Distrofias Musculares
Distrofia Muscular de Duchenne
Distrofia Muscular Animal
Distrofina
Distrofia Miotónica
Distrofia Muscular de Cinturas
Distrofia Muscular Facioescapulohumeral
Ratones Consanguíneos mdx
Distrofia Muscular de Emery-Dreifuss
Sarcoglicanos
Distrofias Hereditarias de la Córnea
Distroglicanos
Distrofia Muscular Oculofaríngea
Utrofina
Músculo Esquelético
Distrofia Endotelial de Fuchs
Timopoyetinas
Distrofias Retinianas
Colágeno Tipo VI
Linaje
Proteínas Asociadas a la Distrofina
Sarcolema
Creatina Quinasa
Proteínas Musculares
Mioblastos
Fibras Musculares Esqueléticas
Cromosomas Humanos Par 4
Lamina Tipo A
Complejo de Proteínas Asociado a la Distrofina
Laminina
Exones
Caveolina 3
Proteína II de Unión a Poli(A)
Mutación
Enfermedades Musculares
Cromosoma X
Enfermedades Neuromusculares
Fenotipo
Proteínas del Citoesqueleto
Distrofias Neuroaxonales
Modelos Animales de Enfermedad
Sarcoglicanopatías
Síndrome de Walker-Warburg
Ligamiento Genético
Calpaína
Cardiomiopatías
Plectina
Terapia Genética
Conectina
Desarrollo de Músculos
Distrofia Simpática Refleja
Proteínas de la Membrana
Debilidad Muscular
Diafragma
Análisis Mutacional de ADN
Fuerza Muscular
Distrofia Macular Viteliforme
Datos de Secuencia Molecular
Heterocigoto
Mapeo Cromosómico
Miostatina
Ratones Consanguíneos C57BL
Laminas
Células Satélite del Músculo Esquelético
Dependovirus
Secuencia de Bases
Mutación Missense
Inmunohistoquímica
Cardiomiopatía Dilatada
Electrorretinografía
Ratones Transgénicos
Atrofia Muscular
Expansión de Repetición de Trinucleótido
Células Musculares
Trastornos Miotónicos
Glicosilación
Manosiltransferasas
Biopsia
N-Acetilglucosaminiltransferasas
Diagnóstico Prenatal
Ratones Noqueados
Degeneración Retiniana
Proteína I de Unión a Poli(A)
Glicoproteínas de Membrana
Morfolinos
Asesoramiento Genético
Reacción en Cadena de la Polimerasa
Oxepinas
Mutación del Sistema de Lectura
Cadenas alfa de Integrinas
Codón sin Sentido
Pregnenodionas
Azul de Evans
Pruebas Genéticas
Retinitis Pigmentosa
Miopatias Distales
Membrana Nuclear
Discapacidad Intelectual
Western Blotting
Eliminación de Gen
Marcadores Genéticos
Secuencia de Aminoácidos
Contracción Muscular
Edad de Inicio
Caveolinas
Epidermólisis Ampollosa Simple
Eliminación de Secuencia
Regulación de la Expresión Génica
Células Cultivadas
Miocardio
Mutación Puntual
Cardiotoxinas
ADN
Fibrosis
Contractura
Técnica del Anticuerpo Fluorescente
Vectores Genéticos
ARN Mensajero
Genotipo
Óxido Nítrico Sintasa de Tipo I
Glicosiltransferasas
Secuencias Repetidas en Tándem
Escala de Lod
Escoliosis
Oligorribonucleótidos Antisentido
Fondo de Ojo
Sarcómeros
Distrofia Corneal Epitelial Juvenil de Meesmann
Haplotipos
Eugenesia
Costameras
Colágeno Tipo VIII
Progresión de la Enfermedad
Perros
Lipodistrofia
Glicerol Quinasa
Oligonucleótidos Antisentido
Efecto Fundador
Las distrofias musculares son un grupo de enfermedades genéticas que se caracterizan por la degeneración y deterioro progresivo del tejido muscular. Estas enfermedades están asociadas con mutaciones en genes que codifican proteínas importantes para la estructura y función normal de los músculos esqueléticos.
Existen varios tipos de distrofias musculares, siendo las más comunes la Distrofia Muscular de Duchenne (DMD) y la Distrofia Muscular de Becker (BMD). La DMD es una enfermedad grave y progresiva que generalmente afecta a niños varones; se caracteriza por debilidad muscular que comienza en las extremidades inferiores y evoluciona hacia el tronco, lo que lleva a pérdida de la capacidad de caminar y eventualmente a afecciones cardíacas y pulmonares graves. La BMD es una forma menos severa de distrofia muscular que también afecta principalmente a niños varones; los síntomas suelen ser menos graves y progresan más lentamente que en la DMD.
Otros tipos de distrofias musculares incluyen la Distrofia Muscular Emery-Dreifuss, la Distrofia Muscular Facioescapulohumeral, la Distrofia Muscular Oculofaríngea y la Distrofia Muscular Distal. Cada tipo de distrofia muscular tiene diferentes patrones de herencia, síntomas y gravedad.
El tratamiento para las distrofias musculares generalmente se centra en el manejo de los síntomas y la prevención de complicaciones. La fisioterapia y la terapia ocupacional pueden ayudar a mantener la fuerza y la movilidad, mientras que los dispositivos de asistencia pueden ser útiles para mejorar la independencia y la calidad de vida. En algunos casos, se pueden considerar opciones de tratamiento más agresivas, como la terapia génica o la terapia celular.
La Distrofia Muscular de Duchenne (DMD, por sus siglas en inglés) es un tipo de distrofia muscular que se caracteriza por una degeneración y necrosis progresiva del tejido muscular esquelético. Es causada por mutaciones en el gen de la distrofina, localizado en el cromosoma X, lo que resulta en una falta o disminución significativa de la proteína distrofina en los músculos.
Esta afección generalmente se manifiesta en la primera infancia, con debilidad muscular simétrica y progresiva que comienza en los músculos proximales de las extremidades inferiores y luego se extiende a otros grupos musculares. Los niños con DMD pueden tener dificultad para caminar, subir escaleras, levantarse del suelo y otras actividades físicas.
Otros síntomas comunes incluyen contracturas articulares, escoliosis, problemas cardíacos y respiratorios, y retraso en el desarrollo cognitivo en algunos casos. La esperanza de vida promedio para los individuos con DMD es de aproximadamente 20 a 30 años, aunque esto ha mejorado en las últimas décadas gracias a los avances en el cuidado y la atención médica.
La Distrofia Muscular de Duchenne se hereda como una enfermedad ligada al cromosoma X, lo que significa que generalmente afecta a los varones, ya que no tienen un segundo cromosoma X para compensar la mutación. Las mujeres que son portadoras del gen anormal tienen un riesgo de transmitir la enfermedad a sus hijos varones y pueden experimentar síntomas más leves o no presentar síntomas en absoluto.
La distrofia muscular en animales es una afección genética que causa debilidad y degeneración progresivas de los músculos esqueléticos. Existen varios tipos de distrofias musculares, cada uno causado por diferentes mutaciones génicas. La más común es la distrofia muscular de Duchenne, que también afecta a los humanos y es causada por una mutación en el gen que produce la proteína distrofina.
En los animales afectados, la falta o disfunción de distrofina conduce a daños en las fibras musculares, lo que provoca inflamación y reemplazo del tejido muscular por tejido cicatricial y grasa. Esto lleva a una progresiva pérdida de fuerza y movilidad, dificultad para respirar, y en algunos casos, problemas cardíacos.
Los síntomas suelen aparecer en la etapa temprana de la vida del animal y empeoran con el tiempo. La distrofia muscular afecta principalmente a perros, pero también se ha descrito en gatos, caballos y otros animales. No existe cura para esta enfermedad, aunque los tratamientos pueden ayudar a aliviar los síntomas y mejorar la calidad de vida de los animales afectados.
La distrofina es una proteína grande y crucial que se encuentra en los músculos esqueléticos. Ayuda a mantener la integridad estructural de las fibras musculares y desempeña un papel importante en el proceso de reparación del tejido muscular después del daño.
La distrofina está codificada por el gen DMD, que se encuentra en el cromosoma X. Las mutaciones en este gen pueden dar lugar a una variedad de trastornos neuromusculares conocidos colectivamente como distrofinopatías. El más grave y común de estos es la distrofia muscular de Duchenne (DMD), una enfermedad progresiva que causa debilidad muscular severa y eventual fallo muscular. Otra afección relacionada es la distrofia muscular de Becker (BMD), que generalmente es menos grave y se desarrolla más lentamente que la DMD.
La proteína distrofina ayuda a unir el citoesqueleto interior del músculo, conocido como filamentos finos de actina, con la membrana celular exterior, o sarcolema. También desempeña un papel en la transducción de señales y puede interactuar con varias otras proteínas para ayudar a mantener la estabilidad y la salud del músculo esquelético.
En las distrofinopatías, como la DMD y la BMD, los niveles de distrofina son bajos o están ausentes en los músculos debido a mutaciones en el gen DMD. Esto puede conducir a una serie de problemas, incluyendo debilidad muscular, rigidez articular, contracturas y problemas cardíacos y respiratorios. El tratamiento para estas afecciones generalmente se centra en la gestión de los síntomas y el mantenimiento de la función muscular y la calidad de vida tanto como sea posible.
La distrofia miotónica es una enfermedad genética hereditaria que se caracteriza por afectar los músculos y otros sistemas corporales. Existen dos tipos principales, conocidos como distrofia miotónica de tipo 1 (DM1) y distrofia miotónica de tipo 2 (DM2), que difieren en su localización genética, severidad y síntomas específicos.
La DM1 es causada por una mutación en el gen DMPK en el cromosoma 19, mientras que la DM2 se debe a una mutación en el gen CNBP (ZNF9) en el cromosoma 3. Ambas mutaciones conllevan a la producción de ARN anormalmente largo y repetitivo, lo que resulta en la alteración de la función celular y la muerte de células musculares.
Los síntomas más comunes de la distrofia miotónica incluyen:
1. Debilidad y atrofia muscular progresiva, especialmente en los músculos faciales, del cuello y de las extremidades superiores e inferiores.
2. Miotonía, que es una rigidez o dificultad para relajar los músculos después de la contracción voluntaria.
3. Problemas cardíacos, como arritmias y disfunción cardiaca.
4. Alteraciones del sistema nervioso autónomo, que pueden causar problemas gastrointestinales, diabetes, hipotensión ortostática y dificultad para regular la temperatura corporal.
5. Problemas respiratorios, especialmente durante el sueño.
6. Cataratas y glaucoma.
7. Disfunción cognitiva leve en algunos casos.
La distrofia miotónica es una enfermedad progresiva, lo que significa que sus síntomas empeoran con el tiempo. El tratamiento se centra en aliviar los síntomas y mejorar la calidad de vida del paciente. No existe cura conocida para esta enfermedad.
La distrofia muscular de cinturas es un grupo de enfermedades genéticas que se caracterizan por la degeneración y debilidad progresivas del músculo esquelético, especialmente en las áreas de la cintura pélvica y la cintura escapular. Estas enfermedades están asociadas con mutaciones en genes que producen proteínas importantes para la estructura y función muscular normal.
Existen varios tipos de distrofia muscular de cinturas, siendo los más comunes la Distrofia Muscular de Duchenne (DMD) y la Distrofia Muscular de Becker (BMD). La DMD es una enfermedad grave y progresiva que generalmente se diagnostica en la infancia y conduce a una discapacidad significativa. Los niños con DMD rara vez viven más allá de los 30 años. Por otro lado, la BMD es una forma menos severa y más variable de la enfermedad, que suele diagnosticarse en la adolescencia o en la edad adulta temprana.
Los síntomas comunes de las distrofias musculares de cinturas incluyen debilidad muscular progresiva, contracturas articulares, dificultad para caminar y subir escaleras, caídas frecuentes, escápulas aladas (omóplatos prominentes), escoliosis y problemas cardíacos y respiratorios en etapas más avanzadas de la enfermedad. El tratamiento suele ser sintomático y de apoyo, e incluye fisioterapia, dispositivos de asistencia, cirugía ortopédica y medicamentos para tratar los problemas cardíacos y respiratorios. No existe cura conocida para estas enfermedades.
La Distrofia Muscular Facioescapulohumeral (FSHD, por sus siglas en inglés) es un trastorno genético que debilita progressivemente los músculos del rostro, los hombros y los brazos. El término "distrofia muscular" se refiere a una serie de enfermedades hereditarias que debilitan progresivamente y dañan los músculos controlados por el sistema nervioso voluntario.
La FSHD es una forma particular de distrofia muscular que afecta principalmente a los músculos faciales (facio), los del hombro (escapular) y los del brazo (humeral). Los síntomas generalmente comienzan en la adolescencia o en la edad adulta temprana, aunque en algunos casos pueden comenzar en la infancia.
La debilidad muscular inicial afecta a los músculos de la cara, lo que puede causar dificultad para cerrar los ojos completamente, una expresión facial inexpresiva y pérdida del arco de las cejas. Con el tiempo, la debilidad se propaga a los músculos de los hombros y los brazos, lo que puede dificultar el levantamiento de objetos, el peinarse el cabello o incluso el levantar los brazos para colocarlos detrás de la cabeza.
La FSHD se hereda de manera autosómica dominante, lo que significa que solo necesita un gen defectuoso de uno de sus padres para heredar la enfermedad. Sin embargo, aproximadamente el 30% de los casos son el resultado de una nueva mutación genética y no tienen antecedentes familiares de la enfermedad.
El tratamiento de la FSHD se centra en la rehabilitación y la terapia física para mantener la fuerza muscular y la movilidad lo más largo posible. También pueden ser útiles los dispositivos de asistencia, como las sillas de ruedas o los andadores, según la gravedad de la enfermedad. No existe una cura conocida para la FSHD, pero se están investigando varios tratamientos experimentales.
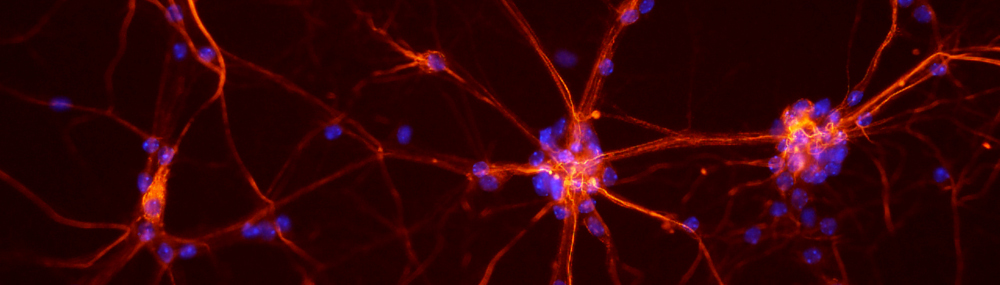
Los ratones consanguíneos mdx son un tipo de ratón de laboratorio que se utiliza en investigación médica y biológica. Este strain de ratón tiene una mutación espontánea en el gen que codifica la distrofina, una proteína importante para la estructura y función de las fibras musculares esqueléticas. La mutación en el gen mdx causa la ausencia de distrofina, lo que lleva a una enfermedad similar a la distrofia muscular de Duchenne (DMD) en humanos.
La distrofia muscular de Duchenne es una enfermedad genética rara y progresiva que causa debilidad y atrofia muscular. Afecta predominantemente a los niños y generalmente se diagnostica antes de los 5 años de edad. La enfermedad es causada por mutaciones en el gen que codifica la distrofina, lo que resulta en una falta o disminución significativa de esta proteína en las células musculares.
Los ratones mdx se utilizan a menudo como modelos animales para estudiar los mecanismos subyacentes de la distrofia muscular y probar posibles tratamientos y terapias. Aunque los ratones mdx no desarrollan la enfermedad tan gravemente como los humanos con DMD, todavía exhiben síntomas similares, como debilidad muscular y fibrosis. Por lo tanto, proporcionan un medio útil para investigar la fisiopatología de la enfermedad y evaluar posibles intervenciones terapéuticas.
La Distrofia Muscular de Emery-Dreifuss es una enfermedad genética y progresiva que afecta los músculos esqueléticos, caracterizada por debilidad y rigidez muscular. Se hereda de manera dominante ligada al cromosoma X o recesiva ligada al cromosoma X, lo que significa que generalmente afecta a los varones.
Los síntomas suelen comenzar en la infancia o adolescencia y pueden incluir:
1. Debilidad muscular que afecta principalmente a los músculos de los hombros, las caderas y los muslos.
2. Contracturas articulares (limitación del movimiento articular) en los codos, rodillas y tobillos.
3. Rigidez muscular progresiva que puede conducir a una postura anormal o encorvada.
4. Anomalías cardíacas, como ritmos cardíacos irregulares (arritmias) o insuficiencia cardíaca congestiva.
5. Problemas de conductividad eléctrica en el corazón, lo que puede aumentar el riesgo de muerte súbita cardiaca.
La Distrofia Muscular de Emery-Dreifuss se asocia con mutaciones en genes específicos, como EMD, LMNA y FHL1. El diagnóstico suele confirmarse mediante análisis genético o biopsia muscular.
El tratamiento actual se centra en la gestión de los síntomas y puede incluir fisioterapia, ortopedia, medicamentos para controlar las arritmias cardíacas y dispositivos como desfibriladores implantables para prevenir la muerte súbita cardiaca. No existe cura conocida para esta enfermedad.
Los sarcoglicanos son una familia de proteínas que se encuentran en la membrana celular de las fibras musculares y otros tejidos. Se unen a otras proteínas, como la distrofina y los dystroglicanes, para formar el complejo de distrofina-glicoproteína (DGC). Este complejo desempeña un papel importante en la estabilidad estructural y la función mecánica de las membranas celulares.
Las mutaciones en los genes que codifican para los diferentes sarcoglicanos pueden causar diversas distrofias musculares, como la distrofia muscular congénita de tipo limb-girdle y la distrofia muscular de Becker. Estas enfermedades se caracterizan por debilidad y atrofia muscular progresivas, que pueden afectar a diferentes grupos musculares dependiendo del tipo de distrofia muscular.
Además de su función estructural, los sarcoglicanos también pueden desempeñar un papel en la señalización celular y la regulación del crecimiento y desarrollo de los tejidos. Sin embargo, aún se está investigando el mecanismo preciso por el cual las mutaciones en estas proteínas causan las distrofias musculares asociadas.
Las distrofias hereditarias de la córnea son un grupo de enfermedades oculares raras y genéticas que afectan la estructura y transparencia de la córnea, la parte frontal transparente del ojo. Estas condiciones se caracterizan por anomalías en el tejido corneal, lo que puede causar visión borrosa, dolor ocular e incluso pérdida de visión en casos graves.
Las distrofias hereditarias de la córnea se clasifican según el patrón de herencia (autosómico dominante, autosómico recesivo o ligado al cromosoma X) y el nivel de afectación en las capas corneales. Algunos tipos comunes incluyen:
1. Distrofia de Epitelio Corneal (DECT): Afecta la capa más externa de la córnea, causando erosiones y dolor ocular severo.
2. Distrofia de Estroma Central Profunda (DSCP): Afecta la capa media de la córnea, provocando opacidades y disminución de la visión.
3. Distrofia Endotelial Fuchs: Afecta la capa más interna de la córnea, causando edema y pérdida de transparencia.
4. Distrofia Macular Corneal (DMC): Afecta la zona central de la córnea, provocando opacidades y disminución de la visión.
Estas enfermedades suelen presentarse en ambos ojos y pueden empeorar gradualmente con el tiempo. El tratamiento puede incluir lentes de contacto especiales, medicamentos para aliviar los síntomas y, en casos graves, trasplante de córnea. La causa subyacente de las distrofias hereditarias de la córnea es una mutación genética que afecta a las proteínas estructurales o funcionales de la córnea. El diagnóstico se realiza mediante un examen oftalmológico completo y, en algunos casos, pruebas adicionales como la tomografía de coherencia óptica (OCT) o la biomicroscopía especular.
Los distroglianos son una familia de proteínas perlejadas en la membrana basal de las uniones neuromusculares y otras sinapsis. Se unen a la laminina, una proteína importante en la matriz extracelular, y ayudan en la formación y mantenimiento de las uniones entre los músculos y los nervios. También desempeñan un papel en el tráfico de vesículas intracelulares y la señalización celular. Mutaciones en los genes que codifican para los distroglianos pueden conducir a diversas formas de distrofia muscular, una enfermedad degenerativa que se caracteriza por debilidad y atrofia muscular progresiva. La forma más común de distrofia muscular asociada con defectos en los distroglianos es la distrofia muscular de Becker. Además, los distroglianos también se han relacionado con algunas formas de demencia y trastornos mentales.
La distrofia muscular oculofaringea (OFPD, por sus siglas en inglés) es un trastorno genético extremadamente raro que afecta principalmente a los músculos que controlan los párpados y la parte superior del ojo, así como los músculos de la garganta y el pecho. Esta afección se caracteriza por debilidad progresiva de los músculos faciales y del cuello, problemas de visión y dificultad para deglutir.
La OFPD es causada por mutaciones en el gen PIEZO2 y se hereda con un patrón autosómico dominante, lo que significa que una copia del gen alterado en cada célula es suficiente para causar la afección. Sin embargo, también puede ocurrir espontáneamente como resultado de una nueva mutación en el gen, lo que se denomina mutación "de novo".
Los síntomas generalmente comienzan en la infancia o adolescencia y pueden incluir:
1. Párpados caídos (ptosis)
2. Ojos inexpresivos
3. Dificultad para mover los ojos hacia arriba (limitación de la elevación del globo ocular)
4. Debilidad en el cuello y los hombros
5. Problemas de deglución (disfagia)
6. Debilidad en los músculos respiratorios, lo que puede provocar neumonías recurrentes
7. Escaso crecimiento y desarrollo
8. Disartria (dificultad para articular palabras)
9. Problemas de audición
10. Anormalidades esqueléticas, como escoliosis o contracturas articulares
El diagnóstico se basa en los síntomas, el examen físico y los resultados de las pruebas genéticas. No existe cura específica para la OFPD, y el tratamiento se centra en aliviar los síntomas y prevenir complicaciones. Esto puede incluir fisioterapia, terapia del habla y la audición, dispositivos de asistencia, como anteojos con elevadores para los párpados caídos, y, en algunos casos, cirugía. El pronóstico varía; algunas personas con OFPD tienen una esperanza de vida normal, mientras que otras pueden experimentar complicaciones graves que afectan su calidad de vida y expectativa de vida.
La utrofina, también conocida como tropomiosina-tropomodulina complejo de unión a actina o TCTC, es una proteína estructural que se encuentra en los músculos esqueléticos y cardíacos. Se une a la actina y regula su interacción con la tropomiosina, una proteína que también se une a la actina y desempeña un papel importante en la regulación de la contracción muscular.
La utrofina está compuesta por varias subunidades, incluyendo la utrofinina-1 (o Tctex1), la utrofinina-2/zynxin y la tropomodulina. La utrofinina-1 se une a la actina y media en la unión de la tropomiosina a la actina, mientras que la utrofinina-2/zynxin se une a varias proteínas y puede desempeñar un papel en la señalización celular. La tropomodulina se une al extremo del filamento de actina y regula su longitud.
La utrofina desempeña un papel importante en la estabilidad estructural de los músculos y puede estar involucrada en el desarrollo y mantenimiento de la fuerza muscular. Las mutaciones en los genes que codifican para las subunidades de la utrofina se han asociado con varias enfermedades musculares, incluyendo la distrofia muscular de Duchenne y la distrofia muscular miotónica de tipo 1.
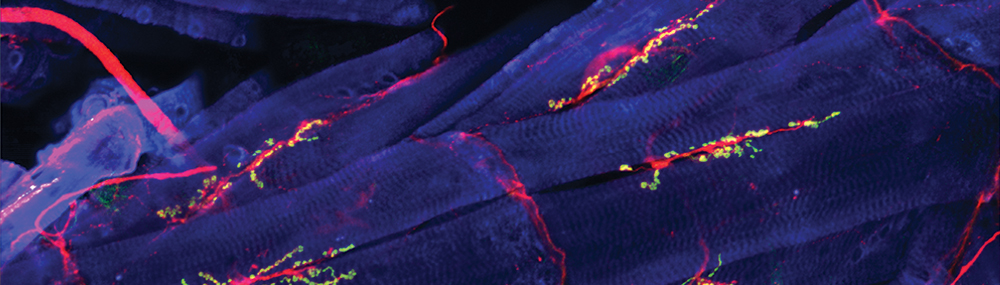
El músculo esquelético, también conocido como striated muscle o musculus voluntarius, está compuesto por tejidos especializados en la generación de fuerza y movimiento. Estos músculos se unen a los huesos a través de tendones y su contracción provoca el movimiento articular.
A diferencia del músculo liso (presente en paredes vasculares, útero, intestinos) o el cardíaco, el esquelético se caracteriza por presentar unas bandas transversales llamadas estrías, visibles al microscopio óptico, que corresponden a la disposición de las miofibrillas, compuestas a su vez por filamentos proteicos (actina y miosina) responsables de la contracción muscular.
El control de la actividad del músculo esquelético es voluntario, es decir, está bajo el control consciente del sistema nervioso central, a través de las neuronas motoras somáticas que inervan cada fibra muscular y forman la unión neuromuscular.
La función principal de los músculos esqueléticos es la generación de fuerza y movimiento, pero también desempeñan un papel importante en el mantenimiento de la postura, la estabilización articular, la respiración, la termorregulación y la protección de órganos internos.
La distrofia endotelial de Fuchs, también conocida como dystrophia epithelialis corneae de Fuchs, es una enfermedad degenerativa progresiva y hereditaria que afecta la parte posterior de la córnea, específicamente al endotelio corneal. El endotelio corneal es una capa celular única y delgada responsable de mantener la transparencia de la córnea y regular el fluido en ella.
La distrofia de Fuchs se caracteriza por la presencia de células endoteliales anormales, engrosamiento y opacidad corneal, y la formación de vesículas subepiteliales (guttae) en el endotelio. Estas guttae son pequeñas elevaciones que contienen material acuoso y pueden eventualmente conducir a una disminución en el número de células endoteliales sanas.
La progresión de la enfermedad puede resultar en edema corneal, lo que provoca visión borrosa y sensibilidad a la luz. En etapas avanzadas, la córnea puede volverse opaca, lo que lleva a una disminución significativa de la agudeza visual y, en algunos casos, incluso a la pérdida de visión.
El tratamiento para la distrofia endotelial de Fuchs generalmente implica el uso de lentes de contacto blandos o duros para aliviar los síntomas y retrasar la progresión de la enfermedad. En etapas más avanzadas, se puede considerar un trasplante de córnea para reemplazar las células endoteliales dañadas.
La timopoyetina es un tipo de fármaco que se encuentra en la categoría de agentes estimulantes de las colonias de granulocitos (G-CSF, por sus siglas en inglés). Estos medicamentos funcionan al unirse a los receptores de G-CSF en la superficie de las células madre de la médula ósea y estimular su diferenciación y multiplicación, lo que conduce a un aumento en el número de glóbulos blancos, específicamente neutrófilos, en la sangre.
Las timopoyetinas se utilizan clínicamente para tratar diversas condiciones en las que los recuentos de neutrófilos están disminuidos, como la neutropenia inducida por quimioterapia, la neutropenia congénita y el síndrome mielodisplásico. Algunos ejemplos de timopoyetinas incluyen filgrastim (Neupogen®) y pegfilgrastim (Neulasta®).
Aunque las timopoyetinas se utilizan comúnmente en la práctica clínica, su uso no está exento de riesgos potenciales. Algunos de estos riesgos incluyen la posibilidad de desarrollar síndrome de liberación de citocinas, trombocitopenia y dolor óseo. Por lo tanto, es importante que los médicos monitoreen cuidadosamente a los pacientes que reciben este tipo de fármacos para minimizar los riesgos y maximizar los beneficios terapéuticos.
Las distrofias retinianas representan un grupo heterogéneo de enfermedades hereditarias que afectan predominantemente a las células fotorreceptoras de la retina, resultando en una progresiva pérdida de visión. Estas enfermedades se caracterizan por una degeneración progresiva de los fotorreceptores, especialmente los bastones y conos, que son responsables de la percepción de la luz y el color.
Existen diferentes tipos de distrofias retinianas, clasificadas según su patrón de herencia (autosómica dominante, autosómica recesiva o ligada al cromosoma X), el tipo de células afectadas (bastones, conos o ambos), la edad de inicio y la velocidad de progresión. Algunos de los tipos más comunes incluyen:
1. Retinitis Pigmentosa (RP): Es el tipo más frecuente de distrofia retiniana, que afecta aproximadamente a 1 en 4000 personas. La RP se caracteriza por la pérdida progresiva de los bastones y, en etapas más avanzadas, también de los conos. Los síntomas iniciales suelen incluir una disminución de la visión nocturna (nictalopía) y la presencia de pigmento en forma de "manchas oscuras" en el fondo de ojo. La enfermedad puede progressar a un estrechamiento del campo visual (túnel visual) y, finalmente, a una pérdida severa de la visión central.
2. Distrofia Macular Progresiva: Este tipo de distrofia retiniana afecta predominantemente a las células fotorreceptoras en la mácula, la zona de la retina responsable de la visión central y detallada. Los síntomas iniciales suelen incluir una disminución progresiva de la agudeza visual y distorsiones en la visión (metamorfopsias). La enfermedad puede progressar a una pérdida severa de la visión central, aunque el campo periférico generalmente se preserva.
3. Distrofia Retiniana de Usher: Este tipo de distrofia retiniana se asocia con sordera y se hereda como un trastorno autosómico recesivo. Existen dos tipos principales, Usher I y Usher II, que difieren en el grado y la edad de inicio de las manifestaciones auditivas y visuales. Las personas afectadas por distrofia retiniana de Usher suelen experimentar una pérdida progresiva de la visión debido a la degeneración de los bastones y, en menor medida, de los conos.
4. Amaurosis Congénita de Leber: Es una forma rara de distrofia retiniana que se caracteriza por una pérdida severa o ausencia completa de la visión al nacer o durante la infancia temprana. La enfermedad se asocia con mutaciones en el gen RPE65 y puede responder al tratamiento con terapia génica.
El diagnóstico de distrofias retinianas generalmente implica una evaluación clínica completa, incluidos exámenes oftalmológicos especializados como la electroretinografía (ERG) y la tomografía de coherencia óptica (OCT). En algunos casos, el diagnóstico molecular puede confirmar el tipo y el pronóstico de la enfermedad. El tratamiento actual se centra en la terapia de reemplazo génico, las terapias celulares y los dispositivos de asistencia visual para mejorar la calidad de vida de las personas afectadas por estas enfermedades.
El colágeno tipo VI es una forma menos común de colágeno que se encuentra en el tejido conectivo del cuerpo humano. Es producido por las células endoteliales, fibroblastos y células musculares lisas. El colágeno tipo VI tiene una estructura única y forma una red tridimensional en la matriz extracelular que desempeña un papel importante en la estabilidad de los tejidos conectivos.
La deficiencia o anormalidades del colágeno tipo VI se han relacionado con varias afecciones médicas, como la distrofia muscular congénita de Ullrich y la miopatía congénita de Bethlem. Estas enfermedades hereditarias están asociadas con debilidad muscular progresiva, contracturas articulares y anomalías esqueléticas.
En resumen, el colágeno tipo VI es una forma especializada de colágeno que desempeña un papel importante en la estabilidad de los tejidos conectivos y puede estar involucrado en varias afecciones médicas cuando hay deficiencias o anormalidades en su producción.
En el contexto de la medicina y la biología, un linaje se refiere a una sucesión o serie de organismos relacionados genéticamente que descienden de un antepasado común más reciente. Puede hacer referencia a una secuencia particular de genes que se heredan a través de generaciones y que ayudan a determinar las características y rasgos de un organismo.
En la genética, el linaje mitocondrial se refiere a la línea de descendencia materna, ya que las mitocondrias, que contienen su propio ADN, se transmiten generalmente de madre a hijo. Por otro lado, el linaje del cromosoma Y sigue la línea paterna, ya que los cromosomas Y se heredan del padre y se mantienen intactos durante la meiosis, lo que permite rastrear la ascendencia masculina.
Estos linajes pueden ser útiles en la investigación genética y antropológica para estudiar la evolución y la migración de poblaciones humanas y otras especies.
Las proteínas asociadas a la distrofina (DAPs, por sus siglas en inglés) son un grupo de proteínas que se encuentran en la membrana celular de los músculos esqueléticos y están relacionadas con la distrofina, una proteína importante en el mantenimiento de la integridad estructural de las fibras musculares.
La distrofina se une a un complejo de proteínas en la membrana celular, incluyendo varias DAPs. Estas proteínas desempeñan diversas funciones importantes en el mantenimiento y la señalización de la membrana muscular, como ayudar a transportar moléculas a través de la membrana, regular la actividad de los canales iónicos y participar en la respuesta al daño muscular.
Las mutaciones en los genes que codifican para estas proteínas se han asociado con varias enfermedades neuromusculares, incluyendo la distrofia muscular de Duchenne y Becker, así como las miopatías congénitas. El estudio de las DAPs y su interacción con la distrofina puede ayudar a entender mejor los mecanismos subyacentes a estas enfermedades y desarrollar nuevas estrategias terapéuticas.
El sarcolema es la membrana celular que rodea las fibras musculares esqueléticas individuales. Es una parte importante de la estructura del músculo esquelético y desempeña un papel crucial en el mantenimiento de la integridad celular, la regulación del transporte de iones y la comunicación celular. El sarcolema está compuesto por dos capas de lípidos y proteínas, y está conectado a la membrana nuclear y los orgánulos intracelulares dentro de la fibra muscular. Las enfermedades del sarcolema pueden causar diversos trastornos neuromusculares, como distrofia muscular y miopatías.
La detección de heterocigotos es un proceso de identificación de individuos que tienen diferentes alelos (variantes genéticas) de un gen en cada uno de sus cromosomas homólogos. Este término se utiliza a menudo en el contexto de la genética médica y la counseleía genética.
En condiciones genéticas recesivas, los individuos que heredan una copia normal y una copia alterada/dañada del gen aún no mostrarán síntomas de la enfermedad, ya que solo se manifiesta cuando ambas copias del gen están alteradas. Sin embargo, estos individuos son portadores de la condición y pueden transmitirla a su descendencia.
La detección de heterocigotos permite identificar a estos portadores asintomáticos, lo que puede ser útil en la planificación familiar y el asesoramiento genético. Por ejemplo, si ambos miembros de una pareja son portadores de una mutación recesiva particular, hay un 25% de probabilidad de que su hijo herede las dos copias alteradas del gen y desarrolle la enfermedad correspondiente.
Es importante tener en cuenta que algunas pruebas genéticas solo detectan mutaciones en uno o ambos alelos, pero no siempre pueden determinar con certeza si un individuo es heterocigoto o no. Por lo tanto, los resultados deben interpretarse con precaución y bajo la guía de profesionales médicos y genéticos calificados.
La creatina quinasa (CK) es una enzima presente en diferentes tejidos corporales, especialmente en el músculo esquelético, cardíaco y cerebral. Su función principal es catalizar la reacción de reversibilidad de la creatina y fosfatos para producir ATP, que es una molécula importante que proporciona energía a las células del cuerpo.
Existen tres tipos principales de creatina quinasa en el cuerpo humano: CK-MM, CK-MB y CK-BB. La CK-MM se encuentra principalmente en el músculo esquelético, la CK-MB se encuentra en el corazón y en menor medida en el músculo esquelético, y la CK-BB se encuentra en el cerebro y otros tejidos.
Los niveles de creatina quinasa en sangre pueden aumentar después de un daño muscular o cardíaco, como durante una lesión muscular, un infarto de miocardio o un derrame cerebral. Por lo tanto, la medición de los niveles séricos de CK se utiliza a menudo como un marcador bioquímico para ayudar en el diagnóstico y el seguimiento del daño tisular en estas condiciones.
En resumen, la creatina quinasa es una enzima importante que desempeña un papel crucial en la producción de energía en las células del cuerpo. Los niveles séricos de CK pueden aumentar después de un daño muscular o cardíaco y se utilizan como marcadores bioquímicos para ayudar en el diagnóstico y seguimiento de estas condiciones.
Las proteínas musculares son específicas proteínas que se encuentran en el tejido muscular y desempeñan un papel crucial en su estructura y función. La proteína más abundante en el músculo es la actina, seguida de la miosina, ambas involucradas en la contracción muscular. Otras proteínas musculares importantes incluyen las troponinas y la tropomiosina, que regulan la interacción entre la actina y la miosina, así como diversos componentes de la matriz extracelular que brindan soporte estructural al tejido muscular. La síntesis y degradación de proteínas musculares están cuidadosamente reguladas y desempeñan un papel importante en el crecimiento, reparación y mantenimiento del músculo esquelético. La disminución de la síntesis de proteínas musculares y el aumento de la degradación están asociados con diversas condiciones patológicas, como la sarcopenia (pérdida de masa muscular relacionada con la edad) y la cachexia (pérdida de peso y debilidad muscular asociadas con enfermedades graves).
Los mioblastos son células musculares embrionarias que se diferencian en fibras musculares maduras durante el desarrollo fetal y la regeneración del tejido muscular. Se originan a partir de las células madre mesenquimales y experimentan una serie de cambios para convertirse en células musculares especializadas. Los mioblastos contienen varias estructuras distintivas, incluyendo miofibrillas, que son cadenas proteicas responsables de la contracción muscular. Estas células también tienen un importante papel en el proceso de reparación y regeneración del tejido muscular dañado en adultos. Cuando un músculo se lesiona, los mioblastos se activan y migran al sitio de la lesión, donde se fusionan para formar nuevas fibras musculares y restaurar la función muscular.
Las fibras musculares esqueléticas, también conocidas como músculos estriados, son tipos de tejido muscular involuntario unidos a los huesos del esqueleto por tendones. Se caracterizan por su estructura estriada o rayada, visible bajo un microscopio, que resulta de la organización regular de las miofibrillas y los sarcómeros dentro de las células musculares.
Estas fibras se contraen y relajan en respuesta a señales nerviosas para producir movimiento y mantener la postura. Están controladas por el sistema nervioso somático, lo que significa que su actividad es voluntaria y conciente.
Las fibras musculares esqueléticas se clasifican en tres tipos principales según sus propiedades funcionales y metabólicas: tipo I (lentas), tipo IIA (rápidas, intermedias) y tipo IIB (rápidas). La fibra tipo I, también llamada fibra roja o resistente a la fatiga, tiene una alta capacidad oxidativa y un suministro sanguíneo rico, lo que le permite funcionar durante períodos de tiempo más largos a bajas intensidades. Por otro lado, las fibras tipo II, también conocidas como fibras blancas o propensas a la fatiga, tienen una alta capacidad para generar fuerza y velocidad pero se cansan rápidamente porque dependen principalmente de los procesos anaeróbicos.
Las fibras musculares esqueléticas están sujetas a entrenamiento y adaptación, lo que significa que pueden cambiar sus propiedades metabólicas e histológicas en respuesta a diferentes formas de ejercicio y entrenamiento.
Los músculos, en términos médicos, se definen como tejidos contráctiles que tienen la capacidad de acortarse y endurecerse bajo el control del sistema nervioso para producir movimientos del cuerpo. También desempeñan un papel importante en mantener la postura, circulación sanguínea y respiración. Los músculos están compuestos por células especializadas llamadas fibras musculares. Hay tres tipos de músculos: esquelético (que se une a los huesos para producir movimiento), cardiaco (que forma parte del corazón) e involuntario liso (que está presente en las paredes de órganos internos como el estómago, útero y vasos sanguíneos).
Los cromosomas humanos del par 4, también conocidos como cromosomas 4, son uno de los 23 pares de cromosomas presentes en cada célula humana. Normalmente, los individuos tienen dos copias de cada cromosoma, una heredada de la madre y otra del padre. Por lo tanto, los cromosomas humanos del par 4 consisten en dos cromosomas 4 idénticos (homólogos), cada uno con aproximadamente 190 millones de pares de bases y conteniendo entre 700 y 900 genes.
Los cromosomas 4 son un poco más grandes que el promedio de los cromosomas humanos y representan alrededor del 6-7% del total del ADN contenido en las células somáticas humanas. Están involucrados en varios procesos biológicos importantes, como el desarrollo del sistema nervioso central, la homeostasis del calcio y la regulación de la expresión génica. Las anomalías cromosómicas en los cromosomas 4 pueden dar lugar a diversas condiciones genéticas y trastornos, como el síndrome de Wolf-Hirschhorn (deleción parcial del brazo corto del cromosoma 4) o la síndrome de cri du chat (deleción parcial del brazo largo del cromosoma 4).
La "lámina tipo A" no es un término médico ampliamente reconocido o utilizado en la literatura médica principal. Sin embargo, en el contexto de la anatomía patológica y la neuropatología, a veces se utiliza el término "lámina basal tipo A" para describir un tipo específico de lámina basal que se encuentra en la unión neuroepitelial-mesenquimal. Esta lámina basal es producida por las células epiteliales y proporciona una matriz extracelular especializada que ayuda en la adhesión, la comunicación y el intercambio de moléculas entre los tejidos epiteliales y mesenquimales.
El término "tipo A" se utiliza a veces para distinguir esta lámina basal de otras variedades, como la lámina basal "tipo B", que se encuentra en las uniones entre los tejidos epiteliales y musculares lisos. Sin embargo, es importante tener en cuenta que estos términos no son universalmente aceptados ni utilizados de manera consistente en toda la literatura médica.
Si está buscando información sobre un término médico diferente o más específico, por favor proporcione más detalles para que podamos ayudarlo mejor.
El complejo de proteínas asociado a la distrofina (DAPK en inglés) es un conjunto de proteínas que están involucradas en el proceso de apoptosis o muerte celular programada. Este complejo se une a la proteína distrofina, que es una proteína importante en la estabilización de las membranas de las fibras musculares y se encuentra disminuida en enfermedades como la distrofia muscular.
El DAPK desempeña un papel crucial en la regulación de la apoptosis inducida por estrés y participa en la transducción de señales que conducen a la muerte celular. La activación del DAPK puede desencadenar una cascada de eventos que llevan a la fragmentación del ADN y, finalmente, a la muerte de la célula.
La disfunción del complejo de proteínas asociado a la distrofina se ha relacionado con diversas enfermedades, incluyendo algunos tipos de cáncer y enfermedades neurodegenerativas.
La laminina es una glicoproteína abundante en la matriz extracelular, que desempeña un papel importante en la adhesión celular, la migración y la diferenciación. Es una molécula de gran tamaño con una estructura compleja, formada por tres cadenas polipeptídicas diferentes (α, β y γ). Las cadenas se unen entre sí para formar una estructura en forma de cruz, que contiene varios dominios reconocidos por receptores celulares.
La laminina es una componente clave de la membrana basal, una estructura especializada de la matriz extracelular que separa los tejidos epiteliales y conectivos. Ayuda a mantener la integridad estructural de los tejidos y desempeña un papel importante en la organización de la arquitectura tisular durante el desarrollo embrionario.
La laminina interactúa con varias moléculas de la matriz extracelular, como la fibronectina, el colágeno y la heparan sulfato proteoglicana, así como con receptores celulares como los integrines y las dystroglycanas. Estas interacciones permiten a la laminina regular una variedad de procesos celulares, incluyendo la proliferación, la supervivencia y la diferenciación celular.
En resumen, la laminina es una glicoproteína importante en la matriz extracelular que desempeña un papel crucial en la adhesión, migración y diferenciación celular, así como en el mantenimiento de la integridad estructural de los tejidos.
En genética, un exón es una sección de una molécula de ARN (ácido ribonucleico) que codifica para una proteína. Después de la transcripción del ADN a ARN, antes del procesamiento posterior del ARN, el transcrito primario contiene tanto exones como intrones. Los intrones son secuencias no codificantes que se eliminan durante el procesamiento del ARN.
Tras la eliminación de los intrones, los exones restantes se unen en una secuencia continua a través de un proceso llamado splicing o empalme. El ARN maduro resultante contiene únicamente los exones, que representan las regiones codificantes para la síntesis de proteínas.
La estructura y organización de los genes en exones e intrones permite una diversidad genética adicional, ya que diferentes combinaciones de exones (un proceso conocido como splicing alternativo) pueden dar lugar a la producción de varias proteínas a partir de un solo gen. Esto amplía el repertorio funcional del genoma y contribuye a la complejidad estructural y funcional de las proteínas en los organismos vivos.
La caveolina-3 es una proteína que se encuentra en las membranas de las caveolas, que son pequeñas invaginaciones o depresiones de la membrana plasmática encontradas en muchas células del cuerpo humano. Las caveolas y las caveolinas desempeñan un papel importante en una variedad de procesos celulares, incluyendo el tráfico intracelular, la señalización celular y la homeostasis lipídica.
La caveolina-3 es específica de los miocitos o células musculares, donde se localiza en las membranas de las caveolas de la superficie sarcolemática y del retículo sarcoplásmico. Se ha demostrado que desempeña un papel importante en la organización y funcionamiento de los canales iónicos y las bombas de la membrana muscular, así como en la regulación de la señalización celular y el metabolismo energético.
Las mutaciones en el gen que codifica para la caveolina-3 se han asociado con diversas enfermedades musculares hereditarias, incluyendo la distrofia muscular de cinturas, las miopatías nemalínicas y las miocardiopatías hipertróficas. Estas mutaciones pueden afectar a la estructura y función de las caveolas y alterar la homeostasis celular, lo que lleva a la debilidad muscular y otros síntomas clínicos.
En términos médicos, una mutación se refiere a un cambio permanente y hereditable en la secuencia de nucleótidos del ADN (ácido desoxirribonucleico) que puede ocurrir de forma natural o inducida. Esta alteración puede afectar a uno o más pares de bases, segmentos de DNA o incluso intercambios cromosómicos completos.
Las mutaciones pueden tener diversos efectos sobre la función y expresión de los genes, dependiendo de dónde se localicen y cómo afecten a las secuencias reguladoras o codificantes. Algunas mutaciones no producen ningún cambio fenotípico visible (silenciosas), mientras que otras pueden conducir a alteraciones en el desarrollo, enfermedades genéticas o incluso cancer.
Es importante destacar que existen diferentes tipos de mutaciones, como por ejemplo: puntuales (sustituciones de una base por otra), deletérreas (pérdida de parte del DNA), insercionales (adición de nuevas bases al DNA) o estructurales (reordenamientos más complejos del DNA). Todas ellas desempeñan un papel fundamental en la evolución y diversidad biológica.
Las enfermedades musculares, también conocidas como miopatías, se refieren a un grupo diverso de condiciones que afectan los músculos esqueléticos y causan debilidad, rigidez, dolor o incapacidad para relajar los músculos. Estas enfermedades pueden ser hereditarias o adquiridas.
Las miopatías hereditarias se deben a mutaciones genéticas que causan alteraciones en las proteínas musculares. Ejemplos de estas enfermedades incluyen la distrofia muscular de Duchenne y Becker, la miopatía nemalínica y la miotonia congénita.
Las miopatías adquiridas pueden ser el resultado de infecciones, trastornos autoinmunitarios, deficiencias nutricionales o efectos secundarios de ciertos medicamentos. Algunos ejemplos son la polimiositis, la dermatomiositis y la miopatía inflamatoria asociada a estatinas.
El tratamiento para las enfermedades musculares depende del tipo específico de miopatía y puede incluir fisioterapia, medicamentos para aliviar los síntomas o, en algunos casos, terapias génicas o de reemplazo de tejidos.
El cromosoma X es uno de los dos cromosomas sexuales en humanos (el otro es el cromosoma Y), que juegan un papel fundamental en la determinación del sexo. Las mujeres tienen dos cromosomas X (llamadas genotipo XX) y los hombres tienen un cromosoma X y un cromosoma Y (genotipo XY).
Los cromosomas X contienen alrededor de 155 millones de pares de bases y representan aproximadamente el 5% del ADN total en las células somáticas. Contiene entre un 1,5 y un 2 por ciento más de genes que el cromosoma Y y codifica para alrededor de 1.500 proteínas diferentes.
El cromosoma X también contiene una gran cantidad de ADN repetitivo y pseudogenes, así como regiones no codificantes reguladoras importantes que controlan la expresión génica. Además, el cromosoma X presenta un fenómeno llamado inactivación del cromosoma X, en el que uno de los dos cromosomas X se comprime y silencia en cada célula somática femenina, lo que garantiza que las mujeres expresen cantidades aproximadamente iguales de genes del cromosoma X que los hombres.
Las mutaciones en los genes del cromosoma X pueden causar una variedad de trastornos genéticos, como la hemofilia, el daltonismo y la distrofia muscular de Duchenne. Estos trastornos se denominan a menudo enfermedades ligadas al cromosoma X porque los hombres, que solo tienen un cromosoma X, tienen más probabilidades de desarrollarlas que las mujeres, quienes tienen dos copias del cromosoma X y por lo tanto una copia de respaldo en caso de que haya una mutación.
Las enfermedades neuromusculares (ENM) se refieren a un grupo diverso de condiciones clínicas que afectan los músculos y los nervios que controlan estos músculos. Estas enfermedades pueden involucrar diferentes sitios a lo largo del sistema neuromuscular, desde la corteza cerebral hasta los músculos esqueléticos periféricos.
Los dos componentes principales del sistema neuromuscular son:
1. El sistema nervioso, que incluye el cerebro, la médula espinal y los nervios periféricos. Este sistema controla y coordina las acciones musculares.
2. Los músculos esqueléticos, que son responsables del movimiento corporal.
Las ENM pueden causar debilidad, rigidez, espasticidad, fasciculaciones (contracciones musculares involuntarias), atrofia (pérdida de masa muscular) y otros síntomas relacionados con el sistema nervioso y muscular. Algunos ejemplos comunes de enfermedades neuromusculares incluyen distrofias musculares, esclerosis lateral amiotrófica (ELA), esclerosis múltiple, miastenia gravis, y neuropatías periféricas. El tratamiento y el pronóstico de estas enfermedades varían ampliamente dependiendo del tipo específico de ENM y la gravedad de la afección.
El término 'fenotipo' se utiliza en genética y medicina para describir el conjunto de características observables y expresadas de un individuo, resultantes de la interacción entre sus genes (genotipo) y los factores ambientales. Estas características pueden incluir rasgos físicos, biológicos y comportamentales, como el color de ojos, estatura, resistencia a enfermedades, metabolismo, inteligencia e inclinaciones hacia ciertos comportamientos, entre otros. El fenotipo es la expresión tangible de los genes, y su manifestación puede variar según las influencias ambientales y las interacciones genéticas complejas.
Las proteínas del citoesqueleto son un tipo de proteína que desempeñan un papel crucial en la estructura y funcionalidad de las células. Forman una red dinámica de filamentos dentro de la célula, proporcionando soporte estructural y manteniendo la forma celular. También participan en procesos celulares importantes como la división celular, el transporte intracelular y la motilidad celular.
Existen tres tipos principales de filamentos de proteínas del citoesqueleto: actina, microtúbulos y intermediate filaments (filamentos intermedios).
- Los filamentos de actina son delgados y polares, y suelen encontrarse en la periferia de la célula. Participan en procesos como el cambio de forma celular, la citocinesis (división celular) y el movimiento intracelular de vesículas y orgánulos.
- Los microtúbulos son los filamentos más grandes y rígidos. Están compuestos por tubulina y desempeñan un papel importante en la estructura celular, el transporte intracelular y la división celular. Además, forman parte de las fibras del huso durante la mitosis y son responsables del movimiento de los cromosomas.
- Los filamentos intermedios son más gruesos que los filamentos de actina pero más delgados que los microtúbulos. Existen seis tipos diferentes de filamentos intermedios, cada uno compuesto por diferentes proteínas. Estos filamentos proporcionan resistencia y rigidez a la célula, especialmente en células expuestas a estrés mecánico como las células musculares y epiteliales.
En resumen, las proteínas del citoesqueleto son un componente fundamental de la arquitectura celular, desempeñando un papel crucial en el mantenimiento de la forma celular, el transporte intracelular y la división celular.
Las distrofias neuroaxonales son un grupo de trastornos neurológicos hereditarios que afectan el crecimiento y la mantención de los axones, que son las prolongaciones nerviosas que transmiten los impulsos nerviosos en el sistema nervioso. Estos trastornos se caracterizan por la degeneración progresiva de los axones y la acumulación anormal de proteínas en ellos.
Existen varios tipos de distrofias neuroaxonales, incluyendo la enfermedad de Infantil Neuroaxonal (IN), la enfermedad de Seitelberger (también conocida como enfermedad de Seitelberger-Syndrom o SCA27), y la enfermedad de Schindler. Cada tipo tiene diferentes características clínicas, pero todos comparten la degeneración axonal y la acumulación proteica anormal.
La IN es el tipo más común de distrofia neuroaxonal y se caracteriza por la aparición en la infancia o en la niñez temprana de problemas neurológicos progresivos, como espasticidad, ataxia, disartria, y deterioro cognitivo. La enfermedad de Seitelberger es una forma rara de distrofia neuroaxonal que se caracteriza por la aparición en la infancia o en la niñez temprana de convulsiones, movimientos involuntarios anormales, y deterioro cognitivo progresivo. La enfermedad de Schindler es una forma muy rara de distrofia neuroaxonal que se caracteriza por la aparición en la infancia o en la niñez temprana de convulsiones, retraso mental, y problemas oculares progresivos.
El diagnóstico de las distrofias neuroaxonales se realiza mediante una combinación de estudios clínicos, neurológicos, genéticos y de neuroimagen. No existe cura para estos trastornos, y el tratamiento se centra en la gestión de los síntomas y en el apoyo a las personas afectadas y a sus familias.
Los Modelos Animales de Enfermedad son organismos no humanos, generalmente mamíferos o invertebrados, que han sido manipulados genéticamente o experimentalmente para desarrollar una afección o enfermedad específica, con el fin de investigar los mecanismos patofisiológicos subyacentes, probar nuevos tratamientos, evaluar la eficacia y seguridad de fármacos o procedimientos terapéuticos, estudiar la interacción gen-ambiente en el desarrollo de enfermedades complejas y entender los procesos básicos de biología de la enfermedad. Estos modelos son esenciales en la investigación médica y biológica, ya que permiten recrear condiciones clínicas controladas y realizar experimentos invasivos e in vivo que no serían éticamente posibles en humanos. Algunos ejemplos comunes incluyen ratones transgénicos con mutaciones específicas para modelar enfermedades neurodegenerativas, cánceres o trastornos metabólicos; y Drosophila melanogaster (moscas de la fruta) utilizadas en estudios genéticos de enfermedades humanas complejas.
Las sarcoglicanopatías son un grupo de enfermedades musculares degenerativas hereditarias que se caracterizan por la deficiencia o ausencia de proteínas de la membrana sarcolemal llamadas sarcoglicanos. Los sarcoglicanos son componentes importantes del complejo de distrofina-glicoproteína, el cual desempeña un papel crucial en la estabilidad y funcionalidad de las fibras musculares esqueléticas y cardiacas.
La deficiencia o ausencia de sarcoglicanos conduce a una disrupción del complejo de distrofina-glicoproteína, lo que resulta en un debilitamiento progresivo de las fibras musculares y la degeneración muscular. Las sarcoglicanopatías incluyen varias formas clínicas, como la distroglicanopatía limb-girdle (LGMD2F), la sarcoglicanopatía limb-girdle (LGMD2C) y la miopatía congénita de tipo Ullrich (UCMD).
Estas enfermedades se caracterizan por debilidad muscular progresiva, contracturas articulares, hiperlordosis y dificultad para caminar o levantarse de una silla. Algunos pacientes también pueden experimentar problemas respiratorios y cardiacos graves. El diagnóstico se realiza mediante análisis genéticos y pruebas de biopsia muscular, que muestran una disminución o ausencia de sarcoglicanos en la membrana sarcolemal.
El tratamiento actual se centra en el manejo de los síntomas y la prevención de complicaciones, como la fisioterapia, la terapia ocupacional y la asistencia respiratoria si es necesario. No existe una cura conocida para las sarcoglicanopatías, pero se están investigando posibles tratamientos dirigidos a reemplazar o aumentar los niveles de sarcoglicanos en el músculo esquelético.
El síndrome de Walker-Warburg es un trastorno genético extremadamente raro que afecta el desarrollo del cerebro y los ojos. Se caracteriza por anomalías congénitas en el cerebro, incluyendo hidrocefalia, agenesis del cuerpo calloso, lisencefalia y hemorragia intracraneal. También se observan malformaciones oculares como cataratas congénitas, glaucoma, retinosquisis y colobomas.
Este síndrome está asociado con anormalidades en la unión neuromuscular y los músculos esqueléticos, lo que resulta en debilidad muscular y retraso en el desarrollo motor. Otras características comunes incluyen rasgos dismórficos faciales, como una frente prominente, puente nasal plano, orejas bajas y separadas, y micrognatia (mandíbula pequeña).
El síndrome de Walker-Warburg es causado por mutaciones en varios genes diferentes involucrados en el desarrollo temprano del cerebro y los ojos. La mayoría de los casos se heredan de forma autosómica recesiva, lo que significa que una persona debe heredar dos copias del gen mutado (una de cada padre) para tener la enfermedad. Sin embargo, algunos casos pueden ser el resultado de nuevas mutaciones en el gen y no haber herencia familiar de la afección.
El pronóstico para las personas con síndrome de Walker-Warburg es generalmente malo, ya que la mayoría de los afectados mueren durante la infancia o la primera edad adulta. La atención médica y de apoyo pueden ayudar a mejorar la calidad de vida de las personas afectadas.
El ligamiento genético, en términos médicos, se refiere al fenómeno en el que dos o más loci (regiones específicas del ADN) en un cromosoma tienden a heredarse juntos durante la reproducción porque están demasiado próximos entre sí para ser separados por el proceso de recombinación genética. La medida de cuán a menudo se heredan juntos se expresa como una unidad llamada "unidades de mapa centimorgan" (cM), que refleja la probabilidad de recombinación entre ellos. Cuanto más cerca estén los loci uno del otro en un cromosoma, mayor será su ligamiento y menor será la probabilidad de recombinación entre ellos. Por lo tanto, el ligamiento genético proporciona información importante sobre la ubicación relativa y la organización de los genes en un cromosoma.
La calpaína es una familia de proteasas, o enzimas que descomponen proteínas, que se encuentran en varios tipos de células en los mamíferos. Hay varios tipos diferentes de calpaínas, y cada uno tiene un papel específico en el cuerpo.
Las calpaínas están involucradas en una variedad de procesos celulares, incluyendo la señalización celular, la remodelación del citoesqueleto y la apoptosis (muerte celular programada). La actividad de las calpaínas se regula cuidadosamente, ya que un desequilibrio en su actividad puede llevar a una variedad de trastornos, incluyendo enfermedades neurodegenerativas y cardiovascularas.
La activación de la calpaína requiere la presencia de calcio, por lo que los niveles elevados de calcio en las células pueden llevar a una mayor actividad de la calpaína. La inhibición de la calpaína se ha considerado como un posible objetivo terapéutico para una variedad de trastornos, incluyendo la enfermedad de Alzheimer y la insuficiencia cardíaca congestiva.
Las cardiomiopatías se refieren a enfermedades del músculo cardíaco (miocardio) que afectan su estructura y función, lo que puede llevar a insuficiencia cardíaca o arritmias. Pueden ser clasificadas en varios tipos según sus características clínicas, etiológicas y patológicas. Algunos de los tipos más comunes incluyen:
1. Cardiomiopatía hipertrófica: Es una enfermedad genética que causa engrosamiento anormal del músculo cardíaco, lo que dificulta el llenado y la eyección de sangre desde el ventrículo izquierdo.
2. Cardiomiopatía dilatada: Es una enfermedad en la cual los ventrículos se agrandan y se debilitan, lo que lleva a un deterioro progresivo de la función cardíaca. Puede ser causada por diversas condiciones, como enfermedades metabólicas, infecciosas o genéticas.
3. Cardiomiopatía restrictiva: Es una enfermedad rara que causa endurecimiento del músculo cardíaco y dificulta el llenado de las cámaras cardíacas. Puede ser causada por enfermedades del tejido conectivo, infiltración de grasa o proteínas anormales.
4. Cardiomiopatía arritmogénica del ventrículo derecho: Es una enfermedad genética que afecta el músculo cardíaco del ventrículo derecho y puede causar arritmias graves y aumentar el riesgo de muerte súbita.
El tratamiento de las cardiomiopatías depende del tipo y la gravedad de la enfermedad, y puede incluir medicamentos, dispositivos médicos como marcapasos o desfibriladores implantables, cirugía o trasplante de corazón.
En genética, los genes recesivos son aquellos que para expresar su fenotipo (característica visible) necesitan que las dos copias del gen (una heredada de cada padre) sean idénticas y exhiben este gen. Si un individuo tiene una sola copia de un gen recesivo, no mostrará el rasgo asociado con ese gen, ya que el gen dominante cubre o encubre la expresión del gen recesivo. Los genes recesivos solo se manifiestan en la ausencia de un gen dominante. Esto significa que ambos padres pueden no mostrar el rasgo fenotípico, pero aún pueden llevar y pasar el gen recesivo a su descendencia. Un ejemplo común de genes recesivos son los asociados con la enfermedad de la fibrosis quística o la anemia falciforme.
La plectina es una proteína estructural que se encuentra en el citoesqueleto de las células. No hay una definición médica específica para "plectina", pero como proteína, a veces se menciona en contextos médicos y biológicos. La plectina desempeña un papel importante en la organización y estabilidad del citoesqueleto, especialmente en la unión de diferentes componentes del citoesqueleto, como los microtúbulos y los filamentos intermedios.
Las mutaciones en el gen que codifica la plectina se han relacionado con diversas afecciones médicas, como la distrofia muscular epidérmica bullosa con acantosis queratósica (EBA-CK), una enfermedad hereditaria rara que afecta la piel y los músculos. Además, se ha sugerido que la plectina puede desempeñar un papel en el desarrollo de diversos tipos de cáncer, aunque este campo necesita más investigación.
En resumen, la plectina es una proteína estructural importante en el citoesqueleto celular que tiene implicaciones médicas en ciertas afecciones hereditarias y posiblemente en el desarrollo de cáncer.
La terapia génica es un enfoque terapéutico que consiste en introducir material genético normal y funcional en células o tejidos para compensar o reemplazar genes defectuosos o ausentes causantes de enfermedades. Esto se realiza generalmente mediante la inserción de un gen sano en un vector, como un virus no patógeno, que luego se introduce en las células del paciente.
El objetivo de la terapia génica es restablecer la expresión correcta de las proteínas necesarias para mantener la función celular normal y, por lo tanto, tratar o incluso prevenir enfermedades genéticas graves. Sin embargo, aún existen desafíos significativos en términos de eficacia, seguridad y entrega del material genético al tejido objetivo. La investigación en terapia génica continúa siendo un área activa y prometedora de la medicina moderna.
La consanguinidad es un término utilizado en genética y medicina que se refiere a la relación de parentesco entre dos personas que descienden de un antepasado común. Cuanto más reciente sea el ancestro común, mayor será el grado de consanguinidad.
La consanguinidad aumenta la probabilidad de que dos personas compartan genes recesivos para ciertas características o enfermedades, ya que tienen una mayor probabilidad de heredar los mismos alelos (variantes de un gen) de su antepasado común.
Cuando dos personas consanguíneas se aparean y tienen hijos, existe un riesgo aumentado de que sus hijos nazcan con trastornos genéticos recesivos, especialmente si los padres están emparentados en grado cercano, como primos o más cercanos.
Es importante destacar que la consanguinidad no es sinónimo de infertilidad o de malformaciones congénitas, sino que simplemente aumenta el riesgo de presentarlas. Además, existen programas de consejo genético y pruebas diagnósticas prenatales que pueden ayudar a identificar y gestionar los riesgos asociados con la consanguinidad.
No he encontrado ninguna definición médica específica o generalmente aceptada para la palabra "Conectina". Es posible que se haya confundido con el término "conectivo", que se refiere a los tejidos conectivos del cuerpo, los cuales son responsables de mantener unidas y proteger las estructuras corporales. También pueden existir otros términos similares en diferentes contextos médicos o científicos, por lo que si puedes proporcionar más información sobre el contexto en el que se utilizó este término, podré darte una respuesta más precisa y adecuada.
El término 'Desarrollo de Músculos' no está claramente definido en la literatura médica, ya que puede referirse a diferentes aspectos relacionados con los músculos. Sin embargo, generalmente se refiere al proceso de fortalecimiento y aumento del tamaño de los músculos esqueléticos a través de ejercicios físicos y entrenamientos de resistencia planificados.
Este proceso involucra la hipertrofia de las fibras musculares, que son células especializadas en la contracción y producción de fuerza. La hipertrofia ocurre cuando las fibras musculares sufren daños microscópicos durante el ejercicio intenso, lo que desencadena una respuesta de reparación y crecimiento en los músculos. Como resultado, los músculos se vuelven más grandes y fuertes.
El desarrollo muscular también puede referirse al proceso normal de maduración y crecimiento de los músculos que ocurre durante el desarrollo fetal y la infancia, así como a la rehabilitación y recuperación funcional de los músculos después de una lesión o enfermedad.
En resumen, el 'Desarrollo de Músculos' se refiere al proceso de fortalecimiento y aumento del tamaño de los músculos esqueléticos a través de ejercicios físicos y entrenamientos de resistencia planificados, así como al crecimiento y desarrollo normales de los músculos durante el desarrollo fetal y la infancia.
La regeneración, en el contexto de la medicina y biología, se refiere al proceso por el cual los tejidos dañados o perdidos en un organismo vivo son reemplazados y restaurados a su estado original y función. Esto es posible gracias a la capacidad de ciertas células de dividirse y diferenciarse en tipos celulares específicos, lo que permite la formación de nuevos tejidos.
Existen diferentes grados de regeneración en los organismos vivos. Algunos animales, como las estrellas de mar y las salamandras, tienen una capacidad excepcional para regenerar partes enteras de su cuerpo, incluyendo extremidades, órganos e incluso tejido nervioso. Por otro lado, los mamíferos, incluido el ser humano, tenemos una capacidad limitada de regeneración, especialmente en tejidos como la piel, hígado y médula ósea.
La regeneración es un área de investigación activa en la medicina regenerativa, con el objetivo de desarrollar estrategias y terapias que promuevan la capacidad natural del cuerpo para repararse a sí mismo y restablecer la función normal de los tejidos dañados o perdidos, especialmente en casos de lesiones graves, enfermedades degenerativas o envejecimiento.
La distrofia simpático-refleja (DSR) es un trastorno del sistema nervioso simpático que puede ocurrir después de una lesión nerviosa periférica. La condición se caracteriza por una serie de síntomas, incluyendo cambios en la sudoración y la temperatura, alteraciones en el flujo sanguíneo y dolor crónico.
La DSR se cree que es el resultado de una disfunción del sistema nervioso simpático, que normalmente ayuda a controlar las respuestas involuntarias del cuerpo, como la frecuencia cardíaca y la presión arterial. Después de una lesión nerviosa periférica, el sistema nervioso simpático puede entrar en un estado de sobreactivación, lo que lleva a los síntomas clásicos de la DSR.
Los síntomas más comunes de la DSR incluyen:
1. Sudoración excesiva o ausencia de sudoración en la zona afectada
2. Cambios en la temperatura de la piel, con enrojecimiento o palidez
3. Dolor crónico, que puede ser descrito como quemante, punzante o dolor neuropático
4. Hipersensibilidad al tacto o al dolor
5. Cambios en la textura y el crecimiento del cabello y las uñas en la zona afectada
6. Disminución del rango de movimiento y rigidez articular
El diagnóstico de DSR se basa en los síntomas clínicos y en pruebas especializadas, como la termografía o la sintomatología por sudoración. El tratamiento de la DSR puede ser complejo y requiere un enfoque multidisciplinario que incluye medicamentos para controlar el dolor y la sobreactivación del sistema nervioso simpático, fisioterapia y terapias complementarias, como la acupuntura o la estimulación nerviosa eléctrica transcutánea.
Las proteínas de membrana son tipos específicos de proteínas que se encuentran incrustadas en las membranas celulares o asociadas con ellas. Desempeñan un papel crucial en diversas funciones celulares, como el transporte de moléculas a través de la membrana, el reconocimiento y unión con otras células o moléculas, y la transducción de señales.
Existen tres tipos principales de proteínas de membrana: integrales, periféricas e intrínsecas. Las proteínas integrales se extienden completamente a través de la bicapa lipídica de la membrana y pueden ser permanentes (no covalentemente unidas a lípidos) o GPI-ancladas (unidas a un lipopolisacárido). Las proteínas periféricas se unen débilmente a los lípidos o a otras proteínas integrales en la superficie citoplásmica o extracelular de la membrana. Por último, las proteínas intrínsecas están incrustadas en la membrana mitocondrial o del cloroplasto.
Las proteínas de membrana desempeñan un papel vital en muchos procesos fisiológicos y patológicos, como el control del tráfico de vesículas, la comunicación celular, la homeostasis iónica y la señalización intracelular. Las alteraciones en su estructura o función pueden contribuir al desarrollo de diversas enfermedades, como las patologías neurodegenerativas, las enfermedades cardiovasculares y el cáncer.
La debilidad muscular, en términos médicos, se refiere a una reducción en la fuerza y potencia de los músculos esqueléticos. Esta condición puede afectar a uno o varios músculos y puede ser el resultado de diversas causas, que van desde problemas neuromusculares hasta enfermedades sistémicas o incluso efectos secundarios de ciertos medicamentos.
La debilidad muscular puede manifestarse como dificultad para levantar objetos, realizar movimientos precisos o mantener una postura durante un período prolongado. También puede provocar fatiga muscular temprana y dolor. En casos graves, puede interferir con las actividades diarias normales e incluso hacer que sea difícil realizar tareas simples como caminar o subir escaleras.
Es importante destacar que la debilidad muscular no debe confundirse con la fatiga, que es una sensación temporal de agotamiento después del ejercicio, aunque ambas condiciones pueden coexistir en algunas afecciones. Si experimenta debilidad muscular persistente o inexplicable, debe buscar atención médica para determinar la causa subyacente y recibir un tratamiento apropiado.
El diafragma es un músculo importante en el sistema respiratorio. Se trata de una delgada pared muscular que divide el tórax de la cavidad abdominal. Durante la inspiración, este músculo se contrae y se desplaza hacia abajo, aumentando así el volumen de la cavidad torácica y disminuyendo la presión dentro de ella. Esta disminución de presión permite que el aire fluya desde los pulmones hacia el exterior. Durante la espiración, el diafragma se relaja y asciende, reduciendo el volumen torácico y aumentando la presión, lo que favorece la entrada de aire en los pulmones. Por lo tanto, el diafragma desempeña un papel crucial en la ventilación pulmonar y en procesos como la tos o el vómito.
El análisis mutacional de ADN es un proceso de laboratorio que se utiliza para identificar cambios o alteraciones en el material genético de una persona. Este análisis puede ayudar a diagnosticar enfermedades genéticas, determinar la susceptibilidad a ciertas condiciones médicas y seguir la evolución del cáncer.
El proceso implica la secuenciación del ADN para identificar cambios en las letras que conforman el código genético. Estos cambios, o mutaciones, pueden ocurrir de forma natural o ser causados por factores ambientales, como la exposición a sustancias químicas o radiación.
El análisis mutacional de ADN puede ser utilizado en una variedad de contextos clínicos y de investigación. Por ejemplo, en oncología, el análisis mutacional de ADN se utiliza para identificar mutaciones específicas que puedan estar conduciendo al crecimiento y desarrollo del cáncer. Esta información puede ayudar a los médicos a seleccionar tratamientos más efectivos y personalizados para cada paciente.
En genética clínica, el análisis mutacional de ADN se utiliza para diagnosticar enfermedades genéticas raras y complejas que pueden ser difíciles de identificar mediante otros métodos. El análisis puede ayudar a determinar si una persona ha heredado una mutación específica que aumenta su riesgo de desarrollar una enfermedad genética.
En resumen, el análisis mutacional de ADN es una técnica de laboratorio que se utiliza para identificar cambios en el material genético de una persona. Este análisis puede ayudar a diagnosticar enfermedades genéticas, determinar la susceptibilidad a ciertas condiciones médicas y seguir la evolución del cáncer.
La fuerza muscular, en términos médicos, se refiere a la capacidad máxima de un músculo o grupo de músculos para ejercer una fuerza contra una resistencia externa en una contracción muscular voluntaria. Se mide generalmente en newtons (N) o libras-fuerza (lbf). Es un componente importante de la fuerza, el poder y la resistencia física. La evaluación de la fuerza muscular es común en la práctica clínica y en la investigación para medir el estado funcional, la salud ósea, el riesgo de caídas y lesiones, y la respuesta al entrenamiento de resistencia. También desempeña un papel fundamental en la realización de las actividades diarias e influye en la calidad de vida general.
La Distrofia Macular Viteliforme es una condición rara y genéticamente heterogénea que afecta a la mácula, la parte central de la retina responsable de la visión aguda y el procesamiento de los detalles visuales. Esta enfermedad ocular se caracteriza por la presencia de una lesión amarillenta, similar a un huevo frito (viteliforme), en la mácula. La distrofia macular viteliforme puede manifestarse en diferentes formas y grados de severidad, dependiendo de la edad del individuo y el tipo específico de distrofia macular viteliforme que tenga.
Existen dos tipos principales de Distrofia Macular Viteliforme:
1. La Forma Juvenil (tipo 2) o Distrofia Macular Viteliforme No-Familial, que generalmente se presenta en la infancia o adolescencia y puede progresar lentamente a lo largo de la vida. Los síntomas pueden incluir visión borrosa, distorsiones visuales y dificultad para leer o reconocer rostros.
2. La Forma Adulta (tipo 1) o Distrofia Macular Viteliforme Familiar, que se presenta en individuos de mediana edad o mayores y puede ser hereditaria. Esta forma es a menudo más leve y progresa más lentamente que la forma juvenil.
La Distrofia Macular Viteliforme se diagnostica mediante un examen oftalmológico completo, incluyendo una evaluación de la agudeza visual, el examen del fondo de ojo y, en algunos casos, pruebas adicionales como la tomografía de coherencia óptica (OCT) y la electroretinografía (ERG). Actualmente no existe un tratamiento específico para esta afección; sin embargo, el seguimiento regular con un oftalmólogo especializado en retina puede ayudar a monitorizar el progreso de la enfermedad y brindar asesoramiento sobre posibles opciones de manejo y tratamientos sintomáticos.
Los Datos de Secuencia Molecular se refieren a la información detallada y ordenada sobre las unidades básicas que componen las moléculas biológicas, como ácidos nucleicos (ADN y ARN) y proteínas. Esta información está codificada en la secuencia de nucleótidos en el ADN o ARN, o en la secuencia de aminoácidos en las proteínas.
En el caso del ADN y ARN, los datos de secuencia molecular revelan el orden preciso de las cuatro bases nitrogenadas: adenina (A), timina/uracilo (T/U), guanina (G) y citosina (C). La secuencia completa de estas bases proporciona información genética crucial que determina la función y la estructura de genes y proteínas.
En el caso de las proteínas, los datos de secuencia molecular indican el orden lineal de los veinte aminoácidos diferentes que forman la cadena polipeptídica. La secuencia de aminoácidos influye en la estructura tridimensional y la función de las proteínas, por lo que es fundamental para comprender su papel en los procesos biológicos.
La obtención de datos de secuencia molecular se realiza mediante técnicas experimentales especializadas, como la reacción en cadena de la polimerasa (PCR), la secuenciación de ADN y las técnicas de espectrometría de masas. Estos datos son esenciales para la investigación biomédica y biológica, ya que permiten el análisis de genes, genomas, proteínas y vías metabólicas en diversos organismos y sistemas.
En genética, un heterocigoto se refiere a un individuo que tiene dos alelos diferentes en un par de genes específicos. Cada persona hereda un alelo de cada uno de sus padres para cada gen, y en el caso de un heterocigoto, esos dos alelos son distintos entre sí.
Esto quiere decir que el individuo tiene una combinación única de características genéticas provenientes de ambos padres. Los heterocigotos pueden manifestar rasgos o enfermedades genéticas dependiendo del tipo de alelos que haya heredado y de cómo interactúen entre sí.
Un ejemplo común es el gen responsable del color de los ojos. Algunas personas pueden ser heterocigotas para este gen, heredando un alelo que determina el color de ojos marrón y otro que determina el color de ojos azul. En este caso, el individuo tendrá los ojos de un color intermedio como verde o avellana.
El mapeo cromosómico es un proceso en genética molecular que se utiliza para determinar la ubicación y orden relativo de los genes y marcadores genéticos en un cromosoma. Esto se realiza mediante el análisis de las frecuencias de recombinación entre estos marcadores durante la meiosis, lo que permite a los genetistas dibujar un mapa de la posición relativa de estos genes y marcadores en un cromosoma.
El mapeo cromosómico se utiliza a menudo en la investigación genética para ayudar a identificar los genes que contribuyen a enfermedades hereditarias y otros rasgos complejos. También se puede utilizar en la medicina forense para ayudar a identificar individuos o determinar la relación entre diferentes individuos.
Existen diferentes tipos de mapeo cromosómico, incluyendo el mapeo físico y el mapeo genético. El mapeo físico implica la determinación de la distancia física entre los marcadores genéticos en un cromosoma, medida en pares de bases. Por otro lado, el mapeo genético implica la determinación del orden y distancia relativa de los genes y marcadores genéticos en términos del número de recombinaciones que ocurren entre ellos durante la meiosis.
En resumen, el mapeo cromosómico es una técnica importante en genética molecular que se utiliza para determinar la ubicación y orden relativo de los genes y marcadores genéticos en un cromosoma, lo que puede ayudar a identificar genes asociados con enfermedades hereditarias y otros rasgos complejos.
La miositis es un término general que se refiere a la inflamación de los músculos, y puede incluir varias condiciones específicas. La forma más común de miositis es la miositis idiopática, que incluye la dermatomiomitis y la polimiositis. Estas afecciones se caracterizan por debilidad muscular progresiva y dolor, y a menudo se asocian con erupciones cutáneas en el caso de la dermatomiositis. Otras formas de miositis incluyen la miositis por cuerpos de inclusión, que afecta predominantemente a los ancianos, y la miositis ocasionalmente asociada con infecciones, medicamentos o traumatismos. El tratamiento de la miositis generalmente implica el uso de corticosteroides o inmunosupresores para controlar la inflamación y prevenir daños adicionales en los tejidos musculares. La fisioterapia y el ejercicio también pueden ser útiles para mantener la fuerza y la movilidad muscular.
La miostatina es una proteína que se produce naturalmente en el cuerpo y actúa como un inhibidor de la formación y crecimiento del músculo esquelético. Es producida por células en los tejidos musculares y envía señales a otras células musculares para evitar que se dividan y crezcan demasiado, manteniendo así el equilibrio entre la formación de músculo y su descomposición.
La miostatina pertenece a una clase de proteínas llamadas factores de transformación del crecimiento beta (TGF-β), que son responsables de regular diversos procesos biológicos, incluyendo el crecimiento celular y la diferenciación. La miostatina es codificada por el gen MSTN en humanos.
Las mutaciones en el gen MSTN pueden dar lugar a un aumento anormal en el crecimiento muscular, lo que ha llevado al interés en la miostatina como una posible diana terapéutica para tratar enfermedades musculares degenerativas y mejorar el rendimiento físico. Sin embargo, también se han planteado preocupaciones éticas sobre el uso de inhibidores de la miostatina para mejorar el rendimiento deportivo.
Los ratones consanguíneos C57BL, también conocidos como ratones de la cepa C57BL o C57BL/6, son una cepa inbred de ratones de laboratorio que se han utilizado ampliamente en la investigación biomédica. La designación "C57BL" se refiere al origen y los cruces genéticos específicos que se utilizaron para establecer esta cepa particular.
La letra "C" indica que el ratón es de la especie Mus musculus, mientras que "57" es un número de serie asignado por el Instituto Nacional de Estándares y Tecnología (NIST) en los Estados Unidos. La "B" se refiere al laboratorio original donde se estableció la cepa, y "L" indica que fue el laboratorio de Little en la Universidad de Columbia.
Los ratones consanguíneos C57BL son genéticamente idénticos entre sí, lo que significa que tienen el mismo conjunto de genes en cada célula de su cuerpo. Esta uniformidad genética los hace ideales para la investigación biomédica, ya que reduce la variabilidad genética y facilita la comparación de resultados experimentales entre diferentes estudios.
Los ratones C57BL son conocidos por su resistencia a ciertas enfermedades y su susceptibilidad a otras, lo que los hace útiles para el estudio de diversas condiciones médicas, como la diabetes, las enfermedades cardiovasculares, el cáncer y las enfermedades neurológicas. Además, se han utilizado ampliamente en estudios de genética del comportamiento y fisiología.
En genética, un gen dominante es aquel que produce y manifesta sus características fenotípicas, incluso si el individuo solo hereda una copia del gen. Esto significa que el gen dominante se expresa en la presencia de al menos una sola copia, ya sea en forma paterna o materna. Un rasgo dominante se manifiesta en la primera generación filial (F1) incluso cuando un individuo portador se apareó con un individuo que no tiene el gen en cuestión.
Un ejemplo clásico de genes dominantes es el gen de la afección conocida como síndrome de Huntington. Si una persona hereda solo una copia del gen defectuoso de este trastorno neurodegenerativo, todavía desarrollará los síntomas asociados con la enfermedad.
Sin embargo, es importante tener en cuenta que el término "dominante" no implica necesariamente que un rasgo sea más fuerte o potente que su contraparte recesiva. Simplemente significa que se necesita solo una copia del gen para expresar el rasgo.
En la anatomía humana, las láminas se refieren a finas placas planas o capas de tejido. Pueden estar compuestas por diferentes tipos de tejidos, dependiendo de su ubicación y función en el cuerpo. Algunos ejemplos comunes de láminas incluyen:
1. Lámina basal: Una delgada capa de tejido conectivo que recubre las células epiteliales en la superficie externa del cuerpo y los órganos internos. Sirve como una barrera protectora y ayuda en la adhesión de las células epiteliales al tejido subyacente.
2. Lámina dura: Una capa densa y dura de tejido conectivo que forma el revestimiento externo del cráneo y los huesos de la oreja. Está compuesta principalmente por fibras de colágeno y minerales como el calcio.
3. Lámina cribosa: Una capa delgada y porosa de tejido conectivo que forma parte de la estructura de los huesos etmoides en el cráneo. Contiene pequeños agujeros a través de los cuales pasan los nervios olfatorios desde la nariz hasta el cerebro.
4. Lámina intermedia: Una capa delgada de tejido conectivo que se encuentra entre los músculos lisos en la pared del tracto gastrointestinal y otras estructuras huecas. Ayuda a dividir los músculos y proporciona soporte estructural.
5. Lámina fascia: Una capa delgada de tejido conectivo que recubre los músculos, tendones, nervios y vasos sanguíneos. Ayuda a reducir la fricción entre estas estructuras y facilita su deslizamiento una sobre otra.
En resumen, las láminas son finas capas o planchas de tejido conectivo que desempeñan diversas funciones en el cuerpo humano, como proporcionar soporte estructural, reducir la fricción y facilitar el paso de nervios y vasos sanguíneos.
Las células satélite del músculo esquelético son células madre adultas que se encuentran en reposo junto a las fibras musculares esqueléticas. Están adheridas a la membrana basal de los miofibrillas y desempeñan un papel crucial en el crecimiento, reparación y regeneración del tejido muscular dañado.
Las células satélite permanecen inactivas durante el desarrollo y mantenimiento normal del músculo; sin embargo, cuando ocurre una lesión o daño en la fibra muscular, se activan y proliferan, diferenciándose posteriormente en mioblastos. Estos mioblastos pueden fusionarse entre sí o con fibras musculares existentes para formar nuevas células musculares y promover la reparación y regeneración del tejido dañado.
La capacidad de las células satélite para dividirse y diferenciarse en células musculares las convierte en un objetivo importante en el campo de la medicina regenerativa y terapias celulares, particularmente en el tratamiento de enfermedades musculares degenerativas y lesiones musculoesqueléticas.
Los Dependovirus, también conocidos como Virus dependientes del gen E6 o E7 de los Papilomavirus humanos (HPV), son un grupo de virus que pertenecen a la familia de los *Parvoviridae* y al género *Dependoparvovirus*. Estos virus requieren la expresión de ciertas proteínas virales, como E6 y E7 en el caso de los HPV, para poder replicarse y sobrevivir. No son capaces de completar su ciclo de vida sin la ayuda de estas proteínas, que a menudo son proporcionadas por otros virus con los que coexisten. Los Dependovirus pueden infectar una variedad de células huésped y tienen el potencial de causar enfermedades en humanos y animales.
La secuencia de bases, en el contexto de la genética y la biología molecular, se refiere al orden específico y lineal de los nucleótidos (adenina, timina, guanina y citosina) en una molécula de ADN. Cada tres nucleótidos representan un codón que especifica un aminoácido particular durante la traducción del ARN mensajero a proteínas. Por lo tanto, la secuencia de bases en el ADN determina la estructura y función de las proteínas en un organismo. La determinación de la secuencia de bases es una tarea central en la genómica y la biología molecular moderna.
Una mutación missense es un tipo específico de mutación en el ADN que causa la sustitución de un solo nucleótido (la unidad básica de los genes), lo que resulta en la producción de un aminoácido diferente en la proteína codificada. Esta alteración puede tener diversos efectos en la función de la proteína, dependiendo de dónde ocurra y cuán crucial sea el aminoácido reemplazado.
En algunos casos, una mutación missense podría no afectar significativamente la función de la proteína, especialmente si el aminoácido original y el nuevo son químicamente similares. Sin embargo, cuando el cambio ocurre en un dominio crucial de la proteína o involucra aminoácidos con propiedades químicas muy diferentes, esto puede conducir a una pérdida total o parcial de la función de la proteína.
Las mutaciones missense pueden asociarse con diversas enfermedades genéticas, dependiendo del gen y la proteína afectados. Por ejemplo, algunas mutaciones missense en el gen BRCA1 aumentan el riesgo de cáncer de mama y ovario hereditario.
La inmunohistoquímica es una técnica de laboratorio utilizada en patología y ciencias biomédicas que combina los métodos de histología (el estudio de tejidos) e inmunología (el estudio de las respuestas inmunitarias del cuerpo). Consiste en utilizar anticuerpos marcados para identificar y localizar proteínas específicas en células y tejidos. Este método se utiliza a menudo en la investigación y el diagnóstico de diversas enfermedades, incluyendo cánceres, para determinar el tipo y grado de una enfermedad, así como también para monitorizar la eficacia del tratamiento.
En este proceso, se utilizan anticuerpos específicos que reconocen y se unen a las proteínas diana en las células y tejidos. Estos anticuerpos están marcados con moléculas que permiten su detección, como por ejemplo enzimas o fluorocromos. Una vez que los anticuerpos se unen a sus proteínas diana, la presencia de la proteína se puede detectar y visualizar mediante el uso de reactivos apropiados que producen una señal visible, como un cambio de color o emisión de luz.
La inmunohistoquímica ofrece varias ventajas en comparación con otras técnicas de detección de proteínas. Algunas de estas ventajas incluyen:
1. Alta sensibilidad y especificidad: Los anticuerpos utilizados en esta técnica son altamente específicos para las proteínas diana, lo que permite una detección precisa y fiable de la presencia o ausencia de proteínas en tejidos.
2. Capacidad de localizar proteínas: La inmunohistoquímica no solo detecta la presencia de proteínas, sino que también permite determinar su localización dentro de las células y tejidos. Esto puede ser particularmente útil en el estudio de procesos celulares y patológicos.
3. Visualización directa: La inmunohistoquímica produce una señal visible directamente en el tejido, lo que facilita la interpretación de los resultados y reduce la necesidad de realizar análisis adicionales.
4. Compatibilidad con microscopía: Los métodos de detección utilizados en la inmunohistoquímica son compatibles con diferentes tipos de microscopía, como el microscopio óptico y el microscopio electrónico, lo que permite obtener imágenes detalladas de las estructuras celulares e intracelulares.
5. Aplicabilidad en investigación y diagnóstico: La inmunohistoquímica se utiliza tanto en la investigación básica como en el diagnóstico clínico, lo que la convierte en una técnica versátil y ampliamente aceptada en diversos campos de estudio.
Sin embargo, la inmunohistoquímica también presenta algunas limitaciones, como la necesidad de disponer de anticuerpos específicos y de alta calidad, la posibilidad de obtener resultados falsos positivos o negativos debido a reacciones no específicas, y la dificultad para cuantificar con precisión los niveles de expresión de las proteínas en el tejido. A pesar de estas limitaciones, la inmunohistoquímica sigue siendo una técnica poderosa y ampliamente utilizada en la investigación y el diagnóstico de diversas enfermedades.
La cardiomiopatía dilatada es una afección del músculo cardíaco (miocardio) en la cual el corazón se agranda y se vuelve más débil. La cavidad del ventrículo izquierdo, la cámara principal de bombeo del corazón, se dilata o se estira y no puede bombear sangre con fuerza suficiente.
Esto puede llevar a insuficiencia cardíaca, en la que el corazón no puede abastecer al cuerpo con la cantidad de sangre y oxígeno necesarios. La cardiomiopatía dilatada también puede desencadenar arritmias (latidos irregulares del corazón) y embolia sistémica, que es cuando un coágulo viaja a otras partes del cuerpo y bloquea una arteria.
La cardiomiopatía dilatada puede ser causada por diversos factores, como enfermedades genéticas, infecciones virales, trastornos metabólicos, uso prolongado de alcohol o cocaína, exposición a tóxicos y estrés extremo. En algunos casos, la causa es desconocida y se denomina idiopática.
El tratamiento puede incluir medicamentos para mejorar la función cardíaca y controlar los síntomas de insuficiencia cardíaca, como diuréticos y inhibidores de la enzima convertidora de angiotensina (IECA). También pueden ser necesarios dispositivos médicos, como un desfibrilador automático implantable (DAI) o un asistente ventricular izquierdo. En casos graves, puede ser necesario un trasplante cardíaco.
La electrorretinografía (ERG) es un procedimiento diagnóstico que mide la respuesta eléctrica de las células fotorreceptoras en la retina (los conos y bastones) cuando son expuestas a una variedad de estímulos luminosos. Esto proporciona información sobre el funcionamiento fisiológico de la retina y puede ayudar a diagnosticar y monitorear diversas condiciones o enfermedades oculares, como la retinopatía diabética, degeneración macular relacionada con la edad (DMAE), distrofias de los conos y bastones, y otras neuropatías ópticas.
Durante el procedimiento, se coloca un electrodo en el ojo del paciente (generalmente después de instilar gotas anestésicas) para registrar la actividad eléctrica generada por las células fotorreceptoras y otras células retinianas. Luego, se presentan diferentes patrones de luz a los ojos del paciente mientras se registra la respuesta eléctrica. Los resultados del ERG se analizan para evaluar la función visual y detectar posibles anomalías.
En definitiva, la electrorretinografía es una herramienta importante en el campo de la oftalmología para evaluar y comprender el funcionamiento de la retina y diagnosticar diversas afecciones oculares.
Los ratones transgénicos son un tipo de roedor modificado geneticamente que incorpora un gen o secuencia de ADN exógeno (procedente de otro organismo) en su genoma. Este proceso se realiza mediante técnicas de biología molecular y permite la expresión de proteínas específicas, con el fin de estudiar sus funciones, interacciones y efectos sobre los procesos fisiológicos y patológicos.
La inserción del gen exógeno se lleva a cabo generalmente en el cigoto (óvulo fecundado) o en embriones tempranos, utilizando métodos como la microinyección, electroporación o virus vectoriales. Los ratones transgénicos resultantes pueden manifestar características particulares, como resistencia a enfermedades, alteraciones en el desarrollo, crecimiento o comportamiento, según el gen introducido y su nivel de expresión.
Estos modelos animales son ampliamente utilizados en la investigación biomédica para el estudio de diversas enfermedades humanas, como cáncer, diabetes, enfermedades cardiovasculares, neurológicas y otras patologías, con el objetivo de desarrollar nuevas terapias y tratamientos más eficaces.
La atrofia muscular es un término médico que se refiere al deterioro y disminución del tamaño de los músculos esqueléticos. Esta afección puede ser causada por una variedad de factores, como la inactividad física prolongada, lesiones nerviosas, enfermedades neuromusculares o trastornos hormonales.
La atrofia muscular se produce cuando los músculos no reciben suficientes estímulos para mantenerse fuertes y saludables. Con el tiempo, los músculos pueden volverse más débiles, flácidos y menos eficaces en su función. Los síntomas de la atrofia muscular pueden incluir debilidad, fatiga, pérdida de tono muscular, movimientos lentos y torpes, y dificultad para realizar actividades cotidianas.
Existen diferentes tipos de atrofia muscular, cada uno con causas y patrones de progresión distintos. Algunos tipos pueden ser reversibles si se tratan a tiempo, mientras que otros pueden ser permanentes o incluso progresivos. El tratamiento de la atrofia muscular depende de su causa subyacente y puede incluir fisioterapia, ejercicios de rehabilitación, terapia ocupacional, medicamentos o cirugía.
La Expansión de Repetición de Trinucleótido (ERT) es un tipo de mutación genética donde una secuencia específica de tres nucleótidos (basados en ADN) se repite y el número de repeticiones es aumentado considerablemente. Este tipo de mutación ocurre más comúnmente en regiones no codificantes del gen, aunque también pueden ocurrir dentro de genes. Cuando estas expansiones ocurren dentro de genes, pueden perturbar la función normal del gen y llevar a una variedad de enfermedades hereditarias. Las ERT se asocian con más de 40 trastornos neurológicos y neuromusculares, incluyendo distrofia miotónica tipo 1 y ataxia espinocerebelosa tipo 1. El tamaño del número de repeticiones puede aumentar de generación en generación, lo que a veces resulta en una edad más temprana de inicio o una gravedad más severa de la enfermedad en individuos descendientes.
Las células musculares, también conocidas como miocitos, son las células especializadas en la contracción y relajación para producir movimiento y fuerza. Existen tres tipos principales de células musculares: esqueléticas, lisas e cardíacas.
1. Células musculares esqueléticas: Son las más grandes y abundantes en el cuerpo humano. Se unen entre sí para formar fascículos y fibras musculares. Están controladas por el sistema nervioso somático y se encargan de los movimientos voluntarios del cuerpo. Cada fibra muscular esquelética contiene varios núcleos celulares y está compuesta por miofibrillas, que son largas estructuras proteicas responsables de la contracción muscular.
2. Células musculares lisas: Se encuentran en la pared de los órganos huecos y tubos del cuerpo, como los vasos sanguíneos, el tracto gastrointestinal, y los bronquios. Estas células musculares son involuntarias, lo que significa que su contracción y relajación no están bajo control consciente. Se encargan de movimientos como la digestión, la circulación sanguínea y la excreción. Las células musculares lisas individuales tienen un solo núcleo celular y son más cortas y delgadas que las fibras musculares esqueléticas.
3. Células musculares cardíacas: Son células involuntarias que forman el músculo cardíaco o miocardio. Se encuentran en el corazón y son responsables de sus latidos regulares y ritmos. Las células musculares cardíacas tienen un solo núcleo celular y están conectadas entre sí por desmosomas y uniones comunicantes, lo que les permite coordinar su actividad contráctil.
En general, las células musculares se caracterizan por presentar miofilamentos (actina y miosina) y sarcolema (membrana celular), además de tener una alta capacidad contráctil gracias a la presencia del sarcómero.
Los trastornos miotónicos son un grupo de afecciones neuromusculares hereditarias que se caracterizan por la presencia de rigidez y debilidad muscular. Estos trastornos reciben su nombre porque involucran una anormalidad en la relajación de los músculos después de la contracción, un fenómeno conocido como miotonía.
La miotonía puede causar síntomas tales como espasmos musculares, rigidez y dificultad para relajar los músculos después de usarlos. Por ejemplo, una persona con un trastorno miotónico podría tener dificultad para soltar un apretón o abrir los dedos después de agarrar un objeto.
Existen varios tipos de trastornos miotónicos, cada uno con diferentes patrones de herencia, síntomas y gravedad. Algunos de los trastornos miotónicos más comunes incluyen la distrofia miotónica de Steiner (también conocida como distrofia miotónica de tipo 1) y la paramiotonía congénita (también conocida como distrofia miotónica de tipo 2).
La distrofia miotónica de Steiner es el trastorno miotónico más común y afecta a aproximadamente 1 de cada 8.000 personas. Los síntomas de este trastorno suelen comenzar en la edad adulta y pueden incluir debilidad muscular, rigidez, miotonía, cataratas, problemas cardíacos y alteraciones endocrinas.
La paramiotonía congénita es un trastorno miotónico menos común que afecta principalmente a los músculos faciales y del cuello. Los síntomas de este trastorno suelen comenzar en la infancia o la adolescencia y pueden incluir rigidez, espasmos musculares y dificultad para mover los músculos faciales y del cuello.
Ambos trastornos miotónicos se heredan de forma autosómica dominante, lo que significa que una copia del gen anormal es suficiente para causar la enfermedad. Si una persona hereda el gen anormal, existe un 50% de probabilidades de que transmita el trastorno a sus hijos.
No existe cura para los trastornos miotónicos, pero se pueden realizar tratamientos para aliviar los síntomas y mejorar la calidad de vida de las personas afectadas. Estos tratamientos pueden incluir fisioterapia, terapia ocupacional, medicamentos para aliviar la rigidez y los espasmos musculares, y cirugía para tratar los problemas cardíacos o las cataratas.
En genética, el término "homocigoto" se refiere a un individuo que ha heredado dos alelos idénticos para un gen determinado, uno de cada padre. Esto significa que ambos alelos de los dos cromosomas homólogos en un par de cromosomas son iguales. Puede ocurrir que esos dos alelos sean la misma variante alélica normal (llamada también wild type), o bien dos copias de una variante alélica patológica (como en una enfermedad genética). El término contrario a homocigoto es heterocigoto, que se refiere a un individuo que ha heredado dos alelos diferentes para un gen determinado.
La glicosilación es un proceso bioquímico fundamental que ocurre en células vivas, donde se agregan cadenas de carbohidratos a proteínas o lípidos. Es el proceso más común de modificación postraduccional de proteínas en células eucariotas y también ocurre en procariotas.
En la glicosilación, los glúcidos (azúcares) se unen a las moléculas de proteína para formar glicoproteínas o a lípidos para formar glicolípidos. Estas modificaciones pueden influir en la estructura tridimensional, la función y la estabilidad de las proteínas, y desempeñan un papel crucial en una variedad de procesos biológicos, como el plegamiento de proteínas, el tráfico intracelular, la reconocimiento celular, la señalización celular y la interacción proteína-proteína.
Hay dos tipos principales de glicosilación: N-glicosilación y O-glicosilación. La N-glicosilación se produce en el grupo amida del carbono α-aspartato o glutamato de un residuo de asparagina (Asn-X-Ser/Thr, donde X no es Pro) en la secuencia de aminoácidos de una proteína. Por otro lado, la O-glicosilación se produce en el grupo hidroxilo (-OH) de los residuos de serina o treonina en las proteínas.
La glicosilación incorrecta o anormal ha sido vinculada a diversas enfermedades, como la fibrosis quística, la enfermedad de Pompe, el síndrome de West y varios trastornos neurodegenerativos y cánceres. Por lo tanto, comprender los mecanismos moleculares de la glicosilación es fundamental para desarrollar estrategias terapéuticas efectivas para tratar tales enfermedades.
Los mioblastos esqueléticos son células musculares embrionarias que se diferencian en miofibroblastos y finalmente en miócitos, que son las células musculares maduras del tejido esquelético. Los mioblastos contienen varios núcleos y, a medida que se diferencian, fusionan sus citoplasmas para formar largas fibras multinucleadas llamadas miofibrillas. Estas miofibrillas son responsables de la capacidad contráctil del tejido muscular esquelético. Los mioblastos también desempeñan un papel importante en la reparación y regeneración del tejido muscular dañado en adultos, dividiéndose y diferenciándose para reemplazar las células musculares dañadas o muertas.
La forma MM de la creatina quinasa (CK) es una isoenzima específica de la enzima creatina kinasa, que se encuentra principalmente en el músculo esquelético. La creatina kinasa es responsable de catalizar la reversible transferencia de fosfato desde ATP a creatina para producir ADP y fosfocreatina. Esta reacción está involucrada en la regeneración de ATP durante períodos de intensa actividad muscular.
La forma MM de la creatina kinasa se compone de dos subunidades idénticas de cadena media (M), y es producida por el gen CKM. Esta isoenzima es particularmente abundante en el músculo esquelético, especialmente en los tipos de fibras musculares de contracción rápida.
Las alteraciones en los niveles séricos de la forma MM de la creatina kinasa pueden utilizarse como un marcador bioquímico para detectar y monitorizar daños musculoesqueléticos, como los que ocurren en lesiones musculares, distrofia muscular y miopatías. Los niveles elevados de esta isoenzima en la sangre pueden indicar una lesión aguda o crónica del músculo esquelético.
Las manosiltransferasas son un tipo específico de enzimas (más concretamente, transferasas) que transfieren un grupo manosa desde un dónor, típicamente una molécula de doliquilo-fosfato manosa, a un aceptor, como puede ser una proteína o un glicano. Estas enzimas desempeñan un papel crucial en la modificación postraduccional de proteínas y en la síntesis de glucanos complejos.
Existen diferentes clases de manosiltransferasas, cada una con su propia especificidad de sustrato y función biológica. Algunas de ellas participan en la formación de estructuras de glicanos en la superficie celular, mientras que otras intervienen en el procesamiento y la calidad del control de proteínas. Las disfunciones en estas enzimas se han relacionado con diversas patologías, incluyendo trastornos congénitos del metabolismo y enfermedades neurodegenerativas.
Una biopsia es un procedimiento médico en el que se extrae una pequeña muestra de tejido corporal para ser examinada en un laboratorio. Este procedimiento se realiza con el fin de evaluar si el tejido extraído presenta signos de enfermedad, como cáncer o inflamación.
Existen diferentes tipos de biopsias, dependiendo de la ubicación y el método utilizado para obtener la muestra de tejido. Algunas de las más comunes incluyen:
1. Biopsia por aspiración con aguja fina (FNA): se utiliza una aguja delgada y hueca para extraer células o líquido del bulto o área sospechosa.
2. Biopsia por punción con aguja gruesa (CNB): se emplea una aguja más grande para obtener una muestra de tejido sólido.
3. Biopsia incisional: se realiza una pequeña incisión en la piel y se extrae una parte del tejido sospechoso.
4. Biopsia excisional: se extirpa todo el bulto o área anormal, junto con una porción de tejido normal circundante.
Los resultados de la biopsia suelen ser evaluados por un patólogo, quien determinará si el tejido muestra signos de enfermedad y, en caso afirmativo, qué tipo de enfermedad es. La información obtenida de una biopsia puede ayudar a guiar el tratamiento médico y proporcionar información importante sobre la gravedad y extensión de la enfermedad.
Las N-acetilglucosaminiltransferasas (GNGTs) son un tipo de enzimas transferasas que desempeñan un papel crucial en la modificación postraduccional de proteínas y la formación de glucanos. Estas enzimas transfieren un residuo de N-acetilglucosamina (GlcNAc) desde una molécula donadora, como el UDP-GlcNAc, a un grupo hidroxilo específico en un aceptor proteico o glúcido.
Existen varios tipos de N-acetilglucosaminiltransferasas, cada uno con su propia función y sustrato preferido. Algunas de estas enzimas participan en la formación del complejo de bridas (o glicanos) en las proteínas, que desempeñan un papel importante en diversos procesos celulares, como la señalización intracelular, el tráfico vesicular y la adhesión celular.
Las alteraciones en la actividad de estas enzimas se han relacionado con varias enfermedades, como el cáncer y los trastornos neurodegenerativos. Por lo tanto, comprender su función y regulación puede ayudar a desarrollar nuevas estrategias terapéuticas para tratar estas condiciones.
Nota: La definición médica puede ser complicada debido al lenguaje técnico utilizado. Si necesita una explicación más sencilla, no dude en preguntar.
El diagnóstico prenatal es un proceso médico que consiste en determinar las condiciones de salud, anomalías congénitas o trastornos cromosómicos del feto antes de su nacimiento. Esto se logra mediante diversas pruebas y procedimientos realizados durante el embarazo. Los métodos más comunes incluyen análisis de sangre materna, ecografías, amniocentesis y muestras de vellosidades coriónicas. El diagnóstico prenatal puede ayudar a los padres a tomar decisiones informadas sobre el curso del embarazo, prepararse para cuidados especiales que pueda necesitar el bebé después del nacimiento o, en casos graves, considerar la interrupción del embarazo. Sin embargo, es importante tener en cuenta que no todos los problemas de salud se pueden detectar antes del nacimiento y que obtener un resultado anormal no siempre significa que el feto está afectado.
En toxicología y farmacología, la frase "ratones noqueados" (en inglés, "mice knocked out") se refiere a ratones genéticamente modificados que han tenido uno o más genes "apagados" o "noqueados", lo que significa que esos genes específicos ya no pueden expresarse. Esto se logra mediante la inserción de secuencias génicas específicas, como un gen marcador y un gen de resistencia a antibióticos, junto con una secuencia que perturba la expresión del gen objetivo. La interrupción puede ocurrir mediante diversos mecanismos, como la inserción en el medio de un gen objetivo, la eliminación de exones cruciales o la introducción de mutaciones específicas.
Los ratones noqueados se utilizan ampliamente en la investigación biomédica para estudiar las funciones y los roles fisiológicos de genes específicos en diversos procesos, como el desarrollo, el metabolismo, la respuesta inmunitaria y la patogénesis de enfermedades. Estos modelos ofrecen una forma poderosa de investigar las relaciones causales entre los genes y los fenotipos, lo que puede ayudar a identificar nuevas dianas terapéuticas y comprender mejor los mecanismos moleculares subyacentes a diversas enfermedades.
Sin embargo, es importante tener en cuenta que el proceso de creación de ratones noqueados puede ser complicado y costoso, y que la eliminación completa o parcial de un gen puede dar lugar a fenotipos complejos y potencialmente inesperados. Además, los ratones noqueados pueden tener diferentes respuestas fisiológicas en comparación con los organismos que expresan el gen de manera natural, lo que podría sesgar o limitar la interpretación de los resultados experimentales. Por lo tanto, es crucial considerar estas limitaciones y utilizar métodos complementarios, como las técnicas de edición génica y los estudios con organismos modelo alternativos, para validar y generalizar los hallazgos obtenidos en los ratones noqueados.
En términos médicos, un síndrome se refiere a un conjunto de signos y síntomas que ocurren juntos y pueden indicar una condición particular o enfermedad. Los síndromes no son enfermedades específicas por sí mismos, sino más bien una descripción de un grupo de características clínicas.
Un síndrome puede involucrar a varios órganos y sistemas corporales, y generalmente es el resultado de una combinación de factores genéticos, ambientales o adquiridos. Algunos ejemplos comunes de síndromes incluyen el síndrome de Down, que se caracteriza por retraso mental, rasgos faciales distintivos y problemas de salud congénitos; y el síndrome metabólico, que implica una serie de factores de riesgo cardiovascular como obesidad, diabetes, presión arterial alta e hiperlipidemia.
La identificación de un síndrome a menudo ayuda a los médicos a hacer un diagnóstico más preciso y a desarrollar un plan de tratamiento apropiado para el paciente.
La degeneración retiniana es un término general que se refiere a un grupo de condiciones o enfermedades que involucran el daño y la muerte progresiva de las células fotorreceptoras en la retina, la parte posterior del ojo responsable de capturar la luz e iniciar el proceso visual. Existen dos tipos principales de células fotorreceptoras: los conos, que son responsables de la visión central y del color, y los bastones, que se encargan de la visión periférica y la visión nocturna.
La degeneración retiniana puede afectar a ambos tipos de células fotorreceptoras o solo a uno de ellos. La forma más común de degeneración retiniana es la enfermedad de déficit de vitamina A, también conocida como deficiencia de retinol, que afecta principalmente a los bastones y puede causar ceguera nocturna.
Sin embargo, el término "degeneración retiniana" se utiliza con mayor frecuencia para referirse a una enfermedad hereditaria progresiva llamada degeneración macular relacionada con la edad (DMAE), que afecta principalmente a las personas mayores de 50 años. La DMAE se caracteriza por el daño y muerte de los fotorreceptores en una pequeña área de la retina llamada mácula, responsable de la visión central y detallada. Esto puede conducir a la pérdida progresiva de la visión central y la distorsión de las líneas rectas, lo que dificulta realizar tareas cotidianas como leer, conducir o reconocer rostros.
Otro tipo de degeneración retiniana es la neuropatía óptica hereditaria de Leber (NOHL), una enfermedad mitocondrial hereditaria que afecta principalmente a los jóvenes y provoca la pérdida repentina e irreversible de la visión central.
La degeneración retiniana puede ser causada por diversos factores, como mutaciones genéticas, envejecimiento, exposición a la luz azul o al humo del tabaco, y el tratamiento depende del tipo y gravedad de la enfermedad. En algunos casos, se pueden utilizar dispositivos de baja visión, terapia con células madre o trasplantes de retina para mejorar la visión.
Las glicoproteínas de membrana son moléculas complejas formadas por un componente proteico y un componente glucídico (o azúcar). Se encuentran en la membrana plasmática de las células, donde desempeñan una variedad de funciones importantes.
La parte proteica de la glicoproteína se sintetiza en el retículo endoplásmico rugoso y el aparato de Golgi, mientras que los glúcidos se adicionan en el aparato de Golgi. La porción glucídica de la molécula está unida a la proteína mediante enlaces covalentes y puede estar compuesta por varios tipos diferentes de azúcares, como glucosa, galactosa, manosa, fucosa y ácido sialico.
Las glicoproteínas de membrana desempeñan un papel crucial en una variedad de procesos celulares, incluyendo la adhesión celular, la señalización celular, el transporte de moléculas a través de la membrana y la protección de la superficie celular. También pueden actuar como receptores para las hormonas, los factores de crecimiento y otros mensajeros químicos que se unen a ellas e inician una cascada de eventos intracelulares.
Algunas enfermedades están asociadas con defectos en la síntesis o el procesamiento de glicoproteínas de membrana, como la enfermedad de Pompe, la enfermedad de Tay-Sachs y la fibrosis quística. El estudio de las glicoproteínas de membrana es importante para comprender su función normal y los mecanismos patológicos que subyacen a estas enfermedades.
Los morfolinos, también conocidos como oligómeros de morfolino fosforodiamidato, son pequeñas moléculas sintéticas de cadena abierta que se utilizan en la investigación biomédica y bioquímica. Están diseñados para unirse específicamente a secuencias de ARNm (ácido ribonucleico mensajero) y bloquear su traducción, es decir, impedir que se convierta en proteínas.
La estructura química de los morfolinos consiste en un esqueleto de cadena de fosforodiamidato con grupos laterales de morfolina unidos a cada átomo de carbono en la cadena principal. Estos grupos laterales pueden ser modificados para conferir propiedades específicas a las moléculas, como una mayor estabilidad o una mayor afinidad de unión al ARNm objetivo.
Los morfolinos se utilizan en diversas aplicaciones, como el estudio de la función génica, el análisis de vías de señalización celular y el desarrollo de terapias antisentido para enfermedades genéticas. Sin embargo, su uso en humanos está limitado debido a preocupaciones sobre su toxicidad y distribución in vivo.
El asesoramiento genético es una comunicación interactiva y educativa entre un profesional de la salud entrenado en genética y un individuo, su familia o grupo social, que se realiza para ayudar a comprender los aspectos genéticos de las enfermedades. El proceso incluye una evaluación médica e historial familiar, educación sobre la enfermedad hereditaria, discusión de las opciones de diagnóstico y manejo, discusión del riesgo de recurrencia o transmisión, y asesoramiento psicológico y social. El objetivo es ayudar a las personas a tomar decisiones informadas sobre su salud y la de sus familias.
La Reacción en Cadena de la Polimerasa, generalmente conocida como PCR (Polymerase Chain Reaction), es un método de bioquímica molecular que permite amplificar fragmentos específicos de DNA (ácido desoxirribonucleico). La técnica consiste en una serie de ciclos de temperatura controlada, donde se produce la separación de las hebras de DNA, seguida de la síntesis de nuevas hebras complementarias usando una polimerasa (enzima que sintetiza DNA) y pequeñas moléculas de DNA llamadas primers, específicas para la región a amplificar.
Este proceso permite obtener millones de copias de un fragmento de DNA en pocas horas, lo que resulta útil en diversos campos como la diagnóstica molecular, criminalística, genética forense, investigación genética y biotecnología. En el campo médico, se utiliza ampliamente en el diagnóstico de infecciones virales y bacterianas, detección de mutaciones asociadas a enfermedades genéticas, y en la monitorización de la respuesta terapéutica en diversos tratamientos.
La oxepina es un compuesto heterocíclico que contiene un anillo de siete miembros con un átomo de oxígeno y seis átomos de carbono. No es una sustancia médica en sí misma, pero forma la base estructural de algunos fármacos y compuestos químicos utilizados en medicina.
En un contexto médico, el término "oxepinas" podría referirse a los fármacos que contienen este anillo heterocíclico en su estructura. Algunos ejemplos de tales fármacos incluyen certaines clases de antibióticos y antifúngicos, como las oxepinonas y las oxepininas, respectivamente. Estos compuestos han demostrado tener actividad biológica y se están investigando para su uso en el tratamiento de diversas afecciones médicas.
Sin embargo, es importante destacar que la definición médica específica de "oxepinas" puede variar dependiendo del contexto y la aplicación, y siempre debe consultarse con un profesional médico o químico especializado para obtener información precisa y actualizada.
La mutación del sistema de lectura no es un término médico ampliamente reconocido o utilizado en la literatura clínica o científica. Sin embargo, en el contexto de la neurobiología y la psicolingüística, se puede referir a una alteración en el sistema cerebral responsable del procesamiento del lenguaje escrito.
Este sistema incluye varias regiones cerebrales que trabajan juntas para permitir la lectura, como las áreas de Broca y Wernicke en el lóbulo temporal y parietal respectivamente. Una mutación en este sistema podría significar una disfunción o alteración en alguna de estas regiones debido a una lesión cerebral, un trastorno neurológico o una enfermedad mental, lo que podría resultar en dificultades para leer.
Sin embargo, es importante destacar que el término 'mutación' generalmente se utiliza en el contexto de la genética para referirse a un cambio en la secuencia de ADN. Por lo tanto, su uso en este contexto podría ser inapropiado o confuso. Los trastornos específicos del procesamiento del lenguaje escrito se describen mejor utilizando términos más precisos y ampliamente aceptados, como dislexia.
Las cadenas alfa de integrinas son un tipo de subunidad proteica que forma parte de la familia de receptores celulares conocidos como integrinas. Las integrinas desempeñan un papel crucial en la adhesión celular, la señalización celular y el tráfico intracelular.
Las cadenas alfa de integrinas se combinan con cadenas beta correspondientes para formar heterodímeros funcionales que reconocen y se unen a diversos ligandos situados en la matriz extracelular, como la fibronectina, la colágena y la laminina. Existen varios tipos de cadenas alfa de integrinas (por ejemplo, α1, α2, α3, etc.) que se combinan con un conjunto más limitado de cadenas beta (por ejemplo, β1, β2, β3) para formar diferentes heterodímeros con diversas especificidades de ligando y funciones.
Las cadenas alfa de integrinas contienen un dominio extracelular que interactúa con el ligando, un dominio transmembrana y una cola citoplasmática que participa en la señalización celular y la interacción con la estructura del citoesqueleto. Las mutaciones o alteraciones en las cadenas alfa de integrinas se han relacionado con diversas afecciones médicas, como trastornos del desarrollo, enfermedades inflamatorias y cáncer.
Un codón sin sentido, también conocido como codón nonsense o codón de terminación, es una secuencia específica de tres nucleótidos en el ARN mensajero (ARNm) que señala el final de la síntesis de una proteína. Durante el proceso de traducción, los ribosomas leen el ARNm en tramos de tres nucleótidos, o codones, y utilizan esta información para unir aminoácidos específicos y sintetizar una cadena polipeptídica. Cuando el ribosoma alcanza un codón sin sentido, como UAG, UAA o UGA, se detiene la traducción y se libera la nueva proteína. Las mutaciones que convierten un codón de sentido en un codón sin sentido pueden resultar en la producción de una proteína truncada o no funcional, lo que a su vez puede causar enfermedades genéticas.
La progesterona es en realidad la hormona steroide principal que se clasifica como pregnenolona. La pregnenolona es una hormona esteroidea precursora que se sintetiza a partir del colesterol en el mitocondria de las células de la glándula suprarrenal y el ovario. Es el precursor común para la biosíntesis de otras hormonas esteroides, incluyendo los glucocorticoides, mineralocorticoides, androgénicos y estrógenos. La progesterona es una hormona importante en el ciclo menstrual femenino y durante el embarazo.
Así que, en resumen, las pregnenolonas son una clase de hormonas esteroides que desempeñan un papel vital en la producción de otras hormonas importantes en el cuerpo humano.
El azul de Evans es un colorante vital utilizado en la medicina para examinar el sistema linfático y los vasos sanguíneos. Se inyecta en el tejido celular subcutáneo y luego se observa la circulación del tinte a través de los vasos linfáticos. El color azul distintivo del tinte ayuda a los médicos a visualizar y evaluar el flujo linfático y la anatomía de los vasos linfáticos. También se puede usar en cirugías reconstructivas para identificar los vasos linfáticos durante la operación. El uso del azul de Evans está aprobado por la FDA y generalmente se considera seguro, aunque pueden ocurrir reacciones alérgicas u otros efectos secundarios adversos en raras ocasiones.
Las pruebas genéticas son procedimientos diagnósticos que examinan los genes, el ADN y el material cromosómico para identificar cambios específicos o variantes relacionadas con enfermedades hereditarias. Estas pruebas pueden ayudar a confirmar un diagnóstico, determinar la probabilidad de desarrollar una enfermedad genética, identificar portadores de determinados rasgos genéticos, establecer el riesgo de transmisión a la descendencia y guiar los planes de tratamiento.
Existen diferentes tipos de pruebas genéticas, como:
1. Pruebas de diagnóstico genético: Se utilizan para identificar cambios específicos en genes o cromosomas que causan o aumentan el riesgo de desarrollar una enfermedad hereditaria. Estas pruebas suelen realizarse después del nacimiento y pueden ayudar a confirmar un diagnóstico clínico.
2. Pruebas prenatales: Se llevan a cabo durante el embarazo para detectar posibles anomalías cromosómicas o genéticas en el feto. Algunas pruebas prenatales, como la amniocentesis y la biopsia de vellosidades coriónicas, analizan directamente las células fetales; otras, como el análisis de ADN fetal libre en sangre materna, detectan fragmentos de ADN fetal presentes en la sangre de la madre.
3. Pruebas predictivas: Se utilizan para identificar variantes genéticas que aumentan el riesgo de desarrollar enfermedades genéticas en personas sin síntomas clínicos. Estas pruebas pueden ayudar a tomar decisiones informadas sobre la prevención, el diagnóstico y el tratamiento temprano.
4. Pruebas de detección de portadores: Se emplean para identificar individuos que no presentan síntomas pero que pueden transmitir una enfermedad genética a sus hijos. Estas pruebas suelen realizarse en parejas que deseen tener hijos y tienen antecedentes familiares de ciertas enfermedades hereditarias.
5. Pruebas farmacogenéticas: Analizan variantes genéticas relacionadas con la respuesta a determinados fármacos, lo que permite personalizar los tratamientos médicos y minimizar los efectos adversos.
En conclusión, existen diferentes tipos de pruebas genéticas que se adaptan a diversas situaciones clínicas y objetivos preventivos. Es fundamental contar con un profesional especializado en genética para interpretar correctamente los resultados y ofrecer una asesoría adecuada a cada paciente.
Retinitis Pigmentosa es un grupo de enfermedades genéticas que afectan a las células fotorreceptoras (bastones y conos) en la retina, la parte luminosa del ojo que contiene células sensibles a la luz que transmiten señales al cerebro para ayudarlo a interpretar imágenes.
La característica distintiva de esta afección es la pérdida progresiva de visión periférica (visión lateral) y la capacidad de ver en condiciones de poca luz, conocidas como "noche ciega". Esto ocurre porque el tipo de células fotorreceptoras más afectadas son los bastones, que nos ayudan a ver en condiciones de poca luz.
La enfermedad generalmente comienza en la infancia o adolescencia y gradualmente empeora con el tiempo, aunque la velocidad de progresión puede variar mucho entre diferentes personas e incluso entre los dos ojos del mismo individuo. En etapas avanzadas, la pérdida de visión central también puede ocurrir, lo que puede conducir a la ceguera total en algunos casos.
Hasta ahora, no existe cura para la retinitis pigmentosa, pero los tratamientos pueden ayudar a retrasar su progresión y mejorar la calidad de vida de las personas afectadas. Estos tratamientos incluyen dispositivos de bajo visionado, terapia con vitaminas A y posibles terapias génicas en investigación.
Las miopatías distales son un grupo de trastornos neuromusculares que se caracterizan por debilidad y atrofia muscular selectiva en los músculos distales, es decir, aquellos más lejos de los troncos nerviosos centrales. A diferencia de las miopatías proximales, que afectan principalmente a los músculos cercanos al torso, como los hombros y las caderas, las miopatías distales afectan a los músculos de las extremidades más distantes, como los antebrazos, las manos, los muslos y los pies.
Las miopatías distales pueden ser hereditarias o adquiridas. Las formas hereditarias suelen presentarse en la infancia o la adolescencia y pueden estar asociadas con mutaciones genéticas que afectan a las proteínas musculares estructurales o metabólicas. Por otro lado, las formas adquiridas pueden ser el resultado de diversas causas, como infecciones, enfermedades autoinmunes o exposición a toxinas.
Los síntomas de las miopatías distales varían según la forma específica de la enfermedad y pueden incluir debilidad muscular progresiva, calambres, rigidez, contracturas y dificultad para realizar movimientos finos con las manos o los pies. El diagnóstico suele requerir una evaluación clínica detallada, pruebas de laboratorio y estudios de imagenología muscular, como la electromiografía y la resonancia magnética nuclear.
El tratamiento de las miopatías distales depende del tipo específico de enfermedad y puede incluir fisioterapia, terapia ocupacional, dispositivos de asistencia y, en algunos casos, medicamentos para aliviar los síntomas o ralentizar la progresión de la enfermedad. La investigación en el campo de las enfermedades neuromusculares sigue avanzando y ofrece esperanza para el desarrollo de nuevas terapias y tratamientos más efectivos en el futuro.
La membrana nuclear, en términos médicos y biológicos, se refiere a la doble capa lipídica que rodea el núcleo de una célula eucariota. Esta estructura compleja desempeña un papel crucial en el control del tráfico molecular entre el núcleo y el citoplasma, ya que regula la entrada y salida de ARN mensajero (ARNm), ARN ribosómico (ARNr), ARN de transferencia (ARNt) y diversas proteínas.
La membrana nuclear está compuesta por dos capas: la membrana externa, que está asociada con el retículo endoplásmico rugoso, y la membrana interna, que está en contacto directo con el nucleoplasma o matriz nuclear. Estas dos membranas están separadas por un espacio llamado espacio perinuclear, donde se encuentran los poros nucleares, que permiten el paso selectivo de moléculas entre el núcleo y el citoplasma.
La membrana nuclear desempeña un papel fundamental en la preservación de la integridad genómica, ya que ayuda a proteger el material genético de los posibles daños o interacciones indeseables con moléculas presentes en el citoplasma. Además, también participa en la organización del cromosoma y la expresión génica al permitir la comunicación entre las diversas estructuras subnucleares y el citoplasma.
La Discapacidad Intelectual, según la American Association of Intellectual and Developmental Disabilities (AAIDD), se define como "una discapacidad caracterizada por limitaciones significativas en las habilidades intelectuales y en los comportamientos adaptativos que cubren muchas habilidades de la vida diaria, tales como la comunicación, la autogestión, las relaciones sociales y la participación escolar o laboral. La discapacidad intelectual origina durante el desarrollo del individuo, antes de los 18 años de edad".
Esta definición incluye tanto aspectos cognitivos como adaptativos, y subraya la importancia de considerar el contexto en el que vive la persona para evaluar y entender sus limitaciones y capacidades. Además, establece que la discapacidad intelectual se manifiesta durante el desarrollo temprano, lo que diferencia esta condición de otras que pueden ocurrir más tarde en la vida como consecuencia de una lesión cerebral adquirida o una enfermedad degenerativa.
La Western blotting, también conocida como inmunoblotting, es una técnica de laboratorio utilizada en biología molecular y bioquímica para detectar y analizar proteínas específicas en una muestra compleja. Este método combina la electroforesis en gel de poliacrilamida (PAGE) con la transferencia de proteínas a una membrana sólida, seguida de la detección de proteínas objetivo mediante un anticuerpo específico etiquetado.
Los pasos básicos del Western blotting son:
1. Electroforesis en gel de poliacrilamida (PAGE): Las proteínas se desnaturalizan, reducen y separan según su tamaño molecular mediante la aplicación de una corriente eléctrica a través del gel de poliacrilamida.
2. Transferencia de proteínas: La proteína separada se transfiere desde el gel a una membrana sólida (generalmente nitrocelulosa o PVDF) mediante la aplicación de una corriente eléctrica constante. Esto permite que las proteínas estén disponibles para la interacción con anticuerpos.
3. Bloqueo: La membrana se bloquea con una solución que contiene leche en polvo o albumina séricade bovino (BSA) para evitar la unión no específica de anticuerpos a la membrana.
4. Incubación con anticuerpo primario: La membrana se incuba con un anticuerpo primario específico contra la proteína objetivo, lo que permite la unión del anticuerpo a la proteína en la membrana.
5. Lavado: Se lavan las membranas para eliminar el exceso de anticuerpos no unidos.
6. Incubación con anticuerpo secundario: La membrana se incuba con un anticuerpo secundario marcado, que reconoce y se une al anticuerpo primario. Esto permite la detección de la proteína objetivo.
7. Visualización: Las membranas se visualizan mediante una variedad de métodos, como quimioluminiscencia o colorimetría, para detectar la presencia y cantidad relativa de la proteína objetivo.
La inmunoblotting es una técnica sensible y específica que permite la detección y cuantificación de proteínas individuales en mezclas complejas. Es ampliamente utilizado en investigación básica y aplicada para estudiar la expresión, modificación postraduccional y localización de proteínas.
La "eliminación de gen" no es un término médico ampliamente reconocido o utilizado en la literatura médica. Sin embargo, dado que en el contexto proporcionado puede referirse al proceso de eliminar o quitar un gen específico durante la investigación genética o la edición de genes, aquí está una definición relacionada:
La "eliminación de gen" o "gen knockout" es un método de investigación genética que involucra la eliminación intencional de un gen específico de un organismo, con el objetivo de determinar su función y el papel en los procesos fisiológicos. Esto se logra mediante técnicas de ingeniería genética, como la inserción de secuencias de ADN que interrumpen o reemplazan el gen diana, lo que resulta en la producción de una proteína no funcional o ausente. Los organismos con genes knockout se utilizan comúnmente en modelos animales para estudiar enfermedades y desarrollar terapias.
Tenga en cuenta que este proceso también puede denominarse "gen knockout", "knocking out a gene" o "eliminación génica".
Los marcadores genéticos, en términos médicos, se definen como segmentos específicos de ADN con características conocidas y heredables que sirven como puntos de referencia en el genoma. A diferencia de los genes, los marcadores genéticos no codifican proteínas ni influyen directamente en los rasgos o características de un individuo.
En su lugar, los marcadores genéticos son útiles para identificar y localizar genes asociados con enfermedades u otras características heredadas. Estos marcadores tienden a encontrarse en regiones cercanas al gen de interés en el cromosoma, por lo que un cambio en el marcador genético puede estar vinculado a un cambio en el gen asociado con una enfermedad particular.
Existen varios tipos de marcadores genéticos, incluyendo polimorfismos de longitud de fragmentos de restricción (RFLP), microsatélites o simple tandem repeats (STRs), y variantes de nucleótido único (SNVs). Estos marcadores se utilizan ampliamente en la investigación genética, como el mapeo genético, la asignación de parentesco y la identificación forense.
La secuencia de aminoácidos se refiere al orden específico en que los aminoácidos están unidos mediante enlaces peptídicos para formar una proteína. Cada proteína tiene su propia secuencia única, la cual es determinada por el orden de los codones (secuencias de tres nucleótidos) en el ARN mensajero (ARNm) que se transcribe a partir del ADN.
Las cadenas de aminoácidos pueden variar en longitud desde unos pocos aminoácidos hasta varios miles. El plegamiento de esta larga cadena polipeptídica y la interacción de diferentes regiones de la misma dan lugar a la estructura tridimensional compleja de las proteínas, la cual desempeña un papel crucial en su función biológica.
La secuencia de aminoácidos también puede proporcionar información sobre la evolución y la relación filogenética entre diferentes especies, ya que las regiones conservadas o similares en las secuencias pueden indicar una ascendencia común o una función similar.
La contracción muscular es el proceso en el que los músculos se acortan y endurecen al contraerse, lo que genera fuerza y produce movimiento. Esta acción es controlada por el sistema nervioso y ocurre cuando las células musculares, conocidas como fibras musculares, se estimulan para que se muevan.
Hay tres tipos principales de contracciones musculares: isotónicas, isométricas y auxotónicas.
1. Las contracciones isotónicas ocurren cuando los músculos se acortan mientras producen fuerza y el objeto que están moviendo cambia de posición. Hay dos tipos de contracciones isotónicas: concéntricas y excéntricas. En una contracción concéntrica, el músculo se acorta y produce movimiento, como cuando levantas una pesa. Por otro lado, en una contracción excéntrica, el músculo se alarga mientras resiste la fuerza, como cuando bajas lentamente la pesa para controlar su descenso.
2. Las contracciones isométricas ocurren cuando los músculos se tensan y producen fuerza sin que haya cambio en la longitud del músculo ni movimiento del objeto. Un ejemplo de esto es empujar contra un objeto inamovible, como una pared.
3. Las contracciones auxotónicas son una combinación de isotónicas y isométricas, en las que el músculo se acorta mientras resiste la fuerza. Un ejemplo de esto es levantar un peso mientras te paras sobre una superficie inestable, como una pelota de equilibrio.
La contracción muscular también puede clasificarse en voluntaria e involuntaria. Las contracciones voluntarias son controladas conscientemente por el cerebro y el sistema nervioso central, mientras que las contracciones involuntarias son automáticas y no requieren control consciente.
La capacidad de los músculos para contraerse y relajarse es fundamental para la movilidad y el funcionamiento adecuado del cuerpo. Las lesiones, enfermedades o trastornos que afectan la contracción muscular pueden causar debilidad, rigidez, dolor y otros síntomas que impacten negativamente en la calidad de vida.
La edad de inicio, en el contexto médico, se refiere a la edad en la que comienzan a presentarse los síntomas de una enfermedad o trastorno por primera vez. Es un término utilizado frecuentemente en pediatría y psiquiatría, donde la edad de inicio puede desempeñar un papel importante en el diagnóstico, el pronóstico y el plan de tratamiento. Por ejemplo, ciertos trastornos del neurodesarrollo, como el autismo o el trastorno por déficit de atención con hiperactividad (TDAH), suelen presentarse en la infancia, por lo que su edad de inicio es antes de los 6 a 12 años. Del mismo modo, algunos trastornos mentales, como la esquizofrenia, pueden manifestarse por primera vez en la adolescencia o al comienzo de la edad adulta, lo que indica una edad de inicio más tardía. Es importante tener en cuenta que la edad de inicio puede variar ampliamente entre los individuos y entre diferentes trastornos o enfermedades.
Las caveolinas son proteínas específicas que se encuentran en la membrana de las caveolas, un tipo especializado de invaginaciones lipídicas presentes en la membrana plasmática de muchas células. Las caveolas tienen un papel importante en diversos procesos celulares, como el tráfico intracelular, la señalización celular y el control del metabolismo.
Las caveolinas son pequeñas proteínas integrales de membrana que se unen a los esfingolípidos y a la cavina, una proteína estructural de las caveolas. Existen tres isoformas de caveolinas en humanos (Cav1, Cav2 y Cav3), cada una con diferentes distribuciones tisulares y funcionales específicas.
La caveolina-1 (Cav1) es la isoforma más estudiada y se expresa en muchos tipos de células, como células endoteliales, fibroblastos y células musculares lisas. Cav1 está involucrada en la formación y estabilidad de las caveolas, así como en la regulación de diversos procesos celulares, como la endocitosis mediada por caveolas, la transducción de señales y el control del crecimiento celular.
La caveolina-2 (Cav2) se expresa principalmente en células epiteliales y neuronales, donde forma heteroóligos con Cav1 para estabilizar las caveolas. La caveolina-3 (Cav3) es específica del músculo esquelético y cardiaco, donde desempeña un papel importante en la organización de las miocitias y el control de la contractilidad muscular.
Las caveolinas también se han relacionado con diversas enfermedades humanas, como el cáncer, las enfermedades cardiovasculares y las neurodegenerativas. Por ejemplo, mutaciones en los genes que codifican para las caveolinas se han asociado con enfermedades musculares hereditarias, como la distrofia muscular de cinturas y la miopatía nemalínica. Además, alteraciones en la expresión y localización de las caveolinas se han observado en diversos tipos de cáncer, lo que sugiere un papel potencial de estas proteínas en el desarrollo y progresión del cáncer.
La Epidermólisis Ampollosa Simple (EAS) es un término general que se utiliza para describir un grupo de trastornos genéticos hereditarios de la piel, caracterizados por una fragilidad cutánea que provoca formación de ampollas con facilidad, especialmente en áreas de roce o fricción. La palabra "simple" se utiliza para distinguir esta forma de Epidermólisis Ampollosa de otras formas más graves y complejas.
La EAS se debe a mutaciones en genes que codifican proteínas importantes para la integridad estructural de la unión dermo-epidérmica, es decir, el punto de unión entre la capa externa de la piel (epidermis) y la capa subyacente (dermis). Estas mutaciones causan una disminución en la adhesión entre estas dos capas, haciendo que la piel sea más frágil y susceptible a las ampollas.
Los síntomas de la EAS suelen presentarse al nacer o durante los primeros meses de vida e incluyen:
1. Formación espontánea o inducida por traumatismos de ampollas en áreas de piel expuesta a fricción, como palmas de las manos, plantas de los pies, rodillas y codos.
2. Ampollas que pueden infectarse y formar costras o úlceras.
3. Cicatrización lenta de las ampollas y cicatrices atróficas (depresiones en la piel).
4. Blanquecinas o translúcidas, sin pigmento ni vellosidad.
5. Pueden haber uñas frágiles, onduladas o congénitamente ausentes.
6. En algunos casos, se pueden presentar complicaciones en los ojos, como sequedad ocular y úlceras corneales.
El diagnóstico de la EAS se realiza mediante una biopsia cutánea y el examen histopatológico de la muestra. El tratamiento consiste en el cuidado de las heridas, la prevención de infecciones y la protección de la piel contra traumatismos. En algunos casos, se pueden utilizar medicamentos para aliviar el dolor o tratar las complicaciones infecciosas. La fisioterapia y la logopedia también pueden ser necesarias en caso de afectación de músculos o articulaciones o dificultades en la alimentación y el habla.
La eliminación en secuencia, también conocida como "sequential elimination" en inglés, no es un término médico específico que se utilice generalmente en el campo de la medicina. Sin embargo, en algunos contextos clínicos especializados, particularmente en estudios de farmacología y toxicología, se puede referir a una serie de pruebas o procedimientos eliminatorios realizados en un orden específico para identificar o descartar la presencia de sustancias tóxicas, fármacos u otras moléculas de interés.
En este contexto, la eliminación secuencial implica el uso de diferentes métodos analíticos y técnicas de prueba, cada uno con diferentes grados de especificidad y sensibilidad, para reducir gradualmente las posibilidades de identificar la sustancia en cuestión. Esto puede ser útil en situaciones en las que se sospecha una intoxicación o exposición a una variedad de sustancias y es necesario priorizar los análisis y las intervenciones terapéuticas.
Sin embargo, fuera de este contexto específico, la eliminación en secuencia no tiene una definición médica generalmente aceptada.
La regulación de la expresión génica en términos médicos se refiere al proceso por el cual las células controlan la activación y desactivación de los genes para producir los productos genéticos deseados, como ARN mensajero (ARNm) y proteínas. Este proceso intrincado involucra una serie de mecanismos que regulan cada etapa de la expresión génica, desde la transcripción del ADN hasta la traducción del ARNm en proteínas. La complejidad de la regulación génica permite a las células responder a diversos estímulos y entornos, manteniendo así la homeostasis y adaptándose a diferentes condiciones.
La regulación de la expresión génica se lleva a cabo mediante varios mecanismos, que incluyen:
1. Modificaciones epigenéticas: Las modificaciones químicas en el ADN y las histonas, como la metilación del ADN y la acetilación de las histonas, pueden influir en la accesibilidad del gen al proceso de transcripción.
2. Control transcripcional: Los factores de transcripción son proteínas que se unen a secuencias específicas de ADN para regular la transcripción de los genes. La activación o represión de estos factores de transcripción puede controlar la expresión génica.
3. Interferencia de ARN: Los microARN (miARN) y otros pequeños ARN no codificantes pueden unirse a los ARNm complementarios, lo que resulta en su degradación o traducción inhibida, disminuyendo así la producción de proteínas.
4. Modulación postraduccional: Las modificaciones químicas y las interacciones proteína-proteína pueden regular la actividad y estabilidad de las proteínas después de su traducción, lo que influye en su función y localización celular.
5. Retroalimentación negativa: Los productos génicos pueden interactuar con sus propios promotores o factores reguladores para reprimir su propia expresión, manteniendo así un equilibrio homeostático en la célula.
El control de la expresión génica es fundamental para el desarrollo y la homeostasis de los organismos. Las alteraciones en este proceso pueden conducir a diversas enfermedades, como el cáncer y las enfermedades neurodegenerativas. Por lo tanto, comprender los mecanismos que regulan la expresión génica es crucial para desarrollar estrategias terapéuticas efectivas para tratar estas afecciones.
Las células cultivadas, también conocidas como células en cultivo o células in vitro, son células vivas que se han extraído de un organismo y se están propagando y criando en un entorno controlado, generalmente en un medio de crecimiento especializado en un plato de petri o una flaska de cultivo. Este proceso permite a los científicos estudiar las células individuales y su comportamiento en un ambiente controlado, libre de factores que puedan influir en el organismo completo. Las células cultivadas se utilizan ampliamente en una variedad de campos, como la investigación biomédica, la farmacología y la toxicología, ya que proporcionan un modelo simple y reproducible para estudiar los procesos fisiológicos y las respuestas a diversos estímulos. Además, las células cultivadas se utilizan en terapias celulares y regenerativas, donde se extraen células de un paciente, se les realizan modificaciones genéticas o se expanden en número antes de reintroducirlas en el cuerpo del mismo individuo para reemplazar células dañadas o moribundas.
Los ratones mutantes son animales de laboratorio que han sufrido alguna alteración en su genoma, provocando así una o más modificaciones en sus características y comportamiento. Estas modificaciones pueden ser espontáneas o inducidas intencionalmente por diversos métodos, como la exposición a radiaciones ionizantes, agentes químicos o mediante técnicas de manipulación genética directa, como el empleo de sistemas de recombinación homóloga o CRISPR-Cas9.
Los ratones mutantes se utilizan ampliamente en la investigación biomédica para entender los mecanismos moleculares y celulares implicados en diversas enfermedades, así como para probar nuevas terapias y fármacos. Un ejemplo clásico es el ratón "knockout", en el que se ha inactivado un gen específico para estudiar su función. De esta forma, los científicos pueden analizar los efectos de la pérdida o ganancia de determinadas funciones génicas en un organismo vivo y obtener información relevante sobre los procesos patológicos y fisiológicos en mamíferos.
El miocardio es el tejido muscular involucrado en la contracción del corazón para impulsar la sangre a través del cuerpo. Es la capa más gruesa y potente del músculo cardíaco, responsable de la función de bombeo del corazón. El miocardio se compone de células musculares especializadas llamadas cardiomiocitos, que están dispuestas en un patrón entrelazado para permitir la contracción sincronizada y eficiente del músculo cardíaco. Las enfermedades que dañan o debilitan el miocardio pueden provocar insuficiencia cardíaca, arritmias u otras afecciones cardiovasculares graves.
Una mutación puntual es un tipo específico de mutación genética que involucra el cambio o alteración de un solo nucleótido (base) en el ADN. Esta pequeña variación puede resultar en un cambio en el aminoácido codificado, lo que se conoce como una sustitución de aminoácidos. Existen dos tipos principales de mutaciones puntuales: las transiciones y las transversiones.
- Transiciones: Son los cambios de una purina (Adenina o Guanina) a otra purina, o de una pirimidina (Timina o Citosina) a otra pirimidina. Por ejemplo, un cambio de A (Adenina) a G (Guanina), o de T (Timina) a C (Citosina).
- Transversiones: Son los cambios de una purina a una pirimidina, o viceversa. Por ejemplo, un cambio de A (Adenina) a T (Timina) o de G (Guanina) a C (Citosina).
Las mutaciones puntuales pueden tener diversos efectos sobre la función y estructura de las proteínas. Algunas no tienen ningún impacto significativo, mientras que otras pueden alterar la actividad enzimática, estabilidad de la proteína o incluso llevar a la producción de una proteína truncada e infuncional. Las mutaciones puntuales son importantes en el estudio de la genética y la evolución, ya que pueden conducir a cambios fenotípicos y ser la base de la divergencia genética entre especies.
La cardiotoxicidad se refiere a la capacidad de una sustancia para dañar el músculo cardíaco y/o el sistema de conducción cardíaca. Las cardiotoxinas son tóxicos que pueden causar esta forma de toxicidad. Pueden provenir de diferentes fuentes, como venenos de animales, toxinas ambientales o fármacos.
Las cardiotoxinas pueden dañar el corazón de diversas maneras, como mediante la disrupción de la función contráctil del músculo cardíaco, la alteración de los canales iónicos que controlan el ritmo cardíaco o la promoción de la formación de especies reactivas de oxígeno que dañan las células cardíacas.
Un ejemplo bien conocido de una cardiotoxina es la digital, un compuesto extraído de la planta Digitalis purpurea, que se ha utilizado durante siglos como medicamento para tratar diversas afecciones cardíacas. Aunque la digital puede ser eficaz en dosis terapéuticas bajas, también puede ser altamente tóxica en dosis más altas y causar arritmias cardíacas potencialmente mortales.
Otro ejemplo son algunos fármacos utilizados en el tratamiento del cáncer, como la doxorrubicina y la daunorrubicina, que pueden causar cardiotoxicidad aguda o crónica en algunos pacientes. La cardiotoxicidad inducida por fármacos es una preocupación importante en la oncología, ya que puede limitar la eficacia del tratamiento y aumentar el riesgo de morbilidad y mortalidad a largo plazo en los sobrevivientes de cáncer.
La deletión cromosómica es un tipo de mutación estructural que involucra la pérdida total o parcial de una sección del cromosoma. Esto sucede cuando una parte del cromosoma se rompe y se pierde durante la división celular, lo que resulta en una copia más corta del cromosoma. La cantidad de material genético perdido puede variar desde un solo gen hasta una gran región que contiene muchos genes.
Las consecuencias de una deletción cromosómica dependen del tamaño y la ubicación de la parte eliminada. Una pequeña deletción en una región no crítica podría no causar ningún problema, mientras que una gran deletión o una deletión en una región importante puede provocar graves anomalías genéticas y desarrollo anormal.
Los síntomas asociados con las deletiones cromosómicas pueden incluir retraso en el desarrollo, discapacidades intelectuales, defectos de nacimiento, problemas de crecimiento, y aumentado riesgo de infecciones o ciertas condiciones médicas. Algunos ejemplos comunes de síndromes causados por deletiones cromosómicas incluyen el Síndrome de Angelman, Síndrome de Prader-Willi, Síndrome de cri du chat y Síndrome de DiGeorge.
Es importante destacar que las deletaciones cromosómicas se pueden heredar o pueden ocurrir espontáneamente durante la formación de los óvulos o espermatozoides, o incluso después de la concepción. Los padres que tienen un hijo con una deletión cromosómica tienen un riesgo ligeramente aumentado de tener otro hijo con la misma condición.
La definición médica de ADN (Ácido Desoxirribonucleico) es el material genético que forma la base de la herencia biológica en todos los organismos vivos y algunos virus. El ADN se compone de dos cadenas de nucleótidos, formadas por una molécula de azúcar (desoxirribosa), un grupo fosfato y cuatro tipos diferentes de bases nitrogenadas: adenina (A), timina (T), guanina (G) y citosina (C). Las dos cadenas se enrollan entre sí para formar una doble hélice, con las bases emparejadas entre ellas mediante enlaces de hidrógeno: A siempre se empareja con T, y G siempre se empareja con C.
El ADN contiene los genes que codifican la mayoría de las proteínas del cuerpo humano, así como información adicional sobre su expresión y regulación. La secuencia específica de las bases en el ADN determina la estructura y función de las proteínas, lo que a su vez influye en los rasgos y características del organismo.
El ADN se replica antes de que una célula se divida, creando dos copias idénticas de cada cromosoma para la célula hija. También puede experimentar mutaciones, o cambios en su secuencia de bases, lo que puede dar lugar a variaciones genéticas y posibles trastornos hereditarios.
La investigación del ADN ha tenido un gran impacto en el campo médico, permitiendo la identificación de genes asociados con enfermedades específicas, el diagnóstico genético prenatal y el desarrollo de terapias génicas para tratar enfermedades hereditarias.
La fibrosis es un proceso patológico que involucra la producción excesiva y acumulación de tejido conectivo fibroso en órganos u otros tejidos. Este tejido fibroso reemplaza gradualmente el tejido normal, lo que puede afectar la función del órgano y, en casos graves, conducir a insuficiencia orgánica. La fibrosis puede ser el resultado de una variedad de condiciones médicas, como enfermedades inflamatorias crónicas, infecciones, lesiones o trastornos genéticos.
El tejido conectivo normalmente contiene células y fibras, como colágeno y elastina, que proporcionan estructura y soporte a los órganos y tejidos. Durante la fibrosis, las células especializadas llamadas fibroblastos producen cantidades excesivas de colágeno y otras proteínas en respuesta a diversos estímulos, como factores de crecimiento, citokinas inflamatorias o daño tisular. Esto conduce a la acumulación de tejido conectivo anormal y fibroso, que puede alterar la arquitectura normal del órgano y restringir su funcionalidad.
La fibrosis puede afectar a varios órganos, como el hígado, los pulmones, el corazón, los riñones y la piel. Algunas enfermedades asociadas con la fibrosis incluyen la cirrosis hepática, la fibrosis quística, la neumoconiosis, la esclerodermia y la hipertensión pulmonar. El tratamiento de la fibrosis generalmente se dirige a la enfermedad subyacente que la causa y puede incluir medicamentos antiinflamatorios, inmunosupresores o terapias dirigidas específicamente a los procesos moleculares involucrados en la fibrosis.
La contractura es una condición médica en la cual un músculo o tejido circundante se acorta o endurece, lo que limita los movimientos y puede causar dolor. Puede ser el resultado de una lesión, enfermedad o simplemente por el uso excesivo o mantenido de una postura. En casos graves, la contractura puede bloquear por completo el rango de movimiento de una articulación. El tratamiento puede incluir fisioterapia, terapia ocupacional, medicamentos para aliviar el dolor y en casos más severos, cirugía.
La Técnica del Anticuerpo Fluorescente, también conocida como Inmunofluorescencia (IF), es un método de laboratorio utilizado en el diagnóstico médico y la investigación biológica. Se basa en la capacidad de los anticuerpos marcados con fluorocromos para unirse específicamente a antígenos diana, produciendo señales detectables bajo un microscopio de fluorescencia.
El proceso implica tres pasos básicos:
1. Preparación de la muestra: La muestra se prepara colocándola sobre un portaobjetos y fijándola con agentes químicos para preservar su estructura y evitar la degradación.
2. Etiquetado con anticuerpos fluorescentes: Se añaden anticuerpos específicos contra el antígeno diana, los cuales han sido previamente marcados con moléculas fluorescentes como la rodaminia o la FITC (fluoresceína isotiocianato). Estos anticuerpos etiquetados se unen al antígeno en la muestra.
3. Visualización y análisis: La muestra se observa bajo un microscopio de fluorescencia, donde los anticuerpos marcados emiten luz visible de diferentes colores cuando son excitados por radiación ultravioleta o luz azul. Esto permite localizar y cuantificar la presencia del antígeno diana dentro de la muestra.
La técnica del anticuerpo fluorescente es ampliamente empleada en patología clínica para el diagnóstico de diversas enfermedades, especialmente aquellas de naturaleza infecciosa o autoinmunitaria. Además, tiene aplicaciones en la investigación biomédica y la citogenética.
En genética, un vector es un agente que transporta un fragmento de material genético, como una plásmido, un fago o un virus, a una célula huésped. El término "vectores genéticos" se utiliza a menudo en el contexto de la ingeniería genética, donde se refiere específicamente a los vehículos utilizados para introducir genes de interés en un organismo huésped con fines de investigación o terapéuticos.
En este sentido, un vector genético típico contiene al menos tres componentes: un marcador de selección, un origen de replicación y el gen de interés. El marcador de selección es una secuencia de ADN que confiere resistencia a un antibiótico específico o alguna otra característica distinguible, lo que permite identificar las células que han sido transfectadas con éxito. El origen de replicación es una secuencia de ADN que permite la replicación autónoma del vector dentro de la célula huésped. Por último, el gen de interés es el fragmento de ADN que se desea introducir en el genoma del huésped.
Es importante destacar que los vectores genéticos no solo se utilizan en la ingeniería genética de bacterias y células animales, sino también en plantas. En este último caso, se utilizan vectores basados en plásmidos o virus para transferir genes a las células vegetales, lo que permite la modificación genética de las plantas con fines agrícolas o industriales.
En resumen, un vector genético es un agente que transporta material genético a una célula huésped y se utiliza en la ingeniería genética para introducir genes de interés en organismos con fines de investigación o terapéuticos.
El ARN mensajero (ARNm) es una molécula de ARN que transporta información genética copiada del ADN a los ribosomas, las estructuras donde se producen las proteínas. El ARNm está formado por un extremo 5' y un extremo 3', una secuencia codificante que contiene la información para construir una cadena polipeptídica y una cola de ARN policitol, que se une al extremo 3'. La traducción del ARNm en proteínas es un proceso fundamental en la biología molecular y está regulado a niveles transcripcionales, postranscripcionales y de traducción.
El genotipo, en términos médicos y genéticos, se refiere a la composición específica del material genético (ADN o ARN) que una persona hereda de sus padres. Más concretamente, el genotipo hace referencia a las combinaciones particulares de alelos (formas alternativas de un gen) que una persona tiene en uno o más genes. Estos alelos determinan rasgos específicos, como el grupo sanguíneo, el color del cabello o los posibles riesgos de desarrollar ciertas enfermedades hereditarias. Por lo tanto, el genotipo proporciona la información inherente sobre los genes que una persona posee y puede ayudar a predecir la probabilidad de que esa persona desarrolle ciertos rasgos o condiciones médicas.
Es importante distinguir entre el genotipo y el fenotipo, ya que este último se refiere al conjunto observable de rasgos y características de un individuo, resultantes de la interacción entre sus genes (genotipo) y los factores ambientales. Por ejemplo, una persona con un genotipo para el color de ojos marrón puede tener fenotipo de ojos marrones, pero si es expuesta a ciertos factores ambientales, como la radiación solar intensa, podría desarrollar unas manchas en los ojos (fenotipo) que no estaban determinadas directamente por su genotipo.
La óxido nítrico sintasa de tipo I, también conocida como NOS-1 o iNOS (del inglés, inducible nitric oxide synthase), es una enzima heterodimérica que cataliza la producción de óxido nítrico (NO) a partir de L-arginina. A diferencia de las otras isoformas de NOS, la NOS-1 se expresa principalmente en el sistema nervioso central y su activación está regulada por diversos factores, incluyendo las concentraciones intracelulares de calcio y los estímulos neurotóxicos. La producción de NO por esta isoforma puede desempeñar un papel importante en la señalización celular, la neurotransmisión y la respuesta inmunitaria, aunque también se ha asociado con diversos procesos patológicos, como la neurodegeneración y el daño tisular.
Las glicosiltransferasas son un tipo de enzimas (más específicamente, transferasas) que desempeñan un papel crucial en la síntesis de glucanos y glicoconjugados. Estas enzimas transfieren un residuo de azúcar, generalmente desde un nucleótido dador de azúcar a un aceptor, lo que resulta en la formación de un enlace glucosídico.
Existen diversos tipos de glicosiltransferasas, cada una con su propia especificidad de sustrato y función particular. Algunas participan en la biosíntesis de oligosacáridos y polisacáridos, mientras que otras desempeñan un papel en la modificación de proteínas y lípidos para formar glicoproteínas y glicolípidos.
Las anormalidades en la actividad de las glicosiltransferasas se han relacionado con diversas afecciones médicas, como enfermedades genéticas y cáncer. Por lo tanto, el estudio y comprensión de estas enzimas pueden tener importantes implicaciones clínicas y terapéuticas.
Las Secuencias Repetidas en Tándem (STR, por sus siglas en inglés) se refieren a un tipo de variación genética que ocurre cuando una determinada secuencia de nucleótidos (los building blocks del ADN) se repite varias veces consecutivas. Estas secuencias repetidas pueden variar en longitud, desde dos hasta cientos de pares de bases, y el número de repeticiones puede variar entre diferentes individuos.
Las STR son comunes en el genoma humano y se encuentran dispersas a lo largo de todos los cromosomas. Suelen localizarse en regiones no codificantes del ADN, es decir, en zonas que no contienen genes y por lo tanto no producen proteínas. Sin embargo, algunas STR se encuentran dentro de genes y pueden influir en su expresión o función.
Las variaciones en el número de repeticiones de estas secuencias pueden asociarse con diversas enfermedades genéticas y neurológicas, como la corea de Huntington, distrofia miotónica, ataxias espinocerebelosas y algunos trastornos del espectro autista. Además, las STR también se utilizan en la identificación forense y en la investigación genética, ya que cada individuo tiene un perfil único de repeticiones en diferentes lugares del genoma.
La Escala de LODD, o la "Escala de Gravedad de Lesiones en el Lugar de los Hechos" (en inglés, "Law Enforcement Officers Killed and Assaulted - LEOKA - Law Enforcement Officer Deadly Force Encounters Scale"), es una herramienta de medición utilizada en medicina forense y ciencias de la salud pública para evaluar y clasificar la gravedad de las lesiones sufridas por los oficiales de la ley durante el desempeño de sus deberes.
La escala se divide en cinco niveles, cada uno de los cuales refleja un mayor grado de gravedad:
1. Sin lesión o lesión menor (sin pérdida de tiempo de trabajo).
2. Lesión que requiere tratamiento médico y pérdida de tiempo de trabajo de menos de una hora.
3. Lesión que requiere tratamiento médico y pérdida de tiempo de trabajo de más de una hora.
4. Lesión grave que requiere hospitalización por un período de hasta tres días.
5. Lesión grave que requiere hospitalización durante más de tres días o lesiones con resultado de discapacidad permanente o muerte.
Esta escala se utiliza a menudo en estudios epidemiológicos y análisis de lesiones relacionadas con el trabajo para evaluar los riesgos y las consecuencias de las diferentes situaciones y tácticas policiales, con el fin de mejorar la seguridad y la eficacia de los oficiales de la ley.
La escoliosis es una afección médica en la columna vertebral donde existe una curvatura lateral anormal de la columna, lo que hace que se desvíe de su posición normal. La curva puede tomar forma de "S" o "C". A menudo, la escoliosis comienza como un problema menor, pero si no se trata, puede provocar posturas encorvadas importantes, especialmente durante el crecimiento rápido.
En la mayoría de los casos, la causa de la escoliosis no se puede identificar y se denomina escoliosis idiopática. Otras causas pueden incluir problemas con los músculos o nervios, anomalías congénitas de la columna vertebral, infecciones o tumores.
La escoliosis a menudo se diagnostica durante las revisiones escolares o durante un examen físico rutinario. Si se sospecha escoliosis, se pueden tomar radiografías para confirmar el diagnóstico y medir la gravedad de la curvatura. El tratamiento puede incluir observación regular, fisioterapia, uso de un corsé ortopédico o, en casos graves, cirugía.
Los oligorribonucleótidos antisentido (ORNas) son moléculas de ARN sintéticas cortas, generalmente compuestas por 15-30 nucleótidos, que están diseñadas para ser complementarias a un ARN mensajero (ARNm) específico. Una vez formada la unión híbrida con su objetivo, el ORN antisentido puede regular la expresión génica de diversas maneras. Por ejemplo, puede bloquear la traducción del ARNm, promover su degradación o modificar su procesamiento y localización celular.
Los ORN antisentido se utilizan en investigación básica para estudiar la función génica y en terapia génica como potenciales fármacos para el tratamiento de diversas enfermedades, incluyendo infecciones virales, cáncer y trastornos genéticos. Sin embargo, su uso clínico está limitado por varios factores, como la dificultad para lograr una especificidad adecuada y la posibilidad de desencadenar respuestas inmunes no deseadas.
La unión neuromuscular, también conocida como la placa motora, es el punto donde los nervios (más específicamente, las terminaciones nerviosas de los axones motores) se conectan y transmiten señales a los músculos esqueléticos. Esta unión es crucial para el control del movimiento ya que es responsable de convertir los impulsos eléctricos generados en el sistema nervioso en una respuesta mecánica en el sistema muscular.
La unión neuromuscular está compuesta por la terminal del axón, que libera neurotransmisores (como acetilcolina) en la hendidura sináptica, un pequeño espacio entre la terminal nerviosa y la membrana muscular. Los receptores de neurotransmisores en la membrana muscular detectan estos neurotransmisores, lo que provoca un cambio en la permeabilidad de la membrana y el inicio de una respuesta eléctrica within the muscle fiber, llamada potencial de acción.
Este proceso desencadena una serie de eventos que finalmente conducen a la contracción del músculo esquelético, permitiendo así el movimiento y la función muscular controlada por el sistema nervioso. Las afecciones que dañan o interfieren con la unión neuromuscular, como las miastenias gravis, pueden causar debilidad muscular y otros síntomas relacionados.
Un transgén es el resultado del proceso de ingeniería genética en el que se inserta un fragmento de ADN extraño o foráneo, conocido como transgen, en el genoma de un organismo receptor. Este transgen contiene normalmente uno o más genes funcionales, junto con los elementos regulatorios necesarios para controlar su expresión.
El proceso de creación de organismos transgénicos implica la transferencia de material genético entre especies que no se aparearían naturalmente. Por lo general, esto se logra mediante técnicas de biología molecular, como la transformación mediada por agente viral o la transformación directa del ADN utilizando métodos físicos, como la electroporación o la gunodisrupción.
Los organismos transgénicos se han convertido en herramientas importantes en la investigación biomédica y agrícola. En el campo médico, los transgenes a menudo se utilizan para producir modelos animales de enfermedades humanas, lo que permite una mejor comprensión de los mecanismos patológicos subyacentes y el desarrollo de nuevas estrategias terapéuticas. En agricultura, las plantas transgénicas se han diseñado para mostrar resistencia a plagas, tolerancia a herbicidas y mayor valor nutricional, entre otros rasgos deseables.
Sin embargo, el uso de organismos transgénicos también ha suscitado preocupaciones éticas y ambientales, lo que ha llevado a un intenso debate sobre su regulación y aplicación en diversas esferas de la vida.
El fondo de ojo, también conocido como examen del fondo de ojo, es un procedimiento médico en oftalmología y optometría que implica el uso de un instrumento especial, llamado oftalmoscopio, para observar la estructura interna del ojo. Específicamente, este examen permite al profesional de la visión mirar directamente a la parte posterior del ojo, incluyendo la retina, el disco óptico (donde se une la nervio óptico a la retina), los vasos sanguíneos y la mácula (zona central de la retina responsable de la visión aguda).
El fondo de ojo puede ayudar a diagnosticar diversas condiciones oftalmológicas y sistémicas, como:
1. Degeneración macular relacionada con la edad
2. Desprendimiento de retina
3. Glaucoma
4. Hipertensión arterial
5. Diabetes mellitus
6. Enfermedades vasculares y otras afecciones sistémicas
Este procedimiento es indoloro y no invasivo, aunque el paciente puede experimentar una leve molestia o sensación de presión durante su realización. Es recomendable realizar exámenes periódicos del fondo de ojo para mantener un seguimiento adecuado de la salud ocular y detectar posibles problemas a tiempo.
Los sarcómeros son estructuras contráctiles en las células musculares esqueléticas y cardíacas. Constituyen la unidad funcional del músculo estriado, donde se produce la contracción y relajación muscular. Un sarcómero se extiende desde una línea Z hasta la siguiente línea Z, abarcando varias miofibrillas.
Está compuesto por filamentos finos de actina y filamentos gruesos de miosina, organizados en una disposición altamente ordenada. Cuando se estimula el músculo, las cabezas de miosina se unen a los sitios de unión de la actina en los filamentos finos, lo que provoca una conformación cambiante que acorta los sarcómeros y, por lo tanto, acorta y engrosa el músculo. Después de que termina la estimulación, los sarcómeros se relajan a su longitud original.
Los defectos en la estructura o función de los sarcómeros pueden dar lugar a diversas patologías musculares, como distrofias musculares y miocardiopatías.
La distrofia corneal epitelial juvenil de Meesmann es una forma rara y hereditaria de distrofia corneal. Se caracteriza por la presencia de pequeñas vesículas (burbujas llenas de líquido) en las células del epitelio corneal, que es la capa más externa de la córnea. Esta afección generalmente afecta a ambos ojos y puede manifestarse desde la infancia hasta la adolescencia.
La distrofia corneal epitelial juvenil de Meesmann es causada por mutaciones en el gen KRT12, que codifica una proteína importante en la estructura y función de las células epiteliales de la córnea. Esta condición se hereda de manera autosómica recesiva, lo que significa que una persona debe heredar dos copias del gen mutado (una de cada padre) para desarrollar la afección.
Los síntomas más comunes de esta distrofia incluyen visión borrosa, lagrimeo excesivo, sensibilidad a la luz y una superficie corneal irregular. En algunos casos, las vesículas pueden romperse y causar erosiones corneales, lo que puede provocar dolor y molestias adicionales.
El tratamiento de la distrofia corneal epitelial juvenil de Meesmann generalmente se centra en aliviar los síntomas y prevenir complicaciones. Esto puede incluir el uso de lágrimas artificiales o lubricantes para mantener la superficie ocular húmeda, y evitar entornos secos o con mucho viento que puedan empeorar los síntomas. En casos graves, se pueden considerar opciones más invasivas, como el trasplante de córnea.
Es importante destacar que la distrofia corneal epitelial juvenil de Meesmann es una afección rara y su pronóstico puede variar ampliamente dependiendo del grado de afectación y la respuesta al tratamiento. En general, se trata de una afección crónica que requiere un seguimiento regular con un especialista en oftalmología para monitorizar su evolución y adaptar el tratamiento a las necesidades individuales del paciente.
Los haplotipos son una serie de variantes genéticas que generalmente se heredan juntas en un solo cromosoma. Están formados por un conjunto de alelos (las diferentes formas en que pueden expresarse los genes) que se encuentran en genes cercanos uno al otro a lo largo de un cromosoma. Debido a que es poco probable que los alelos cambien o intercambien posiciones durante la recombinación genética, los haplotipos tienden a permanecer intactos a través de varias generaciones.
Esta característica hace que los haplotipos sean útiles en la investigación genética, especialmente en el campo de la genética de poblaciones y la medicina personalizada. Por ejemplo, los científicos pueden utilizar haplotipos para rastrear la historia evolutiva de diferentes poblaciones o determinar la predisposición individual a ciertas enfermedades. Además, los haplotipos también se utilizan en las pruebas de paternidad y en los estudios de ascendencia genética.
La eugenesia es un término que proviene del griego y significa "bien nacido". En términos médicos y sociales, se refiere a la creencia y práctica de mejorar las características genéticas de la población mediante la promoción de la reproducción entre personas con rasgos deseables y la prevención o disuasión de la reproducción en aquellos con rasgos indeseables.
Esta idea se basa en la premisa de que ciertos rasgos, como la inteligencia, la salud física y mental, y otras características consideradas positivas, son hereditarias y pueden ser seleccionadas deliberadamente para aumentar su prevalencia en la población.
Históricamente, la eugenesia ha sido objeto de controversia y debate ético, ya que ha llevado a prácticas como el esterilización forzada, el matrimonio selectivo y, en casos extremos, el genocidio y el asesinato masivo de personas consideradas "inferiores". Hoy en día, la mayoría de los profesionales médicos y científicos rechazan la eugenesia como una intervención ética y socialmente aceptable.
Las costameras son estructuras anatómicas especializadas que se encuentran en la interfaz entre los músculos y los huesos. Consisten en engrosamientos de la membrana basal del tejido conectivo que se encuentra entre las fibras musculares y el periostio del hueso, y contienen una alta densidad de receptores sensoriales y mecanorreceptores.
Las costameras desempeñan un papel importante en la transducción de fuerzas mecánicas en señales bioquímicas, lo que permite a los músculos adaptarse a las cargas y mantener la integridad estructural durante la contracción muscular. También pueden desempeñar un papel en el control del crecimiento y desarrollo musculoesquelético, así como en la respuesta al ejercicio y la sobrecarga mecánica.
La investigación sobre las costameras ha contribuido a nuestra comprensión de los procesos fisiológicos subyacentes a la adaptación muscular y la patología asociada con enfermedades neuromusculares y esqueléticas. Sin embargo, aún queda mucho por aprender sobre las funciones específicas y el mecanismo de acción de estas estructuras complejas.
La mioglobinuria es un término médico que describe la presencia de mioglobina en la orina. La mioglobina es una proteína muscular que puede liberarse al torrente sanguíneo cuando los músculos sufren daños o lesiones, como en el caso de una contusión grave, un traumatismo, una isquemia o una enfermedad neuromuscular.
Cuando la mioglobina se filtra a través de los riñones, puede causar irritación y daño renal, lo que lleva a la aparición de mioglobinuria. La mioglobinuria puede detectarse mediante análisis de orina, donde la presencia de mioglobina confiere al líquido una coloración rosada o marrón oscuro, similar a la del te o el café.
La mioglobinuria es un signo de alarma que puede indicar complicaciones graves, como rabdomiolisis, insuficiencia renal aguda y fallo multiorgánico. Por lo tanto, requiere una evaluación médica inmediata y un tratamiento oportuno para prevenir daños adicionales a los músculos y los riñones.
El colágeno tipo VIII es una forma menos común de colágeno que se encuentra en la membrana basal, particularmente en los ojos y los vasos sanguíneos. Es un colágeno de red fine que ayuda a mantener la integridad estructural de los tejidos donde se encuentra. También desempeña un papel importante en la interacción entre las células y la matriz extracelular. Las mutaciones en el gen que codifica para el colágeno tipo VIII se han relacionado con ciertas condiciones oculares hereditarias, como la distrofia corneal de Fuchs y el glaucoma primario abierto.
La progresión de la enfermedad es un término médico que se refiere al curso natural y los cambios en el estado clínico de una enfermedad a lo largo del tiempo. Se caracteriza por la evolución de la enfermedad desde su etapa inicial, incluyendo la progresión de los síntomas, el deterioro de las funciones corporales y la respuesta al tratamiento. La progresión puede ocurrir a diferentes velocidades dependiendo del tipo de enfermedad y otros factores como la edad del paciente, su estado de salud general y los tratamientos recibidos.
La progresión de la enfermedad se mide a menudo mediante el seguimiento de marcadores o biomarcadores específicos de la enfermedad, como el crecimiento del tumor en el caso de un cáncer o la disminución de la función pulmonar en el caso de una enfermedad pulmonar obstructiva crónica. La evaluación de la progresión de la enfermedad es importante para determinar la eficacia del tratamiento, planificar la atención futura y proporcionar información al paciente sobre su pronóstico.
Lo siento, parece que hubo un error en su pregunta. La palabra 'Perros' no está relacionada con ningún término médico específico. Si desea saber sobre el término "perro" desde un punto de vista zoológico o biológico, le informaría que los perros (Canis lupus familiaris) son mamíferos domésticos que pertenecen a la familia Canidae.
Sin embargo, en el campo médico, a veces se hace referencia al término "perro de caza" o "nariz" en relación con los entrenamientos de animales para detectar sustancias químicas, como explosivos o drogas, mediante su agudo sentido del olfato.
Si tuvo la intención de preguntar sobre algo diferente, por favor, proporcione más detalles para que pueda ayudarlo mejor.
La lipodistrofia es un trastorno que afecta la distribución de la grasa corporal, resultando en una pérdida anormal o excesiva de tejido adiposo en ciertas áreas del cuerpo. Existen diferentes tipos de lipodistrofia, y cada uno tiene características específicas:
1. Lipodistrofia congénita generalizada: Es una afección rara que se presenta desde el nacimiento o durante la primera infancia. Las personas con este tipo de lipodistrofia tienen muy poca grasa corporal en todo el cuerpo, lo que puede dar lugar a un aspecto envejecido y desnutrido.
2. Lipodistrofia parcial: Este tipo se caracteriza por la pérdida de grasa en áreas específicas del cuerpo, como los brazos, las piernas y las mejillas, mientras que otras zonas, como el abdomen y el cuello, pueden tener un aumento de la grasa. Puede ser congénita o adquirida más tarde en la vida.
3. Lipodistrofia inducida por medicamentos: Algunos fármacos, especialmente los antirretrovirales utilizados para tratar el VIH, pueden causar lipodistrofia como efecto secundario. Esto puede provocar una acumulación de grasa en el abdomen, el cuello y la espalda, mientras que otras partes del cuerpo pueden perder grasa.
Los síntomas de la lipodistrofia pueden incluir:
- Pérdida de grasa en las mejillas, los brazos y las piernas
- Aumento de grasa en el abdomen, el cuello y la espalda
- Apariencia envejecida prematuramente
- Acumulación de grasa alrededor de los órganos internos (hepatoesplenomegalia)
- Resistencia a la insulina y diabetes
- Anomalías en el colesterol y los triglicéridos
- Dolores articulares y musculares
- Fatiga
- Depresión y ansiedad
El tratamiento de la lipodistrofia dependerá de su causa. Si está causada por medicamentos, es posible que sea necesario cambiar a un fármaco diferente. En otros casos, el manejo puede incluir dieta y ejercicio, terapias hormonales y cirugía estética para abordar los cambios en la distribución de la grasa. Es importante trabajar con un equipo médico que pueda brindar un tratamiento integral y monitorear su progreso.
La glicerol quinasa es una enzima que participa en el metabolismo de los lípidos. Más específicamente, cataliza la fosforilación del glicerol para producir glicerol-3-fosfato, un compuesto importante en la síntesis de triglicéridos y fosfolípidos. La reacción catalizada por esta enzima es:
Glicerol + ATP -> Glicerol-3-fosfato + ADP
Existen diferentes isoformas de glicerol quinasa, expresadas en diversos tejidos y con diferente regulación. Por ejemplo, la glicerol quinasa-5 se expresa principalmente en el músculo esquelético y su actividad está regulada por hormonas como la insulina y el glucagón. La glicerol quinasa desempeña un papel importante en el metabolismo de los carbohidratos y lípidos, y su disfunción se ha relacionado con diversas patologías, incluyendo diabetes e hipertrigliceridemia.
Los oligonucleótidos antisentido son moléculas de ácido nucleico sintéticas, que contienen una secuencia complementaria a un ARNm específico objetivo. Se unen a este ARNm mediante procesos de hibridación, formando dúplex de ARN-ARN o ARN-ADN, lo que impide la traducción del ARNm en proteínas. Esta tecnología se utiliza en terapias génicas y técnicas de diagnóstico, ya que permite regular la expresión de genes específicos. Los oligonucleótidos antisentido pueden ser modificados químicamente para mejorar su estabilidad, especificidad y eficacia terapéutica. Algunos ejemplos de oligonucleótidos antisentido aprobados por la FDA incluyen fomivirsen (Vitravene) para el tratamiento del virus del herpes simple en pacientes con retinitis, y patisiran (Onpattro) para el tratamiento de la amiloidosis familiar sistémica de transtiretina.
El efecto fundador, en el contexto de la genética de poblaciones, se refiere a la pérdida de diversidad genética que ocurre cuando una pequeña población se establece y se separa de una población más grande. Este término fue acuñado por Ernst Mayr, un importante biólogo evolutivo del siglo XX.
Cuando una nueva colonia se establece a partir de un número relativamente pequeño de individuos, las características genéticas de esos individuos fundadores pueden tener un impacto significativo en la composición genética de la población resultante. Debido al azar en la selección de quiénes son los individuos fundadores (llamado deriva genética), ciertos alelos (formas alternativas de un gen) pueden estar sobre o subrepresentados en la nueva población en comparación con la población original.
Este proceso puede conducir a una disminución general de la diversidad genética dentro de la nueva población, ya que algunos alelos raros en la población original pueden no estar representados en absoluto en la nueva población. El efecto fundador también puede resultar en diferencias genéticas distintas entre las poblaciones originales y derivadas, lo que podría eventualmente llevar al aislamiento reproductivo y la especiación.
Es importante notar que el efecto fundador no debe confundirse con la deriva genética, ya que el primero es un tipo específico de deriva genética que ocurre durante la formación de nuevas poblaciones.
Los alelos son diferentes formas de un mismo gen que se encuentran en el mismo locus (ubicación) en los cromosomas homólogos. Cada persona hereda dos alelos, uno de cada progenitor, y pueden ser la misma forma (llamados alelos idénticos) o diferentes (alelos heterocigotos). Los alelos controlan las características heredadas, como el color de ojos o el grupo sanguíneo. Algunos alelos pueden causar enfermedades genéticas cuando una persona hereda dos copias defectuosas del mismo gen (una desde cada progenitor), una situación llamada homocigosis para el alelo anormal.
 Pueraria lobata
Pueraria lobata
Dimension W
Debilidad muscular
Terapia con células madre
Animal modificado genéticamente
Carne cultivada
Miotonía
Guillaume Duchenne de Boulogne
Losartán
Organismo genéticamente modificado
Miostatina
Catepsina
Morfolino
Eddie Rabbitt
Músculo
Transposón
Miopatía
Cavia porcellus
Elecciones al Parlamento Europeo de 2014 (España)
Modificación genética en embriones
Ingeniería genética humana
Genética inversa
Factor de crecimiento insulínico tipo 1
Labrador retriever
Filamento intermedio
Colágeno
Cromosoma X
Ingeniería genética
Hipoterapia
Pie
Pueraria lobata - Wikipedia
 Corrigen por primera vez en un mamífero de mayor tamaño una mutación responsable de la distrofia muscular de Duchenne -...
Corrigen por primera vez en un mamífero de mayor tamaño una mutación responsable de la distrofia muscular de Duchenne -...
 Vamorolone (VBP15), los datos muestran mejoras en la fuerza y la resistencia en los niños con Distrofia Muscular de Duchenne ...
Vamorolone (VBP15), los datos muestran mejoras en la fuerza y la resistencia en los niños con Distrofia Muscular de Duchenne ...
 El riesgo de muerte fetal fue similar durante la 'gripe española' y Covid-19
El riesgo de muerte fetal fue similar durante la 'gripe española' y Covid-19
La distrofia muscular de Duchenne podría ser tratable con un medicamento para el cáncer: Dasatinib - Duchenne Parent Project...
 Científicas Archivos - Page 2 of 5 - Fundación Isabel Gemio
Científicas Archivos - Page 2 of 5 - Fundación Isabel Gemio
 Posibles fármacos para tratar la Distrofia Muscular de Cinturas
Posibles fármacos para tratar la Distrofia Muscular de Cinturas
 Cómo funciona la Terapia Asistida por Animales (TAA)?
Cómo funciona la Terapia Asistida por Animales (TAA)?
 LABRADOR RETRIEVER: Un perro muy leal y amistoso | Animaleros
LABRADOR RETRIEVER: Un perro muy leal y amistoso | Animaleros
 Veterinarios
Veterinarios
 Listado de proyectos - Fundación Ramón Areces
Listado de proyectos - Fundación Ramón Areces
 Morgan Hoffmann, el talento del PGA Tour que busca la cura a la distrofia muscular en Costa Rica - Tengolf
Morgan Hoffmann, el talento del PGA Tour que busca la cura a la distrofia muscular en Costa Rica - Tengolf
 Distrofia muscular ligada al cromosoma x | Actualizado diciembre 2023
Distrofia muscular ligada al cromosoma x | Actualizado diciembre 2023
 Venta de dianabol en uruguay | Larkspur Logistics
Venta de dianabol en uruguay | Larkspur Logistics
 La batalla jurídica por la titularidad de los derechos sobre la herramienta de edición del genoma CRISPR-Cas9
La batalla jurídica por la titularidad de los derechos sobre la herramienta de edición del genoma CRISPR-Cas9
 Ayuda a Terminar con los Experimentos en Animales en las Facultades - PETA Latino
Ayuda a Terminar con los Experimentos en Animales en las Facultades - PETA Latino
Programa Alimentación Animal Archives
Energizantes - Baldini
Donde descargar archivos jar de selenio (2020)
 Comprar Nekton E 35gr
Comprar Nekton E 35gr
Química y Salud: ELEMENTOS MINERALES: Hierro, Zinc, Cobre, Yodo, Selenio, Fluoruro, Cromo, Molibdeno, Manganeso, Elementos...
 Principios básicos de nutrición para vacuno de carne - BM Editores
Principios básicos de nutrición para vacuno de carne - BM Editores
 Vitamina E promueve la reparación de la membrana celular ~ Expresión Genética
Vitamina E promueve la reparación de la membrana celular ~ Expresión Genética
 HOME | Lopapharma
HOME | Lopapharma
 Terapia ecuestre | Consumer
Terapia ecuestre | Consumer
 La célula. 4. Núcleo. Envuelta nuclear. Atlas de Histología Vegetal y Animal
La célula. 4. Núcleo. Envuelta nuclear. Atlas de Histología Vegetal y Animal
Cristina Cifuentes - Fundación Isabel Gemio
 Búsqueda | BVS CLAP/SMR-OPS/OMS
Búsqueda | BVS CLAP/SMR-OPS/OMS
 Neurology Up To Date 19ª edición 2023 | MASS+ Formación Médica
Neurology Up To Date 19ª edición 2023 | MASS+ Formación Médica
 El equipo de Izpisúa consigue revertir los signos del envejecimiento
El equipo de Izpisúa consigue revertir los signos del envejecimiento
Fibras musculares3
- Al final de un periodo de 32 semanas, edad murina que equivadría a los primeros años de la adultez humana, los animales que había recibido el cargado con microdistrofia tenían una fuerza muscular significativamente mayor y sustancialmente más fibras musculares productoras de distrofina. (geomedica.com.ar)
- Cuando usted se mueve, sus fibras musculares se contraen y se estiran, lo que ejerce presión sobre el músculo. (healthychildren.org)
- Sin embargo, con la distrofia muscular, las fibras musculares tienen una estructura o un funcionamiento anormal, e incluso la actividad física de rutina puede lesionar los músculos y los músculos no sanan como deberían. (healthychildren.org)
Duchenne y Becker1
- Sus formas más comunes en los niños, las distrofias musculares de Duchenne y Becker, por sí solas afectan aproximadamente 1 de cada 3,500 a 5,000 niños, o entre 400 y 600 nacimientos de varones vivos cada año en los Estados Unidos. (nih.gov)
Pacientes5
- Debido a que la distrofia muscular de Duchenne es poco frecuente y el fármaco se dirige sólo un pequeño subconjunto de las variantes genéticas responsables de la enfermedad, la contratación de los pacientes elegibles no fue fácil. (medicalpress.es)
- Ya se está utilizando en enfermedades humanas con base genética (Como distrofias musculares que impiden a los pacientes cualquier movilidad, diversos tipos de cáncer, la enfermedad de Alzheimer, Parkinson, etc.) así como también en alimentos de origen animal, en cultivos de cereales, en la producción de frutas y hortalizas, en tratamientos farmacológicos, en investigación para conocer los efectos de cada gen, etc. (unizar.es)
- VYONDYS 53 se utiliza para tratar a pacientes con distrofia muscular de Duchenne (DMD) que tienen una mutación confirmada en el gen de la distrofina que puede ser tratada mediante la omisión del exón 53. (vyondys53.com)
- Si bien los orígenes de dolor crónico son muchos, el cannabis es un tratamiento a menudo recomendado por médicos, y la causa más frecuente entre los pacientes que usan cannabis medicinal. (medicinaalternativamexico.com)
- Donde comprar hydrochlorothiazide en panama aunque la mayoría de los pacientes dicen que no hay dolor durante su procedimiento LASIK, es posible que estos resultados no se informen a las autoridades de salud pública. (telewizjarzeczjasna.pl)
Becker2
- ReveraGen tiene IND abiertos para el desarrollo de vamorolone tanto en la distrofia muscular de Duchenne, como en la distrofia muscular de Becker. (duchenne-spain.org)
- Los tipos más comunes de distrofias musculares en niños son la distrofia muscular de Duchenne (DMD) y la distrofia muscular de Becker (BMD) . (healthychildren.org)
Enfermedad21
- El primer relato histórico de distrofia muscular apareció en 1830, cuando Sir Charles Bell escribió un ensayo sobre una enfermedad que causaba debilidad progresiva en niños varones. (blogspot.com)
- En la década siguiente, el neurólogo francés Guillaume Duchenne presentó un relato completo de 13 niños con la forma más común y más grave de la enfermedad (que ahora lleva el nombre de distrofia muscular de Duchenne). (blogspot.com)
- Revierte la enfermedad del músculo blanco, distrofias o disfunciones musculares. (richmondvet.com.ar)
- Está indicado como curativo y preventivo en la distrofia muscular nutricional (enfermedad del músculo blanco STD/Selenium - Tocopherol Deficiency Syndrome) en corderos y terneros. (richmondvet.com.ar)
- Los estudios de vamorolone en modelos animales de múltiples enfermedades inflamatorias crónicas (esclerosis múltiple, asma, enfermedad inflamatoria intestinal, artritis y otras), han demostrado una eficacia similar a los corticosteroides y un perfil de seguridad mejorado. (duchenne-spain.org)
- Hoy sabemos que por lo menos un 15 % de las razas actuales de perros desarrollan alguna enfermedad congénita, más o menos grave, que puede comprometer la vida del animal. (naukas.com)
- Hipogonadismo produccion natural insuficiente debido a Enfermedad testicular primaria Disfuncion en el eje hipotalamo-hipofisis-testiculo HHT secundaria Terapia de reemplazo hormonal TRH en adultos mayores con niveles bajos de testosterona indicacion medica controvertida Esteroides anabolicos y abuso de androgenos en los deportes para mejorar el rendimiento no es una indicacion medica, pero se utilizan o se abusan con frecuencia, fumeur pays europe. (vira-design.ir)
- La artrosis canina es una enfermedad de las articulaciones de carácter progresivo (los síntomas se agudizan con el tiempo), degenerativo y que puede ser congénita. (kirdalia.es)
- La babesiosis en perros, es una enfermedad parasitaria de animales domésticos, causada por un hematozoario de ciclo indirecto:Babesia spp, y transmitida por garrapatas.La babesiosis en el perro es causada por Babesia canis. (kirdalia.es)
- Nadia Nerea es una niña afectada de tricotiodistrofia, una enfermedad rara. (hipertextual.com)
- La tricotiodistrofia es una enfermedad rara, muy poco frecuente. (hipertextual.com)
- Por qué es importante un diagnóstico de la enfermedad de Duchenne? (vyondys53.com)
- Dado que la enfermedad de Duchenne es progresiva, es importante que el médico de su hijo confirme un diagnóstico e identifique la mutación genética específica para guiar la atención y el tratamiento de su hijo. (vyondys53.com)
- La depresión es una enfermedad psiquiátrica que afecta a millones de personas en el mundo. (medicinaalternativamexico.com)
- El glaucoma es una enfermedad en el nervio óptico que a menudo puede resultar en ceguera. (medicinaalternativamexico.com)
- La EM es una enfermedad degenerativa que afecta al sistema nervioso central y puede causar discapacidad permanente. (medicinaalternativamexico.com)
- La ELA es una enfermedad debilitante del sistema nervioso que ataca a la médula espinal y las neuronas del cerebro. (medicinaalternativamexico.com)
- El Parkinson o Enfermedad de Parkinson es una enfermedad crónica, neurodegenerativa e invalidante que afecta en España a más de 160.000 personas, en la que uno de cada cinco afectados es menor de 50 años. (fundacion-alborada.org)
- La enfermedad de Parkinson afecta, por tanto, a aquellas zonas encargadas de coordinar la actividad, el tono muscular y los movimientos. (fundacion-alborada.org)
- Un diagnóstico certero tendrá en cuenta los tres ámbitos de actuación de la enfermedad: sus manifestaciones motoras, las no motoras y las premotoras, observando que la progresión del párkinson es, como se ha apuntado, muy variable y no todos los síntomas concurren en todas las personas. (fundacion-alborada.org)
- La herencia autosómica recesiva es una anomalía genética que se puede transmitir al hijo solamente si ambos padres son portadores del mismo gen defectuoso (por ejemplo, la fibrosis quística , la enfermedad de Tay-Sachs o la anemia drepanocítica). (healthychildren.org)
Modelos5
- 4] En modelos de experimentación animal se ha probado su posible utilidad para la prevención del Alzheimer. (wikipedia.org)
- En el siguiente podcast se explica la relevancia de los modelos de experimentación animal para llevar a cabo estudios pre-clínicos relacionados con las enfermedades raras. (onero.org)
- Intervienen Lluís Montoliu , investigador del CNB-CSIC y el CIBERER y experto en aspectos éticos de la experimentación animal, y Cecilia Jiménez , investigadora del Institut de Recerca Sant Joan de Déu y del CIBERER, que estudia las enfermedades neuromusculares en modelos pre-clínicos. (onero.org)
- Los modelos animales han sido herramientas críticas desde los primeros días del descubrimiento científico. (lifelegacyphotography.com)
- Labome identificó 4 publicaciones utilizando ratones A / J como modelos animales (Tabla 1). (lifelegacyphotography.com)
Provoca3
- La creatina eleva el contenido de agua dentro de las células musculares, lo que provoca un efecto de voluminización celular que puede desempeñar un papel en el crecimiento muscular. (saludissimo.com)
- Con el tiempo, esto provoca una progresiva pérdida de masa muscular y debilidad. (healthychildren.org)
- Se achaca a deficiencias de vitamina E (Provoca distrofia muscular, pericarditis con fluido gelatinoso y múltiples hemorragias en epicardio y endocardio con mortalidad elevada de hasta el 20% en lechones destetados y cerdos en crecimiento. (bmeditores.mx)
Debilidad muscular7
- Evita debilidad muscular y disminuye fatiga celular. (todovet.com.ar)
- Las distrofias musculares son trastornos musculares genéticos que provocan una debilidad muscular progresiva. (healthychildren.org)
- De manera similar, la debilidad muscular puede progresar a diferentes ritmos. (healthychildren.org)
- La BMD generalmente tiene una debilidad muscular menos grave, un inicio más tardío y una progresión más lenta y menos predecible. (healthychildren.org)
- en la LGMD, la debilidad muscular comienza en los músculos más cercanos al centro del cuerpo, incluidas las caderas, la pelvis, los hombros, la parte superior de los brazos y las piernas. (healthychildren.org)
- Esto a menudo mejora con el tiempo antes de que desarrollen una mayor debilidad muscular en los últimos años de la adolescencia y dificultad para relajar los músculos (miotonía). (healthychildren.org)
- Las personas con distrofia miotónica de inicio en la infancia podrían tener problemas cognitivos o de aprendizaje como síntoma principal y luego desarrollar debilidad muscular y miotonía. (healthychildren.org)
Tipos3
- Algunos tipos de distrofia muscular también afectan al corazón, el sistema gastrointestinal, las glándulas endocrinas, la columna, los ojos, el cerebro y otros órganos. (nih.gov)
- Nunca es tarde para mencionar que la piel es el órgano más grande del cuerpo, medicamentos esteroides tipos. (dodgyozies.com)
- Cuáles son los diferentes tipos de distrofia muscular? (healthychildren.org)
Humanos7
- En los humanos, una mutación de G por A es muy común. (agenciasinc.es)
- Los delfines son animales muy inteligentes, tanto que en muchas ocasiones esa inteligencia es comparada con la de los seres humanos. (eduplanetamusical.es)
- Cuando se crearon a la par de la humanidad, animales y plantas no cabe duda alguna que a los segundos también se les otorgó de ciertas características que los distinguen a unos de otros, bien sean perceptibles por los humanos o no lo que no quiere decir que no las tengan. (eduplanetamusical.es)
- La artritis no sólo se produce en los seres humanos, sino que es común en los perros y otros animales también. (kirdalia.es)
- El sistema CRISPR-Cas9 permite a los científicos realizar cambios precisos en el ADN de diversos organismos, incluyendo plantas, animales e incluso humanos. (cienciaparatodos.org)
- Después de pasar los tests en animales, un fármaco empieza a ser testado en humanos. (addarevista.org)
- Cultivo de tejidos (*22): Consisten en introducir en recipientes de cultivo fragmentos tisulares animales (humanos y no humanos) o vegetales. (addarevista.org)
Afecta3
- La distrofia muscular se produce en todo el mundo y afecta a todas las razas. (nih.gov)
- Cómo afecta la distrofia muscular a los músculos? (blogspot.com)
- Son una muestra la distrofia muscular, la parálisis de patas traseras que a menudo afecta a los golden retriever y labradores, y la sordera unilateral o bilateral que desarrollan los perros dálmata. (naukas.com)
Tono muscular1
- Los bebés recién nacidos con distrofia miotónica tienen bajo tono muscular y debilidad al nacer, por lo que normalmente necesitan apoyo para respirar y alimentarse. (healthychildren.org)
Discapacidad5
- Al final pudieron aligerar la marcha con ayuda de dos caballos, uno para los dos hermanos con discapacidad -metidos en grandes alforjas y atados a los costados del animal- y otro para las pesadas sillas. (amnesty.org)
- Para personas con discapacidad como nosotros -comentó Alan-, lograr cruzar las fronteras es como un milagro' . (amnesty.org)
- Para personas con discapacidad como nosotros , lograr cruzar las fronteras es como un milagro . (amnesty.org)
- lo complicado que es esto con una persona con discapacidad. (amnesty.org)
- Es común que presenten discapacidad intelectual. (healthychildren.org)
Casos6
- Muchos casos de distrofia muscular se producen de mutaciones espontáneas que no se encuentran en los genes de ninguno de los padres, y este defecto puede trasmitirse a la siguiente generación. (nih.gov)
- Estas alteraciones están implicadas en casos de crisis epilépticas parciales, distrofia muscular de Duchenne y párkinson. (agenciasinc.es)
- En concreto, España ya es el segundo país de Europa con mayor tasa de incidencia de diabéticos sobre la población adulta y suma 400.000 nuevos casos cada año. (biocrucesbizkaia.org)
- Pero hay un obstáculo: si le das un gen que es una receta para una proteína normal a alguien con una versión defectuosa del gen, cuyo cuerpo nunca fabricó la proteína normal antes, el sistema inmune de esa persona experimentará una reacción, en algunos casos letal, para la proteína normal, como ocurriría con cualquier proteína extraña. (geomedica.com.ar)
- El 90% de los casos de párkinson son formas esporádicas, es decir, no se deben a una alteración genética concreta. (fundacion-alborada.org)
- La muerte provocada no es la solución a los conflictos de la sociedad, ni en los casos de eutanasia ni de aborto" - Luis Argüello, obispo auxiliar de Valladolid. (ermes-electronics.com)
Tratar5
- 7] Se utiliza para tratar acufenos, vértigo, y el síndrome Wei (distrofia muscular). (wikipedia.org)
- La revista Science revela una nueva herramienta, llamada REPAIR, que edita ARN en lugar de ADN y es capaz de tratar patologías sin modificar permanentemente el genoma. (agenciasinc.es)
- Al permitir la corrección de genes defectuosos, se podría utilizar para tratar enfermedades como la fibrosis quística, la distrofia muscular, la anemia de células falciformes y muchas otras enfermedades hereditarias. (cienciaparatodos.org)
- La azatioprina (Imuran) es un medicamento que se utiliza para tratar ciertas afecciones autoinmunitarias (enfermedades en las que el sistema natural de. (dodgyozies.com)
- Se ha demostrado que el cannabis es muy efectivo para tratar los síntomas que sobrevienen al estrés que genera haber vivido alguna situación traumática. (medicinaalternativamexico.com)
Plantas1
- Las ventajas de la técnica CRISPR-Cas9 incluyen su alta precisión y eficiencia en la edición genética, su relativa simplicidad en comparación con otras técnicas anteriores, su amplio rango de aplicaciones potenciales y su capacidad para realizar modificaciones en diversos organismos, desde bacterias hasta plantas y animales. (cienciaparatodos.org)
Nacidos2
- Para generar cualquier raza de perros estos deben cruzarse entre sí, aprovechando animales nacidos espontáneamente con alguna alteración anatómica, para amplificarla y preservarla. (naukas.com)
- la distrofia miotónica es la distrofia muscular más común en adultos, pero también puede afectar a bebés recién nacidos y niños. (healthychildren.org)
Causa6
- La distrofia muscular se refiere a un grupo de más de 30 enfermedades genéticas que causa debilidad y degeneración progresivas de los músculos esqueléticos usados durante el movimiento voluntario. (nih.gov)
- Qué causa la distrofia muscular? (nih.gov)
- Los investigadores clínicos en la UC Davis Medical Center y un puñado de otros centros de investigación de todo el país están probando un medicamento de alta tecnología diseñada para corregir el defecto genético subyacente que causa la disminución progresiva muscular que se observa en niños con Duchenne. (medicalpress.es)
- En los Estados Unidos, la degeneración macular relacionada con la edad es la principal causa de pérdida de la vista entre las personas de edad avanzada. (medlineplus.gov)
- La mutación del gen LRRK2 es la causa más frecuente conocida de párkinson y supone un 40% de las causas genéticas. (fundacion-alborada.org)
- Además, es importante que los contactos cercanos de niños con enfermedades neurológicas, tales como los padres, hermanos, miembros del hogar y las personas encargadas de los cuidados, como niñeras, médicos, personal de enfermería y maestros, se vacunen para evitar contraer influenza o contagiar a estos niños o a personas con mayor riesgo de presentar complicaciones graves a causa de la influenza . (cdc.gov)
Formas2
- Todas las formas de distrofia muscular empeoran a medida que los músculos degeneran y se debilitan progresivamente. (nih.gov)
- Las afecciones ligadas al cromosoma X son anomalías genéticas que ocurren sobre todo en los varones (por ejemplo, hemofilia, daltonismo, formas de distrofia muscular). (healthychildren.org)
Preventivo1
- Selenio: elemento preventivo y terapéutico de las miopatías y las distrofias musculares. (todovet.com.ar)
Nutricional2
- Suplemento nutricional de última generación, el cual combina tres componentes esenciales para los animales en producción: Selenio, Vitamina A y Vitamina E. Es un desarrollo original y propio de Laboratorios Richmond Veterinaria S.A. (richmondvet.com.ar)
- Puede repetirse el tratamiento a los 15 días dependiendo del estado nutricional del animal. (comitedeganaderosdelhuila.org)
Utiliza1
- Qué es CRISPR-Cas9 y para qué se utiliza? (cienciaparatodos.org)
Afecciones2
- Hay otras afecciones parecidas a la distrofia muscular? (blogspot.com)
- Es particularmente importante que se vacunen los niños con afecciones neurológicas, ya que corren mayor riesgo de presentar complicaciones si contraen la influenza. (cdc.gov)
Esteroides4
- Por lo tanto, si toma el medicamento, es probable que coma menos de lo que está acostumbrado y esto ayuda significativamente a la pérdida de grasa, que esteroides comprar steroide wo kaufen forum. (lcrearthworkengineering.net)
- Com es el mejor lugar para comprar esteroides con tarjetas de credito! (sourceofwonder.com)
- Es posible que escuche otras palabras para los medicamentos esteroides, como corticosteroides, glucocorticoides o cortisona. (sourceofwonder.com)
- Si prefieres salir más tarde, Ouigo dispone de varios horarios para el trayecto Barcelona-Zaragoza hasta las 20h45 , que es cuando sale el último tren del día, medicamentos esteroides y hormonas testosterona. (dodgyozies.com)
Disminuye1
- Disminuye la muerte perinatal y eleva las defensas celulares y humorales en animales jóvenes. (richmondvet.com.ar)
Causada2
- Distrofia muscular de Duchenne es causada por mutaciones genéticas en el gen para la proteína muscular distrofina. (medicalpress.es)
- La meningitis micótica , o causada por hongos, es un problema raro, pero muy grave y peligroso. (noticierodiario.com)
Fuerza2
- La fuerza es un componente crucial en el Ciclismo, pues, no en vano, constituye la capacidad de producir potencia. (lcrearthworkengineering.net)
- Lo que hace que la HGH sea popular entre los culturistas es su capacidad para promover la fuerza muscular y aumentar el rendimiento. (dodgyozies.com)
Cultivos1
- Los cultivos Bt , por ejemplo, están diseñados para contener una forma natural de pesticida derivado de bacterias, lo que significa que no es necesario rociarlos con pesticidas químicos. (jorgecarlosfernandez.com)
Masa muscular2
- También puede ayudar a aumentar la masa muscular ( 1 ). (saludissimo.com)
- Puede aumentar la masa muscular total al reducir la degradación muscular. (saludissimo.com)
Noelia1
- También contamos con Noelia , presidenta de laFundación Noelis y madre de Adrià, un niño con distrofia muscular por déficit de colágeno VI . (onero.org)
Mutaciones2
- La capacidad de corregir las mutaciones que causan enfermedades es uno de los principales objetivos de la edición del genoma", explica Zhang. (agenciasinc.es)
- No es una mejora a la hora de corregir irreversiblemente las mutaciones, tal y como podemos hacer ahora con la edición de ADN, pero si permitirá hacer modificaciones transitorias, es decir, no permanentes, para un efecto terapéutico durante un tiempo limitado", puntualiza Montoliu. (agenciasinc.es)
Distrofina1
- La terapia génica para la distrofia de Duchenne suele basarse en la introducción de una fracción del gen que codifica la distrofina -demasiado grande para poder insertarse en el vector- que se acopla a un virus para que lo incorpore al organismo. (geomedica.com.ar)
Dificultades2
- Sin embargo, el geógrafo tiene dificultades en las mas normales habilidades físicas, sentar, correr o trepar árboles están muy llejos de lo que puede hacer, e incluso caminar es difícil. (medicalpress.es)
- Su duración es más limitada debido a estas dificultades (*36). (addarevista.org)
Problemas2
- Creo que es un paso más para que los políticos y la sociedad sean conscientes de los problemas que dan los pesticidas», subrayó. (fundacion-alborada.org)
- Cuando un niño nace sin 46 cromosomas , o cuando algunas partes de los cromosomas faltan o están duplicadas, es posible que se vea y se comporte de manera diferente a los demás niños de su edad y que tenga graves problemas de salud (por ejemplo. (healthychildren.org)
Ratones3
- Por ejemplo, la diferencia de comportamiento entre cepas/cepas de ratones es común. (lifelegacyphotography.com)
- Una de sus hipótesis de trabajo era que el cáncer tenía un fuerte componente hereditario y, para poder estudiarlo, necesitaba una cepa de ratones … También es posible inducir en los ratones un gran número de enfermedades humanas manipulando sus genes por medio de técnicas de ingeniería genética. (lifelegacyphotography.com)
- Todavía es temprano -al fin y al cabo, esto es un experimento con ratones-, pero parece que podemos inducir tolerancia a una gran variedad de proteínas que antes eran inmunogénicas si insertamos el gen de la proteína que nos interesa en el plásmido", considera Steinman. (geomedica.com.ar)
Crecimiento2
- Porcinos y ovinos al preparto, preservicio en machos y hembras, animales jóvenes en crecimiento y engorde. (richmondvet.com.ar)
- Permite más trabajo total o volumen en una sola sesión de entrenamiento, un factor clave en el crecimiento muscular a largo plazo. (saludissimo.com)
CRISPR11
- La técnica de edición genética CRISPR es uno de los mayores avances en la medicina actual. (agenciasinc.es)
- CRISPR se compone de una secuencia sintetizada de ARN guía que coincide con una secuencia de ADN objetivo, es decir, la porción de ADN que se modificará, y una enzima Cas. (jorgecarlosfernandez.com)
- El último avance en Biotecnología, el CRISPR ha revolucionado los campos de la Medicina, de la Veterinaria, de la Agricultura y, en general, de todos aquellos aspectos relacionados con la Genética o lo que es lo mismo con el material genético de los seres vivos que poblamos este Planeta. (unizar.es)
- A partir de unos genes que poseen las bacterias (el gen llamado CRISPR ) se ha adaptado a todos los demás ADN de otros organismos, y en particular de nuestro genoma y de las especies de interés en nuestra alimentación, como frutas, hortalizas, cereales y productos de origen animal, como los derivados lácteos. (unizar.es)
- 1. CRISPR-Cas9 es una revolucionaria tecnología de edición genética que ha ganado una gran atención en los últimos años. (cienciaparatodos.org)
- CRISPR-Cas9 es una tecnología de edición genética que permite realizar modificaciones precisas en el ADN de organismos vivos. (cienciaparatodos.org)
- Cuando una bacteria es atacada por un virus, el sistema CRISPR "guarda" una copia del ADN viral como una especie de huella digital. (cienciaparatodos.org)
- Qué es la tecnología CRISPR? (cienciaparatodos.org)
- La tecnología CRISPR es un conjunto de herramientas que se basa en el sistema CRISPR-Cas9 para editar el ADN de manera precisa. (cienciaparatodos.org)
- Sin embargo, el sistema CRISPR-Cas9 es el más utilizado y conocido debido a su facilidad de uso y su versatilidad en la edición genética. (cienciaparatodos.org)
- CRISPRpedia es el recurso estilo libro de texto de IGI para todo lo relacionado con CRISPR. (innovativegenomics.org)
Desarrollo2
- El desarrollo es constantemente evaluado por los padres, el personal de la escuela y los médicos que siguen a los niños. (msdmanuals.com)
- Es imposible acelerar significativamente el desarrollo motor aumentando la estimulación. (msdmanuals.com)
Prueba2
- Para evaluar las capacidades físicas de cada niño y el progreso, los participantes completar una prueba de marcha de seis minutos específicamente diseñado y validado por un equipo de la Universidad de California Davis, que incluyó McDonald y Erik Henricson, investigador de la Universidad de California Davis distrofia muscular. (medicalpress.es)
- Por qué es importante la prueba genética? (vyondys53.com)
Funcionamiento1
- Las distrofias musculares son causadas por anormalidades en las proteínas que son importantes para la estructura y el funcionamiento del músculo. (healthychildren.org)
Aparece1
- Su caso lo contó hace unos días el periodista Pedro Simón en El Mundo , aunque no es la primera vez que la pequeña aparece en los medios de comunicación. (hipertextual.com)
Tratamiento3
- Hoy en día, Jacob es uno de los 51 niños que participan en un ensayo clínico a nivel nacional para un nuevo tipo de tratamiento que pueda ofrecer ayuda a las personas que padecen devastadoras enfermedades neuromusculares. (medicalpress.es)
- En el tratamiento de los procesos autoinmunes en perros, es la adaptación del fármaco y dosis, en cada paciente. (kirdalia.es)
- Es eficaz el tratamiento? (vyondys53.com)
Vida7
- Muchas de estas razas comprometen la calidad de vida de los animales y nunca habrían existido de no ser por la intervención humana. (naukas.com)
- Este parece ser el caso de los bulldogs ingleses, cuyos pliegues en la piel y morro achatado llevan a los perros de esta raza a desarrollar patologías favorecidas por la selección brutal a favor de estos rasgos que algunas personas consideran estéticamente interesantes, pero que ponen la vida de estos animales en riesgo. (naukas.com)
- Cifra que es previsible que crezca en los próximos años, debido al envejecimiento de la población y el aumento de la esperanza de vida. (fundacion-alborada.org)
- 8. Frente a la vieja idea de que existe la vida y después la muerte, lo real es que van juntas, que la muerte es la sombra de la vida, que nacemos muriendo y que, por tanto, somos vida-muerte. (ermes-electronics.com)
- Ningún ser humano es dueño de la vida, todos tenemos derecho a la vida: Derecho Natural, Derecho Humano de la ONU, Derecho Constitución Española. (ermes-electronics.com)
- La eutanasia es una acción deliberada que toma un médico u otra parte y que, a sabiendas, tiene como resultado el final de la vida de una persona. (ermes-electronics.com)
- Todos merecen un orgasmo' es una manera justa de expresar la actitud de Diana de Vegh hacia la vida. (noticierodiario.com)
Demostrado1
- Un equipo de investigadores de la Facultad de Medicina de la Universidad de Stanford así lo ha demostrado en un modelo de ratón que reproduce la distrofia muscular de Duchenne. (geomedica.com.ar)
Medio1
- Una mirada curiosa es lo que caracteriza los ojos de esta raza, los cuales son de tamaño medio y en forma de almendra. (piensosloboazul.com)
Inicio1
- El Ártico es el lugar donde tiene la base de inicio esta raza canina, donde las tribus existentes la criaron, primeramente, alrededor del siglo XIX. (piensosloboazul.com)
Personas4
- El objetivo del proyecto solidario Mediolanum Aproxima es convertirse en el nexo de unión entre ONG con sede en nuestro entorno más cercano y las personas que vivimos en él, porque muchas veces nos gustaría apoyarlas, pero no sabemos cómo hacerlo. (migranodearena.org)
- La patología es en realidad un grupo heterogéneo de enfermedades de origen genético detectadas en 36 personas de todo el mundo, de acuerdo con lo que dice el padre de Nadia a Hipertextual. (hipertextual.com)
- El procedimiento que se suele seguir es ofertar una compensación económica a cambio de poder realizar el test en aquellas personas que se presten. (addarevista.org)
- Consulte la lista completa de las personas para quienes la vacunación es especialmente importante . (cdc.gov)
Saber2
- Dado que los genomas de los productos PairWise están modificados, es posible que algunos consumidores deseen saber si las frutas y verduras están clasificadas como organismos genéticamente modificados (OGM). (jorgecarlosfernandez.com)
- Por otro lado, es un alivio saber que puedo hacer cosas con seguridad con la ayuda de mi andador que no podría hacer sin él. (ataxias-galicia.org)