho de una persona a otra. Algunas personas con NF1 tienen solo síntomas leves, mientras que otras pueden experimentar problemas más graves relacionados con el desarrollo del cerebro, los huesos y los órganos internos.
La NF1 se hereda de manera autosómica dominante, lo que significa que solo necesita un gen anormal para desarrollar la enfermedad. Sin embargo, aproximadamente la mitad de los casos surgen debido a una mutación espontánea en el gen NF1.
El tratamiento de la NF1 generalmente se centra en controlar los síntomas y prevenir complicaciones. Los neurofibromas benignos suelen ser retirados quirúrgicamente solo si causan problemas estéticos o funcionales significativos. Es importante que las personas con NF1 reciban atención médica regular para controlar el crecimiento y la progresión de los tumores y detectar cualquier complicación temprano.
La neurofibromatosis 2 (NF2) es una condición genética hereditaria que afecta al sistema nervioso, específicamente al crecimiento y desarrollo de los nervios del oído interno. Es causada por mutaciones en el gen NF2 en el cromosoma 22.
NF2 se caracteriza por la aparición de tumores benignos (no cancerosos) llamados schwannomas, que se desarrollan en los nervios que conectan el oído interno con el cerebro (nervios auditivos). Estos tumores pueden causar pérdida de audición, zumbidos en los oídos (tinnitus), problemas de equilibrio y dificultad para hablar.
Además de los schwannomas, las personas con NF2 también pueden desarrollar meningiomas (tumores en las membranas que rodean el cerebro y la médula espinal) y ependimomas (tumores en el revestimiento de los conductos del líquido cefalorraquídeo).
Los síntomas de NF2 suelen aparecer en la adolescencia o al comienzo de la edad adulta. La condición se diagnostica mediante una combinación de exámenes físicos, pruebas de audición y resonancia magnética (RM) del cerebro y la médula espinal. El tratamiento puede incluir cirugía para extirpar los tumores, radioterapia o terapia con medicamentos para controlar el crecimiento de los tumores y aliviar los síntomas.
La neurofibromatosis 1 (NF1) es una enfermedad genética hereditaria que afecta al desarrollo y función del sistema nervioso. Es causada por mutaciones en el gen NF1, localizado en el brazo largo del cromosoma 17 (17q11.2). Este gen proporciona instrucciones para la producción de una proteína llamada neurofibromina, que actúa como un supresor tumoral y regula la actividad de la vía de señalización RAS/MAPK involucrada en el crecimiento celular y diferenciación.
Las mutaciones en el gen NF1 conllevan a una disminución o ausencia de neurofibromina, lo que provoca un aumento en la actividad de la vía RAS/MAPK y, en última instancia, lleva al desarrollo de tumores benignos (no cancerosos) y otros síntomas asociados con la NF1. Los tipos más comunes de tumores observados en esta enfermedad son los neurofibromas, que se forman a partir de células nerviosas y tejido conectivo.
La NF1 se hereda siguiendo un patrón autosómico dominante, lo que significa que una copia alterada del gen NF1 en cualquiera de los padres es suficiente para causar la enfermedad. Sin embargo, aproximadamente el 50% de los casos surgen debido a nuevas mutaciones en el gen y no tienen antecedentes familiares de la enfermedad. Los síntomas y la gravedad de la NF1 pueden variar ampliamente entre las personas afectadas. Algunas de las características clínicas más comunes incluyen:
1. Manchas café con leche: manchas planas de color marrón claro o beige que aparecen en la piel, especialmente en la parte superior del tronco y los brazos.
2. Lentigines: pequeñas manchas pigmentadas oscuras que se encuentran en la piel alrededor de los ojos, la boca y el área genital.
3. Neurofibromas: tumores benignos que crecen debajo de la piel o dentro de los nervios. Pueden ser de diferentes tamaños y formas, desde pequeñas protuberancias hasta grandes masas.
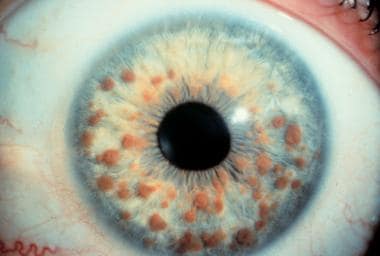
4. Lisch nódulos: pequeños depósitos de pigmento en el iris del ojo.
5. Displasia esponjosa ósea: huesos frágiles y propensos a las fracturas.
6. Protuberancias anormales en la piel (papilomas cutáneos) alrededor de los pliegues corporales, como axilas e ingles.
7. Retraso del desarrollo o retraso mental leve en algunos casos.
8. Problemas de aprendizaje y déficit de atención.
9. Mayor riesgo de ciertos tipos de cáncer, como el glioma y el feocromocitoma.
Es importante tener en cuenta que no todos los individuos con neurofibromatosis tipo 1 desarrollarán todos estos signos o síntomas, y algunas personas pueden experimentar solo una o dos de estas características. El diagnóstico y el seguimiento médico adecuados son esenciales para garantizar un tratamiento y manejo óptimos de la enfermedad.
La neurofibromatosis 2 (NF2) es una condición genética hereditaria que afecta al sistema nervioso, específicamente al crecimiento y desarrollo de los nervios del oído interno. Es causada por mutaciones en el gen NF2, localizado en el brazo corto del cromosoma 22 (22q12.2). Este gen proporciona instrucciones para la producción de una proteína llamada merlina o schwannomin, que ayuda a regular el crecimiento y la división celular en los tejidos conectivos que recubren los nervios (la vaina de mielina).
Las mutaciones en el gen NF2 pueden dar lugar a una producción reducida o ausente de la proteína merlina, lo que lleva al crecimiento descontrolado de células que forman tumores benignos llamados schwannomas. Estos tumores se desarrollan en los nervios del oído interno y pueden causar pérdida auditiva, zumbido en los oídos (acúfenos) y problemas de equilibrio. Los schwannomas también pueden crecer en otros nervios craneales y espinales, provocando diversos síntomas neurológicos.
La neurofibromatosis 2 se hereda con un patrón autosómico dominante, lo que significa que una copia alterada del gen NF2 en cualquiera de los padres es suficiente para causar la enfermedad. Sin embargo, aproximadamente el 50% de los casos surgen como resultado de nuevas mutaciones en el gen y no tienen antecedentes familiares de la afección.
La neurofibromatosis es un trastorno genético que afecta el crecimiento y desarrollo del sistema nervioso. Existen tres tipos principales: neurofibromatosis tipo 1 (NF1), neurofibromatosis tipo 2 (NF2) y schwannomatosis.
La NF1, también conocida como enfermedad de Von Recklinghausen, es la forma más común. Se caracteriza por la aparición de manchas café con leche en la piel, lunares anormales y tumores benignos (neurofibromas) que se desarrollan a lo largo de los nervios debajo de la piel y en ocasiones dentro del cráneo o la médula espinal. Los síntomas generalmente aparecen durante la infancia o la adolescencia.
La NF2, por otro lado, se manifiesta principalmente por la presencia de tumores benignos (schwannomas) que crecen en los nervios auditivos, lo que puede causar sordera. También pueden desarrollarse meningiomas (tumores de las membranas que rodean el cerebro y la médula espinal) y neurofibromas. Los síntomas suelen comenzar en la adolescencia o al comienzo de la edad adulta.
Schwannomatosis es el menos común de los tres tipos y se caracteriza por la aparición de múltiples schwannomas, pero generalmente no afecta los nervios auditivos. Los síntomas pueden incluir dolor crónico e inestabilidad.
Estos trastornos son causados por mutaciones en diferentes genes (NF1, NF2 y SMARCB1 o LZTR1 para schwannomatosis). Pueden heredarse de los padres o pueden ocurrir espontáneamente. El tratamiento depende del tipo y gravedad de los síntomas y puede incluir cirugía, radioterapia o monitoreo regular.
La neurofibromina 1 es un tipo de proteína que está codificada por el gen NF1 y desempeña un papel crucial en la supresión del crecimiento y división celular. Esta proteína ayuda a regular células nerviosas y otras células del cuerpo, previniendo así el crecimiento excesivo de tejido que podría conducir al desarrollo de tumores benignos o malignos.
Las mutaciones en el gen NF1 pueden dar lugar a la enfermedad neurofibromatosis tipo 1 (NF1), una afección genética hereditaria que se caracteriza por la aparición de tumores benignos llamados neurofibromas en los nervios y manchas cutáneas pigmentadas llamadas lunares café con leche. La proteína neurofibromina 1 también participa en la conducción de señales químicas dentro de las células, especialmente aquellas involucradas en el crecimiento y desarrollo celular.
La deficiencia o disfunción de la neurofibromina 1 puede provocar una serie de problemas de salud, como dificultades de aprendizaje, retrasos del desarrollo, presión arterial alta, dolor crónico y un mayor riesgo de cáncer. El tratamiento para los trastornos relacionados con la neurofibromina 1 generalmente se centra en el control de los síntomas y la prevención de complicaciones graves, como el crecimiento de tumores malignos.
La neurofibromina 2, también conocida como proteína de merlín o schwannomin, es una proteína suprimiedora de tumores que desempeña un papel crucial en la regulación del crecimiento y la diferenciación celular. Se codifica por el gen NF2 localizado en el cromosoma 22q12. La neurofibromina 2 ayuda a inhibir la actividad de las vías de señalización que promueven la proliferación celular y la supervivencia, como la vía Ras/MAPK y la vía PI3K/AKT.
Las mutaciones en el gen NF2 están asociadas con el síndrome de neurofibromatosis tipo 2 (NF2), una enfermedad hereditaria autosómica dominante que se caracteriza por la aparición de tumores benignos, como schwannomas vestibulares, meningiomas y neurofibromas. Estos tumores pueden causar diversas complicaciones clínicas, como pérdida auditiva, vértigo, debilidad muscular, dolor y problemas visuales. El diagnóstico de NF2 se basa en los criterios clínicos y genéticos, y el manejo suele implicar la observación clínica, la cirugía y, en algunos casos, la radioterapia.
Un neurofibroma plexiforme es un tipo raro y más complejo de neurofibroma, que es un tumor benigno (no canceroso) que se desarrolla a partir de los nervios periféricos. Los neurofibromas plexiformes se caracterizan por el crecimiento anormal de tejido nervioso y células conectivas en los haces nerviosos, lo que puede provocar una masa tumoral dolorosa y deforme.
A diferencia de los neurofibromas más comunes, que suelen localizarse justo debajo de la piel y tienen un tamaño relativamente pequeño, los neurofibromas plexiformes pueden crecer dentro y alrededor de los nervios importantes en todo el cuerpo, incluidos los nervios craneales, espinales y periféricos. Pueden crecer gradualmente a lo largo del tiempo y afectar a grandes áreas del sistema nervioso periférico.
Los neurofibromas plexiformes suelen estar asociados con el síndrome de neurofibromatosis tipo 1 (NF1), una enfermedad genética hereditaria que afecta al desarrollo y crecimiento del sistema nervioso y causa la aparición de tumores benignos en todo el cuerpo. Sin embargo, también pueden ocurrir de forma esporádica, sin antecedentes familiares de NF1.
El tratamiento de los neurofibromas plexiformes puede ser complicado y depende del tamaño, la ubicación y los síntomas asociados. En algunos casos, se pueden extirpar quirúrgicamente, pero esto puede ser difícil si el tumor está cerca de nervios importantes o órganos vitales. La radioterapia y la terapia con fármacos también pueden utilizarse en algunos casos para reducir el tamaño del tumor o aliviar los síntomas asociados.
Un neurofibroma es un tipo de tumor benigno que se origina en los nervios. Se desarrolla a partir de las células de la vaina de mielina, que son las envolturas protectoras de los nervios. Los neurofibromas pueden crecer a lo largo del nervio (neurofibroma plexiforme) o como una masa única y bien definida (neurofibroma discreto).
Estos tumores suelen presentarse en la piel o justo debajo de ella, donde se pueden sentir como bultos firmes y a menudo dolorosos al tacto. Los neurofibromas también pueden crecer dentro del cuerpo, en cuyo caso pueden comprimir los órganos cercanos y causar problemas de salud.
La aparición de neurofibromas está asociada con una afección genética llamada neurofibromatosis tipo 1 (NF1), que se caracteriza por la presencia de múltiples tumores benignos en todo el cuerpo. Sin embargo, también pueden ocurrir de forma esporádica, sin un historial familiar de NF1.
El tratamiento de los neurofibromas depende de su tamaño, ubicación y síntomas asociados. En general, se recomienda la observación y el monitoreo regulares si los tumores son pequeños y no causan problemas. Si los neurofibromas crecen grandes, causan dolor o interfieren con las funciones corporales, pueden requerir tratamiento, que puede incluir cirugía para extirparlos.
Las manchas café con leche, también conocidas como nevus depigmentosus o nevo pigmentoso, son manchas planas y de color blanco lechoso u ocre que aparecen en la piel. Suelen ser pequeñas, aunque pueden crecer hasta alcanzar tamaños considerables. Estas manchas suelen presentarse desde el nacimiento o durante la infancia y se localizan con mayor frecuencia en el tronco, los brazos y las piernas.
Aunque generalmente no representan un problema médico importante, las manchas café con leche pueden asociarse a determinadas condiciones genéticas o síndromes, como el síndrome de neurofibromatosis tipo 1 (NF1). Cuando se presentan más de cinco manchas café con leche en el cuerpo y tienen un diámetro superior a 0,5 cm en los niños o 1,5 cm en los adultos, es recomendable realizar una evaluación adicional para descartar la presencia del síndrome de NF1 u otras afecciones subyacentes.
En resumen, las manchas café con leche son manchas planas y pigmentadas que aparecen en la piel y pueden asociarse a determinados síndromes genéticos. Aunque generalmente no representan un problema médico grave, es importante vigilarlas y realizar un seguimiento médico si se presentan más de cinco manchas café con leche o tienen un diámetro superior al normal.
Un glioma del nervio óptico es un tipo raro de tumor cerebral que se origina en el tejido glial del nervio óptico, que es la vía de transmisión de las señales visuales desde el ojo hasta el cerebro. Los gliomas son generalmente tipos de tumores que surgen del tejido de soporte del sistema nervioso central (SNC), llamado glia.
Los gliomas del nervio óptico pueden ser benignos (no cancerosos) o malignos (cancerosos). Los gliomas benignos tienden a crecer lentamente y rara vez se diseminan más allá del lugar donde se originaron. Por otro lado, los gliomas malignos crecen y se propagan más rápidamente, invadiendo tejidos circundantes y otros órganos.
Los síntomas de un glioma del nervio óptico pueden variar dependiendo del tamaño y la ubicación del tumor, así como de su velocidad de crecimiento. Algunos de los síntomas más comunes incluyen:
1. Pérdida de visión progresiva en uno o ambos ojos.
2. Dolor de cabeza.
3. Protuberancia o hinchazón en la región del ojo afectado.
4. Dificultad para ver los colores o distinguir entre ellos.
5. Pupilas anormales o desiguales.
6. Movimientos oculares incontrolables (nistagmo).
7. Disminución del campo visual (pérdida de visión periférica).
El tratamiento de un glioma del nervio óptico dependerá del tipo, tamaño y localización del tumor, así como de la edad y el estado general de salud del paciente. La extirpación quirúrgica, la radioterapia y la quimioterapia son algunas de las opciones de tratamiento que se pueden considerar, individual o en combinación, según cada caso particular. En algunos casos, se puede optar por un seguimiento periódico sin tratamiento inmediato si el tumor no está causando síntomas graves o si su crecimiento es lento.
Las neoplasias de la vaina del nervio, también conocidas como tumores de la vaina del nervio, se refieren a un crecimiento anormal de tejido en la vaina de un nervio. La vaina del nervio es el revestimiento protector que rodea a los nervios y ayuda en la transmisión de los impulsos nerviosos.
Existen dos tipos principales de neoplasias de la vaina del nervio: el Schwannoma y el Neurofibroma.
1. Schwannoma: Este tipo de tumor se desarrolla a partir de las células de Schwann, que producen la mielina, la sustancia grasa aislante que recubre los axones de los nervios. La mayoría de los schwannomas son benignos (no cancerosos), pero en raras ocasiones pueden ser malignos (cancerosos). Por lo general, crecen lentamente y suelen manifestarse como una masa única y bien definida.
2. Neurofibroma: Este tipo de tumor se origina a partir de las células nerviosas y los tejidos conectivos que rodean los nervios. Los neurofibromas pueden ser benignos o malignos. A diferencia de los schwannomas, que suelen formarse como una masa única, los neurofibromas a menudo se presentan como tumores múltiples y difusos.
Ambos tipos de neoplasias de la vaina del nervio pueden causar diversos síntomas dependiendo de su tamaño, localización y crecimiento. Los síntomas más comunes incluyen dolor, entumecimiento, hormigueo o debilidad en el área afectada por el tumor. En algunos casos, los tumores pueden comprimir estructuras adyacentes, como vasos sanguíneos y órganos, lo que puede dar lugar a complicaciones más graves.
El tratamiento de las neoplasias de la vaina del nervio depende del tipo de tumor, su tamaño, localización y los síntomas asociados. Las opciones de tratamiento incluyen la observación, la extirpación quirúrgica, la radioterapia y la quimioterapia. En algunos casos, se puede recurrir a técnicas más avanzadas, como la ablación por radiofrecuencia o la crioterapia.
El neurilemoma, también conocido como schwannoma o neurofibroma del nervio vestibular, es un tipo de tumor benigno que se origina en la vaina de mielina de los nervios periféricos. Está compuesto por células de Schwann, que producen la mielina que recubre y protege los axones de los nervios.
Este tipo de tumor puede crecer a lo largo del nervio, pero rara vez invade el tejido circundante. Los neurilemomas suelen ser pequeños y asintomáticos, aunque en algunos casos pueden crecer lo suficiente como para comprimir los nervios adyacentes y causar dolor, entumecimiento o debilidad muscular.
Los neurilemomas pueden aparecer a cualquier edad, pero son más comunes en adultos de mediana edad. A menudo se encuentran en el cuello, la cabeza, las extremidades y la columna vertebral. En casos raros, los neurilemomas pueden desarrollarse en el sistema nervioso central o en los órganos internos.
El tratamiento de un neurilemoma depende del tamaño y la ubicación del tumor. En muchos casos, se puede observar el tumor con seguimiento periódico mediante resonancia magnética o tomografía computarizada. Si el tumor causa síntomas o crece con el tiempo, se puede considerar la extirpación quirúrgica. La radioterapia y la quimioterapia no suelen ser efectivas en el tratamiento de los neurilemomas.
Las neoplasias del nervio óptico se refieren a tumores benignos o malignos que se desarrollan en el nervio óptico, la estructura que transmite las señales visuales desde el ojo al cerebro. Estos tumores pueden causar diversos síntomas, dependiendo de su tamaño, ubicación y grado de invasividad.
Los tumores benignos, como los gliomas del nervio óptico, suelen crecer lentamente y raramente representan una amenaza para la vida. Sin embargo, pueden provocar pérdida de visión progresiva o incluso ceguera si comprimen el nervio óptico o las estructuras circundantes.
Por otro lado, los tumores malignos, como los glioblastomas del nervio óptico, crecen más rápidamente y tienen un mayor potencial de invasión y diseminación a otras partes del cuerpo. Estos tumores suelen requerir un tratamiento más agresivo, que puede incluir cirugía, radioterapia y quimioterapia.
El diagnóstico de las neoplasias del nervio óptico suele basarse en la historia clínica del paciente, el examen físico y los resultados de pruebas de imagenología avanzada, como la resonancia magnética nuclear (RMN) o la tomografía computarizada (TC). En algunos casos, se puede necesitar una biopsia para confirmar el diagnóstico y determinar el tipo y grado de tumor.
El tratamiento de estas neoplasias depende del tipo y grado del tumor, su localización y extensión, y la edad y estado de salud general del paciente. En algunos casos, se puede optar por una observación cuidadosa y un seguimiento periódico en lugar de un tratamiento agresivo. Sin embargo, en otros casos, el tratamiento puede ser necesario para prevenir la pérdida de visión o la propagación del tumor a otras partes del cuerpo.
El neuroma acústico, también conocido como schwannoma vestibular, es un tipo específico de tumor benigno que se origina en el nervio auditivo o nervio vestibular (que son parte del octavo par craneal). Este tumor está compuesto por células de Schwann, las cuales producen la mielina, la sustancia grasa que recubre y protege los nervios.
El neuroma acústico generalmente se desarrolla en el ganglio de Scarpa, la zona donde el nervio vestibular se divide en sus dos ramas: el nervio coclear y el nervio vestibular propiamente dicho. A medida que el tumor crece, puede comprimir los nervios adyacentes, lo que puede causar diversos síntomas.
Los síntomas más comunes del neuroma acústico incluyen:
1. Pérdida auditiva unilateral (en un solo oído) gradual y progresiva.
2. Zumbido o pitidos en el oído afectado (tinnitus).
3. Sensación de plenitud o presión en el oído afectado.
4. Problemas de equilibrio y coordinación.
5. Mareos o vértigos, especialmente al cambiar de posición rápidamente.
6. Debilidad facial leve o parálisis facial en casos avanzados.
El diagnóstico de un neuroma acústico se realiza mediante una combinación de pruebas, como la audiometría, la resonancia magnética nuclear (RMN) y, en algunos casos, la biopsia del tumor. El tratamiento puede incluir la observación periódica, la radioterapia o la cirugía para extirpar el tumor, dependiendo del tamaño, el crecimiento y los síntomas asociados.
Las neoplasias del sistema nervioso periférico (SNP) se refieren a un crecimiento anormal y descontrolado de tejidos en el sistema nervioso periférico, que es el componente del sistema nervioso que se encuentra fuera del cerebro y la médula espinal. Estos tumores pueden ser benignos o malignos (cancerosos).
Los tipos más comunes de neoplasias en el SNP incluyen:
1. Schwannomas: estos son tumores que se desarrollan a partir de las células de Schwann, que recubren los nervios periféricos. Los schwannomas suelen ser benignos, pero raramente pueden volverse cancerosos. El más conocido es el neurinoma del acústico o schwannoma vestibular, que afecta al nervio auditivo.
2. Neurofibromas: estos tumores se originan en las células de los tejidos conectivos que rodean los nervios periféricos. La mayoría son benignos, pero aproximadamente el 5-10% pueden transformarse en malignos, especialmente en personas con neurofibromatosis tipo 1 (NF1), una enfermedad genética hereditaria.
3. Mielomas multiples: estas son neoplasias malignas de las células plasmáticas, que se encuentran en la médula ósea y producen anticuerpos. Los mielomas multiples pueden causar lesiones en los huesos y afectar el funcionamiento del SNP.
4. Ganglioneuromas: estos son tumores benignos que surgen de las células nerviosas del sistema nervioso simpático, localizadas principalmente en la cavidad torácica y abdominal.
5. Sarcomas: estos son tumores malignos que se desarrollan a partir de tejidos conectivos como músculos, tendones, ligamentos, vasos sanguíneos o grasa. Algunos ejemplos incluyen el sarcoma de Ewing, el sarcoma sinovial y el rabdomiosarcoma.
6. Linfomas: estas neoplasias malignas surgen de los linfocitos, un tipo de glóbulo blanco que forma parte del sistema inmunológico. Los linfomas pueden afectar los ganglios linfáticos y otros órganos, incluyendo el SNP.
El tratamiento de las neoplasias del SNP depende del tipo de tumor, su localización, el estadio de la enfermedad y la salud general del paciente. Las opciones de tratamiento pueden incluir cirugía, radioterapia, quimioterapia, terapia dirigida o inmunoterapia. En algunos casos, se puede considerar un trasplante de células madre para tratar tumores malignos agresivos. El pronóstico y la supervivencia varían ampliamente dependiendo del tipo de neoplasia y el estadio en el momento del diagnóstico.
El neurofibrosarcoma es un tipo raro y agresivo de cáncer que se origina en las células nerviosas periféricas o en los tejidos que rodean los nervios. Este tipo de sarcoma se caracteriza por el crecimiento anormal y descontrolado de células tumorales en los tejidos blandos, como músculos, tendones, ligamentos, grasa y vasos sanguíneos.
Neurofibrosarcomas generalmente afectan a adultos mayores de 30 años, aunque también pueden ocurrir en niños y adolescentes. Los síntomas iniciales pueden incluir hinchazón, dolor o sensibilidad en el área afectada, debilidad muscular y dificultad para moverse. A medida que el tumor crece, puede comprimir los nervios cercanos y causar entumecimiento, hormigueo o parálisis en las extremidades.
El diagnóstico de neurofibrosarcoma generalmente se realiza mediante una biopsia del tejido afectado, seguida de pruebas de imagenología, como resonancia magnética (RM) o tomografía computarizada (TC), para determinar la extensión y localización del tumor. El tratamiento puede incluir cirugía para extirpar el tumor, radioterapia y quimioterapia para destruir las células cancerosas restantes. La tasa de supervivencia a largo plazo para los pacientes con neurofibrosarcoma es relativamente baja, especialmente si el cáncer se ha diseminado a otras partes del cuerpo.
Las neoplasias de los nervios craneales se refieren a tumores benignos o malignos que se desarrollan en los nervios craneales, que son los nervios que emergen directamente del tronco encefálico y la base del cráneo y suministran funciones sensoriales, motoras y autónomas a la cabeza y el cuello.
Existen doce pares de nervios craneales, numerados del I al XII. Cada uno de estos nervios puede verse afectado por neoplasias primarias o secundarias (metastásicas). Las neoplasias primarias son relativamente raras y pueden ser benignas (como schwannomas y neurinomas) o malignas (como los neurofibrosarcomas y los astrocitomas periféricos).
Las neoplasias de los nervios craneales pueden causar diversos síntomas, dependiendo del nervio afectado y la ubicación del tumor. Estos síntomas pueden incluir dolor de cabeza, debilidad o parálisis de los músculos faciales, pérdida de sensibilidad en la cara, trastornos del equilibrio y la audición, dificultad para tragar o hablar, y problemas visuales.
El tratamiento de las neoplasias de los nervios craneales depende del tipo y el tamaño del tumor, así como de su localización y la extensión de la enfermedad. La cirugía es a menudo el pilar del tratamiento, con o sin radioterapia adyuvante o quimioterapia. En algunos casos, la radioterapia o la quimioterapia pueden ser las opciones de tratamiento preferidas, especialmente si el tumor es inoperable o se ha diseminado a otras partes del cuerpo.
Las neoplasias del sistema nervioso se refieren a un crecimiento anormal y descontrolado de células en el tejido cerebral, medular o de los nervios periféricos. Pueden ser benignas (no cancerosas) o malignas (cancerosas). Las neoplasias benignas suelen crecer más lentamente, tienen límites bien definidos y raramente se diseminan a otras partes del cuerpo. Por otro lado, las neoplasias malignas crecen más rápidamente, invaden los tejidos circundantes y pueden diseminarse o metastatizar a otras partes del cuerpo.
Existen diversos tipos de neoplasias del sistema nervioso, dependiendo de la región afectada y del tipo de células involucradas en el proceso tumoral. Algunos ejemplos incluyen gliomas (que surgen de las células gliales que soportan y protegen a las neuronas), meningiomas (que se originan en las membranas que recubren el cerebro y la médula espinal), neuromas (que se desarrollan a partir de los nervios periféricos) y meduloblastomas (tumores malignos primarios del cerebro que comúnmente afectan a niños).
El tratamiento de estas neoplasias dependerá del tipo, localización y grado de malignidad. Puede incluir cirugía, radioterapia, quimioterapia o una combinación de estos enfoques terapéuticos. La pronóstico también varía ampliamente dependiendo del tipo y estadio de la neoplasia.
Un meningioma es un tipo específico de tumor cerebral que se origina en las membranas que rodean el cerebro y la médula espinal, conocidas como meninges. Más concretamente, este tipo de tumores suelen desarrollarse en las capas más externas de las meninges, llamadas aracnoides.
La mayoría de los meningiomas son benignos, lo que significa que no son cancerosos y crecen lentamente. Sin embargo, incluso los meningiomas benignos pueden causar problemas importantes si aumentan de tamaño y presionan estructuras vitales dentro del cráneo o la columna vertebral, como el cerebro, los nervios craneales o la médula espinal.
En algunos casos, los meningiomas pueden ser malignos (cancerosos) o presentar un comportamiento atípico, lo que significa que crecen y se diseminan más rápidamente en comparación con los meningiomas benignos. Estos tipos de meningiomas tienen un mayor riesgo de recurrencia después del tratamiento y pueden ser más difíciles de tratar.
Los síntomas asociados con los meningiomas varían ampliamente, dependiendo de su tamaño, ubicación y crecimiento. Algunos síntomas comunes incluyen dolores de cabeza, convulsiones, problemas de visión, pérdida de audición, déficits neurológicos (como debilidad o entumecimiento en los brazos o las piernas), cambios cognitivos y trastornos del equilibrio. El tratamiento puede incluir la observación cuidadosa, la cirugía para extirpar el tumor, la radioterapia o una combinación de estas opciones.
Los Implantes Auditivos de Tronco Encefálico (IATE) son dispositivos médicos implantados quirúrgicamente que estimulan directamente el nervio auditivo en el tronco encefálico. Estos implantes están diseñados para personas con sorderas profundas o completas, especialmente aquellas que no han obtenido beneficios de los implantes cocleares tradicionales.
El sistema IATE consta de dos partes principales: un conjunto de electrodos que se coloca directamente en el nervio auditivo y una unidad externa que contiene el procesador de señal y la batería. La unidad externa captura los sonidos del entorno, los convierte en impulsos eléctricos y luego los transmite a través de la piel hasta el conjunto de electrodos. Estos impulsos estimulan directamente el nervio auditivo, lo que permite que las señales se transmitan al cerebro y se interpreten como sonido.
Los IATE pueden ser una opción para aquellas personas con sorderas causadas por lesiones en el tronco encefálico, tumores cerebrales o infecciones, entre otras afecciones. Sin embargo, debido a su naturaleza invasiva y al alto riesgo de complicaciones asociadas con la cirugía, los IATE generalmente se consideran solo después de que otros tratamientos hayan fallado.
La meningocele es una afección congénita del sistema nervioso central donde una porción de las membranas que recubren el cerebro o la médula espinal (meninges) sobresale a través de un defecto en el cráneo o la columna vertebral. Esto crea una bolsa llena de líquido cefalorraquídeo bajo la piel. A diferencia de la espina bífida, que también es un trastorno del cierre del tubo neural, la meningocele no involucra las estructuras nerviosas mismas, aunque aún puede causar problemas neurológicos si el saco afecta los nervios circundantes. El tratamiento generalmente implica una cirugía para corregir el defecto y prevenir complicaciones como infecciones o daño nervioso adicional.
Un hamartoma es un crecimiento benigno (no canceroso) que está compuesto de un exceso de células y tejidos normales, pero que están organizados de manera desordenada. Se encuentran comúnmente en la piel, el pulmón, el hígado y el cerebro. A menudo se ven como una masa o tumoración, aunque no son cancerosos. Su crecimiento tiende a detenerse después de un tiempo, pero en algunos casos pueden seguir creciendo lentamente. La mayoría de los hamartomas no causan síntomas y no requieren tratamiento, a menos que interfieran con las funciones corporales o provoquen molestias.
La pseudartrosis es una condición en la que ocurre una unión anormal entre los extremos de un hueso roto que no ha sanado correctamente mediante un proceso normal de curación llamado osteogénesis. En cambio, se forma tejido fibroso o cicatricial en lugar de hueso verdadero en el sitio de la fractura. Esta unión es inestable y puede causar dolor, debilidad o incluso conducir a una deformidad si no se trata. La pseudartrosis puede ser congénita (presente desde el nacimiento) o adquirida (desarrollada más tarde en la vida), y puede ser el resultado de una fractura mal curada, una infección, una enfermedad ósea subyacente o un tratamiento deficiente. El tratamiento puede incluir cirugía para promover la formación de hueso verdadero y estabilizar la extremidad afectada.
Los cromosomas humanos del par 17, generalmente denotados como chromosome pairs 17 o simplemente 17, son una de las 23 parejas de cromosomas homólogos que se encuentran en el núcleo de cada célula somática humana. Los cromosomas humanos normales están presentes en la mayoría de los tejidos corporales en una cantidad diploide, es decir, 46 en total, incluidos dos cromosomas 17.
Cada cromosoma 17 contiene miles de genes y varios centenares de miles de pares de bases de ADN, que codifican gran parte del genotipo individual y determinan muchas características fenotípicas. Los cromosomas 17 son acrocéntricos, lo que significa que tienen un brazo corto (p) y un brazo largo (q). El brazo corto contiene alrededor de 35 millones de pares de bases de ADN y el brazo largo contiene aproximadamente 75 millones de pares de bases.
El brazo corto del cromosoma 17 contiene genes importantes asociados con enfermedades genéticas, como la neurofibromatosis tipo 1 (NF1), el síndrome de Marfan y la enfermedad de Huntington. El brazo largo contiene genes relacionados con varios cánceres, incluido el cáncer de mama y el cáncer colorrectal hereditario sin poliposis (HNPCC).
Las anomalías numéricas o estructurales en los cromosomas 17 se han relacionado con diversas afecciones genéticas y síndromes, como la trisomía 17 mosaico, que se ha asociado con retraso mental y rasgos dismórficos. Las alteraciones estructurales del cromosoma 17, como las deleciones o duplicaciones, también pueden causar diversas enfermedades genéticas, dependiendo de la región afectada y el tamaño de la reestructuración.
Las células de Schwann, también conocidas como neurilemmas o células de la vaina de mielina, son células gliales que revisten y protegen los axones de las neuronas en el sistema nervioso periférico. Su función principal es producir y mantener la mielina, una capa aislante grasa que rodea los axones y permite una conducción rápida y eficiente de los impulsos nerviosos.
Cada célula de Schwann envuelve únicamente a un segmento de axón, formando múltiples capas de membrana plasmática enrolladas en espiral alrededor del axón, dando lugar a la formación de los nódulos de Ranvier. Estos nódulos son zonas sin mielina donde se producen las sinapsis eléctricas entre las células de Schwann y los axones, lo que facilita una conducción saltatoria de los impulsos nerviosos a lo largo del axón.
Las células de Schwann desempeñan un papel crucial en la regeneración y reparación de los nervios periféricos dañados, ya que pueden proliferar y migrar hacia las zonas lesionadas para promover el crecimiento axonal y facilitar la reconstrucción de las vías nerviosas. Además, también participan en la respuesta inmunitaria al eliminar los fragmentos de mielina desmielinizados y presentar antígenos a los linfocitos.
Anomalías en el desarrollo o funcionamiento de las células de Schwann pueden dar lugar a diversas patologías, como la neuropatía diabética, la enfermedad de Charcot-Marie-Tooth y los tumores neurilemmatos.
Los Trastornos del Aprendizaje (TDA) son un grupo de trastornos neurológicos que se manifiestan en la dificultad para adquirir y utilizar habilidades académicas necesarias para el progreso escolar adecuado a la edad, como lo son la lectura, la escritura y las matemáticas. Estos trastornos se caracterizan por un desfase significativo entre el desarrollo intelectual general del individuo y su capacidad para aprender o comprender ciertas habilidades académicas.
Según el Manual Diagnóstico y Estadístico de los Trastornos Mentales (DSM-5), publicado por la Asociación Americana de Psiquiatría, existen tres tipos principales de trastornos del aprendizaje:
1. Trastorno de la Lectura (Dislexia): Dificultad en el reconocimiento y manipulación de los sonidos de las palabras (conciencia fonológica), que afecta negativamente a la capacidad de desencriptar palabras, leer con fluidez o comprender el lenguaje escrito.
2. Trastorno de la Expresión Escrita (Disgrafía y Disortografía): Dificultad en la expresión escrita, que se manifiesta en problemas para organizar y escribir palabras, frases y párrafos coherentes y adecuados a la edad. Incluye dificultades en la ortografía, gramática y puntuación.
3. Trastorno Matemático (Dyscalculia): Dificultad en el procesamiento de números y operaciones matemáticas básicas, como contar, identificar símbolos numéricos o resolver problemas aritméticos sencillos.
Los trastornos del aprendizaje suelen aparecer durante la infancia y pueden persistir en la edad adulta. A menudo, están asociados a otros trastornos como el Trastorno por Déficit de Atención con Hiperactividad (TDAH), dislexia o trastornos del lenguaje. Es importante identificar y abordar los trastornos del aprendizaje lo antes posible, ya que un diagnóstico y tratamiento precoces pueden mejorar significativamente el rendimiento académico y la calidad de vida de las personas afectadas.
El síndrome de Noonan es un trastorno genético que afecta el crecimiento y desarrollo. Se caracteriza por rasgos faciales distintivos, problemas de corazón al nacer (cardiopatías congénitas), retrasos en el desarrollo y baja estatura.
Las características faciales pueden incluir ojos ampliamente espaciados, cejas prominentes, orejas grandes y redondeadas que se doblan en la punta, nariz ancha con un puente plano, labio superior delgado y boca pequeña. También puede haber una protuberancia en el pecho (pecho protuberante) o una curvatura de la columna vertebral (escoliosis).
Las cardiopatías congénitas más comunes asociadas con este síndrome son las estenosis pulmonar, defectos del tabique interventricular y defectos del septo atrial. Algunos individuos también pueden tener problemas de coagulación sanguínea.
El retraso en el desarrollo puede variar desde leve hasta severo e incluye retrasos en el habla, la capacidad para coordinar movimientos (motricidad fina) y el aprendizaje. La estatura final tiende a ser por debajo del promedio.
El síndrome de Noonan se hereda de una manera autosómica dominante, lo que significa que solo necesita una copia del gen anormal para tener la enfermedad. Los genes asociados con este síndrome son PTPN11, SOS1, RAF1, BRAF, MEK1, KRAS y NRAS. Estos genes proporcionan instrucciones para hacer proteínas que controlan las vías de señalización celular importantes para el crecimiento y desarrollo normales. Las mutaciones en estos genes pueden conducir a un crecimiento y desarrollo anormales, causando el síndrome de Noonan.
Neoplasia es un término general que se utiliza para describir el crecimiento anormal de células o tejidos, lo que puede dar lugar a la formación de tumores. Cuando se utiliza en el contexto de 'neoplasias faciales', se refiere específicamente al crecimiento anormal de células en las estructuras faciales.
Las neoplasias faciales pueden ser benignas (no cancerosas) o malignas (cancerosas). Las neoplasias benignas suelen crecer lentamente y raramente se diseminan a otras partes del cuerpo. Sin embargo, incluso si son benignas, pueden causar problemas importantes según su localización, tamaño y crecimiento. Por ejemplo, un crecimiento benigno en la nariz o los ojos podría interferir con la visión o la respiración.
Por otro lado, las neoplasias malignas faciales, también conocidas como cánceres de cabeza y cuello, pueden ser muy agresivas y crecer rápidamente. Pueden invadir los tejidos circundantes y los ganglios linfáticos cercanos, y algunos tipos incluso se pueden diseminar (metastatizar) a otras partes del cuerpo.
Los diferentes tipos de neoplasias faciales dependen del tipo de tejido en el que se originan. Algunos ejemplos comunes incluyen:
1. Carcinoma basocelular: es el tipo más común de cáncer de piel y a menudo se desarrolla en áreas expuestas al sol, como la cara. Se caracteriza por crecimientos rojos, escamosos o úlceras que no cicatrizan.
2. Carcinoma espinocelular: es el segundo tipo más común de cáncer de piel y también se asocia con la exposición al sol. Puede causar lesiones cutáneas ásperas, escamosas o engrosadas que pueden sangrar fácilmente.
3. Melanoma: es un tipo menos común pero más agresivo de cáncer de piel que se origina en las células productoras de pigmento (melanocitos). Puede aparecer como un lunar cambiante o una nueva mancha oscura en la piel.
4. Linfoma: es un tipo de cáncer que afecta al sistema inmunológico y puede desarrollarse en los ganglios linfáticos faciales o en las glándulas salivales. Puede causar hinchazón, dolor e inflamación en la cara.
5. Sarcoma: es un tipo raro de cáncer que se origina en el tejido conectivo, como los músculos, los tendones o los vasos sanguíneos. Puede causar masas dolorosas o hinchazón en la cara.
El tratamiento de las neoplasias faciales depende del tipo y el estadio del cáncer, así como de la edad y el estado general de salud del paciente. Las opciones de tratamiento pueden incluir cirugía, radioterapia, quimioterapia o terapia dirigida. En algunos casos, se puede combinar más de un enfoque para lograr los mejores resultados posibles.
La prevención es siempre la mejor estrategia para reducir el riesgo de desarrollar cáncer. Las medidas preventivas incluyen evitar el tabaco y el alcohol, mantener una dieta saludable y equilibrada, hacer ejercicio regularmente, protegerse del sol y someterse a exámenes médicos regulares para detectar cualquier signo temprano de cáncer. Si tiene alguna preocupación sobre su riesgo de desarrollar cáncer o si ha notado algún cambio inusual en su cuerpo, consulte a un médico lo antes posible.
El mosaicismo, en el contexto médico y genético, se refiere a un estado en el que una persona tiene células con diferentes composiciones cromosómicas o génicas en su cuerpo. Esto ocurre cuando hay una variación estructural o numérica del material genético que no está presente en todas las células del individuo.
El mosaicismo puede deberse a diversas causas, como errores durante la división celular temprana en el desarrollo embrionario, lo que resulta en diferentes líneas celulares con distintos patrones genéticos. También puede ser el resultado de recombinaciones genéticas o mutaciones espontáneas (de novo) que ocurren después de la fecundación.
El grado y la extensión del mosaicismo varían ampliamente, dependiendo del momento en que ocurra el evento genético desencadenante y de cuántas células se vean afectadas. En algunos casos, el mosaicismo puede involucrar solo un pequeño porcentaje de células y no causar ningún síntoma visible o efecto adverso sobre la salud. Sin embargo, en otros casos, el mosaicismo puede afectar significativamente a varios tejidos y órganos, dando lugar a diversas manifestaciones clínicas y trastornos genéticos.
El diagnóstico y la evaluación del mosaicismo generalmente requieren análisis citogenéticos o pruebas moleculares especializadas, como el análisis de ADN en tejidos específicos o el muestreo de vellosidades coriónicas en el caso de embriones en desarrollo. El manejo y el asesoramiento médico dependen del tipo y la gravedad del mosaicismo, así como de los posibles riesgos y complicaciones asociados con el trastorno genético subyacente.
Las Enfermedades del Desarrollo Óseo (EDO) son un grupo de trastornos que afectan el crecimiento y desarrollo normal de los huesos. Estas condiciones pueden ser presentes desde el nacimiento o adquirirse más tarde en la vida, y pueden ser causadas por una variedad de factores, incluyendo genéticos, hormonales, nutricionales y ambientales.
Las EDO se caracterizan por anomalías en la forma, tamaño, densidad y fuerza de los huesos. Pueden afectar a uno o más huesos y pueden causar una variedad de síntomas, dependiendo de la gravedad y la ubicación de la afección. Algunos de los síntomas más comunes incluyen dolor óseo, deformidades esqueléticas, crecimiento anormal, fracturas espontáneas o repetidas, y discapacidad física.
Algunos ejemplos de EDO incluyen la enfermedad de Paget del hueso, la osteogénesis imperfecta, la displasia fibrosa poliestóptica, el síndrome de Marfan, y el síndrome de Hurler. El tratamiento de estas condiciones depende del tipo y la gravedad de la afección, pero puede incluir medicamentos, terapia física, cirugía ortopédica y cambios en el estilo de vida.
Los cromosomas humanos del par 22, también conocidos como cromosoma 22, son uno de los 23 pares de cromosomas humanos. Los cromosomas son las estructuras en las que reside el material genético hereditario de todos los seres vivos. El par 22 está compuesto por dos cromosomas idénticos, cada uno de los cuales mide aproximadamente 0.67-0.85 micrómetros de largo.
El cromosoma 22 es el segundo cromosoma más pequeño en términos de longitud y contiene alrededor del 1,5% del total de ADN presente en una célula humana. Contiene entre 400 y 500 genes, que son secuencias específicas de ADN que contienen la información necesaria para producir proteínas y otros productos genéticos importantes.
El cromosoma 22 es particularmente conocido por albergar el gen NF1, que está asociado con el neurofibromatosis tipo 1, una enfermedad hereditaria que afecta al sistema nervioso y causa tumores benignos. También contiene el gen EGFR, que se ha relacionado con ciertos tipos de cáncer, como el cáncer de pulmón y el cáncer de mama.
Además, una región específica del brazo corto del cromosoma 22, conocida como region 11.22, se ha asociado con un trastorno genético llamado síndrome de DiGeorge, que se caracteriza por anomalías cardiovasculares, retrasos en el desarrollo y deficiencias inmunológicas. Esta región contiene una serie de genes importantes para el desarrollo normal del cuerpo humano.
Neoplasias Primarias Múltiples (NPM) es un término médico que se refiere a la presencia simultánea o sucesiva de más de un cáncer primario en el organismo, es decir, dos o más tumores malignos independientes en diferentes localizaciones anatómicas, que no guardan relación entre sí y no son metástasis del mismo.
Las NPM pueden ser sincrónicas, cuando los tumores se diagnostican al mismo tiempo, o metacrónicas, cuando hay un intervalo de tiempo entre el diagnóstico de cada uno de ellos. Las NPM pueden deberse a diferentes factores de riesgo, como la predisposición genética, la exposición a radiaciones o determinados agentes químicos y ambientales, o hábitos tóxicos como el consumo de tabaco y alcohol.
El diagnóstico y tratamiento de las NPM requieren una evaluación multidisciplinar y un enfoque personalizado, ya que cada tumor puede presentar diferentes características biológicas y clínicas, y precisar de un tratamiento específico. Además, es importante establecer un seguimiento a largo plazo para detectar precozmente la aparición de nuevos tumores y mejorar el pronóstico y la supervivencia de los pacientes.
El piebaldismo es una condición genética rara que afecta el patrón de distribución de pigmento en la piel y el cabello. La palabra "piebald" significa manchas blancas o parches desprovistos de color. Esta afección está presente desde el nacimiento y se caracteriza por parches de piel sin pigmento (piel blanca) en contraste con áreas normales de pigmentación. Los parches suelen estar localizados en la frente, mejillas, pecho y extremidades. El cabello también puede tener parches desprovistos de pigmento, lo que resulta en un patrón distintivo de mechones de cabello blanco o rubio pálido junto a mechones morenos o negros.
El piebaldismo es causado por mutaciones en el gen KIT, el cual desempeña un papel importante en la migración y supervivencia de los melanocitos, las células responsables de producir melanina, el pigmento que da color a la piel, el cabello y los ojos. Las mutaciones en este gen impiden que los melanocitos migren a sus posiciones correctas durante el desarrollo fetal, resultando en parches sin pigmentación.
A diferencia de otras afecciones de pigmentación de la piel, el piebaldismo no empeora con el tiempo y generalmente no presenta otros síntomas sistémicos. Sin embargo, algunas personas con piebaldismo pueden tener problemas de audición asociados si los melanocitos ausentes también afectan la formación del oído interno. El tratamiento suele ser cosmético y puede incluir el uso de maquillaje para cubrir las manchas blancas o procedimientos quirúrgicos como injertos de piel para rellenar los parches desprovistos de pigmento.
El feocromocitoma es un tipo raro de tumor que se forma en las glándulas suprarrenales, que son glándulas endocrinas situadas por encima de los riñones. Estos tumores producen catecolaminas, especialmente adrenalina y noradrenalina, hormonas que ayudan al cuerpo a prepararse para responder a situaciones estresantes. La sobreproducción de estas sustancias puede causar hipertensión arterial (tanto sostenida como paroxística), taquicardia, sudoración, temblores, ansiedad, dolores de cabeza y náuseas, entre otros síntomas. Aproximadamente el 90% de los feocromocitomas son benignos, pero el 10% pueden ser malignos y diseminarse a otras partes del cuerpo. El diagnóstico se realiza mediante pruebas especializadas como la determinación de metanefrinas en plasma o orina y la imagenología médica, como TAC, RMN o escintigrafía con meta-iodobencilguanidina (MIBG). El tratamiento suele consistir en la extirpación quirúrgica del tumor.
La Imagen por Resonancia Magnética (IRM) es una técnica de diagnóstico médico no invasiva que utiliza un campo magnético potente, radiaciones ionizantes no dañinas y ondas de radio para crear imágenes detalladas de las estructuras internas del cuerpo. Este procedimiento médico permite obtener vistas en diferentes planos y con excelente contraste entre los tejidos blandos, lo que facilita la identificación de tumores y otras lesiones.
Durante un examen de IRM, el paciente se introduce en un túnel o tubo grande y estrecho donde se encuentra con un potente campo magnético. Las ondas de radio se envían a través del cuerpo, provocando que los átomos de hidrógeno presentes en las células humanas emitan señales de radiofrecuencia. Estas señales son captadas por antenas especializadas y procesadas por un ordenador para generar imágenes detalladas de los tejidos internos.
La IRM se utiliza ampliamente en la práctica clínica para evaluar diversas condiciones médicas, como enfermedades del cerebro y la columna vertebral, trastornos musculoesqueléticos, enfermedades cardiovasculares, tumores y cánceres, entre otras afecciones. Es una herramienta valiosa para el diagnóstico, planificación del tratamiento y seguimiento de la evolución de las enfermedades.
Las neoplasias meníngeas se refieren a tumores que se originan en las meninges, las membranas protectoras que recubren el cerebro y la médula espinal. Estos tumores pueden ser benignos o malignos (cancerosos) y su crecimiento puede comprimir estructuras vitales del sistema nervioso central, lo que provoca una variedad de síntomas neurológicos.
Existen varios tipos de neoplasias meníngeas, incluyendo meningiomas, hemangioblastomas, neurinomas, sarcomas y linfomas, entre otros. El tratamiento dependerá del tipo y grado del tumor, así como de su localización y extensión. Puede incluir cirugía, radioterapia y quimioterapia.
Somatostatinoma es un tipo raro de tumor neuroendocrino que se origina en las células delta del páncreas, que producen somatostatina, una hormona que ayuda a regular la producción y secreción de otras hormonas digestivas. Este tipo de tumor suele ser no canceroso (benigno) pero en algunos casos puede ser canceroso (maligno) y diseminarse a otros órganos.
Los síntomas de un somatostatinoma pueden incluir diabetes, diarrea, dolor abdominal, pérdida de apetito y peso, anemia, venas dilatadas en la parte superior del cuerpo, y en algunos casos, ictericia. El diagnóstico se realiza mediante análisis de sangre para medir los niveles de somatostatina y otras hormonas, así como por imágenes médicas como TAC o RMN para localizar el tumor.
El tratamiento puede incluir cirugía para extirpar el tumor, quimioterapia y radioterapia para reducir su tamaño y evitar la propagación del cáncer, y terapia de reemplazo hormonal para controlar los síntomas. La esperanza de vida y el pronóstico dependen del tamaño y localización del tumor, si se ha diseminado o no, y de la respuesta al tratamiento.
La descalcificación patológica, también conocida como osteoporosis inducida por medicamentos o enfermedad renal crónica, es una afección que se caracteriza por la pérdida excesiva de calcio y otros minerales importantes en los huesos. Esto hace que los huesos se vuelvan más frágiles y propensos a sufrir fracturas. La descalcificación patológica puede ser causada por ciertos medicamentos, como corticosteroides o anticonvulsivos, enfermedades intestinales graves que impiden la absorción adecuada de calcio y vitamina D, o enfermedad renal crónica.
En la descalcificación patológica, el cuerpo no puede mantener suficiente calcio en los huesos, lo que lleva a una disminución de la densidad mineral ósea y un aumento del riesgo de fracturas. Los síntomas pueden incluir dolor óseo, pérdida de altura y curvatura de la columna vertebral. El diagnóstico se realiza mediante una densitometría ósea, que mide la densidad mineral ósea en las caderas y la columna vertebral.
El tratamiento de la descalcificación patológica puede incluir cambios en la dieta para aumentar la ingesta de calcio y vitamina D, ejercicio regular, terapia hormonal y medicamentos que ayuden a ralentizar la pérdida ósea. Es importante trabajar con un médico para desarrollar un plan de tratamiento individualizado que aborde las causas subyacentes de la enfermedad y reduzca el riesgo de fracturas.
En términos médicos, una mutación se refiere a un cambio permanente y hereditable en la secuencia de nucleótidos del ADN (ácido desoxirribonucleico) que puede ocurrir de forma natural o inducida. Esta alteración puede afectar a uno o más pares de bases, segmentos de DNA o incluso intercambios cromosómicos completos.
Las mutaciones pueden tener diversos efectos sobre la función y expresión de los genes, dependiendo de dónde se localicen y cómo afecten a las secuencias reguladoras o codificantes. Algunas mutaciones no producen ningún cambio fenotípico visible (silenciosas), mientras que otras pueden conducir a alteraciones en el desarrollo, enfermedades genéticas o incluso cancer.
Es importante destacar que existen diferentes tipos de mutaciones, como por ejemplo: puntuales (sustituciones de una base por otra), deletérreas (pérdida de parte del DNA), insercionales (adición de nuevas bases al DNA) o estructurales (reordenamientos más complejos del DNA). Todas ellas desempeñan un papel fundamental en la evolución y diversidad biológica.
Los trastornos de la pigmentación se refieren a condiciones médicas que afectan la producción, transporte o almacenamiento de melanina, el pigmento responsable del color de la piel, cabello y ojos. Estos trastornos pueden causar hiperpigmentación (piel más oscura de lo normal), hipopigmentación (piel más clara de lo normal) o una combinación de ambas.
La producción de melanina está controlada por un grupo de células conocidas como melanocitos. Los trastornos de la pigmentación pueden ser el resultado de anomalías en estas células, los genes que controlan su funcionamiento, o factores externos como la exposición a la luz solar, quemaduras o inflamaciones de la piel.
Ejemplos comunes de trastornos de la pigmentación incluyen el vitiligo, en el cual las áreas de la piel pierden melanocitos y se vuelven blancas; el melasma, que causa manchas oscuras en la cara y otras áreas expuestas al sol; y el albinismo, una condición genética que impide la producción de melanina.
El tratamiento de los trastornos de la pigmentación depende del tipo y gravedad de la afección. Puede incluir cremas despigmentantes, protectores solares, terapia con láser o, en casos graves, trasplante de piel.
Un tumor glómico, también conocido como glomangioma, es un tipo raro de tumor benigno que se origina en los glomus, que son pequeñas estructuras situadas en la dermis profunda de la piel, especialmente en las zonas distales de los miembros, como los dedos de las manos y los pies. Los glomus participan en el control de la circulación sanguínea local, regulando la temperatura y el flujo sanguíneo.
Los tumores glómicos suelen presentarse como nódulos pequeños, bien circunscritos, de color rojizo o azulado, que se encuentran típicamente en los dedos, aunque también pueden aparecer en otras partes del cuerpo. Estos tumores pueden causar dolor intenso, especialmente al ser estimulados por el frío, el tacto o la presión, y a menudo interfieren con las actividades diarias de los pacientes.
La mayoría de los tumores glómicos son benignos y crecen lentamente; sin embargo, en algunos casos pueden transformarse en malignos (sarcoma glómico), lo que puede provocar una invasión local o metástasis a distancia. El tratamiento preferido para los tumores glómicos benignos es la extirpación quirúrgica, mientras que el tratamiento de los sarcomas glómicos implica cirugía followed by radiotherapy or chemotherapy.
El quiasma óptico es un punto anatómico en el sistema visual donde las fibras nerviosas de los nervios ópticos se cruzan. Más específicamente, se refiere al lugar justo en la parte frontal del cerebro donde los dos nervios ópticos se encuentran y comparten información.
Este punto es crucial porque las mitades internas (nasales) de ambos nervios ópticos, que transportan señales visuales desde cada lado izquierdo y derecho de la parte más externa (temporal) de nuestro campo visual, se cruzan en el quiasma óptico. Por lo tanto, las fibras nerviosas que provienen de la mitad nasal de un ojo se cruzan y continúan hacia el lado opuesto del cerebro, mientras que las fibras nerviosas de la mitad temporal viajan sin cruzar al mismo lado del cerebro desde donde originaron.
Esta configuración permite que nuestro cerebro procese correctamente la información visual recibida, ya que los estímulos provenientes del lado izquierdo de nuestro campo visual se procesen en el hemisferio derecho del cerebro y viceversa. Anomalías en el quiasma óptico pueden conducir a diversas condiciones visuales, como la pérdida de visión en ciertas áreas del campo visual (hemianopsia).
Las neoplasias de las glándulas suprarrenales se refieren a un crecimiento anormal de tejido en las glándulas suprarrenales, que pueden ser benigno (no canceroso) o maligno (canceroso). Las glándulas suprarrenales son glándulas endocrinas pequeñas ubicadas encima de los riñones que producen varias hormonas importantes, como cortisol, aldosterona, y adrenalina.
Existen varios tipos de neoplasias de las glándulas suprarrenales, incluyendo:
1. Adenoma: es el tipo más común de tumor benigno de la glándula suprarrenal. Por lo general, no causa síntomas y se descubre accidentalmente durante exámenes de imagenología realizados por otras razones. Sin embargo, algunos adenomas pueden producir demasiadas hormonas, causando síndromes paraneoplásicos como el síndrome de Cushing o el síndrome de Conn.
2. Feocromocitoma: es un tumor que se origina en las células cromafines de la glándula suprarrenal y produce demasiadas cantidades de catecolaminas, como la adrenalina y la noradrenalina. Los síntomas pueden incluir hipertensión arterial, taquicardia, sudoración, dolores de cabeza y ansiedad. Aproximadamente el 10% de los feocromocitomas son malignos.
3. Carcinoma suprarrenal: es un tumor maligno que se origina en las glándulas suprarrenales. Puede producir hormonas suprarrenales y causar síntomas relacionados con los niveles elevados de hormonas. Los síntomas pueden incluir hipertensión arterial, debilidad, pérdida de peso y dolor abdominal.
4. Neuroblastoma: es un tumor maligno que se origina en los ganglios nerviosos simpáticos y puede producir catecolaminas. Se presenta principalmente en niños menores de 5 años y puede metastatizar a otros órganos.
El diagnóstico de los tumores suprarrenales se realiza mediante la combinación de estudios de imagenología, como la tomografía computarizada o la resonancia magnética, y pruebas bioquímicas para evaluar la producción hormonal. El tratamiento depende del tipo y grado de malignidad del tumor y puede incluir cirugía, quimioterapia, radioterapia o terapias dirigidas.
Las neoplasias cutáneas, también conocidas como crecimientos anormales o tumores de la piel, se refieren a un amplio espectro de condiciones donde las células de la piel proliferan de manera descontrolada. Estas lesiones pueden ser benignas (no cancerosas) o malignas (cancerosas).
Las neoplasias cutáneas benignas incluyen diversos tipos de lunares, verrugas, fibromas y quistes. Por lo general, crecen lentamente, permanecen localizadas y rara vez representan un peligro para la vida si se diagnostican y tratan a tiempo.
Por otro lado, las neoplasias cutáneas malignas más comunes son el carcinoma basocelular, el carcinoma escamoso y el melanoma. Estos tipos de cáncer de piel pueden invadir los tejidos circundantes e incluso diseminarse a otras partes del cuerpo (metástasis), lo que puede poner en peligro la vida del paciente.
El diagnóstico y el tratamiento oportunos son cruciales para garantizar una buena evolución clínica de los pacientes con neoplasias cutáneas. La prevención, mediante la protección adecuada contra los rayos ultravioleta (UV) del sol y el reconocimiento precoz de las lesiones sospechosas, juegan un papel fundamental en la reducción de la incidencia y mortalidad asociadas con estas afecciones.
Las neoplasias torácicas se refieren a un crecimiento anormal de células en el sistema torácico, que incluye los pulmones, la pleura (membrana que recubre los pulmones), el mediastino (espacio entre los pulmones) y el tejido circundante. Estos crecimientos pueden ser benignos (no cancerosos) o malignos (cancerosos).
Los tipos más comunes de neoplasias torácicas son:
1. Neoplasias pulmonares: Estas incluyen cánceres de pulmón, como el carcinoma de células no pequeñas y el carcinoma de células pequeñas. También pueden incluir tumores benignos, como hamartomas y adenomas.
2. Neoplasias pleurales: Estos incluyen mesotelioma, que es un cáncer raro pero agresivo que se origina en la pleura, y tumores benignos como lipomas y hemangiomas.
3. Neoplasias mediastinales: El mediastino contiene varios órganos y tejidos, incluyendo el corazón, los grandes vasos sanguíneos, el timo, los ganglios linfáticos y el esófago. Las neoplasias mediastinales pueden ser cánceres o tumores benignos y pueden originarse en cualquiera de estos órganos o tejidos. Algunos ejemplos incluyen timomas, linfomas, sarcomas y quistes broncogénicos.
4. Neoplasias de la pared torácica: Estos incluyen tumores benignos y malignos que se originan en los músculos, huesos, nervios o tejidos conectivos de la pared torácica. Algunos ejemplos son el osteosarcoma, el condrosarcoma y el sarcoma de Ewing.
El tratamiento de las neoplasias torácicas depende del tipo de cáncer o tumor, su localización, su tamaño y si se ha diseminado a otras partes del cuerpo. Los tratamientos pueden incluir cirugía, radioterapia, quimioterapia, terapia dirigida o inmunoterapia.
En el contexto de la medicina y la biología, un linaje se refiere a una sucesión o serie de organismos relacionados genéticamente que descienden de un antepasado común más reciente. Puede hacer referencia a una secuencia particular de genes que se heredan a través de generaciones y que ayudan a determinar las características y rasgos de un organismo.
En la genética, el linaje mitocondrial se refiere a la línea de descendencia materna, ya que las mitocondrias, que contienen su propio ADN, se transmiten generalmente de madre a hijo. Por otro lado, el linaje del cromosoma Y sigue la línea paterna, ya que los cromosomas Y se heredan del padre y se mantienen intactos durante la meiosis, lo que permite rastrear la ascendencia masculina.
Estos linajes pueden ser útiles en la investigación genética y antropológica para estudiar la evolución y la migración de poblaciones humanas y otras especies.
Un tumor mixto maligno (TMM) es un tipo raro de cáncer que contiene dos o más tipos diferentes de células cancerosas. Se caracteriza por la presencia de células epiteliales y mesenquimales malignas, creciendo juntas en una lesión única. Los TMM pueden ocurrir en varias partes del cuerpo, pero son más comunes en glándulas como las glándulas salivales y mamarias.
Los tumores mixtos malignos también se conocen como carcinomas sarcomatosos o tumores de células mixtas. Su comportamiento clínico y su tratamiento dependen del tipo y la proporción de células cancerosas presentes en el tumor. Por lo general, requieren un tratamiento agresivo, que puede incluir cirugía, radioterapia y quimioterapia.
Es importante mencionar que los TMM son diferentes a los carcinomas de células mixtas, que son tumores que contienen dos o más tipos de células epiteliales malignas. Los TMM también se distinguen de los sarcomas, que son tumores malignos del tejido conectivo y otros tejidos blandos.
Un astrocitoma es un tipo de tumor cerebral que se origina en las células gliales del sistema nervioso central, específicamente en los astrocitos, que son un tipo de célula glial que proporciona soporte y protección a las neuronas. Los astrocitomas pueden ser benignos o malignos, y se clasifican según su grado de malignidad.
Los astrocitomas de grado bajo crecen lentamente y suelen ser menos invasivos, mientras que los de grado alto crecen rápidamente y son más agresivos, invadiendo el tejido circundante y extendiéndose a otras partes del cerebro. Los síntomas de un astrocitoma pueden variar dependiendo de su tamaño y ubicación, pero pueden incluir dolores de cabeza, convulsiones, náuseas, vómitos, cambios en la visión, el habla o el comportamiento, y debilidad o entumecimiento en un lado del cuerpo.
El tratamiento para los astrocitomas depende del tipo y grado del tumor, así como de su localización y del estado de salud general del paciente. Puede incluir cirugía, radioterapia y quimioterapia. En algunos casos, se puede optar por un enfoque de observación y solo se interviene si el tumor cambia o causa síntomas.
El síndrome de Proteo es un trastorno genético extremadamente raro que involucra una serie de características físicas y cambios en el desarrollo que varían drásticamente de una persona a otra. Se caracteriza por la presencia de anomalías esqueléticas, distintas deformidades faciales, problemas cutáneos e incluso alteraciones en los órganos internos.
Este síndrome recibe su nombre del dios griego Proteo, quien era conocido por su capacidad de cambiar de forma a voluntad. Esto es apropiado ya que las personas con síndrome de Proteo a menudo exhiben una amplia gama de signos y síntomas únicos e individualizados.
El síndrome de Proteo se hereda de manera autosómica dominante, lo que significa que solo necesita recibir una copia del gen anormal (de su padre o su madre) para tener el trastorno. Aproximadamente en dos tercios de los casos, este síndrome es el resultado de una nueva mutación genética y no se hereda de un progenitor afectado.
Los signos y síntomas del síndrome de Proteo pueden variar mucho incluso entre miembros de la misma familia. Algunas personas pueden tener solo algunas características leves, mientras que otras pueden verse gravemente afectadas. Los rasgos comunes incluyen:
1. Deformidades faciales: Las orejas y el puente nasal a menudo están deformados. La mandíbula puede ser más pequeña de lo normal (micrognatia) o sobresalir (prognatismo). Los ojos pueden estar más separados de lo normal (hipertelorismo), y las cejas pueden ser prominentes o escasas.
2. Anomalías esqueléticas: Las personas con síndrome de Proteo a menudo tienen extremidades desiguales en longitud o grosor, dedos adicionales (polidactilia) o dedos fusionados (sindactilia). La columna vertebral puede curvarse anormalmente (escóliosis), y las costillas pueden ser de diferentes longitudes.
3. Problemas cutáneos: Las personas con síndrome de Proteo a menudo tienen manchas cutáneas irregulares, marcas de nacimiento o lunares grandes. La piel puede estar engrosada en ciertas áreas (piel de elefante).
4. Problemas cardiovasculares: Algunas personas con síndrome de Proteo tienen defectos cardíacos congénitos, como válvulas cardíacas anormales o tabiques interventriculares agrandados.
5. Problemas renales: Los riñones pueden ser anormalmente grandes o pequeños, y algunas personas con síndrome de Proteo tienen quistes renales.
6. Problemas neurológicos: Algunas personas con síndrome de Proteo tienen retrasos en el desarrollo, convulsiones u otras anomalías cerebrales.
7. Problemas auditivos y visuales: Las personas con síndrome de Proteo pueden tener problemas de audición o visión, como cataratas congénitas o pérdida de la audición.
El tratamiento del síndrome de Proteus depende de los síntomas específicos que presente cada persona. El manejo puede incluir cirugía para corregir defectos cardiovasculares, renales o ortopédicos; terapia física y ocupacional para ayudar con el desarrollo y la movilidad; y medicamentos para controlar convulsiones u otras afecciones médicas. La educación especial también puede ser necesaria para las personas con retrasos en el desarrollo.
El pronóstico del síndrome de Proteus varía ampliamente, dependiendo de la gravedad de los síntomas y de si se pueden controlar adecuadamente. Algunas personas con síndrome de Proteus tienen una esperanza de vida normal, mientras que otras pueden morir antes de los 20 años debido a complicaciones médicas graves.
En conclusión, el síndrome de Proteus es una enfermedad rara y compleja que afecta a varios sistemas corporales y puede causar una variedad de síntomas graves. Aunque no existe cura para esta enfermedad, el tratamiento puede ayudar a controlar los síntomas y mejorar la calidad de vida de las personas afectadas. El pronóstico varía ampliamente, dependiendo de la gravedad de los síntomas y del manejo médico adecuado.
La leucemia mielomonocítica juvenil (LMM) es un trastorno maligno poco frecuente de las células sanguíneas y de la médula ósea. Se caracteriza por un crecimiento excesivo e incontrolado de los monocitos, un tipo de glóbulos blancos que ayudan a combatir las infecciones.
En la LMM, estas células anormales se acumulan en la médula ósea y pueden interferir con la producción normal de otras células sanguíneas, lo que lleva a una disminución de los glóbulos rojos (lo que puede causar anemia), los plaquetas (lo que aumenta el riesgo de sangrado) y los neutrófilos (lo que aumenta el riesgo de infecciones).
La LMM se diagnostica más a menudo en niños menores de 4 años, aunque también puede ocurrir en adultos. Los síntomas pueden incluir fiebre, fatiga, moretones y sangrado fáciles, infecciones recurrentes, pérdida de peso y dolor óseo. El tratamiento generalmente implica quimioterapia, trasplante de células madre o ambos.
La tomografía computarizada por rayos X, también conocida como TC o CAT (por sus siglas en inglés: Computerized Axial Tomography), es una técnica de diagnóstico por imágenes que utiliza radiación para obtener detalladas vistas tridimensionales de las estructuras internas del cuerpo. Durante el procedimiento, el paciente se coloca sobre una mesa que se desliza dentro de un anillo hueco (túnel) donde se encuentran los emisores y receptores de rayos X. El equipo gira alrededor del paciente, tomando varias radiografías en diferentes ángulos.
Las imágenes obtenidas son procesadas por un ordenador, el cual las combina para crear "rebanadas" transversales del cuerpo, mostrando secciones del tejido blando, huesos y vasos sanguíneos en diferentes grados de claridad. Estas imágenes pueden ser visualizadas como rebanadas individuales o combinadas para formar una representación tridimensional completa del área escaneada.
La TC es particularmente útil para detectar tumores, sangrado interno, fracturas y otras lesiones; así como también para guiar procedimientos quirúrgicos o biopsias. Sin embargo, su uso está limitado en pacientes embarazadas debido al potencial riesgo de daño fetal asociado con la exposición a la radiación.
Las neoplasias del iris son un tipo de crecimiento anormal de tejido en el iris, que es la parte coloreada del ojo. Pueden ser benignas (no cancerosas) o malignas (cancerosas). Las neoplasias benignas más comunes del iris son los nevus y los hemangiomas. Los nevus son manchas pigmentadas que se desarrollan a partir de células pigmentarias llamadas melanocitos. Por otro lado, los hemangiomas son crecimientos anormales de vasos sanguíneos en el iris.
Las neoplasias malignas del iris son mucho menos comunes y más agresivas que las benignas. La forma más común de cáncer del iris es el melanoma del iris, que se desarrolla a partir de células pigmentarias anormales en el iris. Los síntomas del melanoma del iris pueden incluir cambios en el color o tamaño del iris, dolor ocular, visión borrosa y glaucoma.
El tratamiento de las neoplasias del iris depende del tipo y gravedad de la afección. Las lesiones benignas pueden no requerir tratamiento, mientras que las lesiones malignas pueden requerir cirugía, radioterapia o quimioterapia. Es importante buscar atención médica si se sospecha una neoplasia del iris para un diagnóstico y tratamiento precoces.
La angiomatosis es una afección médica rara en la que se forman grupos anormales de vasos sanguíneos o linfáticos en órganos y tejidos. Estos grupos se denominan angiomas y pueden crecer y multiplicarse, reemplazando gradualmente el tejido normal del órgano afectado.
La angiomatosis puede afectar a cualquier parte del cuerpo, pero es más común en la piel, el hígado, el bazo y los pulmones. Los síntomas varían dependiendo de la ubicación y el tamaño de las lesiones. En algunos casos, la angiomatosis puede no causar ningún síntoma y ser descubierta accidentalmente durante exámenes o procedimientos médicos.
En otros casos, los síntomas pueden incluir dolor, hinchazón, sangrado, infecciones recurrentes o problemas de funcionamiento del órgano afectado. El tratamiento depende de la ubicación y el tamaño de las lesiones y puede incluir cirugía, radioterapia o terapia médica.
Es importante buscar atención médica si se sospecha de angiomatosis, ya que esta afección puede ser un signo de otras condiciones médicas más graves, como cánceres o trastornos genéticos.
La arachnoide es una membrana del sistema nervioso central que forma parte de los meninges, las membranas protectoras que recubren el cerebro y la médula espinal. Más específicamente, la arachnoide es la membrana intermedia entre la duramadre (la membrana más externa) y la piamadre (la membrana más interna que está en contacto directo con el tejido cerebral).
La aracnoides tiene una consistencia similar a la de un filtro de papel, es muy delgada y frágil. Está llena de espacio y contiene líquido cefalorraquídeo (LCR), que proporciona protección y amortiguación a los tejidos cerebrales y medulares. La aracnoides está adherida a la duramadre, pero se separa de la piamadre por el espacio subaracnoideo, donde circula el LCR.
La aracnoiditis es una enfermedad que involucra la inflamación y la irritación de la membrana aracnoidea, lo que puede causar dolor, rigidez, debilidad muscular e incluso problemas neurológicos graves. La aracnoiditis puede ser causada por diversos factores, como infecciones, traumatismos, cirugías o la exposición a sustancias químicas tóxicas.
El ligamiento genético, en términos médicos, se refiere al fenómeno en el que dos o más loci (regiones específicas del ADN) en un cromosoma tienden a heredarse juntos durante la reproducción porque están demasiado próximos entre sí para ser separados por el proceso de recombinación genética. La medida de cuán a menudo se heredan juntos se expresa como una unidad llamada "unidades de mapa centimorgan" (cM), que refleja la probabilidad de recombinación entre ellos. Cuanto más cerca estén los loci uno del otro en un cromosoma, mayor será su ligamiento y menor será la probabilidad de recombinación entre ellos. Por lo tanto, el ligamiento genético proporciona información importante sobre la ubicación relativa y la organización de los genes en un cromosoma.
 Neurofibromatosis tipo 1
Neurofibromatosis tipo 1
Neurofibromina 1
Secuencia Alu
Cáncer de páncreas
HOXC8
Neurofibromatosis
Francis Collins
Cromosoma 17 (humano)
Feocromocitoma
Síndrome de Beckwith-Wiedemann
Mutación
Ras
Rasopatía
Cromosoma 22 (humano)
Expresividad (genética)
Astrocitoma pilocítico
Enfermedad genética
Sarcoma sinovial
Meduloblastoma
Tumor cerebral
Imperio parto
Rabdomiosarcoma
Examen genético
Escoliosis
Glioblastoma
Dermatología
Enfermedad de moyamoya
Color de ojos
Cáncer
Neurofibromatosis tipo 1 - Wikipedia
 https://www.cancer.gov/espanol/tipos/sarcoma-de-tejido-blando/paciente/tratamiento-tegi-pdq
https://www.cancer.gov/espanol/tipos/sarcoma-de-tejido-blando/paciente/tratamiento-tegi-pdq
Miocardiopatía hipertrófica obstructiva y neurofibromatosis de tipo 1 | Anales de Pediatría
Cromosoma 17 (humano) - Wikipedia
 Biblioteca de Salud: Neurofibromatosis
Biblioteca de Salud: Neurofibromatosis
 Neurofibromatosis - Salud infantil - Manual MSD versión para público general
Neurofibromatosis - Salud infantil - Manual MSD versión para público general
 Otros síndromes que pueden asociarse a predisposición al cáncer - Health in Code
Otros síndromes que pueden asociarse a predisposición al cáncer - Health in Code
 Nódulos subcutáneos en paciente trasplantado renal: lipomatosis familiar múltiple | Actas Dermo-Sifiliográficas
Nódulos subcutáneos en paciente trasplantado renal: lipomatosis familiar múltiple | Actas Dermo-Sifiliográficas
 Neurofibromatosis Tipo 2 (NF)
Neurofibromatosis Tipo 2 (NF)
 neurofibromatosis
neurofibromatosis
 Centros de Investigación
Centros de Investigación
 TALLA ALTA.- - Competencia intermedia
TALLA ALTA.- - Competencia intermedia
Tratamiento de los astrocitomas infantiles (PDQ®) - NCI
 Neurofibromatosis - Dermatly.com - El sitio de tu piel
Neurofibromatosis - Dermatly.com - El sitio de tu piel
 La neurofibromatosis
La neurofibromatosis
 Métodos de diagnóstico genético de las enfermedades renales hereditarias. El consejo genético | Nefrología
Métodos de diagnóstico genético de las enfermedades renales hereditarias. El consejo genético | Nefrología
 3.2 Genéticos
3.2 Genéticos
 Día Mundial del Sarcoma: Signos y síntomas de los sarcomas de tejidos blandos | Noticias de México | EL IMPARCIAL
Día Mundial del Sarcoma: Signos y síntomas de los sarcomas de tejidos blandos | Noticias de México | EL IMPARCIAL
 Cáncer de mama - Médicos y departamentos - Mayo Clinic
Cáncer de mama - Médicos y departamentos - Mayo Clinic
 Búsqueda | BVS CLAP/SMR-OPS/OMS
Búsqueda | BVS CLAP/SMR-OPS/OMS
Health Library Categories Afecciones genéticas, cromosómicas y metabólicas para padres | Rady Children's Hospital
herenciageneticayenfermedad: Leucemia linfoblástica aguda infantil (PDQ®)-Versión para pacientes - National Cancer Institute
Cáncer de páncreas - Wikipedia, la enciclopedia libre
 Hipomelanosis de Ito y otros síndromes neurocutáneos | NeuroPedWikia
Hipomelanosis de Ito y otros síndromes neurocutáneos | NeuroPedWikia
 Síndrome cardiofaciocutáneo (para Padres) - Nemours KidsHealth
Síndrome cardiofaciocutáneo (para Padres) - Nemours KidsHealth
 Neurofibromatosis - CECR
Neurofibromatosis - CECR
 Panel de Enfermedades Autoinflamatorias | Mendelics
Panel de Enfermedades Autoinflamatorias | Mendelics
 Genética y defectos congénitos: MedlinePlus en español
Genética y defectos congénitos: MedlinePlus en español
 Escoliosis - Cirugía cerebral asequible en México, Puerto Vallarta
Escoliosis - Cirugía cerebral asequible en México, Puerto Vallarta
 DeCS 2020 - versión 23 de junio de 2020
DeCS 2020 - versión 23 de junio de 2020
Enfermedad36
- La neurofibromatosis tipo 1 (también denominada NF-1) es una enfermedad compleja multisistema causada por la mutación de un gen en el cromosoma 17 que es responsable de la producción de la proteína neurofibromina 1 que es necesaria para el normal funcionamiento en muchos tipos de células humanas. (wikipedia.org)
- NF-1 es un trastorno autosómico dominante, lo que significa que la mutación o eliminación de un alelo del gen NF1 es suficiente para el desarrollo de NF-1, aunque la presentación clínica de la enfermedad es muy variable. (wikipedia.org)
- 1] NF-1 es una enfermedad del desarrollo producida por mutaciones en la línea germinal en el gen de la neurofibromina, que está involucrado en la vía RAS (es por tanto una rasopatía). (wikipedia.org)
- NF-1 se conocía anteriormente como la enfermedad de von Recklinghausen, en honor al investigador Friedrich Daniel von Recklinghausen, quien documentó por primera vez el trastorno. (wikipedia.org)
- Un tumor de estroma gastrointestinal es una enfermedad en la que se forman células anormales en los tejidos del tubo gastrointestinal. (cancer.gov)
- Osteogénesis imperfecta Síndrome de Papillon-Lefèvre Síndrome de Li-Fraumeni Pseudogenes mitocondriales Neurofibromatosis tipo 1 enfermedad del alzheimer Síndrome Smith Mageniss Biason-Lauber A. (wikipedia.org)
- La neurofibromatosis tipo 1 (o NF1, también conocida como enfermedad de von Recklinghausen) afecta aproximadamente a 1 de cada 2500 - 3000 personas. (msdmanuals.com)
- tampoco los genes NF1 (neurofibromatosis), SPRED 1 (síndrome de Legius) o PTEN (enfermedad de Cowden). (actasdermo.org)
- La neurofibromatosis tipo 2 ( NF2 , OMIM: 101000) ó neurofibromatosis acústica es una enfermedad caracterizada por la presencia de neurinomas en los nervios auditivos , generalmente bilaterales. (cibic.com.ar)
- La gravedad de la afectación cutánea en la NF1 no es un indicador de la extensión de la enfermedad, ya que las manifestaciones internas son frecuentes y, a menudo, más graves. (dermatly.com)
- En genética médica se denominan mutaciones patogénicas a las variantes de secuencia que causan enfermedad, las cuales generalmente se encuentran en una frecuencia inferior al 1% en la población general. (revistanefrologia.com)
- Actualmente, el diagnóstico genético es aplicable principalmente a las enfermedades monogénicas , que son aquellas en las que una mutación patogénica en un único gen (de unos 25.000 genes que contiene nuestro genoma) es suficiente para causar la enfermedad. (revistanefrologia.com)
- En cambio, las enfermedades poligénicas , también denominadas multifactoriales o complejas, son causadas tanto por factores genéticos, múltiples variantes de secuencia en distintos genes que proporcionan un riesgo genético o predisposición a desarrollar la enfermedad, como por factores ambientales. (revistanefrologia.com)
- En las enfermedades monogénicas recesivas existe una estrecha correlación genotipo-fenotipo , lo que indica que el fenotipo de la enfermedad es determinado de forma prácticamente exclusiva por la/s mutación/es patogénica/s en el gen causante (penetrancia completa), con un alto poder predictivo del análisis genético. (revistanefrologia.com)
- Descrita por primera vez en 1882 por Friedrich Daniel von Recklinghausen, la neurofibromatosis es una enfermedad (o, más bien, grupo de enfermedades) que aparece en uno de cada 2.000 nacimientos. (genotipia.com)
- La neurofibromatosis es una enfermedad genética , autosómica (se encuentra en los cromosomas que no están relacionados con el sexo) y dominante (heredar una sola copia mutada del gen es suficiente para que se manifieste la enfermedad). (genotipia.com)
- Según el tipo de neurofibromatosis la mutación que causa la enfermedad se encuentra en un gen diferente. (genotipia.com)
- Además, la neurofibromatosis no es una enfermedad tiquismiquis: no distingue entre razas ni géneros. (genotipia.com)
- La neurofibromatosis tipo 1 (enfermedad de von Recklinghausen) se asocia con múltiples neurofibromas con manchas en café con leche, gliomas ópticos, astrocitomas, tumores derivados de la cresta neural, germinales, etc. (biocancer.com)
- Es una de las neoplasias más agresivas debido a su pronta difusión, su falta de síntomas específicos tempranos y su diagnóstico tardío (en el momento del diagnóstico, los pacientes por lo general tienen la enfermedad localmente avanzada e incluso metastásica , lo que impide la cirugía curativa). (wikipedia.org)
- La hipomelanosis de Ito, también conocida como incontinencia pigmenti achromianses, es una enfermedad multisistémica que presenta de forma característica lesiones curvilineas hipopigmentadas en la piel asociadas o no a manifestaciones neurológicas y oculares, así como malformaciones dentales y musculoesqueléticas. (neuropedwikia.es)
- Para algunos autores la hipomelanosis de Ito es un fenotipo o una descripción clínica de un mosaicismo cromosómico más que una enfermedad en sí misma. (neuropedwikia.es)
- El principal factor de riesgo de la neurofibromatosis (NF1 y NF2) es la presencia de un familiar afectado, es una enfermedad de herencia autosómica dominante con un 50 % de probabilidad de heredar la mutación genética. (enfermeriadeciudadreal.es)
- Denominada también como Neurofibromatosis generalizada o enfermedad de Von Recklinghausen. (enfermeriadeciudadreal.es)
- a veces la escoliosis puede ocurrir como parte de otra enfermedad, incluyendo la neurofibromatosis y el síndrome de Marfan. (neurosurgeryinmexico.com)
- Hasta la fecha se han descrito m s de 100 casos en la literatura m dica y se cree que la enfermedad es m s com n en mujeres que en hombres. (orpha.net)
- El s ndrome puede estar asociado a la enfermedad de Moyamoya, s ndrome de PHACES, s ndrome de Aicardi, neurofibromatosis tipo 2, malformaci n de Arnold-Chiari tipo I, s ndrome de CHARGE y s ndrome de Poland, entre otros. (orpha.net)
- El programa de subvenciones de PTC tiene como objetivo aumentar los esfuerzos de investigación en ensayos clínicos de Fase I y Fase II dirigidos a la enfermedad de Alzheimer y demencias relacionadas a nivel internacional, lo más probable es que tenga psoriasis en placas. (cejop.cz)
- En este sentido, la oncología médica es una de las especialidades más relevantes en el diagnóstico y tratamiento del cáncer , una enfermedad que ha ido en aumento en los últimos años. (mirial.es)
- Bourgeron asegura que es muy difícil identificar los genes relacionados con el autismo si no están vinculados con una enfermedad genética bien definida, como el síndrome del X frágil, la esclerosis tuberosa o la neurofibromatosis. (genysi.es)
- El término "enfermedad rara"(1,2) hace alusión a aquella enfermedad que afecta a un pequeño número absoluto de personas o a una proporción reducida de la población, explica el Dr. Jesús Jurado-Palomo , especialista en Alergología del Hospital General Nuestra Señora del Prado de Talavera de la Reina. (integrasaludtalavera.com)
- En la Unión Europea se considera "enfermedad rara" a aquella que afecta a 1 de cada 2.000 personas. (integrasaludtalavera.com)
- se define así a un trastorno o enfermedad que sufren menos de 200.000 personas, mientras que en Japón es aquella que afecta a menos de 50.000 personas. (integrasaludtalavera.com)
- Medicamento Huérfano es aquel destinado para el diagnóstico, prevención o tratamiento de una enfermedad rara que ponga en peligro la vida o conlleve una incapacidad grave y crónica. (integrasaludtalavera.com)
- Glaucoma El glaucoma es una enfermedad ocular que causa pérdida de visión. (merckmanuals.com)
- Las anomalías adquiridas en la enfermedad recurrente parecen afectar sobre todo a genes asociados con la regulación del ciclo celular y el desarrollo de las células B. Además, en casi la mitad de los casos, la leucemia recurrente no se derivaba de las células leucémicas que predominaban en el diagnóstico. (blogspot.com)
Mutaciones12
- En marzo del 2009 se publicó en la revista "The American Journal of Human Genetics"[1] un estudio en el que descubren la relación entre mutaciones en el gen CBX2 contenido en este cromosoma con la identidad sexual humana. (wikipedia.org)
- La otra mitad de los casos de NF1 y NF2 son producto de nuevas mutaciones (cambios) en los genes causantes (1) . (marchofdimes.org)
- Aparentemente, la mayoría de los casos (aproximadamente el 85 por ciento) son causados por nuevas mutaciones (1,5) . (marchofdimes.org)
- La neurofibromatosis está causada por mutaciones en ciertos genes. (msdmanuals.com)
- El gen neurofibromina 2 (NF2, OMIM: 607379) localizado en el brazo largo del cromosoma 22 (22q12.2) es el único gen en el que se han identificado mutaciones asociadas a NF2 (1). (cibic.com.ar)
- En la actualidad, se sabe que es debido a mutaciones de los genes SMARCB1 y LZTR1, que suprimen los tumores, están asociadas con este tipo de neurofibromatosis. (enfermeriadeciudadreal.es)
- Se podría, asimismo, describir numerosas enfermedades raras que afectan a unos 250 millones de personas en el mundo, y cuyas causas son desconocidas, aunque en su gran mayoría se debe a mutaciones de genes. (cesarpazymino.com)
- Se consiguió por los siguientes motivos: - una mejora en la metodología - inversiones de la Sociedad de Distrofia Muscular ( de los Amish americanos, ya que en ellos es muy frecuente ) - porque este gen poseía características que facilitaban su clonaje: · por su forma de herencia ya se sabía que estaba ligado al X, por lo que sólo había que buscar en este cromosoma · sus mutaciones son grandes delecciones, fáciles de encontrar. (aprenderly.com)
- Los factores genéticos o ambientales pueden influir en los genes para que sufran mutaciones. (autoridadconsejo.com)
- En 2003, el mismo equipo de investigadores había descubierto en un pequeño número de individuos con autismo, mutaciones en dos de los genes que codifican las neuroliginas (el NLG3 y el NLGN4), unas proteínas que desempeñan un papel crucial en la arquitectura de la sinapsis. (genysi.es)
- Por eso, los individuos que presentan mutaciones que afectan a los genes NLGN o al SHANK3 tienen alterado el desarrollo de las conexiones neuronales esenciales para la comunicación social y el lenguaje. (genysi.es)
- Para que estos genes supresores adquieran su capacidad oncogénica, necesitan sufrir mutaciones independientes en ambos alelos, de manera que pierdan completamente su capacidad funcional. (biocancer.com)
Type6
- The heart in neurofibromatosis type 1: An echocardiographic study. (analesdepediatria.org)
- Expression of two new protein isoforms of the neurofibromatosis type 1 gene product, neurofibromin, in muscle tissues. (analesdepediatria.org)
- Targeted disruption of the neurofibromatosis type 1 gene leads to developmental abnormalities in heart and various neural crest derived tissues. (analesdepediatria.org)
- Mosaicism in neurofibromatosis type 2: an update of risk based on uni/bilaterality of vestibular schwannoma at presentation and sensitive mutation analysis including multiple ligation-dependent probe amplification. (cibic.com.ar)
- Neurofibromatosis type 1 (NF1), is a haploinsufficient and multisystemic disease, caused by inherited or sporadic mutations in the NF1 gene. (unachi.ac.pa)
- Lower Extremity Skin Ulcer Associated With Neurofibromatosis Type 1: A Case Report. (iacs.es)
Enfermedades7
- Las enfermedades monogénicas son enfermedades raras o minoritarias, es decir, con una prevalencia menor de 5 casos por cada 10.000 habitantes, mientras que las enfermedades poligénicas son comunes en la población adulta. (revistanefrologia.com)
- Ver mais ación (NGS) de 22 genes relacionados con enfermedades autoinflamatorias. (mendelics.com.br)
- Este examen es una poderosa herramienta para diagnosticar miles de enfermedades genéticas. (mendelics.com.br)
- Dra Cervera Las distrofias musculares son una serie de enfermedades genéticas con unas características comunes: 1. (aprenderly.com)
- Antiguamente estas enfermedades se clasificaban más bien de una forma clínica: 1. (aprenderly.com)
- Los genes no explican la mayor parte del problema, aunque existe la presencia de condiciones genéticas ligadas al autismo como es el X frágil, Neurofibromatosis, el Síndrome de Rett, las enfermedades metabólicas del recién nacido, entre otras. (dralydaperez.com)
- Cómo es la formación sobre Enfermedades Raras en España (4,5,6)? (integrasaludtalavera.com)
Tipos3
- Hasta la fecha se han descrito más de 50 genes relacionados con diferentes tipos de cáncer hereditario y de síndromes genéticos de predisposición a cáncer asociados a distintos patrones de herencia. (healthincode.com)
- Sarcoma es el término general para un amplio grupo de tipos de cáncer que se origina en los huesos y en los tejidos blandos (también llamados conectivos) del cuerpo (sarcoma de tejido blando). (elimparcial.com)
- Existen tres tipos: neurofibromatosis tipo 1 (NF1), neurofibromatosis tipo 2 (NF2) y schwannomatosis. (enfermeriadeciudadreal.es)
Cromosoma 176
- El cromosoma 17 es uno de los 23 pares de cromosomas del cariotipo humano. (wikipedia.org)
- El cromosoma 17 posee probablemente entre 1200 y 1500 genes. (wikipedia.org)
- También contiene el clúster genético homeobox B. En su estudio contra el cáncer la proteína p53 es producida por el gen TP53 del cromosoma 17, la cual es un gen supresor tumoral, ya que existiendo daño en el ADN, este detiene el ciclo celular e induce a apoptosis o muerte celular programada. (wikipedia.org)
- El gen de la NF1 se encuentra en el cromosoma 17 y los genes de la NF2 y la schwannomatosis en el cromosoma 22. (marchofdimes.org)
- La NF1 es causada por mutación en la neurofibromina gene ubicado en la región pericentromérica de cromosoma 17. (dermatly.com)
- En este caso el gen alterado es NF1 , que se encuentra en el cromosoma 17. (genotipia.com)
Principales genes1
- Estos genes se encuentran incluidos en el panel general de cáncer hereditario o pueden seleccionarse los principales genes involucrados citados a continuación. (healthincode.com)
Trastornos4
- La NF-1 es uno de los trastornos genéticos más comunes y se da independientemente de la raza o el sexo de la persona. (wikipedia.org)
- 5][6] La progresión de la condición es aproximadamente la siguiente:[7] Los trastornos musculoesqueléticos congénitos pueden o no estar presentes. (wikipedia.org)
- Las neurofibromatosis (NF) son trastornos genéticos del sistema nervioso que causan el crecimiento de tumores en los nervios. (marchofdimes.org)
- La neurofibromatosis es un grupo de trastornos genéticos en el que se forman tumores blandos y carnosos de tejido nervioso (neurofibromas) bajo la piel y en otras partes del cuerpo, y en el que a menudo se desarrollan en la piel manchas no protuberantes de color café con leche (manchas café-au-lait). (msdmanuals.com)
Supresores5
- Alteración de los genes supresores de tumores, como ocurre en la neurofibromatosis tipo 1, en el síndrome de Bannayan-Riley-Ruvalcaba y en el síndrome de Cowden. (exam-10.com)
- La mayor incidencia de tumores del SNC en diversos síndromes hereditarios es expresión de la mutación de genes supresores: sis, myc, src, p53, etc. (biocancer.com)
- Ya hemos comentado que en la regulación de la proliferación celular intervienen genes que estimulan el proceso, denominados protooncogenes, y genes que controlan el ciclo celular evitando el crecimiento excesivo, a los que denominamos genes supresores. (biocancer.com)
- Los genes supresores inhiben el crecimiento celular en condiciones normales. (biocancer.com)
- Los mecanismos por los cuales se puede alterar la expresión de los genes supresores son similares a los descritos para los oncogenes. (biocancer.com)
Mutations1
- Screening for large mutations of the NF2 gene. (cibic.com.ar)
Conocida3
- La condición es a veces conocida como "joroba" en el caso de una curva severa. (neurosurgeryinmexico.com)
- Ahora sabemos que la acondroplasia es causada por un cambio en la estructura (una mutación) en el gen que contiene la información química para la producción de la proteína conocida como receptor del factor de crecimiento de fibroblastos 3 (FGFR3) (1,2) (Figura 1). (treatingachondroplasia.com)
- Es por eso la frase conocida en el campo de la Medicina funcional " Los genes cargan el arma y los estilos de vida son los que detonan el gatillo" …sin embargo los genes no son el final de la historia. (dralydaperez.com)
Tuberosa1
- Es la tercera facomatosis en frecuencia después de la neurofibromatosis tipo 1 y de la esclerosis tuberosa . (neuropedwikia.es)
Riesgo3
- Algunos ejemplos de síndromes que aumentan el riesgo de sarcoma incluyen el retinoblastoma familiar y la neurofibromatosis tipo 1. (elimparcial.com)
- El riesgo de celulitis orbitaria después de la fractura orbitaria es bastante bajo, pero se asentará a una altura natural poco después de la cirugía. (cejop.cz)
- Con exactitud no podría hablar de peso de cada causa para determinar la incidencia, hasta el 2017 los estudios evidencian un 50-60 % de génesis asociada a la multicausalidad y a factores de riesgo ambiental, en mi práctica clínica podría decir que es cercano a un 80 % aunque soy muy consciente de la falta de ahondar en estudios avanzados a la hora de determinar la causa. (dralydaperez.com)
Causada2
- La NF2 es causada por una mutación en el cromosoma 22. (dermatly.com)
- El linfedema es una inflamación causada por una acumulación de líquido linfático que se produce cuando el sistema linfático está bloqueado o dañado. (elimparcial.com)
Signos3
- la mayoría de los signos de NF-1 son visibles después del nacimiento (durante la infancia), pero muchos síntomas de NF-1 aparece a medida que la persona envejece y tiene cambios hormonales. (wikipedia.org)
- Según los Institutos Nacionales de Salud (National Institutes of Health, NIH), la NF1 presenta siete signos típicos (1) . (marchofdimes.org)
- Cuáles son los signos y síntomas de la neurofibromatosis? (dermatly.com)
Schwannomatosis1
- La schwannomatosis es una forma rara de neurofibromatosis que se ha identificado recientemente. (enfermeriadeciudadreal.es)
Ocurre2
- Todos los síntomas se explican con la aparición de tumores: en la neurofibromatosis tipo 1 los bultos en la piel son tumores que se han formado en los nervios del tacto y la deformación ósea ocurre por la presión de tumores adyacentes. (genotipia.com)
- Accidente cerebrovascular (ictus) Un accidente cerebrovascular es un problema cerebral repentino que ocurre cuando un vaso sanguíneo de su cerebro se obstruye o se abre y sangra. (merckmanuals.com)
Asociados3
- Anales de Pediatría es una revista de acceso abierto que actúa como el Órgano de Expresión Científica de la Asociación y constituye el vehículo a través del cual se comunican los asociados. (analesdepediatria.org)
- También se han descrito casos asociados a traslocación del Xp11 (hipomelanosis de Ito y la incontinencia pigmenti podrían representar un síndrome de genes contiguos). (neuropedwikia.es)
- Ver mais neración (NGS) de genes asociados con la anemia genética, incluyendo: Anemia de Células Falciformes, Beta-talasemia, Deficiencia de G6PD, Síndrome de Shwachman-Bodian-Diamond y Anemia Megaloblástica Sensible a la Tiamina. (mendelics.com.br)
Herencia1
- El patrón de herencia determina el grado de causalidad genética (tabla 1). (revistanefrologia.com)
Tipo de neurofibromatosis1
- Este tipo de neurofibromatosis se caracteriza por la ocurrencia de cambios en la piel (manchas de color café con leche y pecas bajo la axila) y bultos del tamaño de un guisante por la superficie del cuerpo, llamados neurofibromas. (genotipia.com)
Salud4
- El órgano rector de los planes de salud es el Ministerio de Salud Pública, por tanto el diseño de políticas públicas para diagnóstico, tratamiento, prevención, apoyo, debe provenir de este organismo, pero aún hay vacíos y lentitud. (cesarpazymino.com)
- Todo el tiempo estamos haciendo epigenética, aún desde la preconcepción, las experiencias de nuestras generaciones previas, la salud del padre y de la madre impactan en la expresión de los genes del ser que viene en camino, esto confiere la susceptibilidad a responder a insultos o estresores del medio de una manera u otra. (dralydaperez.com)
- El plan de ejercicios de Mercola en combinación con los demás pilares para la salud, es la fórmula perfecta hacia una buena salud. (mercola.com)
- La información en este sitio de internet no tiene como objetivo reemplazar la relación uno a uno con un profesional del cuidado de la salud calificado y no es una opinión médica. (mercola.com)
Tumor4
- Schwannoma vestibular Un schwannoma vestibular (también llamado como neuroma acústico) es un tumor no canceroso (benigno) que se origina en las células que rodean el nervio vestibular (células de Schwann). (msdmanuals.com)
- Cualquier caso de predisposición al cáncer, así como cualquier tumor sólido, es susceptible de beneficiarse de nuestros test genéticos. (healthincode.com)
- el tumor es inoperable, pues invade el tronco celíaco o los vasos mesentéricos superiores. (wikipedia.org)
- Qué es el tumor carcinoide? (autoridadconsejo.com)
Cromosomas2
- De transmisión autosómica dominante, los genes alterados están en los cromosomas 9 y 16. (biocancer.com)
- Genes y cromosomas Sus genes son los códigos químicos que controlan cómo funciona su cuerpo, cómo está constituido y qué aspecto tiene. (merckmanuals.com)
Cambios3
- Este campo es un campo de validación y debe quedar sin cambios. (healthincode.com)
- CARACTERÍSTICAS MOLECULARES: - La distrofina es una proteína muscular que se encarga de dar estabilidad a la célula durante la contracción - relajación, para que pueda soportar los cambios de longitud durante las mismas. (aprenderly.com)
- Cuando hablamos de epigenética hacemos referencia a los cambios que ocurren en nuestros genes por la exposición a experiencias previas, cambios que no modifican en si la secuencia del DNA, es como si le cambiaran solo unos ingredientes a la receta, son variaciones que ocurren en los peldaños o bases nitrogenadas de nuestros genes. (dralydaperez.com)
Relacionados2
- Ver mais ación (NGS) de genes relacionados con la Anemia de Fanconi. (mendelics.com.br)
- Son muchos los genes conocidos relacionados con los desórdenes de espectro autista. (genysi.es)
Encuentran1
- Como estos genes se expresan en neuronas y otras células nerviosas, los pacientes con neurofibromatosis desarrollarán tumores en estructuras y órganos donde se encuentran estas células, como el cerebro, la médula espinal, o en cualquier nervio. (genotipia.com)
Rara1
- esta es la variante más rara de neurofibromatosis, y sólo se ha descubierto recientemente. (genotipia.com)
Distrofia muscular2
- en el caso de la Distrofia muscular de Duchenne el aumento de CPK es de 60 veces y en portadoras de 3-4 veces. (aprenderly.com)
- 1.- DISTROFIA MUSCULAR DE DUCHENNE - Se trata de una distrofia muscular progresiva de la musculatura proximal por degeneración de las fibras musculares y sustitución por infiltración grasa y tejido conectivo. (aprenderly.com)
Tratamiento2
- El curso de la LFM es completamente benigno, sin haberse descrito degeneración maligna, y no requiere tratamiento. (actasdermo.org)
- La Oncología Médica es una especialidad médica, una rama de la medicina que deriva del medicina interna, y que se encarga del diagnóstico y tratamiento del cáncer. (mirial.es)
Secuencia1
- En cambio, se denominan polimorfismos o variantes neutras a las variantes de secuencia que per se no son patogénicas y que están presentes en una frecuencia superior al 1% en la población. (revistanefrologia.com)
Alterados1
- Genes alterados en cromosoma 3. (biocancer.com)
Causas3
- 3] La gravedad de NF-1 varía ampliamente y se desconocen las causas de que un paciente tenga un caso leve o severo. (wikipedia.org)
- Neurofibromatosis, ¿cuáles son sus causas? (cinfa.com)
- No es claro aún cuáles son las causas de la mayoría de los sarcomas . (elimparcial.com)
Caracteriza1
- La neurofibromatosis se caracteriza por la aparición de tumores benignos (bultos) a lo largo de nervios y debajo de la piel. (genotipia.com)
Afecta3
- La neurofibromatosis (NF) es una genético trastorno que afecta al hueso, Tejido suave , piel y sistema nervioso. (dermatly.com)
- Hoy en día el TEA afecta a más de 70 millones de personas en el mundo, con una predilección mayor por el sexo masculino en una relación hombre: mujer de 5 a 1. (dralydaperez.com)
- El síndrome de Sturge-Weber es un defecto congénito que afecta a los vasos sanguíneos pequeños. (merckmanuals.com)
Afectados1
- La escoliosis focal y la cifosis son las manifestaciones esqueléticas más comunes de NF-1, que ocurren en el 20 % de los pacientes afectados. (wikipedia.org)
Bilateral1
- También conocido como bilateral neurofibromatosis acústica o neurofibromatosis central. (dermatly.com)
Causa7
- Un síndrome genético es un conjunto de síntomas o afecciones que se presentan juntos y cuya causa habitual son genes anormales . (cancer.gov)
- Qué causa la neurofibromatosis? (dermatly.com)
- en la mayoría de los casos, la causa de la escoliosis es desconocida, especialmente entre los niños y adolescentes (escoliosis de aparición temprana, escoliosis idiopática del adolescente. (neurosurgeryinmexico.com)
- Esta es la causa más común de escoliosis en adultos, especialmente si está asociada con la osteoporosis. (neurosurgeryinmexico.com)
- El vitíligo es una pérdida del pigmento, o color, de la piel que causa manchas blancas o puntos blancos en la piel. (appsonly.website)
- Esto es importante desde el punto de vista del desarrollo de medicamentos, debido a que uno de los temas que el investigador de los nuevos medicamentos debe responder es si el objetivo identificado (en este caso, el FGFR3) realmente tiene una relación de causa y efecto con la condición clínica. (treatingachondroplasia.com)
- En otras palabras, tenemos que responder si hay pruebas claras de que el FGFR3 es la causa de la acondroplasia. (treatingachondroplasia.com)
Neurofibromas2
- Los neurofibromas cutáneos y subcutáneos no son específicos de neurofibromatosis, pero los neurofibromas plexiformes solo se observan en NF1. (dermatly.com)
- Los neurofibromas subcutáneos son similares a los neurofibromas cutáneos, excepto que su origen es más profundo. (dermatly.com)
Genoma1
- Una investigación del Instituto Pasteur de París (Francia) ha descubierto una pieza del enorme puzzle que es el ''genoma autista'', el gen SHANK3. (genysi.es)
Expresan1
- Cuando se produce una mutación en estos genes, sus proteínas no se expresan o dan lugar a proteínas no funcionantes, favoreceriendo la aparición del proceso de carcinogénesis, al no existir un control de la proliferación celular. (biocancer.com)
Generalizada1
- La exploración mostraba más de 100 nódulos subcutáneos bien definidos, de 1 a 6 cm de diámetro, consistencia gomosa, móviles, con distribución generalizada pero respetando cabeza, cuello y hombros ( fig. 1 ). (actasdermo.org)
Explican2
- En la neurofibromatosis tipo 2 las pérdidas en los sentidos se explican por la formación de schwannomas (hinchazón de las células de Schwann, que rodean las neuronas con una vaina protectora de proteína para acelerar la señal nerviosa) alrededor del nervio que comunica con los ojos y oídos. (genotipia.com)
- Las ER explican el 35% de muertes antes del año de vida, el 10% entre 1 y 5 años, y el 12% entre 5 y 15 años. (cesarpazymino.com)
Individuos2
- El examen indentifica la alteración genética más frecuente en individuos con Hemofilia A grave, inversiones en los intrones 1 y 22 del gen F8. (mendelics.com.br)
- La prevalencia total del s ndrome es desconocida, pero se estima en 1 / 38.500 en individuos de edades comprendidas entre los 2 y 19 a os en Suecia. (orpha.net)
Manchas3
- Debido a su rareza y al hecho de que el diagnóstico genético se ha utilizado solo en los últimos años, en el pasado el NF-1 se confundió en algunos casos con el Síndrome de Legius, que cursa con síntomas vagamente similares, incluidas las manchas café con leche. (wikipedia.org)
- Seis o más manchas de color marrón claro en la piel, conocidas como manchas �café con leche� de más de 1/5 pulg. (marchofdimes.org)
- Presentar más de seis manchas de color café con leche es un fuerte indicio de NF1. (enfermeriadeciudadreal.es)
Nacidos vivos2
- Presenta una prevalencia de 1/10.000-20.000 recién nacidos vivos (García-Peñas et al 2008). (neuropedwikia.es)
- La prevalencia estimada es de 1/3.000 nacidos vivos. (enfermeriadeciudadreal.es)
Siguientes1
- 4] Los síntomas de la neurofibromatosis tipo 1 son los siguientes. (wikipedia.org)
Sarcoma1
- Hoy se celebra el Día Mundial de Sarcoma, que es un tipo de cáncer que puede ocurrir en distintas partes del cuerpo. (elimparcial.com)
Celular1
- Una hipótesis aceptada por algunos investigadores es que estas reacciones químicas hacen que los condrócitos entren en un estado de parálisis, llamado senescencia celular, cuando la célula no muere, pero deja de llevar a cabo la mayor parte de sus funciones normales (6). (treatingachondroplasia.com)
Crecimiento5
- Exceso de secreción de hormona de crecimiento, como se produce en el gigantismo hipofisiario, en el síndrome de McCune-Albright y en el síndrome MEN-1. (exam-10.com)
- Esta proteína reactiva (así también llamada enzima) es responsable por la reducción de la capacidad de un tipo particular de célula, los condrocitos de las placas de crecimiento cartilaginosas , de proliferar (multiplicar) y de hipertrofiar (madurar) (3). (treatingachondroplasia.com)
- La placa de crecimiento es un tejido delgado y altamente especializado, localizada en ambos extremos de los huesos largos y es responsable por el crecimiento óseo (4) (Figura 2). (treatingachondroplasia.com)
- El FGFR3 es un factor clave para el crecimiento de los huesos, pero no es el único: hay un montón de otros factores químicos y mecánicos que actúan positiva o negativamente para modular el ritmo de desarrollo de los huesos en el cuerpo en crecimiento (5). (treatingachondroplasia.com)
- La principal excepción es exactamente el condrocito de la placa de crecimiento. (treatingachondroplasia.com)
Asocia2
- En los últimos años, algunos estudios han sugerido que NF-1 se asocia con un problema primario en la función muscular (miopatía). (wikipedia.org)
- La neurofibromatosis tipo 2 se asocia con schwanomas y neurinomas del acústico, ependimomas y meningiomas. (biocancer.com)
Cuerpo1
- Resonancia magnética nuclear (RMN) La resonancia magnética nuclear (RMN) es una prueba que utiliza un aparato con un potente imán para generar imágenes del interior de su cuerpo. (merckmanuals.com)