Amiloidosis
Amiloide
Proteína Amiloide A Sérica
Prealbúmina
Cadenas Ligeras de Inmunoglobulina
Fiebre Mediterránea Familiar
Neuropatías Amiloides
Rojo Congo
Neuropatías Amiloides Familiares
Cardiomiopatías
Microglobulina-2 beta
Componente Amiloide P Sérico
Síndrome Nefrótico
Paraproteinemias
Enfermedades de la Tráquea
Macroglosia
Proteínas Amiloidogénicas
Melfalán
Proteína de Bence Jones
Biopsia
Acinonyx
Cadenas lambda de Inmunoglobulina
Gelsolina
Deficiencia del Factor X
Enfermedades Cutáneas Genéticas
Resultado Fatal
Mieloma Múltiple
Enciclopedias como Asunto
Dermatología
Crassulaceae
MedlinePlus
Síndrome de Williams
La amiloidosis es una enfermedad rara pero grave que ocurre cuando se acumulan proteínas anormales llamadas amiloide en diferentes órganos y tejidos del cuerpo. Estas proteínas se pliegan incorrectamente y forman fibrillas, lo que lleva a la formación de depósitos de amiloide.
Existen varios tipos de amiloidosis, cada uno causado por un tipo diferente de proteína amiloide. Los más comunes son:
1. Amiloidosis AL (inmunoglobulina ligada a la leve): Esta forma es causada por la producción excesiva de una proteína inmunoglobulina anormal por células plasmáticas malignas o benignas en la médula ósea.
2. Amiloidosis AA (proteína serica asociada a la amiloidosis secundaria): Esta forma es causada por una respuesta inflamatoria crónica, como la que se observa en enfermedades como la artritis reumatoide o la tuberculosis.
3. Amiloidosis ATTR (transtirretina relacionada con la amiloidosis hereditaria y senil): Esta forma es causada por mutaciones genéticas en el gen de la transtirretina, una proteína producida principalmente en el hígado.
Los síntomas y signos de la amiloidosis dependen del tipo y del órgano o tejido afectados. Los depósitos de amiloide pueden dañar los órganos y tejidos, lo que lleva a disfunción orgánica e insuficiencia orgánica progresiva. Las manifestaciones clínicas comunes incluyen:
- Insuficiencia cardíaca congestiva
- Arritmias cardíacas
- Neuropatía periférica (entumecimiento, hormigueo y debilidad en las extremidades)
- Hiperplasia de la lengua
- Síndrome del túnel carpiano
- Insuficiencia renal
- Disfunción hepática
- Infiltración vascular (pérdida de la visión, piel engrosada y frágil)
El diagnóstico de la amiloidosis se realiza mediante una biopsia del tejido afectado, seguida de un examen histopatológico para confirmar la presencia de depósitos de amiloide. Se pueden utilizar tinciones especiales, como la tinción de rojo congo, y técnicas inmunohistoquímicas o inmunofluorescencia para identificar el tipo de proteína de amiloide presente.
El tratamiento de la amiloidosis depende del tipo y del grado de afectación orgánica. El objetivo principal es eliminar o reducir la producción de la proteína precursora de amiloide y estabilizar o reemplazar los órganos afectados. Los tratamientos disponibles incluyen:
- Quimioterapia con agentes alquilantes, como melphalan, y esteroides
- Terapias dirigidas contra la proteína precursora de amiloide, como bortezomib o lenalidomida
- Trasplante de células madre autólogo o alogénico
- Diálisis o trasplante renal en casos de insuficiencia renal grave
- Trasplante hepático en casos de infiltración vascular grave
- Terapias de soporte, como la administración de fluidos y el control de los síntomas
La supervivencia varía según el tipo y el grado de afectación orgánica. Los pacientes con amiloidosis AL tienen una supervivencia media de 1 a 2 años, mientras que aquellos con amiloidosis AA o TTR tienen una supervivencia media de 5 a 10 años. El pronóstico ha mejorado en los últimos años gracias al desarrollo de nuevas terapias dirigidas contra la proteína precursora de amiloide y al trasplante de células madre.
La amiloidosis familiar es una afección genética rara en la que se acumulan proteínas anormales llamadas amiloides en los tejidos y órganos del cuerpo. Estas proteínas se producen como resultado de mutaciones en genes específicos, lo que hace que las proteínas se plieguen incorrectamente y formen fibrillas amiloides insolubles.
La acumulación de estas fibrillas puede dañar los tejidos y órganos, afectando su función normal. La enfermedad puede afectar a varios órganos y sistemas corporales, como el corazón, los riñones, el hígado, los nervios y el sistema gastrointestinal.
Los síntomas de la amiloidosis familiar pueden variar ampliamente dependiendo del órgano o sistema afectado. Algunos de los síntomas más comunes incluyen:
* Hinchazón en las piernas y los pies
* Fatiga
* Pérdida de apetito y peso
* Dolor articular y muscular
* Náuseas, vómitos y diarrea
* Disfunción eréctil
* Cambios en el pensamiento y la memoria
* Insuficiencia cardíaca congestiva
El diagnóstico de amiloidosis familiar puede ser desafiante y requiere una evaluación médica exhaustiva, que incluye pruebas genéticas y de tejidos. El tratamiento de la enfermedad generalmente implica el control de los síntomas y la prevención de la progresión de la enfermedad.
La terapia dirigida a las proteínas tau y la inmunoterapia son algunos de los tratamientos emergentes para la amiloidosis familiar. En casos graves, se puede considerar un trasplante de células madre o de órganos. La prevención de la enfermedad implica la identificación y el asesoramiento genético de las personas en riesgo.
En la medicina, el término "amiloide" se refiere a un tipo anormal de proteína que puede acumularse en los tejidos y órganos del cuerpo. Esta acumulación se conoce como amiloidosis. Existen diferentes tipos de proteínas amiloides, y cada uno tiene su propio nombre y síntomas asociados. Algunos de los tipos más comunes incluyen:
* Amiloide A: Se produce en personas con trastornos crónicos del hígado y se acumula principalmente en el bazo, el hígado y la médula ósea.
* Amiloide TTR (transtiretina): Puede depositarse en varios órganos y tejidos, incluyendo el corazón, los riñones, el sistema nervioso periférico y los ojos. Existen dos formas hereditarias de amiloidosis TTR: la ATTR familial amiodarona cardiomiopatía y neuropatía y la ATTR V30M familiarla polineuropatía, ambos causados por mutaciones en el gen TTR. También existe una forma adquirida de amiloidosis TTR llamada ATTR senil, que se produce espontáneamente en personas mayores de 60 años y afecta principalmente al corazón.
* Amiloide AL (inmunoglobulina ligera kappa o lambda): Se produce en personas con trastornos de las células plasmáticas, como el mieloma múltiple y los tumores de células B, y se acumula principalmente en el corazón, los riñones, el hígado y los tejidos blandos.
* Amiloide Aβ (beta-amiloide): Se produce en personas con la enfermedad de Alzheimer y se acumula en forma de placas en el cerebro.
La acumulación de amiloide puede causar diversos síntomas, dependiendo del tipo y la ubicación de la acumulación. Los síntomas pueden incluir hinchazón, dolor e insensibilidad en las manos y los pies; problemas cardíacos, como latidos irregulares o insuficiencia cardíaca congestiva; problemas renales, como proteinuria y hematuria; y problemas neurológicos, como demencia, confusión y pérdida de memoria. El diagnóstico de la amiloidosis puede ser difícil y requiere una combinación de pruebas clínicas, de laboratorio e histopatológicas. El tratamiento depende del tipo y la gravedad de la enfermedad y puede incluir terapias dirigidas a reducir la producción o acelerar la eliminación del amiloide, como quimioterapia, trasplante de células madre y terapia con anticuerpos monoclonales.
La proteína amiloide A sérica, también conocida como SAA o proteinada amiloide A sérico-aguda, es un tipo de proteína producida principalmente en el hígado en respuesta a la inflamación aguda en el cuerpo. Forma parte de las proteínas de fase aguda, que son proteínas sintetizadas por el hígado y su concentración en sangre se incrementa durante los procesos inflamatorios.
La proteína amiloide A sérica es una precursora de la proteína amiloide que se encuentra en las placas amiloides, depósitos anormales de proteínas plegadas incorrectamente que se acumulan en los tejidos y órganos del cuerpo. La presencia de estas placas está asociada con diversas enfermedades, como la amiloidosis sistémica y las enfermedades cardiovasculares.
Es importante destacar que un aumento sostenido de los niveles de proteína amiloide A sérica en sangre puede indicar un mayor riesgo de desarrollar enfermedades cardiovasculares, especialmente aterosclerosis y enfermedad coronaria. Además, la determinación de los niveles de SAA se utiliza como marcador de inflamación aguda en diagnósticos clínicos y seguimiento de diversas patologías.
La prealbúmina, también conocida como transtiretina, es una proteína plasmática de bajo peso molecular producida principalmente por el hígado. Es soluble en suero y se utiliza como un marcador de la síntesis proteica hepática. Tiene una vida media corta de aproximadamente 2 días, lo que permite su uso como un indicador rápido de cambios en la producción de proteínas.
La prealbúmina está compuesta por cuatro subunidades idénticas y se une reversiblemente a la tiroxina (T4) y la retinol-binding protein (RBP), desempeñando un papel en el transporte de estas moléculas en el cuerpo.
En la práctica clínica, los niveles séricos de prealbúmina se utilizan a menudo como un indicador del estado nutricional y de la función hepática. Los niveles bajos de prealbúmina pueden estar asociados con desnutrición, enfermedad hepática, inflamación crónica y algunas enfermedades renales. Sin embargo, su utilidad como marcador nutricional puede verse afectada por factores como la edad, el sexo, la obesidad y las enfermedades crónicas.
Las cadenas ligeras de inmunoglobulina son proteínas que forman parte de la estructura de los anticuerpos, también conocidos como inmunoglobulinas. Existen dos tipos principales de cadenas ligeras: kappa (κ) y lambda (λ). Cada molécula de anticuerpo está compuesta por dos cadenas pesadas y dos cadenas ligeras, que se unen entre sí mediante enlaces disulfuro para formar un tetramero.
Las cadenas ligeras están formadas por dos dominios: el dominio variable (V) y el dominio constante (C). El dominio variable es responsable de la especificidad antigénica del anticuerpo, mientras que el dominio constante participa en la unión con otras proteínas y células del sistema inmune.
Las cadenas ligeras se sintetizan en el retículo endoplásmico rugoso de las células plasmáticas y son secretadas al torrente sanguíneo o a la superficie de las células B como parte de los anticuerpos. En condiciones patológicas, como en los trastornos linfoproliferativos, pueden acumularse cadenas ligeras sin unirse a cadenas pesadas, lo que puede dar lugar a la formación de depósitos anormales de proteínas en tejidos y órganos, como en la amiloidosis sistémica.
La Fiebre Mediterránea Familiar (FMF) es una enfermedad genética poco común, inflamatoria y recurrente. Se caracteriza por ataques periódicos de fiebre alta, dolor abdominal, dolor torácico y erupción cutánea. Los ataques pueden durar desde unas horas hasta varios días. La enfermedad es causada por mutaciones en el gen MEFV, que codifica para la proteína pirogenóxica inducida por bacterias (PSTPIP1 o PYD), involucrada en la respuesta inflamatoria del cuerpo.
La FMF afecta predominantemente a personas de origen mediterráneo, especialmente aquellos de ascendencia turca, armenia, árabe y judía sefardí. Sin embargo, también se han reportado casos en otras poblaciones. El diagnóstico generalmente se realiza clínicamente y se confirma mediante pruebas genéticas. El tratamiento suele implicar el uso de medicamentos antiinflamatorios no esteroideos (AINE) durante los ataques y, en casos más graves, fármacos inmunomoduladores como la colchicina, que se utiliza para prevenir los ataques y reducir el riesgo de complicaciones a largo plazo, como la amiloidosis renal.
La neuropatía amiloide es un tipo de neuropatía periférica (una afección que daña los nervios fuera del cerebro y la médula espinal) causada por la acumulación anormal de una proteína llamada "amiloide" en los tejidos nerviosos. Esta acumulación puede dañar los nervios y provocar diversos síntomas, que pueden incluir entumecimiento, hormigueo, dolor, debilidad o pérdida de movimiento en las manos y los pies.
Existen varios tipos de neuropatías amiloides, cada una con causas diferentes. Algunas formas son hereditarias y están asociadas con mutaciones genéticas específicas que hacen que el cuerpo produzca versiones anormales de proteínas que se depositan como amiloide. Otras formas pueden ser adquiridas y están relacionadas con enfermedades subyacentes, como la enfermedad de Alzheimer, la diabetes o la enfermedad de Hodgkin.
El tratamiento de la neuropatía amiloides depende del tipo y de la gravedad de la afección. Puede incluir el control de los síntomas con medicamentos, fisioterapia o cambios en el estilo de vida, así como el tratamiento de las causas subyacentes si es posible. En algunos casos, un trasplante de células madre o un trasplante de médula ósea pueden ser opciones de tratamiento.
El término "Rojo Congo" no está reconocido en la medicina como un término específico o una afección médica. Sin embargo, "Rojo Congo" se refiere a un tipo de bacteria que puede causar infecciones. La bacteria específica es Serratia marcescens y se encuentra comúnmente en el medio ambiente, incluidos los suelos, las plantas y el agua.
Las infecciones por Serratia marcescens pueden ocurrir en diversas partes del cuerpo, como la sangre, los pulmones, la vejiga y la piel. Los síntomas de una infección por esta bacteria dependen de la ubicación de la infección en el cuerpo. Por ejemplo, una infección en la vejiga puede causar micción dolorosa o frecuente, mientras que una infección en los pulmones puede causar tos y dificultad para respirar.
Las personas con sistemas inmunológicos debilitados, como aquellas con enfermedades crónicas o aquellas que están tomando medicamentos inmunosupresores, pueden tener un mayor riesgo de desarrollar una infección por Serratia marcescens. El tratamiento de las infecciones por esta bacteria generalmente implica la administración de antibióticos.
Las neuropatías amiloides familiares (NAF) son un grupo de trastornos neurológicos hereditarios raros caracterizados por la acumulación de proteínas anormales llamadas amiloides en los nervios periféricos. Estas proteínas se producen como resultado de mutaciones genéticas y forman depósitos que dañan y destruyen los nervios, causando diversos síntomas neurológicos.
Existen varios tipos de NAF, cada uno asociado con diferentes mutaciones genéticas y patrones de presentación clínica. Algunos de los tipos más comunes incluyen:
1. La neuropatía amiloidótica familiar tipo 1 (NAF1 o TTR-FAP): Esta forma es causada por mutaciones en el gen transtiretina (TTR) y se caracteriza por la acumulación de amiloides en los nervios periféricos, el corazón y otros tejidos. Los síntomas neurológicos pueden incluir entumecimiento, hormigueo, dolor, debilidad y atrofia muscular en las extremidades, así como problemas de visión y audición.
2. La neuropatía amiloidótica familiar tipo 2 (NAF2 o AGel-FAP): Esta forma es causada por mutaciones en el gen apolipoproteína A1 (ApoA1) y se caracteriza por la acumulación de amiloides principalmente en los nervios periféricos. Los síntomas neurológicos suelen ser más graves que en la NAF1, con una progresión rápida hacia la discapacidad y la muerte.
3. La neuropatía amiloidótica familiar tipo 3 (NAF3 o GSN-FAP): Esta forma es causada por mutaciones en el gen gelsolina (GSN) y se caracteriza por la acumulación de amiloides en los nervios periféricos, el corazón y otros tejidos. Los síntomas neurológicos pueden incluir entumecimiento, hormigueo, dolor, debilidad y atrofia muscular en las extremidades, así como problemas de visión y audición.
4. Otras formas de neuropatía amiloidótica familiar: Existen otras mutaciones genéticas menos comunes que pueden causar neuropatías amiloidóticas familiares, como las mutaciones en el gen fibrinógeno (FGA), factor XII (Hageman) (F12) y transtiretina (TTR).
El tratamiento de la neuropatía amiloidótica familiar depende del tipo y la gravedad de los síntomas. Puede incluir medicamentos para aliviar el dolor, fisioterapia, terapia ocupacional y, en algunos casos, cirugía para tratar complicaciones como úlceras o infecciones. En los últimos años, se han desarrollado nuevos tratamientos dirigidos a reducir la producción de proteínas anormales que causan la acumulación de amiloide en los nervios, como la terapia con anticuerpos monoclonales y la terapia génica. Estos tratamientos pueden ayudar a ralentizar o incluso detener el progreso de la enfermedad en algunos pacientes.
Las cardiomiopatías se refieren a enfermedades del músculo cardíaco (miocardio) que afectan su estructura y función, lo que puede llevar a insuficiencia cardíaca o arritmias. Pueden ser clasificadas en varios tipos según sus características clínicas, etiológicas y patológicas. Algunos de los tipos más comunes incluyen:
1. Cardiomiopatía hipertrófica: Es una enfermedad genética que causa engrosamiento anormal del músculo cardíaco, lo que dificulta el llenado y la eyección de sangre desde el ventrículo izquierdo.
2. Cardiomiopatía dilatada: Es una enfermedad en la cual los ventrículos se agrandan y se debilitan, lo que lleva a un deterioro progresivo de la función cardíaca. Puede ser causada por diversas condiciones, como enfermedades metabólicas, infecciosas o genéticas.
3. Cardiomiopatía restrictiva: Es una enfermedad rara que causa endurecimiento del músculo cardíaco y dificulta el llenado de las cámaras cardíacas. Puede ser causada por enfermedades del tejido conectivo, infiltración de grasa o proteínas anormales.
4. Cardiomiopatía arritmogénica del ventrículo derecho: Es una enfermedad genética que afecta el músculo cardíaco del ventrículo derecho y puede causar arritmias graves y aumentar el riesgo de muerte súbita.
El tratamiento de las cardiomiopatías depende del tipo y la gravedad de la enfermedad, y puede incluir medicamentos, dispositivos médicos como marcapasos o desfibriladores implantables, cirugía o trasplante de corazón.
La microglobulina-2 beta, también conocida como Beta-2-microglobulina (β2M), es un componente proteico pequeño y ligero de los complejos mayor de histocompatibilidad de clase I (MHC de clase I). Los MHC de clase I son moléculas que presentan antígenos en la superficie celular y desempeñan un papel crucial en el sistema inmunitario adaptativo. La β2M se une a las cadenas pesadas alpha del MHC de clase I para formar un heterotrímero estable que participa en la presentación de péptidos endógenos al receptor de células T.
La β2M también se encuentra como componente de otras proteínas no relacionadas con el MHC, como las neonatales Fc receptores (FcRn), que participan en la homeostasis de las inmunoglobulinas y el transporte de péptidos a través de células.
La concentración sérica de β2M se utiliza como un marcador bioquímico de la disfunción renal, especialmente en la enfermedad renal crónica (ERC). Esto se debe a que el riñón es responsable de eliminar la β2M del torrente sanguíneo. Por lo tanto, un aumento en los niveles séricos de β2M puede indicar una disminución en la función renal o una sobrecarga antigénica. Además, altos niveles de β2M se asocian con un peor pronóstico y supervivencia en pacientes con ERC.
La proteína sérica amiloide A (SAA, por sus siglas en inglés) es una proteína de fase aguda producida principalmente en el hígado como respuesta a la inflamación. Cuando la SAA se acumula y no es completamente degradada, puede formar depósitos insolubles y anormales llamados componentes amiloides P (AP, por sus siglas en inglés). La amiloide P se asocia con una variedad de trastornos inflamatorios crónicos y enfermedades autoinmunes.
La acumulación de AP puede causar daño a los órganos y tejidos, lo que lleva a complicaciones graves y potencialmente fatales. Las manifestaciones clínicas de la amiloidosis por componentes P dependen del tipo y la ubicación de los depósitos amiloides. Los síntomas pueden incluir hinchazón, insuficiencia cardíaca, disfunción renal, neuropatía periférica e infiltración hepática.
El diagnóstico de la enfermedad por componentes amiloide P se realiza mediante una biopsia y el examen histológico del tejido afectado. El componente amiloide P puede identificarse mediante tinción con rojo congo o inmunohistoquímica específica para SAA. La evaluación de la carga de amiloides y la extensión de los depósitos se realiza mediante técnicas de imagen como la resonancia magnética nuclear o la tomografía computarizada.
El tratamiento de la enfermedad por componentes amiloide P implica el control de la inflamación subyacente y la prevención de la acumulación adicional de depósitos amiloides. Los medicamentos antiinflamatorios, como los corticosteroides, se utilizan a menudo para tratar la inflamación asociada con la enfermedad por componentes P. En algunos casos, se pueden considerar terapias específicas dirigidas contra el componente amiloide P, como la colestiramina o los anticuerpos monoclonales.
La prognosis de la enfermedad por componentes amiloide P depende de la extensión y la localización de los depósitos amiloides, así como del grado de disfunción orgánica asociada. La enfermedad puede ser progresiva y potencialmente mortal, especialmente si afecta al corazón o a los riñones. Sin embargo, con un diagnóstico temprano y un tratamiento apropiado, es posible controlar la enfermedad y mejorar la calidad de vida del paciente.
El Síndrome Nefrótico es un trastorno renal caracterizado por una serie de anomalías en la función glomerular que conllevan a una pérdida excesiva de proteínas en la orina (proteinuria severa, generalmente más de 3.5 g/día), disminución de las proteínas séricas, particularmente albumina (hipoalbuminemia), hinchazón o edema generalizado debido a la retención de líquidos y a menudo aumento de los lípidos en la sangre (hiperlipidemia).
Este síndrome puede ser causado por diversas enfermedades renales subyacentes, como glomerulonefritis, nefropatía diabética o enfermedad renal vascular entre otras. La presencia de este síndrome indica daño severo en los glomérulos, las estructuras intrarrenales responsables de filtrar los desechos líquidos del torrente sanguíneo.
La combinación de estas anormalidades puede llevar a complicaciones como infecciones, trombosis y falla renal progresiva si no se trata adecuadamente. El tratamiento generalmente involucra medidas para reducir la proteinuria, controlar los niveles de lípidos en sangre, prevenir las complicaciones y managear la enfermedad subyacente.
La paraproteinemia es un término médico que se refiere a la presencia de niveles anormalmente altos de proteínas anormales o "paraproteínas" en la sangre. Estas proteínas son producidas por células plasmáticas anormales, que son un tipo de glóbulo blanco del sistema inmunológico.
Las paraproteinemias pueden ser causadas por diversas afecciones, como el mieloma múltiple, un cáncer de las células plasmáticas que produce una gran cantidad de paraproteínas, y el macroglobulinemia de Waldenström, otro tipo de cáncer de células plasmáticas. También pueden estar presentes en enfermedades benignas como la gammapatía monoclonal de significado incierto (MGUS, por sus siglas en inglés), donde las paraproteínas se producen en niveles bajos y no suelen causar síntomas.
Es importante destacar que la presencia de paraproteinemias no siempre indica la existencia de una enfermedad grave, pero sí requiere un seguimiento médico cuidadoso, ya que puede indicar la presencia de afecciones subyacentes más graves.
Las enfermedades de la tráquea se refieren a un grupo diverso de condiciones que afectan la tráquea, que es el conducto membranoso y rígido situado justo debajo de la laringe y above the bronchial tubes. Su función principal es permitir que el aire fluya hacia los pulmones durante la inspiración y escape de los pulmones durante la espiración.
Algunas enfermedades comunes de la tráquea incluyen:
1. Estenosis traqueal: un estrechamiento anormal de la luz traqueal que puede ocurrir como resultado de una variedad de causas, como infecciones, trauma, tumores o enfermedades inflamatorias crónicas. Puede causar dificultad para respirar, especialmente durante el ejercicio.
2. Tráquea: Una tráquea dilatada o anormalmente grande que puede ser presente desde el nacimiento (congénita) o adquirida más tarde en la vida. Puede causar síntomas como tos, sibilancias y dificultad para respirar.
3. Tráqueitis: inflamación de la tráquea que puede ser causada por infecciones virales o bacterianas. Puede causar síntomas como tos, dolor de garganta y dificultad para respirar.
4. Tumores traqueales: crecimientos anormales en la tráquea que pueden ser benignos o malignos. Los tumores malignos pueden invadir los tejidos circundantes y diseminarse a otras partes del cuerpo, lo que puede causar una variedad de síntomas graves.
5. Tráqueo-bronquiectasia: una enfermedad pulmonar caracterizada por la dilatación anormal y la inflamación de los bronquios y las vías respiratorias más pequeñas. Puede causar síntomas como tos crónica, producción de esputo purulento y dificultad para respirar.
El tratamiento de estas afecciones dependerá de la causa subyacente y puede incluir medicamentos, terapia de oxígeno, cirugía o radioterapia. Si experimenta síntomas que puedan estar relacionados con una afección de la tráquea, es importante buscar atención médica lo antes posible.
Las Enfermedades Renales se refieren a cualquier condición o trastorno que cause daño a uno o ambos riñones y disminuya su capacidad para funcionar correctamente. Los riñones desempeñan un papel vital en mantener la salud general del cuerpo, ya que ayudan a filtrar los desechos y líquidos sobrantes de la sangre, producen hormonas importantes y regulan los niveles de electrolitos.
Existen diversas categorías de enfermedades renales, incluyendo:
1. Enfermedad Renal Aguda (ERA): Ocurre cuando los riñones sufren un daño repentino e intensivo, lo que puede llevar a una disminución grave o falla total de la función renal. La ERA puede ser reversible si se diagnostica y trata a tiempo. Algunas causas comunes incluyen infecciones severas, deshidratación, trauma, insuficiencia cardíaca congestiva, obstrucción del tracto urinario y exposición a ciertos medicamentos tóxicos.
2. Enfermedad Renal Crónica (ERC): Se caracteriza por un deterioro gradual y progresivo de la función renal durante un período prolongado, generalmente meses o años. La ERC puede resultar de diversas afecciones subyacentes, como diabetes, hipertensión arterial, enfermedades glomerulares, enfermedades poliquísticas renales y pielonefritis recurrente. A medida que la enfermedad avanza, los riñones pueden perder su capacidad de filtrar adecuadamente los desechos y líquidos, lo que puede conducir a complicaciones graves, como insuficiencia renal, anemia, hiperpotasemia e hiperfosfatemia.
3. Enfermedades Glomerulares: Estas enfermedades afectan los glomérulos, unidades funcionales del riñón responsables de la filtración de sangre. Las enfermedades glomerulares pueden ser primarias (afectar exclusivamente al riñón) o secundarias (resultado de otras afecciones sistémicas). Algunos ejemplos incluyen la nefropatía diabética, la glomerulonefritis rápidamente progresiva y el síndrome nefrótico.
4. Enfermedades Renales Hereditarias: Existen varias enfermedades renales hereditarias que pueden causar daño renal progresivo, como la enfermedad poliquística autosómica dominante (ADPKD), la enfermedad poliquística autosómica recesiva (ARPKD) y la nefropatía hereditaria de von Hippel-Lindau.
5. Enfermedades Renales Infecciosas: Las infecciones del tracto urinario (ITU) son comunes y, en la mayoría de los casos, se pueden tratar con éxito con antibióticos. Sin embargo, las ITU recurrentes o complicadas pueden provocar daño renal permanente. Otras infecciones renales incluyen la pielonefritis y la glomerulonefritis postinfecciosa.
6. Enfermedades Renales Inmunológicas: Las enfermedades renales inmunológicas son causadas por una respuesta anormal del sistema inmunitario que daña el riñón. Algunos ejemplos incluyen la glomerulonefritis membranosa, la glomerulonefritis membrano-proliferativa y la nefropatía lúpica.
7. Enfermedades Renales Toxicas: La exposición a sustancias tóxicas, como los medicamentos nefrotóxicos o el envenenamiento por metales pesados, puede causar daño renal agudo o crónico.
8. Enfermedades Renales Vasculares: Las enfermedades renales vasculares afectan el suministro de sangre al riñón y pueden ser causadas por hipertensión arterial, diabetes mellitus o enfermedades del tejido conectivo. Algunos ejemplos incluyen la nefropatía diabética, la nefrosclerosis y la glomeruloesclerosis focal segmentaria.
9. Enfermedades Renales Congénitas: Las enfermedades renales congénitas son aquellas que están presentes al nacer y pueden incluir anomalías estructurales, como el riñón poliquístico o la agenesia renal.
10. Enfermedades Renales Neoplásicas: Las enfermedades renales neoplásicas son aquellas que involucran el crecimiento anormal de células cancerosas en el riñón. Algunos ejemplos incluyen el carcinoma renal, el sarcoma renal y el linfoma renal.
En conclusión, existen diversas causas de enfermedades renales que pueden afectar la función renal y provocar complicaciones graves si no se tratan a tiempo. Es importante conocer los factores de riesgo y acudir al médico regularmente para detectar cualquier problema renal a tiempo.
La macroglosia es un término médico que se refiere al aumento del tamaño de la lengua. Puede variar en gravedad, desde una lengua ligeramente más grande de lo normal hasta una lengua excesivamente grande que interfiere con la función normal de habla, masticación, deglución y respiración. La macroglosia puede ser un rasgo distintivo de ciertas condiciones genéticas o adquiridas, como el síndrome de Down, el síndrome de Beckwith-Wiedemann, la amiloidosis y los tumores de la lengua. El tratamiento depende de la causa subyacente y puede incluir terapia del habla, cirugía o radioterapia.
Las proteínas amiloidogénicas son un tipo de proteína que tiene la capacidad de formar depósitos anormales y fibrilares conocidos como amiloides en diversos tejidos y órganos del cuerpo. Estas acumulaciones de amiloides pueden interferir con la función normal de los órganos y conducir a una variedad de enfermedades, llamadas generalmente trastornos amiloides sistémicos.
Existen diferentes tipos de proteínas amiloidogénicas, y cada una se asocia con un tipo específico de enfermedad del sistema amiloide. Algunos ejemplos incluyen:
1. Proteína precursora de amyloid-β (Aβ): Asociada con la enfermedad de Alzheimer y otras demencias relacionadas.
2. Proteína transtirretina (TTR): Vinculada con la polineuropatía amiloidótica familiar y la amiloidosis cardiaca senil.
3. Proteína ligasa de cadenas ligeras de inmunoglobulinas (AL, también conocida como proteína de M componente kappa o lambda): Relacionada con el mieloma múltiple y los trastornos relacionados con la producción monoclonal de inmunoglobulinas.
4. Proteína A de serum (SAA): Implicada en la amiloidosis reactiva asociada con diversas condiciones inflamatorias crónicas.
5. Proteína ATTR: Otra forma de transtirretina que puede causar amiloidosis cardiaca y neuropatía periférica.
La formación de amiloides se produce cuando las proteínas amiloidogénicas se pliegan incorrectamente y agreguen, formando fibrillas insolubles y estructuras anormales que dañan los tejidos circundantes. El diagnóstico y el tratamiento de los trastornos amiloidóticos dependen del tipo específico de proteína involucrada y de la gravedad y extensión de la enfermedad.
Melfalán es un agente quimioterapéutico alquilante que se utiliza en el tratamiento de ciertos tipos de cáncer. Su fórmula molecular es N-(fenilbutirato de sulfonilo)-N-cloruro de metiltrietilamina. Es un agente antineoplásico que funciona interfiriendo con la replicación del ADN del tumor, lo que inhibe su crecimiento y provoca la muerte celular.
Se utiliza en el tratamiento de cánceres como el mieloma múltiple y el sarcoma de Kaposi asociado al sida (SIDA). También se puede usar antes de un trasplante de células madre para disminuir la cantidad de células cancerosas en el cuerpo.
Como con cualquier forma de quimioterapia, el tratamiento con melfalán puede causar efectos secundarios graves, como náuseas, vómitos, diarrea, pérdida del apetito y debilidad. También puede afectar la médula ósea, disminuyendo la producción de células sanguíneas y aumentando el riesgo de infecciones, anemia y hemorragias. Además, en algunos casos, puede causar daño a los tejidos sanos, especialmente al sistema nervioso, los riñones y el tracto urinario.
Es importante que la administración y dosificación de este medicamento sea supervisada por un profesional médico capacitado, ya que una dosis demasiado alta puede causar graves daños en el paciente.
La proteína de Bence Jones es un tipo de proteína monoclonal de cadena ligera producida por algunos tipos de cánceres de células plasmáticas, como el mieloma múltiple y el macroglobulinemia de Waldenstrom. Se encuentra a menudo en la orina de los pacientes con estas enfermedades. Las proteínas de Bence Jones se llaman así por el médico británico Henry Bence Jones, quien las describió por primera vez en 1847. Estas proteínas pueden ser dañinas porque pueden unirse y formar depósitos en los riñones y otros tejidos, lo que puede causar daño renal y otros problemas de salud.
Una biopsia es un procedimiento médico en el que se extrae una pequeña muestra de tejido corporal para ser examinada en un laboratorio. Este procedimiento se realiza con el fin de evaluar si el tejido extraído presenta signos de enfermedad, como cáncer o inflamación.
Existen diferentes tipos de biopsias, dependiendo de la ubicación y el método utilizado para obtener la muestra de tejido. Algunas de las más comunes incluyen:
1. Biopsia por aspiración con aguja fina (FNA): se utiliza una aguja delgada y hueca para extraer células o líquido del bulto o área sospechosa.
2. Biopsia por punción con aguja gruesa (CNB): se emplea una aguja más grande para obtener una muestra de tejido sólido.
3. Biopsia incisional: se realiza una pequeña incisión en la piel y se extrae una parte del tejido sospechoso.
4. Biopsia excisional: se extirpa todo el bulto o área anormal, junto con una porción de tejido normal circundante.
Los resultados de la biopsia suelen ser evaluados por un patólogo, quien determinará si el tejido muestra signos de enfermedad y, en caso afirmativo, qué tipo de enfermedad es. La información obtenida de una biopsia puede ayudar a guiar el tratamiento médico y proporcionar información importante sobre la gravedad y extensión de la enfermedad.
'Acinonyx' es un género taxonómico que incluye a una sola especie de felino, el guepardo (*Acinonyx jubatus*). A diferencia de otros felinos, el guepardo no puede retraer sus garras completamente y tiene una estructura corporal adaptada para la velocidad máxima, con patas largas y una cola balanceadora. Es el félido más rápido, capaz de alcanzar velocidades de hasta 105 km/h (65 mph). A pesar de sus habilidades físicas, el guepardo es vulnerable a la extinción, principalmente debido a la pérdida de hábitat y la caza furtiva.
La definición médica de "cadenas lambda de inmunoglobulina" se refiere a un tipo específico de cadena proteínica que forma parte de las moléculas de inmunoglobulina, también conocidas como anticuerpos. Las cadenas lambda son uno de los dos tipos de cadenas ligeras que se unen a las cadenas pesadas para formar un anticuerpo funcional. El otro tipo de cadena ligera es la cadena kappa.
Cada molécula de inmunoglobulina está compuesta por dos cadenas pesadas y dos cadenas ligeras, que se unen entre sí mediante enlaces disulfuro para formar una estructura Y. Las cadenas lambda e kappa difieren en su secuencia de aminoácidos y en la región variable de la cadena, lo que les permite reconocer y unirse a una variedad de diferentes antígenos.
En humanos, aproximadamente el 60% de los anticuerpos contienen cadenas lambda, mientras que el resto contiene cadenas kappa. La presencia de cadenas lambda o kappa en un anticuerpo se utiliza a menudo como marcador para determinar la clonalidad de una población de células B, lo que puede ser útil en el diagnóstico y monitoreo de enfermedades hematológicas y linfoproliferativas.
La gelsolina es una proteína actina-binding que desempeña un papel crucial en la reorganización y el control del esqueleto de actina, un componente importante de la arquitectura celular. La gelsolina se une e incapacita para la polimerización los extremos "plus" (+) de los filamentos de actina F, pero también puede cortar los filamentos existentes en fragmentos más cortos y promover así la nucleación y el crecimiento de nuevos filamentos.
La localización subcelular y las funciones específicas de la gelsolina están determinadas por su regulación postraduccional, especialmente por la fosforilación y la unión a lípidos. La gelsolina se ha relacionado con diversos procesos celulares, como la migración y adhesión celular, el transporte vesicular, la endocitosis y la exocitosis, así como con enfermedades como el cáncer y las enfermedades neurodegenerativas.
La deficiencia del Factor X es una afección rara de la coagulación sanguínea que se hereda de manera autosómica recesiva. Esto significa que para desarrollar la afección, una persona necesita heredar dos copias del gen anormal, una de cada padre. Los padres generalmente no presentan síntomas de la enfermedad (portadores), ya que solo tienen una copia del gen anormal.
El Factor X es una proteína necesaria para la cascada de coagulación sanguínea, el proceso mediante el cual la sangre forma coágulos para detener el sangrado. Cuando está deficiente o no funciona correctamente, el proceso de coagulación se ralentiza y pueden producirse hemorragias prolongadas e incontroladas.
Existen dos tipos principales de deficiencia del Factor X:
1. Deficiencia del Factor X tipo I: Es una forma más común en la que tanto la cantidad como la función del Factor X están disminuidas.
2. Deficiencia del Factor X tipo II: En este tipo, la cantidad de Factor X es normal, pero su función está alterada.
Los síntomas de la deficiencia del Factor X pueden variar desde leves hasta graves y pueden incluir moretones fáciles, sangrado prolongado después de una lesión o cirugía, hemorragias nasales frecuentes, sangrado menstrual abundante en las mujeres y, en casos más graves, hemorragias internas que pueden poner en peligro la vida. El diagnóstico generalmente se realiza mediante pruebas de coagulación especializadas, y el tratamiento puede incluir infusiones de concentrado de complejo protrombínico activado o Factor X recombinante para controlar los episodios de sangrado.
Las enfermedades cutáneas genéticas se refieren a un grupo diverso de trastornos de la piel que son causados por defectos o mutaciones en los genes. Estas condiciones se heredan, generalmente de manera autosómica dominante o recesiva, y pueden afectar el desarrollo, la función o la apariencia de la piel.
Los síntomas y signos varían ampliamente dependiendo del tipo específico de enfermedad cutánea genética. Algunas condiciones causan lesiones cutáneas visibles en el nacimiento o durante la infancia, mientras que otras no se manifiestan hasta la edad adulta. Los síntomas pueden incluir erupciones cutáneas, sequedad extrema de la piel, hiperqueratosis (engrosamiento de la piel), ampollas, pigmentación anormal, pérdida de pigmento, aumento de la sensibilidad a la luz solar y crecimientos anormales de la piel.
Algunos ejemplos comunes de enfermedades cutáneas genéticas incluyen la enfermedad de Darier, el síndrome de Ehlers-Danlos, el síndrome de Goltz, el síndrome de Ichthyosis, la enfermedad de KID, el síndrome de Marfan, el síndrome de Neurofibromatosis, el síndrome de Sturge-Weber y la enfermedad de Tuberosa.
El tratamiento de las enfermedades cutáneas genéticas depende del tipo específico de trastorno y puede incluir medidas para aliviar los síntomas, como cremas hidratantes, protectores solares y medicamentos tópicos o sistémicos. En algunos casos, la terapia génica o el trasplante de células madre pueden ser consideraciones de tratamiento.
En términos médicos, un "resultado fatal" se refiere a un desenlace desfavorable de un diagnóstico, condición de salud, procedimiento o tratamiento que resulta en la muerte del paciente. Es un término formal y objetivo utilizado para describir una situación en la cual los esfuerzos terapéuticos no han podido revertir el curso de una enfermedad grave o lesión, y desafortunadamente conduce al fallecimiento del individuo.
Es importante mencionar que este término se utiliza con precaución y respeto, dada la naturaleza delicada y sensible de la situación. La comunicación de un resultado fatal a los familiares o cuidadores del paciente suele ser una parte difícil del trabajo médico, y se realiza siempre con empatía y compasión.
El mieloma múltiple es un tipo de cáncer que se origina en las plasmocitos, un tipo de glóbulos blancos presentes en la médula ósea. Los plasmocitos son células que producen anticuerpos para ayudar a combatir infecciones. En el mieloma múltiple, las células cancerosas acumulan en la médula ósea, donde desplazan a las células sanas y provocan una sobreproducción de un tipo de anticuerpo anormal llamado paraproteína M.
Esta acumulación de células cancerosas y la producción excesiva de paraproteínas M pueden llevar a diversas complicaciones, como:
1. Daño en los huesos: Las células cancerosas interfieren con la capacidad del cuerpo para mantener los huesos fuertes, lo que puede causar lesiones óseas y dolor.
2. Insuficiencia renal: La paraproteína M puede acumularse en los riñones y dificultar su funcionamiento, provocando insuficiencia renal.
3. Infecciones recurrentes: Los niveles bajos de glóbulos blancos sanos aumentan el riesgo de infecciones.
4. Anemia: La sobreproducción de células cancerosas desplaza a las células responsables de producir glóbulos rojos, lo que puede causar anemia y fatiga.
El mieloma múltiple se diagnostica mediante análisis de sangre, orina y médula ósea, y su tratamiento puede incluir quimioterapia, terapia dirigida, trasplante de células madre y radioterapia. El pronóstico y el plan de tratamiento dependen del estadio y la gravedad de la enfermedad, así como de la salud general del paciente.
No existe una definición médica específica para "Enciclopedias como Asunto" ya que esta frase parece ser una expresión coloquial o un título en lugar de un término médico. Sin embargo, si nos referimos al término "enciclopedia" desde un punto de vista educativo o del conocimiento, podríamos decir que se trata de una obra de consulta que contiene información sistemática sobre diversas áreas del conocimiento, organizadas alfabética o temáticamente.
Si "Enciclopedias como Asunto" se refiere a un asunto médico en particular, podría interpretarse como el estudio o la investigación de diferentes aspectos relacionados con las enciclopedias médicas, como su historia, desarrollo, contenido, estructura, impacto en la práctica clínica y la educación médica, entre otros.
Sin un contexto más específico, es difícil proporcionar una definición médica precisa de "Enciclopedias como Asunto".
La Dermatología es una rama especializada de la medicina que se ocupa del diagnóstico, tratamiento y prevención de las enfermedades de la piel, membranas mucosas, cabello y uñas. También incluye cosmetología médica, cirugía dermatológica, inmunodermatología e investigación en dermatología. Los dermatólogos son los médicos especialistas capacitados para manejar condiciones que varían desde acné, eccema, psoriasis hasta cáncer de piel y enfermedades inmunológicas graves de la piel.
La familia botánica Crassulaceae, también conocida como la familia de las plantas de grasa o las plantas de piedra, contiene alrededor de 1.400 especies en 33 géneros. Estas plantas suculentas se caracterizan por tener hojas y tallos engrosados que almacenan agua, lo que les permite sobrevivir en condiciones secas. Muchas de las especies son originarias de regiones áridas o semiáridas del mundo.
Las Crassulaceae incluyen plantas populares como las sedums, los echeverias, los sempervivums y los kalanchoes. La mayoría de estas plantas tienen flores pequeñas y discretas que se agrupan en inflorescencias compactas. Las flores suelen tener cuatro o cinco sépalos y pétalos, y pueden ser tubulares, en forma de campana o en forma de estrella.
Las Crassulaceae son interesantes desde el punto de vista médico porque algunas especies contienen compuestos bioactivos que tienen propiedades medicinales. Por ejemplo, los extractos de algunas especies de Sedum se han utilizado en la medicina tradicional china para tratar diversas afecciones, como la tos y el asma. Además, algunos estudios han sugerido que los compuestos presentes en las Crassulaceae pueden tener propiedades antiinflamatorias, antimicrobianas y anticancerígenas.
Sin embargo, es importante tener en cuenta que la investigación sobre las propiedades medicinales de las Crassulaceae es limitada y que se necesitan más estudios para confirmar sus beneficios y determinar sus posibles efectos secundarios. Por lo tanto, antes de utilizar cualquier planta de esta familia con fines medicinales, se recomienda consultar a un profesional médico capacitado.
MedlinePlus es un servicio de información de salud proporcionado por la Biblioteca Nacional de Medicina de EE. UU., que forma parte de los Institutos Nacionales de Salud (NIH). Ofrece información confiable y de alta calidad sobre enfermedades, condiciones y wellness, así como temas de salud para el consumidor. La información está disponible en inglés y español y es escrita en un lenguaje fácil de entender. También proporciona acceso a los recursos de salud de la National Library of Medicine, incluidos artículos médicos revisados por profesionales en PubMed, ensayos clínicos y estudios de salud, así como herramientas interactivas para ayudar a las personas a comprender mejor su salud.
El síndrome de Williams es un trastorno genético causado por la eliminación de una pequeña parte del cromosoma 7. Esto provoca problemas con varios aspectos del desarrollo y la salud, incluyendo un aspecto facial distintivo, problemas cardiovasculares (generalmente estenosis supravalvular aórtica), dificultades de aprendizaje leves a moderadas, retrasos en el desarrollo, problemas de comportamiento y habilidades sociales deficientes.
Las personas con síndrome de Williams también pueden tener una variedad de otros problemas de salud, como problemas renales, problemas de visión, problemas dentales y baja estatura. El síndrome de Williams afecta aproximadamente a 1 en cada 10.000 personas. No existe cura para el síndrome de Williams, pero los tratamientos pueden ayudar a controlar muchos de los problemas de salud asociados con él. La mayoría de las personas con síndrome de Williams llevarán una vida relativamente normal con un apoyo y educación adecuados.
Transtiretina21
- antes conocida como amiloidosis sistemica senil o ASS): causada por un mal plegamiento y agregación del tipo salvaje de transtiretina ( TTR ). (msdmanuals.com)
- Se trata de la Amiloidosis Hereditaria por Transtiretina. (infobae.com)
- En México cerca de 8 millones de personas padece este tipo de enfermedades2 como en el caso de la Amiloidosis Heredofamiliar, padecimiento que tiene más de 25 proteínas causantes3, entre ellas, la Asociada a la Transtiretina (hATTR), que afecta a unas 50,000 personas a nivel mundial4. (sobre-t.com)
- En junio se conmemora el Día Mundial de la Amiloidosis Hereditaria por Transtiretina , una enfermedad rara de origen genético que es crónica, discapacitante y fatal. (globedia.com)
- Una persona con Amiloidosis Hereditaria por Transtiretina, tiene el 50% de riesgo de heredar la enfermedad a sus hijos, dado su origen genético. (globedia.com)
- Por esto pensamos que para los médicos tratantes podría ser útil una pequeña guía con los puntos clave en el diagnóstico y tratamiento de la amiloidosis cardiaca por transtiretina . (medscape.com)
- A pesar de que en este posteo nos vamos a enfocar en las formas por transtiretina (TTR), es importante conocer el resto de las variantes. (medscape.com)
- la transtiretina es una proteína tetramérica producida normalmente por el hígado. (medscape.com)
- Con el advenimiento de terapias efectivas para la amiloidosis por transtiretina, existe una creciente necesidad de reconocer los primeros signos de enfermedad e identificar las poblaciones en riesgo para el screening . (medscape.com)
- Los primeros signos de amiloidosis por transtiretina en las formas salvajes a menudo son extracardiacos y preceden al diagnóstico clínico de miocardiopatía entre 5 y 7 años. (medscape.com)
- La rotura del tendón del bíceps y la estenosis espinal lumbar también pueden estar asociados con la miocardiopatía amiloidosis por transtiretina en las formas salvajes. (medscape.com)
- Además de estos primeros signos, hay grupos con tasas más altas de miocardiopatía en amiloidosis por transtiretina, incluidos los hombres con más de 65 y la mujeres con más de 70 años, los pacientes de ascendencia afro-caribeña (para las forma V122I de amiloidosis por transtiretina en formas hereditarias). (medscape.com)
- La presencia de insuficiencia cardiaca y / o hipertrofia ventricular en estos grupos, debe levantar sospechas de miocardiopatía amiloidosis por transtiretina e impulsar una investigación más a fondo. (medscape.com)
- Se han sido identificado casos de enfermedad familiar hereditaria y otros casos asociados al proceso de envejecimiento que son causados por una proteína particular conocida como transtiretina (TTR). (galenusrevista.com)
- La identificación de la transtiretina como causante común de casos de amiloidosis cardiaca primaria es de suma importancia, ya que se han logrado desarrollar tratamientos farmacológicos que actúan específicamente en la estabilización de la estructura de esta proteína, evitando su deformación. (galenusrevista.com)
- Los webinars van dirigidos a personas cuidadoras de pacientes con Amiloidosis Hereditaria por Transtiretina y Síndrome de Quilomicronemia Familiar. (andradebalear.es)
- Las dos proteínas fundamentales que dan lugar a la amiloidosis cardíaca son fragmentos de cadenas ligeras que constituyen la amiloidosis AL y fragmentos de una proteína fabricada en el hígado denominada transtiretina cuya amiloidosis se llama amiloidosis ATTR. (fundacionfic.es)
- Esto se consigue mediante quimioterapia en caso de la amiloidosis AL y, en el caso de la amiloidosis por transtiretina, se están desarrollando nuevos tratamientos que consiguen impedir que se generen fragmentos de la transtiretina o bien bloquear la fabricación de esta sustancia directamente", afirma el Dr. Pavía. (fundacionfic.es)
- La polineuropatía amiloidótica familiar (PAF), enfermedad de Andrade o amiloidosis hereditaria relacionada con la transtiretina, es una enfermedad genética neurodegenerativa grave. (genosalut.com)
- Acto seguido veamos que tipo de amiloidosis es: Un examen genético daria esta información para ello la muestra de sangre viajó a Estados Unidos y los resultados irrefutables AMILOIDOSIS FAMILIAR POR TRANSTIRETINA en ese momento con la nomenclatura Val30Met. (worldamyloidosisday.org)
- Considerado durante mucho tiempo como un sueño imposible para las grandes farmacéuticas, las terapias de ARNi finalmente llegaron a la mayoría de edad el verano pasado cuando Alnylam se convirtió en la primera compañía en obtener un medicamento de esta clase aprobado por la FDA, con su Onpattro (patisiran) para la rara enfermedad amiloidosis hereditaria por transtiretina. (femexer.org)
Tratamiento de la amiloidosis2
- La terapia combinada con venetoclax, citrato de ixazomib y dexametasona puede ser eficaz en el tratamiento de la amiloidosis de cadenas ligeras en recaída o refractaria. (oneamyloidosisvoice.com)
- Gracias a los avances en el diagnóstico y tratamiento de la amiloidosis cardiaca en los últimos años, el número de pacientes diagnosticados se ha incrementado exponencialmente. (medscape.com)
Cardiaca10
- La amiloidosis cardiaca es una enfermedad compleja y muchas veces su diagnóstico se retrasa mucho tiempo. (amiloidosis.es)
- Los pacientes con amiloidosis cardiaca a menudo tienen síndrome del túnel carpiano. (misaludmemueve.com.co)
- Los pacientes con amiloidosis cardiaca a menudo tienen síndrome del túnel carpiano, que aparece tras las manifestaciones cardiacas después de varios años. (misaludmemueve.com.co)
- 1. ¿Cuántas formas de amiloidosis cardiaca existen? (medscape.com)
- Entre estas condiciones, se ha observado con mayor predominancia la amiloidosis cardiaca. (galenusrevista.com)
- Para poder comprender la magnitud de la amiloidosis cardiaca es importante que recordemos la función de las proteínas en nuestro organismo. (galenusrevista.com)
- La amiloidosis cardiaca puede presentarse como resultado de una enfermedad sistémica o puede desarrollarse como una condición primaria del corazón. (galenusrevista.com)
- La presentación clínica de la amiloidosis cardiaca suele ser muy similar a la presentación de un paciente con insuficiencia cardiaca por otras causas. (galenusrevista.com)
- El diagnóstico de amiloidosis cardiaca puede ser un reto para el médico especialista. (galenusrevista.com)
- Amiloday es un programa formativo desarrollado en el Hospital Puerta de Hierro desde 2018", explica el Dr. Pablo García Pavía, director de la Unidad de Insuficiencia Cardiaca y Cardiopatías Familiares del Hospital Puerta de Hierro y co-director del curso. (fundacionfic.es)
Amiloide14
- La amiloidosis heredofamiliar es una condición genética hereditaria que se caracteriza por el depósito sistémico o localizado de material amiloide en los tejidos del cuerpo humano. (wikipedia.org)
- Los síntomas de la amiloidosis dependerán del subtipo de la enfermedad y del órgano que esté afectado por el depósito de amiloide. (amiloidosis.es)
- Los depósitos de amiloide se tiñen de rosa con hematoxilina y eosina, contienen constituyentes de hidratos de carbono que se tiñen con ácido peryódico Schiff o con azul Alcián, pero lo más característico es su birrefringencia verde manzana con microscopia de luz polarizada después de la tinción con rojo Congo. (msdmanuals.com)
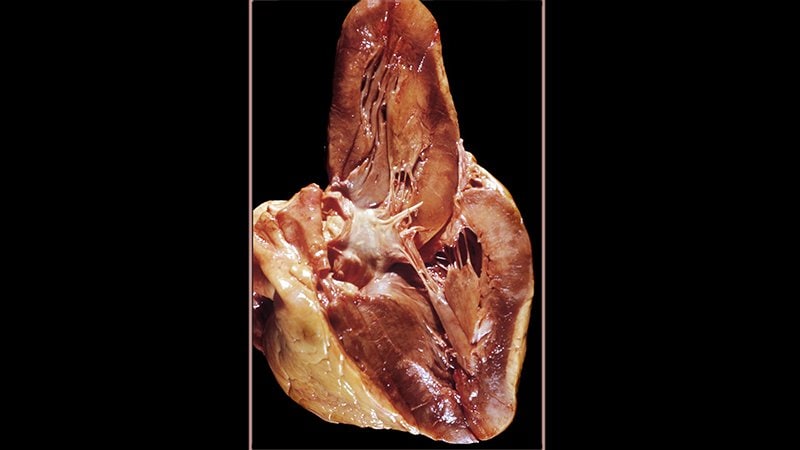
- Micrografía revelando material amiloide (mancha rojiza) tras teñir tejido cardíaco con rojo Congo en un caso de amiloidosis cardíaca . (wikipedia.org)
- Amiloidosis es un término genérico, utilizado para hacer referencia a un grupo de enfermedades de etiología diversa y pronóstico y tratamiento variables, con una característica común: todas ellas están causadas por el depósito extracelular de un material, denominado material amiloide . (wikipedia.org)
- El material amiloide es de origen proteico, insoluble y resistente a la proteólisis . (wikipedia.org)
- Puede teñirse con diversas técnicas, como la de hematoxilina-eosina y la tioflavina T, pero la característica más típica del amiloide es su tinción con rojo Congo , tras la cual adquiere birrefringencia verde manzana al ser expuesto a la luz polarizada. (wikipedia.org)
- Estas características relacionadas con la tinción se deben a la estructura química adquirida por los precursores proteicos que forman el amiloide, independientemente de su naturaleza y es, por lo tanto, común a todos ellos. (wikipedia.org)
- Cualquier proteína que adquiera ese plegamiento y esas características físico-químicas, independientemente de la secuencia de aminoácidos, es amiloide. (wikipedia.org)
- La amiloidosis es una enfermedad rara en la que unas proteínas plegadas de manera anormal forman unos agregados denominados fibrillas de amiloide que se acumulan en diversos tejidos y órganos, lo que en ocasiones da lugar a un deterioro del funcionamiento normal de los órganos, insuficiencia orgánica y muerte. (merckmanuals.com)
- Es un trastorno provocado por depósitos de una proteína anormal (amiloide) en el tejido cardíaco que dificulta el trabajo apropiado del corazón. (misaludmemueve.com.co)
- El síndrome del túnel carpiano puede servir como una señal de advertencia temprana muchos años antes del inicio de la amiloidosis cardíaca, brindando a los médicos amplias oportunidades para detectar y monitorear el desarrollo de amiloide. (misaludmemueve.com.co)
- La amiloidosis es una enfermedad rara y progresiva que ocurre cuando una proteína anormal llamada amiloide se acumula en los órganos e impide que el corazón, los riñones, el hígado, el bazo, el sistema nervioso y el tracto digestivo, funcionen correctamente1. (sobre-t.com)
- La amiloidosis es una condición producida por la acumulación de un material proteico conocido como fibrillas de amiloide, las cuales son producidas por la agregación de proteínas que han sido deformadas en su estructura. (galenusrevista.com)
Enfermedad rara5
- Aunque se habla de la amiloidosis cardíaca como una enfermedad rara, los médicos dicen que no lo es tanto. (telecinco.es)
- La familia del general retirado anunció el pasado mes de junio el ingreso del líder paquistaní en el Hospital Americano de Dubái por un agravamiento de la amiloidosis que padecía, una enfermedad rara que ocurre cuando una proteína anormal se acumula en los órganos e interfiere con las funciones normales del cuerpo. (diariopalentino.es)
- Si esto sumamos que es una enfermedad rara o minoritaria y en consecuencia no se sabe mucho sobre la misma, esto pone todavía más problemas al diagnóstico final. (chiquipedia.com)
- Siendo una enfermedad rara de difícil diagnóstico y tratamiento costoso en el año 2016 se crea la Asociación Venezolana de Pacientes con Amiloidosis Familiar AVPAF cuyo objetivo principal es informar sobre esta dolencia y acercar pacientes con el mismo diagnóstico. (worldamyloidosisday.org)
- Akcea / Ionis también tienen Waylivra (volanesorsen) aprobado en Europa para el síndrome de quilomicronemia familiar, una enfermedad rara aunque ha sido rechazado por la FDA. (femexer.org)
Primaria5
- me gustaria contactar con alguien que le hayan diagnosticado amiloidosis primaria tengo a mi marido con esta enfermedad y no encuentro a nadie con quien compartir mis inquietudes por favor si alguien quiere dejar algun comentario sobre esta enfermedad estamos pasando un calvario y ademas tenemos ni os peque os. (portalesmedicos.com)
- Antiguamente se la llamaba amiloidosis primaria. (medscape.com)
- Su causa primaria más frecuente es el mieloma múltiple, pero no la única. (medscape.com)
- La identificación y el tratamiento de la condición primaria causante de la amiloidosis se consideran como la primera línea en el manejo de estos pacientes (ej. (galenusrevista.com)
- la fiebre mediterránea familiar es poco conocida para los compañeros de Atención Primaria, pero no para los pediatras. (noticiasensalud.com)
Rara2
- Hay una forma hereditaria rara de amiloidosis beta-2-microglobulina debida a una mutación en el gen relevante. (msdmanuals.com)
- A día de hoy pensamos que no es tan rara como creíamos antes, aunque no es habitual", puntualiza este doctor. (telecinco.es)
Cadenas1
- La Alfa Talasemia es una hemoglobinopatía hereditaria caracterizada por un fallo en la síntesis de las cadenas de globina-alfa, que da lugar a un cuadro clínico variable dependiendo del número de alelos afectados. (ceifer.com)
Desarrollar amiloidosis2
- Para desarrollar amiloidosis, además de la producción de las proteínas amiloidogénicas, es probable que también exista un fracaso de los mecanismos de eliminación normales para este tipo de proteínas mal plegadas. (msdmanuals.com)
- según las guías de tratamiento vigentes publicadas por la Liga Europea contra las enfermedades reumáticas (EULAR, en sus siglas en inglés) el tratamiento de elección de la fiebre mediterránea familiar siempre debe ser la colchicina, ya que este fármaco ha demostrado disminuir la frecuencia y la intensidad de los brotes, controlando la enfermedad, así como reducir el riesgo de desarrollar amiloidosis AA, la principal complicación de esta patología. (noticiasensalud.com)
Trastornos3
- La amiloidosis incluye un grupo de trastornos dispares caracterizados por el depósito extracelular de fibrillas insolubles compuestas por proteínas agrupadas irregularmente. (msdmanuals.com)
- La amiloidosis puede presentarse de novo o ser secundaria a varias infecciones, trastornos inflamatorios o enfermedades malignas. (msdmanuals.com)
- Estas denominadas proteopatías incluyen trastornos neurológicos tales como enfermedad de Alzheimer, enfermedad de Parkinson y enfermedad de Huntington, así como trastornos sistémicos diversos, incluyendo las amiloidosis. (patentados.com)
Tipos2
- Una serie de proteínas normales (de tipo salvaje) y mutantes son susceptibles de presentar ese plegamiento y agregación anormales (proteínas amiloidogénicas), lo que explica la gran variedad de causas y tipos de amiloidosis. (msdmanuals.com)
- Todos los tipos de amiloidosis implican una proteína que se pliega de forma anormal. (merckmanuals.com)
Antecedentes familiares2
- Síndrome del túnel carpiano y/o antecedentes familiares del mismo, dolor de espalda crónico, ruptura del tendón del músculo bíceps. (globedia.com)
- Para ese entonces había sufrido varias crisis y fue una de ellas la que propició mi hospitalización, punción lumbar, interrogatorios, busqueda de antecedentes familiares y otras preguntas relevantes. (worldamyloidosisday.org)
Pacientes y familiares1
- El Consejo de CN recomienda la utilización de barbijo quirúrgico en todos los pacientes y familiares durante su estadía en las instalaciones. (sac.org.ar)
Secundaria3
- Esta enfermedad se puede complicar, siendo una de las peores complicaciones la amiloidosis secundaria. (chiquipedia.com)
- Y por supuesto, gracias a que las inflamaciones son menos recurrentes y agudas, esto ayuda que problemas secundarios como la amiloidosis secundaria no sean un problema. (chiquipedia.com)
- Antiguamente llamada amiloidosis secundaria. (medscape.com)
Denominada2
- 1] Polineuropatía amiloidótica familiar provocada por una proteína anómala denominada transtirretina. (wikipedia.org)
- Tuberculosis (TB) La tuberculosis es una infección contagiosa crónica causada por una bacteria, transportada por el aire, denominada Mycobacterium tuberculosis . (merckmanuals.com)
Enfermedades Raras3
- Es una de las Enfermedades Poco Frecuentes que afecta el sistema nervioso, corazón, tracto gastrointestinal ", explicó, en diálogo con Infobae , el doctor César Crespi ( MP115.409 / MN 124 765), Médico Hepatólogo del Centro de Referencia en Enfermedades Raras y de Dificultoso Diagnóstico (CERyD) del Hospital San Juan de Dios, de La Plata. (infobae.com)
- Y es aquí donde surge el problema para el diagnóstico de las enfermedades raras, ya que los cuadros clínicos para las mismas: son poco conocidos, son complicados por su número y diversidad o incluso no existen (si se trata de una enfermedad todavía no descrita). (genosalut.com)
- Encuesta mundial: ¿Es necesario tamizar a los recién nacidos con enfermedades raras? (femexer.org)
Causas1
- Esto es lo que debe saber sobre las causas, los síntomas, el diagnóstico y el tratamiento de este trastorno poco común. (colgate.com)
Formas4
- Las formas localizadas de amiloidosis parecen ser causadas por la producción local y el depósito de una proteína amiloidogénica (con mayor frecuencia inmunoglobulinas de cadena ligera) en el órgano afectado en lugar del depósito de proteínas circulantes. (msdmanuals.com)
- Caben varias formas de clasificación de la amiloidosis. (wikipedia.org)
- Hay muchas formas de amiloidosis, y se requieren otras pruebas para identificar la forma y su causa. (merckmanuals.com)
- En la facultad de Medicina nos enseñaron que existen múltiples formas de amiloidosis con extraños nombres y codificación. (medscape.com)
Salud3
- Si usted es un médico u otro profesional de la salud calificado, no debe ofrecer ningún consejo o tratamiento médico en nuestros Sitios, ni debe permitir que el contenido de nuestros Sitios sustituya su propio criterio médico. (oneamyloidosisvoice.com)
- Su salud y la amiloidosis hereditaria por TTR pdf pág 6. (sobre-t.com)
- La finalidad de este artículo es fomentar la comprensión y el conocimiento de temas generales de salud oral. (colgate.com)
Riesgo2
- Los estudios demuestran que los pacientes con síndrome de túnel del carpo, tenían un riesgo elevado de diagnóstico futuro de amiloidosis, con una media de tiempo desde la cirugía hasta el diagnóstico de 3,1 años. (misaludmemueve.com.co)
- El objetivo de este documento es orientar a los laboratorios de cardiología nuclear, sobre las mejores prácticas a desarrollar durante la pandemia COVID-19, para mitigar el riesgo de transmisión y contagio. (sac.org.ar)
Debida1
- Amiloidosis corneal y encefálica finlandesa debida a una gelsolina anómala. (wikipedia.org)
Tipo6
- Según el tipo hablamos de un tipo de amiloidosis u otro. (amiloidosis.es)
- El tratamiento depende del tipo de amiloidosis. (msdmanuals.com)
- Hay que recordar que este problema no es exclusivo de esta enfermedad, sino que viene dado por el mal control de cualquier enfermedad que provoque inflamaciones de cualquier tipo. (chiquipedia.com)
- Es de gran importancia poder definir el tipo de amiloidosis ya que, de esta manera, podríamos ofrecer alternativas de tratamiento a la persona afectada. (galenusrevista.com)
- Sin tratamiento, la esperanza de vida de los pacientes se sitúa entre el año y los dos años y medio dependiendo del tipo de amiloidosis. (fundacionfic.es)
- El Mieloma Múltiple es un tipo de cáncer que afecta a las células sanguíneas en el interior del hueso. (geomedica.com.ar)
Puede ser2
- 3 A su vez, el corazón es uno de los órganos donde se depositan las proteínas anormales que causan la amiloidosis cardíaca, una enfermedad que puede ser adquirida, pero también puede ser hereditaria. (misaludmemueve.com.co)
- Con un adecuado tratamiento, en cambio, la tasa de supervivencia puede ser mucho mayor y ascender hasta los 5-10 años o más, por lo que un correcto diagnóstico y seguimiento es fundamental para alargar la vida de estas personas. (fundacionfic.es)
Caso1
- En caso de que sea profunda, y estos dispositivos no sean eficaces, aprender el lenguaje de signos en la infancia es muy importante. (ceifer.com)
Cursa1
- Amiloidosis cerebrovascular que cursa con hemorragia cerebral. (wikipedia.org)
Causar1
- Otra afección que se puede presentar es la leucopenia (escasez de glóbulos blancos normales), lo cual puede causar problemas para combatir infecciones . (geomedica.com.ar)
Hipotiroidismo3
- También puede estar relacionada con algunas afecciones adquiridas, como ciertas enfermedades metabólicas y del sistema endocrino, como el hipotiroidismo o la amiloidosis. (colgate.com)
- Por ejemplo, si se determina que la causa es el hipotiroidismo, el tratamiento de este también podría ayudar a tratar la macroglosia. (colgate.com)
- Algunas afecciones médicas, como el hipotiroidismo, la acromegalia, la amiloidosis, el síndrome de Cushing y el síndrome de Down, también se han asociado a la apnea obstructiva del sueño. (medscape.com)
Diversas1
- Los trasplantes de médula ósea consiguen aportar sus beneficios a individuos que presentan diversas enfermedades de origen cancerosas, (es decir malignas) como también de aquellas enfermedades que no son cancerosas (es decir benignas). (tucuerpohumano.com)
20231
- En el Congreso Internacional 2023 de la European Respiratory Society (ERS) , que tuvo lugar en Milán, Italia, la Dra, Cláudia Sofia De Almeida Vicente Ferreira, médica familiar de Coimbra, Portugal, y coordinadora del Respiratory Diseases Interest Group (GRESP) de la Portuguese Association of General and Family Medicine (APMGF), destacó los retos que plantea el diagnóstico de la apnea obstructiva del sueño. (medscape.com)
Nomenclatura2
- Clásicamente, se hacía una clasificación de las amiloidosis atendiendo a sus características clínicas y, por tanto, la nomenclatura seguía esta tendencia. (wikipedia.org)
- Esta clasificación pronto se reveló insuficiente e incómoda a la hora de manejar a los pacientes con esta enfermedad, por lo que en 1998 , las normas para la clasificación y nomenclatura de la amiloidosis fueron revisadas por el Nomenclature Committee of the International Society of Amyloidosis . (wikipedia.org)
Llama1
- Se llama amiloidosis cardíaca . (telecinco.es)
Reto2
- La amiloidosis un reto de diagnóstico. (sobre-t.com)
- más que un reto terapéutico que, por supuesto lo es, la FMF supone un reto diagnóstico. (noticiasensalud.com)
Dolor3
- Artritis reumatoide La artritis reumatoide es una artritis inflamatoria en la que las articulaciones, entre las que se suelen incluir las de manos y pies, se inflaman, dando lugar a hinchazón, dolor y frecuentemente. (merckmanuals.com)
- Cuando la presión dentro del túnel es muy alta y altera la función normal del nervio, aparecen rigidez, hormigueo y dolor en la mano y los dedos. (misaludmemueve.com.co)
- Uno de los síntomas más comunes es el dolor abdominal, vómitos, estreñimientos y la peritonitis inflamatoria no infecciosa. (chiquipedia.com)
Fragmentos1
- La amiloidosis cardíaca es una grave patología mediante la cual fragmentos de proteínas se depositan en el corazón, engrosando sus paredes e impidiendo su normal funcionamiento. (fundacionfic.es)
Casos3
- Al ser una enfermedad poco frecuente, su diagnóstico puede demorar hasta 4 años en promedio.6 "Incluso, se estima que en México la prevalencia de hATTR es de 0.89 casos por cada 100 mil habitantes y los estados con más casos son Morelos, Guerrero, Ciudad de México y Guanajuato"7, comentó la Dra. (sobre-t.com)
- La literatura sobre este tema no es muy abundante y la encontrada no aborda sobre epidemiología de la enfermedad, pues en la mayoría solo se limita a discutir los casos presentados. (sld.cu)
- En gran parte de los casos, previamente al procedimiento de trasplante es requerido, eliminar aquella médula ósea , perteneciente al receptor, a través de tratamientos como la radioterapia o la quimioterapia e incluso ambas a la vez. (tucuerpohumano.com)
Medicina3
- Diana Sánchez, especialista en genética clínica y medicina familiar. (globedia.com)
- Sin embargo, el diagnóstico correcto es indispensable en medicina para poder seguir actuando. (genosalut.com)
- La fiebre mediterránea familiar (FMF), la artritis idiopática juvenil de inicio sistémico y la enfermedad de Still del adulto son enfermedades sistémicas, que tocan distintas esferas del organismo y que, en tiempos de medicina de precisión-medicina personalizada y dianas terapéuticas, son objeto de una alta exigencia en la obtención del control-remisión y de la excelente calidad de vida. (noticiasensalud.com)
Respuesta1
- La respuesta es sí, la fiebre mediterránea familiar es una enfermedad hereditaria que se puede transmitir sin problemas de padres a hijos. (chiquipedia.com)
Artritis1
- La AIJ sistémica/enfermedad de Still del adulto es una patología autoinflamatoria en la que predominan, además de la fiebre, los síntomas sistémicos y la artritis. (noticiasensalud.com)
Menudo1
- Estas tecnologías permiten determinar la causa en una proporción considerable (hasta el 40%) de pacientes no diagnosticados, y lo que es más importante, a menudo con hallazgos que permiten tomar medidas terapéuticas para hacer frente a la enfermedad. (genosalut.com)
Afectar1
- La amiloidosis es capaz de afectar esencialmente cualquier tejido de nuestro cuerpo y es usualmente causada por algún proceso inflamatorio, por infecciones, por malignidades y por enfermedades autoinmunes. (galenusrevista.com)
Frecuente1
- Es una enfermedad genética poco frecuente, progresiva y de difícil diagnóstico que afecta a los adultos ", resumió Crespi. (infobae.com)
Conocida1
- Debes saber que la Fiebre Mediterránea familia es también conocida bajo las siglas (FMF). (chiquipedia.com)
Personas7
- Sólo alrededor del 10 al 20% de las personas con mieloma múltiple desarrollan amiloidosis AL. (merckmanuals.com)
- Es difícil saber cuántas personas la padecen en España, aunque se estima que " alrededor de 2.000 personas" . (telecinco.es)
- Es una enfermedad de personas mayores. (telecinco.es)
- Es posible que algunas personas no presenten síntomas. (misaludmemueve.com.co)
- Por su origen hereditario, el llamado es a que las personas que presenten síntomas, especialmente en las regiones más afectadas, acudan oportunamente y sean valorados por un médico especialista que pueda enviar las pruebas diagnósticas para confirmar la enfermedad y así iniciar un manejo multidisciplinario donde el neurólogo juega un papel fundamental. (globedia.com)
- Noviembre es el mes de las personas cuidadoras y para celebrarlo Akcea, Ionis y Partners for Patients han organizado unos webinars gratuitos. (andradebalear.es)
- Es posible que las personas que padecen esta afección ronquen o emitan sonidos sibilantes al respirar. (colgate.com)
Unidad3
- Esa sustancia se deposita en determinados órganos y provoca que esos órganos funcionen mal", explica a NIUS Pablo García-Pavía, jefe de la Unidad de Cardiopatías Familiares del Hospital Universitario Puerta de Hierro e investigador del CIBERCV. (telecinco.es)
- La nefrona es la unidad funcional , existiendo aproximadamente un millón en cada riñón. (fdocuments.es)
- Web de la Unidad de Cardiopatías Familiares del Hospital Universitario Puerta de Hierro de Majadahonda. (cardiopatiasfamiliares.es)
Tratamientos3
- Existe una asociación para la defensa de los pacientes con amiloidosis , Amiloidosis Visible , que ha reclamado ante Sanidad la financiación de los tratamientos contra su enfermedad. (telecinco.es)
- Revise detenidamente la información provista en nuestros Sitios antes de decidir si alguno de los productos, servicios o tratamientos en los mismos es adecuado para usted o para otros. (oneamyloidosisvoice.com)
- Los tratamientos médicos se pueden poner en práctica cuando la causa es tanto identificable como tratable. (colgate.com)
Cadena1
- Amiloidosis renal y hepática por una anomalía de la cadena alfa del fibrinógeno, se presenta en México y EE. (wikipedia.org)
Mundial1
- Este mes de septiembre se celebra el Día Mundial del Corazón, el objetivo de esta fecha es generar conciencia sobre las enfermedades. (saludyvida.tips)
Alteraciones1
- La causa de esta enfermedad es un defecto en el gen ESAM, que se manifiesta en alteraciones la barrera hematoencefálica. (genosalut.com)
Examen2
- Este examen es una poderosa herramienta para diagnosticar miles de enfermedades genéticas. (mendelics.com.br)
- De acuerdo con la NORD, para confirmar un diagnóstico de macroglosia su médico podrá preguntarle por su historial clínico familiar y realizar un examen físico. (colgate.com)
Padre1
- Tito era un hombre familiar, con su padre de más de noventa y su hermano solían salir a pescar con un bote que lanzaban en Magalluf donde tenía con su esposa un apartamento en el que pasaba la temporada estival. (mallorcadiario.com)
Madre1
- El trasplante de médula ósea, es un método médico que se lleva a cabo con el fin de quitar o eliminar la médula ósea del paciente la que se encuentra dañada o incluso destruida y sustituirla por células madre perteneciente a la médula ósea sana. (tucuerpohumano.com)
Tejido2
- La médula ósea es el tejido blando que se encuentra dentro de los huesos. (geomedica.com.ar)
- En lo que respecta a la médula ósea, tal es un tejido que además de grasoso, es asimismo blando que se halla localizada en los huesos . (tucuerpohumano.com)
Importante3
- Como he comentado antes, la probabilidad de sufrir la enfermedad si alguno de los padres la sufre es importante, debido a que es una enfermedad hereditaria. (chiquipedia.com)
- Esta semana es muy importante para nosotros. (andradebalear.es)
- Desde ABEA sabemos lo importante que es sentirse acompañado cuando te diagnostican una enfermedad. (andradebalear.es)
Hipertensa1
- Si, es Hipertensa y Diabética. (worldamyloidosisday.org)
Ambas1
- Túnel del carpo en ambas manos y palpitaciones: ¿podrían ser síntomas de amiloidosis cardíaca? (misaludmemueve.com.co)
Manejo2
- Teniendo en cuenta lo anterior, es indispensable conocer sus signos y síntomas para hacer un diagnóstico y manejo oportuno[1]. (globedia.com)
- Nos enfocamos en esta forma porque es la que depende casi en exclusividad del manejo cardiológico. (medscape.com)