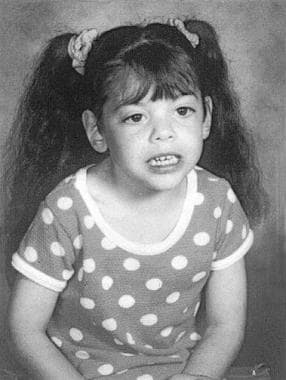Smith-Lemli-Opitz Syndrome
Hypertelorism
Hypospadias
Nuclear Proteins
Transcription Factors
Anus, Imperforate
Genetic Diseases, X-Linked
Abnormalities, Multiple
Down Syndrome
Metabolic Syndrome X
Microtubules
Nephrotic Syndrome
Sjogren's Syndrome
Turner Syndrome
Myelodysplastic Syndromes
Cushing Syndrome
Acute Coronary Syndrome
Polycystic Ovary Syndrome
Williams Syndrome
Oxidized derivatives of 7-dehydrocholesterol induce growth retardation in cultured rat embryos: a model for antenatal growth retardation in the Smith-Lemli-Opitz syndrome. (1/122)
7-Dehydrocholesterol accumulates in fetuses affected by the Smith-Lemli-Opitz syndrome as a result of a deficit in the ultimate step of cholesterol synthesis catalyzed by Delta7 reductase. Rat embryos explanted at gestation day 10 and cultured for 48 h in the presence of the Delta7 reductase inhibitor AY 9944 were used as a model to discriminate between the beneficial effect of supplementation with cholesterol and the deleterious effect of supplementation with 7-dehydrocholesterol. Cholesterol supplementation in the form of mixed cholesterol/lecithin liposomes added to serum serving as the culture medium restores the growth of embryos which is markedly decreased in the presence of the inhibitor. 7-Dehydrocholesterol under identical conditions does not restore growth and impairs the beneficial effect of cholesterol added simultaneously. UV-photooxidation of 7-dehydrocholesterol-supplemented culture medium enhances its embryotoxicity, which suggests uptake by the embryo of toxic by-products formed from 7-dehydrocholesterol. By contrast photooxidation of cholesterol-supplemented culture medium does not induce embryotoxicity. alpha-Tocopherol reduces the toxicity of photooxidized 7-dehydrocholesterol supplementing the culture medium. We conclude that 7-dehydrocholesterol does not fulfill the cholesterol requirement of the developing embryos and exerts an additional embryotoxic effect probably via oxidized by-products. This could explain the antenatal growth retardation of SLOS by a blockage of the maternal compensatory cholesterol influx. (+info)The Opitz syndrome gene product, MID1, associates with microtubules. (2/122)
Opitz syndrome (OS) is a genetically heterogeneous disorder characterized by defects of the ventral midline, including hypertelorism, cleft lip and palate, heart defects, and mental retardation. We recently identified the gene responsible for X-linked OS. The ubiquitously expressed gene product, MID1, is a member of the RING finger family. These proteins are characterized by an N-terminal tripartite protein-protein interaction domain and a conserved C terminus of unknown function. Unlike other RING finger proteins for which diverse cellular functions have been proposed, the function of MID1 is as yet undefined. By using the green fluorescent protein as a tag, we show here that MID1 is a microtubule-associated protein that influences microtubule dynamics in MID1-overexpressing cells. We confirm this observation by demonstrating a colocalization of MID1 and tubulin in subcellular fractions and the association of endogenous MID1 with microtubules after in vitro assembly. Furthermore, overexpressed MID1 proteins harboring mutations described in OS patients lack the capability to associate with microtubules, forming cytoplasmic clumps instead. These data give an idea of the possible molecular pathomechanism underlying the OS phenotype. (+info)Cholesterol biosynthesis from lanosterol. Molecular cloning, tissue distribution, expression, chromosomal localization, and regulation of rat 7-dehydrocholesterol reductase, a Smith-Lemli-Opitz syndrome-related protein. (3/122)
The cDNA encoding the 471-amino acid rat 7-dehydrocholesterol reductase (DHCR), an enzyme that has been implicated in both cholesterol biosynthesis and developmental abnormalities (e.g. Smith-Lemli-Opitz syndrome) in mammals, has been cloned and sequenced, and the primary structure of the enzyme has been deduced. The DHCR gene was mapped to chromosome 8q2.1 by fluorescence in situ hybridization. Rat DHCR, calculated molecular mass of 54.15-kDa polypeptide, shares a close amino acid identity with mouse and human DHCRs (96 and 87%, respectively) as compared with its other related proteins (e.g. fungal sterol Delta14-reductase) and exhibits high hydrophobicity (>68%) with 9 transmembrane domains. Five putative sterol-sensing domains were predicted to be localized in transmembrane domains 4-8, which are highly homologous to those found in 3-hydroxymethylglutaryl-CoA reductase, sterol regulatory element-binding protein cleavage-activating protein, and patched protein. The polypeptide encoded by DHCR cDNA was expressed in yeast as a 55.45-kDa myc-tagged fusion protein, which was recognized with anti-myc monoclonal antibody 9E10 and shown to possess full DHCR activity with respect to dependence on NADPH and sensitivity to DHCR inhibitors. Northern blot analysis indicates that the highest expression of DHCR mRNA was detected in liver, followed by kidney and brain. In rat brains, the highest level of mRNA encoding DHCR was detected in the midbrain, followed by the spinal cord and medulla. Feeding rats 5% cholestyramine plus 0.1% lovastatin in chow resulted in both approximately a 3-fold induction of DHCR mRNA and a 5-fold increase of the enzymic activity in the liver. When rats were fed 0.1% (w/w) AY-9944 (in chow) for 14-days, a complete inhibition of DHCR activity and a significant reduction in serum total cholesterol level were observed. However, the level of hepatic DHCR mRNA fell only slightly, suggesting that AY-9944 may act more rapidly at the protein level than at the level of transcription of the DHCR gene under these conditions. (+info)Marked alteration of sterol metabolism and composition without compromising retinal development or function. (4/122)
PURPOSE: To evaluate the consequences of altering retinal sterol metabolism and composition on the development, histologic organization, and electrophysiological function of the retina, under conditions that mimic the biochemical hallmarks of the Smith-Lemli-Opitz (SLO) syndrome. METHODS: Pregnant Sprague-Dawley rats were fed cholesterol-free chow containing AY9944 (treated group), an inhibitor of 3beta-hydroxysterol delta7-reductase, from gestational day 6 through postnatal day (P)28. Control animals were fed the same chow, but without AY9944. In addition, progeny in the treated group were injected subcutaneously every other day from birth to P28 with an olive oil emulsion containing AY9944; control animals received olive oil emulsion alone. At various postnatal times, tissues from treated and control animals were harvested, and their sterol profiles were analyzed by reversed-phase high-performance liquid chromatography. Companion eyes from animals of both groups were examined histologically at P1. At P28, animals were evaluated by electroretinography; tissues were then harvested for biochemical analysis and companion eyes were subjected to histologic and ultrastructural analyses. RESULTS: Treatment of developing rats with AY9944 caused markedly abnormal accumulation of 7-dehydrosterols and severely reduced cholesterol levels in all tissues examined, relative to control animals. Despite this, treated animals exhibited normal retinal development and had no overt ocular defects or decrease in electroretinographic function, up to P28. CONCLUSIONS: These results were unexpected, given the known biophysical effects of such sterol alterations on membrane properties and the profound dysmorphic and cognitive abnormalities associated with genetic defects in 3beta-hydroxysterol delta7-reductase that have been linked to the SLO syndrome. The results suggest that 7-dehydrosterols can substitute functionally for cholesterol in the retina or perhaps can act synergistically with subthreshold levels of residual cholesterol to allow normal cellular structure and function to be achieved. (+info)Functional characterization of the Opitz syndrome gene product (midin): evidence for homodimerization and association with microtubules throughout the cell cycle. (5/122)
Opitz syndrome (OS) is a multiple congenital anomaly manifested by abnormal closure of midline structures. The gene responsible for the X-linked form of this disease, MID1, encodes a protein (midin) that contains a RING, two B-boxes, a coiled-coil (the so-called tripartite motif) and an RFP-like domain. The tripartite motif is characteristic of a family of proteins, named the B-box family, involved in cell proliferation and development. Since the subcellular compartmentalization and the ability to form multiprotein structures both appear to be crucial for the function of this family of proteins, we have studied these properties on the wild-type and mutated forms of midin. We found that endogenous midin is associated with microtubules throughout the cell cycle, co-localizing with cytoplasmic fibres in interphase and with the mitotic spindle and midbodies during mitosis and cytokinesis. Immunoprecipitation experiments demonstrated the ability of the tripartite motif to mediate midin homodimerization, consistent with the evidence, obtained by gel filtration analysis, that midin exists in the form of large protein complexes. Functional characterization of altered forms of midin, resulting from mutations found in OS patients, revealed that association with microtubules is compromised, while the ability to homodimerize and form multiprotein complexes is retained. We suggest that midin is involved in the formation of multiprotein structures acting as anchor points to microtubules and that impaired association with these cytoskeletal structures causes OS developmental defects. (+info)MID2, a homologue of the Opitz syndrome gene MID1: similarities in subcellular localization and differences in expression during development. (6/122)
The B-box family is an expanding new family of genes encoding proteins involved in diverse cellular functions such as developmental patterning and oncogenesis. A member of this protein family, MID1, is the gene responsible for the X-linked form of Opitz G/BBB syndrome, a developmental disorder characterized by defects of the midline structures. We now report the identification of MID2, a new transcript closely related to MID1. MID2 maps to Xq22 in human and to the syntenic region on the mouse X chromosome. The two X-linked genes share the same domains, the same exon-intron organization, a high degree of similarity at the protein level and the same subcellular localization, both being confined to the cytoplasm in association to micro-tubular structures. The expression pattern studied by RNA in situ hybridization in mouse revealed that Mid2 is expressed early in development and the highest level of expression is detected in the heart, unlike Mid1 for which no expression was detected in the developing heart. Together, these data suggest that midin and MID2 have a similar biochemical function but a different physiological role during development. (+info)Bile acid synthesis in the Smith-Lemli-Opitz syndrome: effects of dehydrocholesterols on cholesterol 7alpha-hydroxylase and 27-hydroxylase activities in rat liver. (7/122)
The Smith-Lemli-Opitz syndrome (SLOS) is a congenital birth defect syndrome caused by a deficiency of 3beta-hydroxysterol Delta(7)-reductase, the final enzyme in the cholesterol biosynthetic pathway. The patients have reduced plasma and tissue cholesterol concentrations with the accumulation of 7-dehydrocholesterol and 8-dehydrocholesterol. Bile acid synthesis is reduced and unnatural cholenoic and cholestenoic acids have been identified in some SLOS patients. To explore the mechanism of the abnormal bile acid production, the activities of key enzymes in classic and alternative bile acid biosynthetic pathways (microsomal cholesterol 7alpha-hydroxylase and mitochondrial sterol 27-hydroxylase) were measured in liver biopsy specimens from two mildly affected SLOS patients. The effects of 7- and 8-dehydrocholesterols on these two enzyme activities were studied by using liver from SLOS model rats that were treated with the Delta(7)-reductase inhibitor (BM15.766) for 4 months and were comparable with more severe SLOS phenotype in plasma and hepatic sterol compositions. In the SLOS patients, cholesterol 7alpha-hydroxylase and sterol 27-hydroxylase were not defective. In BM15.766-treated rats, both enzyme activities were lower than those in control rats and they were competitively inhibited by 7- and 8-dehydrocholesterols. Rat microsomal cholesterol 7alpha-hydroxylase did not transform 7-dehydrocholesterol or 8-dehydrocholesterol into 7alpha-hydroxylated sterols. In contrast, rat mitochondrial sterol 27-hydroxylase catalyzed 27-hydroxylation of 7- and 8-dehydrocholesterols, which were partially converted to 3beta-hydroxycholestadienoic acids. Addition of microsomes to the mitochondrial 27-hydroxylase assay mixture reduced 27-hydroxydehydrocholesterol concentrations, which suggested that 27-hydroxydehydrocholesterols were further metabolized by microsomal enzymes. These results suggest that reduced normal bile acid production is characteristic of severe SLOS phenotype and is caused not only by depletion of hepatic cholesterol but also by competitive inhibition of cholesterol 7alpha-hydroxylase and sterol 27-hydroxylase activities by accumulated 7- and 8-dehydrocholesterols. Unnatural bile acids are synthesized mainly by the alternative pathway via mitochondrial sterol 27-hydroxylase in SLOS. (+info)Smith-Lemli-Opitz syndrome: a treatable inherited error of metabolism causing mental retardation. (8/122)
Smith-Lemli-Opitz syndrome, a syndrome of multiple malformations and mental retardation that for years was relegated to the atlases of genetic esoterica, was recently found to be a relatively common inborn error of metabolism. The underlying defect is absent or deficient activity of 7-dehydrocholesterol- delta 7-reductase, the enzyme catalysing the final step of cholesterol synthesis. The discovery of the biochemical defect causing Smith-Lemli-Opitz syndrome has resulted in the development of a diagnostic test and a potentially beneficial treatment (dietary cholesterol supplementation). Infants and young children with the syndrome have shown marked improvement in growth, behaviour and general health after receiving cholesterol therapy; older children and adults have shown some improvement in development and intellectual functioning. Despite the excitement these developments have elicited among geneticists and biochemists, this syndrome remains relatively unknown to many primary care physicians. Increased awareness of Smith-Lemli-Opitz syndrome is needed to identify affected patients so that they and their families can benefit from appropriate treatment and genetic counselling. (+info)Microtubule proteins are a class of structural proteins that make up the microtubules, which are key components of the cytoskeleton in eukaryotic cells. The main microtubule protein is tubulin, which exists in two forms: alpha-tubulin and beta-tubulin. These tubulins polymerize to form heterodimers, which then assemble into protofilaments, which in turn aggregate to form hollow microtubules. Microtubules are dynamic structures that undergo continuous assembly and disassembly, and they play crucial roles in various cellular processes, including intracellular transport, cell division, and maintenance of cell shape. Other microtubule-associated proteins (MAPs) also bind to microtubules and regulate their stability, dynamics, and interactions with other cellular structures.
Smith-Lemli-Opitz syndrome (SLOS) is a genetic disorder that affects the development of multiple body systems. It is caused by a deficiency in the enzyme 7-dehydrocholesterol reductase, which is needed for the production of cholesterol in the body.
The symptoms of SLOS can vary widely in severity, but often include developmental delays, intellectual disability, low muscle tone (hypotonia), feeding difficulties, and behavioral problems. Physical abnormalities may also be present, such as cleft palate, heart defects, extra fingers or toes (polydactyly), and genital abnormalities in males.
SLOS is an autosomal recessive disorder, which means that an individual must inherit two copies of the mutated gene (one from each parent) in order to develop the condition. It is typically diagnosed through genetic testing and biochemical analysis of blood or body fluids. Treatment for SLOS may include cholesterol supplementation, special education services, and management of associated medical conditions.
Hypertelorism is a medical term that refers to an ocular condition where the distance between two eyes (interpupillary distance) is abnormally increased. It's typically defined as an interpupillary distance that measures more than 2 standard deviations beyond the mean for a given age, gender, and race.
This condition can be associated with various genetic syndromes or conditions such as craniosynostosis (premature fusion of skull sutures), fetal alcohol syndrome, and certain chromosomal abnormalities like Down syndrome. Hypertelorism may also occur in isolation without any other associated anomalies.
It's important to note that hypertelorism can have cosmetic implications, particularly if the distance between the eyes is significantly increased, as it may affect the overall symmetry and appearance of the face. However, in most cases, this condition does not directly impact vision unless there are other related structural abnormalities of the eye or orbit.
Hypospadias is a congenital condition in males where the urethral opening (meatus), which is the end of the urethra through which urine exits, is not located at the tip of the penis but instead appears on the underside of the penis. The severity of hypospadias can vary, with some cases having the meatus located closer to the tip and others further down on the shaft or even at the scrotum or perineum (the area between the scrotum and the anus). This condition affects about 1 in every 200-250 male newborns. The exact cause of hypospadias is not fully understood, but it's believed to be a combination of genetic and environmental factors. Surgical correction is usually recommended during infancy or early childhood to prevent complications such as difficulty urinating while standing, problems with sexual function, and psychological issues related to body image.
A syndrome, in medical terms, is a set of symptoms that collectively indicate or characterize a disease, disorder, or underlying pathological process. It's essentially a collection of signs and/or symptoms that frequently occur together and can suggest a particular cause or condition, even though the exact physiological mechanisms might not be fully understood.
For example, Down syndrome is characterized by specific physical features, cognitive delays, and other developmental issues resulting from an extra copy of chromosome 21. Similarly, metabolic syndromes like diabetes mellitus type 2 involve a group of risk factors such as obesity, high blood pressure, high blood sugar, and abnormal cholesterol or triglyceride levels that collectively increase the risk of heart disease, stroke, and diabetes.
It's important to note that a syndrome is not a specific diagnosis; rather, it's a pattern of symptoms that can help guide further diagnostic evaluation and management.
Nuclear proteins are a category of proteins that are primarily found in the nucleus of a eukaryotic cell. They play crucial roles in various nuclear functions, such as DNA replication, transcription, repair, and RNA processing. This group includes structural proteins like lamins, which form the nuclear lamina, and regulatory proteins, such as histones and transcription factors, that are involved in gene expression. Nuclear localization signals (NLS) often help target these proteins to the nucleus by interacting with importin proteins during active transport across the nuclear membrane.
Transcription factors are proteins that play a crucial role in regulating gene expression by controlling the transcription of DNA to messenger RNA (mRNA). They function by binding to specific DNA sequences, known as response elements, located in the promoter region or enhancer regions of target genes. This binding can either activate or repress the initiation of transcription, depending on the properties and interactions of the particular transcription factor. Transcription factors often act as part of a complex network of regulatory proteins that determine the precise spatiotemporal patterns of gene expression during development, differentiation, and homeostasis in an organism.
Imperforate anus is a congenital condition in which the opening of the anus is absent or abnormally closed or narrowed, preventing the normal passage of stool. This results in a blockage in the digestive tract and can lead to serious health complications if not treated promptly.
The anus is the external opening of the rectum, which is the lower end of the digestive tract. During fetal development, the rectum and anus normally connect through a canal called the anal canal or the recto-anal canal. In imperforate anus, this canal may be completely closed or narrowed, or it may not form properly.
Imperforate anus can occur as an isolated condition or as part of a genetic syndrome or other congenital abnormalities. The exact cause is not fully understood, but it is believed to result from a combination of genetic and environmental factors.
Treatment for imperforate anus typically involves surgery to create an opening in the anus and restore normal bowel function. In some cases, additional procedures may be necessary to correct related abnormalities or complications. The prognosis for individuals with imperforate anus depends on the severity of the condition and any associated abnormalities. With prompt and appropriate treatment, most people with imperforate anus can lead normal lives.
X-linked genetic diseases refer to a group of disorders caused by mutations in genes located on the X chromosome. These conditions primarily affect males since they have only one X chromosome and therefore don't have a second normal copy of the gene to compensate for the mutated one. Females, who have two X chromosomes, are typically less affected because they usually have one normal copy of the gene on their other X chromosome.
Examples of X-linked genetic diseases include Duchenne and Becker muscular dystrophy, hemophilia A and B, color blindness, and fragile X syndrome. Symptoms and severity can vary widely depending on the specific condition and the nature of the genetic mutation involved. Treatment options depend on the particular disease but may include physical therapy, medication, or in some cases, gene therapy.
'Abnormalities, Multiple' is a broad term that refers to the presence of two or more structural or functional anomalies in an individual. These abnormalities can be present at birth (congenital) or can develop later in life (acquired). They can affect various organs and systems of the body and can vary greatly in severity and impact on a person's health and well-being.
Multiple abnormalities can occur due to genetic factors, environmental influences, or a combination of both. Chromosomal abnormalities, gene mutations, exposure to teratogens (substances that cause birth defects), and maternal infections during pregnancy are some of the common causes of multiple congenital abnormalities.
Examples of multiple congenital abnormalities include Down syndrome, Turner syndrome, and VATER/VACTERL association. Acquired multiple abnormalities can result from conditions such as trauma, infection, degenerative diseases, or cancer.
The medical evaluation and management of individuals with multiple abnormalities depend on the specific abnormalities present and their impact on the individual's health and functioning. A multidisciplinary team of healthcare professionals is often involved in the care of these individuals to address their complex needs.
Down syndrome is a genetic disorder caused by the presence of all or part of a third copy of chromosome 21. It is characterized by intellectual and developmental disabilities, distinctive facial features, and sometimes physical growth delays and health problems. The condition affects approximately one in every 700 babies born in the United States.
Individuals with Down syndrome have varying degrees of cognitive impairment, ranging from mild to moderate or severe. They may also have delayed development, including late walking and talking, and may require additional support and education services throughout their lives.
People with Down syndrome are at increased risk for certain health conditions, such as congenital heart defects, respiratory infections, hearing loss, vision problems, gastrointestinal issues, and thyroid disorders. However, many individuals with Down syndrome live healthy and fulfilling lives with appropriate medical care and support.
The condition is named after John Langdon Down, an English physician who first described the syndrome in 1866.
Metabolic syndrome, also known as Syndrome X, is a cluster of conditions that increase the risk of heart disease, stroke, and diabetes. It is not a single disease but a group of risk factors that often co-occur. According to the American Heart Association and the National Heart, Lung, and Blood Institute, a person has metabolic syndrome if they have any three of the following five conditions:
1. Abdominal obesity (waist circumference of 40 inches or more in men, and 35 inches or more in women)
2. Triglyceride level of 150 milligrams per deciliter of blood (mg/dL) or greater
3. HDL cholesterol level of less than 40 mg/dL in men or less than 50 mg/dL in women
4. Systolic blood pressure of 130 millimeters of mercury (mmHg) or greater, or diastolic blood pressure of 85 mmHg or greater
5. Fasting glucose level of 100 mg/dL or greater
Metabolic syndrome is thought to be caused by a combination of genetic and lifestyle factors, such as physical inactivity and a diet high in refined carbohydrates and unhealthy fats. Treatment typically involves making lifestyle changes, such as eating a healthy diet, getting regular exercise, and losing weight if necessary. In some cases, medication may also be needed to manage individual components of the syndrome, such as high blood pressure or high cholesterol.
Microtubules are hollow, cylindrical structures composed of tubulin proteins in the cytoskeleton of eukaryotic cells. They play crucial roles in various cellular processes such as maintaining cell shape, intracellular transport, and cell division (mitosis and meiosis). Microtubules are dynamic, undergoing continuous assembly and disassembly, which allows them to rapidly reorganize in response to cellular needs. They also form part of important cellular structures like centrioles, basal bodies, and cilia/flagella.
Nephrotic syndrome is a group of symptoms that indicate kidney damage, specifically damage to the glomeruli—the tiny blood vessel clusters in the kidneys that filter waste and excess fluids from the blood. The main features of nephrotic syndrome are:
1. Proteinuria (excess protein in urine): Large amounts of a protein called albumin leak into the urine due to damaged glomeruli, which can't properly filter proteins. This leads to low levels of albumin in the blood, causing fluid buildup and swelling.
2. Hypoalbuminemia (low blood albumin levels): As albumin leaks into the urine, the concentration of albumin in the blood decreases, leading to hypoalbuminemia. This can cause edema (swelling), particularly in the legs, ankles, and feet.
3. Edema (fluid retention and swelling): With low levels of albumin in the blood, fluids move into the surrounding tissues, causing swelling or puffiness. The swelling is most noticeable around the eyes, face, hands, feet, and abdomen.
4. Hyperlipidemia (high lipid/cholesterol levels): The kidneys play a role in regulating lipid metabolism. Damage to the glomeruli can lead to increased lipid production and high cholesterol levels in the blood.
Nephrotic syndrome can result from various underlying kidney diseases, such as minimal change disease, membranous nephropathy, or focal segmental glomerulosclerosis. Treatment depends on the underlying cause and may include medications to control inflammation, manage high blood pressure, and reduce proteinuria. In some cases, dietary modifications and lifestyle changes are also recommended.
Sjögren's syndrome is a chronic autoimmune disorder in which the body's immune system mistakenly attacks its own moisture-producing glands, particularly the tear and salivary glands. This can lead to symptoms such as dry eyes, dry mouth, and dryness in other areas of the body. In some cases, it may also affect other organs, leading to a variety of complications.
There are two types of Sjögren's syndrome: primary and secondary. Primary Sjögren's syndrome occurs when the condition develops on its own, while secondary Sjögren's syndrome occurs when it develops in conjunction with another autoimmune disease, such as rheumatoid arthritis or lupus.
The exact cause of Sjögren's syndrome is not fully understood, but it is believed to involve a combination of genetic and environmental factors. Treatment typically focuses on relieving symptoms and may include artificial tears, saliva substitutes, medications to stimulate saliva production, and immunosuppressive drugs in more severe cases.
Turner Syndrome is a genetic disorder that affects females, caused by complete or partial absence of one X chromosome. The typical karyotype is 45,X0 instead of the normal 46,XX in women. This condition leads to distinctive physical features and medical issues in growth, development, and fertility. Characteristic features include short stature, webbed neck, low-set ears, and swelling of the hands and feet. Other potential symptoms can include heart defects, hearing and vision problems, skeletal abnormalities, kidney issues, and learning disabilities. Not all individuals with Turner Syndrome will have every symptom, but most will require medical interventions and monitoring throughout their lives to address various health concerns associated with the condition.
Myelodysplastic syndromes (MDS) are a group of diverse bone marrow disorders characterized by dysplasia (abnormal development or maturation) of one or more types of blood cells or by ineffective hematopoiesis, resulting in cytopenias (lower than normal levels of one or more types of blood cells). MDS can be classified into various subtypes based on the number and type of cytopenias, the degree of dysplasia, the presence of ring sideroblasts, and cytogenetic abnormalities.
The condition primarily affects older adults, with a median age at diagnosis of around 70 years. MDS can evolve into acute myeloid leukemia (AML) in approximately 30-40% of cases. The pathophysiology of MDS involves genetic mutations and chromosomal abnormalities that lead to impaired differentiation and increased apoptosis of hematopoietic stem and progenitor cells, ultimately resulting in cytopenias and an increased risk of developing AML.
The diagnosis of MDS typically requires a bone marrow aspiration and biopsy, along with cytogenetic and molecular analyses to identify specific genetic mutations and chromosomal abnormalities. Treatment options for MDS depend on the subtype, severity of cytopenias, and individual patient factors. These may include supportive care measures, such as transfusions and growth factor therapy, or more aggressive treatments, such as chemotherapy and stem cell transplantation.
Cushing syndrome is a hormonal disorder that occurs when your body is exposed to high levels of the hormone cortisol for a long time. This can happen due to various reasons such as taking high doses of corticosteroid medications or tumors that produce cortisol or adrenocorticotropic hormone (ACTH).
The symptoms of Cushing syndrome may include:
* Obesity, particularly around the trunk and upper body
* Thinning of the skin, easy bruising, and purple or red stretch marks on the abdomen, thighs, breasts, and arms
* Weakened bones, leading to fractures
* High blood pressure
* High blood sugar
* Mental changes such as depression, anxiety, and irritability
* Increased fatigue and weakness
* Menstrual irregularities in women
* Decreased fertility in men
Cushing syndrome can be diagnosed through various tests, including urine and blood tests to measure cortisol levels, saliva tests, and imaging tests to locate any tumors. Treatment depends on the cause of the condition but may include surgery, radiation therapy, chemotherapy, or adjusting medication dosages.
Acute Coronary Syndrome (ACS) is a term used to describe a range of conditions associated with sudden, reduced blood flow to the heart muscle. This reduction in blood flow, commonly caused by blood clots forming in coronary arteries, can lead to damage or death of the heart muscle and is often characterized by symptoms such as chest pain, shortness of breath, and fatigue.
There are three main types of ACS:
1. Unstable Angina: This occurs when there is reduced blood flow to the heart muscle, causing chest pain or discomfort, but the heart muscle is not damaged. It can be a warning sign for a possible future heart attack.
2. Non-ST Segment Elevation Myocardial Infarction (NSTEMI): This type of heart attack occurs when there is reduced blood flow to the heart muscle, causing damage or death of some of the muscle cells. However, the electrical activity of the heart remains relatively normal.
3. ST Segment Elevation Myocardial Infarction (STEMI): This is a serious and life-threatening type of heart attack that occurs when there is a complete blockage in one or more of the coronary arteries, causing extensive damage to the heart muscle. The electrical activity of the heart is significantly altered, which can lead to dangerous heart rhythms and even cardiac arrest.
Immediate medical attention is required for anyone experiencing symptoms of ACS, as prompt treatment can help prevent further damage to the heart muscle and reduce the risk of complications or death. Treatment options may include medications, lifestyle changes, and procedures such as angioplasty or bypass surgery.
Polycyctic Ovary Syndrome (PCOS) is a complex endocrine-metabolic disorder characterized by the presence of hyperandrogenism (excess male hormones), ovulatory dysfunction, and polycystic ovaries. The Rotterdam criteria are commonly used for diagnosis, which require at least two of the following three features:
1. Oligo- or anovulation (irregular menstrual cycles)
2. Clinical and/or biochemical signs of hyperandrogenism (e.g., hirsutism, acne, or high levels of androgens in the blood)
3. Polycystic ovaries on ultrasound examination (presence of 12 or more follicles measuring 2-9 mm in diameter, or increased ovarian volume >10 mL)
The exact cause of PCOS remains unclear, but it is believed to involve a combination of genetic and environmental factors. Insulin resistance and obesity are common findings in women with PCOS, which can contribute to the development of metabolic complications such as type 2 diabetes, dyslipidemia, and cardiovascular disease.
Management of PCOS typically involves a multidisciplinary approach that includes lifestyle modifications (diet, exercise, weight loss), medications to regulate menstrual cycles and reduce hyperandrogenism (e.g., oral contraceptives, metformin, anti-androgens), and fertility treatments if desired. Regular monitoring of metabolic parameters and long-term follow-up are essential for optimal management and prevention of complications.
Williams Syndrome is a rare genetic disorder caused by the deletion of a small portion of chromosome 7. This results in various developmental and medical problems, which can include:
1. Distinctive facial features such as a broad forehead, wide-set eyes, short nose, and full lips.
2. Cardiovascular disease, particularly narrowed or missing blood vessels near the heart.
3. Developmental delays and learning disabilities, although most people with Williams Syndrome have an IQ in the mild to moderate range of intellectual disability.
4. A unique pattern of strengths and weaknesses in cognitive skills, such as strong language skills but significant difficulty with visual-spatial tasks.
5. Overly friendly or sociable personality, often displaying a lack of fear or wariness around strangers.
6. Increased risk of anxiety and depression.
7. Sensitive hearing and poor depth perception.
8. Short stature in adulthood.
Williams Syndrome affects about 1 in every 10,000 people worldwide, regardless of race or ethnic background. It is not an inherited disorder, but rather a spontaneous genetic mutation.
 Smith-Lemli-Opitz syndrome
Smith-Lemli-Opitz syndrome
7-Dehydrocholesterol reductase
Sterol-sensing domain
Smoothened
7-Dehydrocholesterol
Syndromic autism
Steven J. Fliesler
Addison's disease
Lathosterolosis
C-5 sterol desaturase
Dysmorphic feature
Young-Madders syndrome
Hirschsprung's disease
7-Dehydrodesmosterol
Unibrow
Acyl-CoA oxidase deficiency
Dubowitz syndrome
Adrenal insufficiency
Cholesterol
Steroidogenesis inhibitor
Hugo Latulippe
Hydrolethalus syndrome
John M. Opitz
The Resilience Project
Hypoplastic left heart syndrome
List of University of Utah people
List of MeSH codes (C16)
Alphée of the Stars
Breech birth
Photosensitivity in humans
David Weyhe Smith
Smith-Lemli-Opitz syndrome - Wikipedia
 Smith-Lemli-Opitz Syndrome: Practice Essentials, Pathophysiology, Epidemiology
Smith-Lemli-Opitz Syndrome: Practice Essentials, Pathophysiology, Epidemiology
Smith-Lemli-Opitz syndrome: a variable clinical and biochemical phenotype. | Journal of Medical Genetics
 SciELO - Brazil - Smith-Lemli-Opitz syndrome: clinical and biochemical findings in Brazilian patients Smith-Lemli-Opitz...
SciELO - Brazil - Smith-Lemli-Opitz syndrome: clinical and biochemical findings in Brazilian patients Smith-Lemli-Opitz...
Smith-Lemli-Opitz Syndrome | Profiles RNS
Anesthetic considerations in Smith-Lemli-Opitz syndrome - McMaster Experts
 Smith-Lemli-Opitz Syndrome - Clinical test - NIH Genetic Testing Registry (GTR) - NCBI
Smith-Lemli-Opitz Syndrome - Clinical test - NIH Genetic Testing Registry (GTR) - NCBI
 Good Laboratory Practices for Biochemical Genetic Testing and Newborn Screening for Inherited Metabolic Disorders
Good Laboratory Practices for Biochemical Genetic Testing and Newborn Screening for Inherited Metabolic Disorders
 Med Journal 360 | Vitamin D metabolism gene polymorphisms may play role in in Smith-Lemli-Opitz syndrome
Med Journal 360 | Vitamin D metabolism gene polymorphisms may play role in in Smith-Lemli-Opitz syndrome
 Polydactyly: MedlinePlus Medical Encyclopedia
Polydactyly: MedlinePlus Medical Encyclopedia
 Simvastatin Treatment Highlights a New Role for the Isoprenoid/Cholesterol Biosynthetic Pathway in the Modulation of Emotional...
Simvastatin Treatment Highlights a New Role for the Isoprenoid/Cholesterol Biosynthetic Pathway in the Modulation of Emotional...
Clinical and biochemical spectrum of patients with rsh/smith-lemli-opitz syndrome and abnormal cholesterol metabolism<...
A Smith Lemli Opitz syndrome patient diagnosed with mild symptoms Hafif klinik belirtilerle tani alan bir Smith Lemli Opitz...
 Operation Equine Registration Form
Operation Equine Registration Form
 Robert Blassberg | Crick
Robert Blassberg | Crick
IEP suggestions
 Expanded Carrier Screening | Thermo Fisher Scientific - US
Expanded Carrier Screening | Thermo Fisher Scientific - US
 Event Detail
-
Association for Behavior Analysis International
Event Detail
-
Association for Behavior Analysis International
 Human Metabolome Database: Showing metabocard for Acetoacetyl-CoA (HMDB0001484)
Human Metabolome Database: Showing metabocard for Acetoacetyl-CoA (HMDB0001484)
 Dehydrocholesterols | Harvard Catalyst Profiles | Harvard Catalyst
Dehydrocholesterols | Harvard Catalyst Profiles | Harvard Catalyst
 Publications
Publications
 Litteratur og lenker - Frambu
Litteratur og lenker - Frambu
 Morbid Anatomy: August 2014
Morbid Anatomy: August 2014
Orphanet: Search a disease
 Recombinant Human Wnt-3a Protein (5036-WN): Novus Biologicals
Recombinant Human Wnt-3a Protein (5036-WN): Novus Biologicals
 Gabriella Uhercsák - Articles - Scientific Research Publishing
Gabriella Uhercsák - Articles - Scientific Research Publishing
 Shailendra B. Patel
Shailendra B. Patel
 Special Care Clinic | Children's Hospital Colorado
Special Care Clinic | Children's Hospital Colorado
 Race for a Cause
Race for a Cause
 Faculty | Howard University College of Medicine
Faculty | Howard University College of Medicine
SLOS8
- Smith-Lemli-Opitz syndrome (SLOS) is a multiple congenital anomaly (MCA)/ intellectual disability (ID) syndrome caused by a defect in cholesterol synthesis. (medscape.com)
- Smith-Lemli-Opitz syndrome (SLOS) or RSH syndrome comprises multiple congenital anomalies and mental retardation. (scielo.br)
- Smith-Lemli-Opitz (SLOS) or RSH syndrome (MIM 270400) comprises multiple congenital anomalies, mental retardation and an autosomal recessive inborn error of cholesterol metabolism. (scielo.br)
- PURPOSE: Smith-Lemli-Opitz syndrome (SLOS) is an autosomal recessive syndrome characterized by congenital anomalies affecting the airway, cardiorespiratory, gastrointestinal, genitourinary, and central nervous systems. (mcmaster.ca)
- Smith Lemli Opitz Syndrome (SLOS) is an autosomal recessive cholesterol biosynthesis defect which is characterized by both multiple kongenital anomaly and mental retardation. (lokmanhekim.edu.tr)
- Through medical conferences, doctors referred Aryanna to the National Institutes of Health (NIH) to determine if Aryanna might have a rare metabolic disorder called Smith-Lemli-Opitz Syndrome (SLOS). (childrensinn.org)
- Smith-Lemli-Opitz syndrome (SLOS) is a rare variable genetic disorder that is characterized by slow growth before and after birth, small head (microcephaly), mild to moderate intellectual disability and multiple birth defects. (vumc.org)
- According to several medical resources, Smith-Lemli-Opitz Syndrome (SLOS) is congenital and caused by mutation, e.g. (stackexchange.com)
Deficiency4
- In the case of Smith-Lemli-Opitz syndrome, the precursor 7DHC is potentially toxic in high concentrations, and cholesterol deficiency is almost certainly detrimental. (medscape.com)
- [ 6 ] A deficiency of the microsomal enzyme DHCR7, which reduces the 7-8 double bond of 7DHC to form cholesterol in the final step of the cholesterol synthetic pathway, was hypothesized and later proven to cause Smith-Lemli-Opitz syndrome. (medscape.com)
- This syndrome is characterized by multiple CONGENITAL ABNORMALITIES, growth deficiency, and INTELLECTUAL DISABILITY. (rush.edu)
- RSH/Smith-Lemli-Opitz (RSH/SLO) syndrome is an autosomal recessive malformation syndrome recently shown to be associated with a severe deficiency of cholesterol biosynthesis and markedly elevated plasma and tissue levels of 7-dehydrocholesterol (7DHC), the immediate precursor of cholesterol in the Kandutsch-Russell biosynthetic pathway. (johnshopkins.edu)
Biochemical4
- Smith-Lemli-Opitz syndrome is usually suspected clinically, but the diagnosis must be confirmed by biochemical and/or molecular genetic studies. (medscape.com)
- Postnatally, the syndrome is usually suspected clinically, but biochemical and/or genetic testing is necessary for diagnosis. (medscape.com)
- Measurement of plasma sterols, including, at a minimum, cholesterol and 7DHC, is the biochemical test for Smith-Lemli-Opitz syndrome. (medscape.com)
- Smith-Lemli-Opitz syndrome: a variable clinical and biochemical phenotype. (bmj.com)
Clinical4
- [ 5 ] The clinical characteristics of Smith-Lemli-Opitz syndrome have been well established over the past 4 decades. (medscape.com)
- As demonstrated in other clinical syndromes when redefined biochemically, we have found a wider range of clinical expression of RSH/SLO than previously recognized. (johnshopkins.edu)
- 1%) of individuals with clinical findings of the 22q11.2 deletion syndrome have chromosomal rearrangements involving 22q11.2, such as a translocation between chromosome 22 and another chromosome. (22q.org)
- Clinical and genetic analysis of a Chinese pedigree affected with Smith-Lemli-Opitz syndrome]. (nih.gov)
Metabolism2
- Smith-Lemli-Opitz syndrome, cholesterol metabolism, 7-dehydrocholesterol. (scielo.br)
- The role of vitamin D metabolism gene polymorphisms (vitDMGPs) in various health conditions, including Smith-Lemli-Opitz syndrome, is critical, according to a comprehensive bibliometric study. (medjournal360.com)
Phenotype2
- Smith-Lemli-Opitz syndrome: phenotype, natural history, and epidemiology. (harvard.edu)
- Most of these genes are also responsible for Joubert syndrome, leading to the concept that MKS is the extreme lethal phenotype of Joubert syndrome. (orpha.net)
Autosomal recessive syndrome1
- We report here the identification of mutations in sterol-C4-methyl oxidase-like gene (SC4MOL) as the cause of an autosomal recessive syndrome in a human patient with psoriasiform dermatitis, arthralgias, congenital cataracts, microcephaly, and developmental delay. (jci.org)
Congenital anomalies1
- D. W. Smith, L. Lemli and J. M. Opitz, "A Newly Recognized Syndrome of Multiple Congenital Anomalies," The Journal of Pediatrics, Vol. 64, 1964, pp. 210-217. (scirp.org)
Gene3
- It is an autosomal recessive, multiple malformation syndrome caused by a mutation in the enzyme 7-Dehydrocholesterol reductase encoded by the DHCR7 gene. (wikipedia.org)
- As a postdoctoral research associate at Brown University, Dr. Csoka was instrumental in the identification of the gene that causes Hutchinson-Gilford Progeria Syndrome (progeria), a disease with many features of "accelerated aging. (howard.edu)
- These individuals may have a change in a gene within the regions, such as TBX1, or a different condition such as CHARGE syndrome. (22q.org)
Inborn1
- Smith-Lemli-Opitz syndrome is an inborn error of cholesterol synthesis. (wikipedia.org)
Trisomy 131
- Differential diagnoses include trisomy 13, Bardet-Biedl syndrome, Hydrolethalus, and Smith-Lemli-Opitz Syndrome. (orpha.net)
Genetic2
- Smith, Lemli, and Opitz initially described Smith-Lemli-Opitz syndrome (while working at the University of Wisconsin) as a genetic MCA/ID syndrome, in 1964. (medscape.com)
- His work in that section has focused on understanding two rare genetic disorders involving cholesterol, Smith-Lemli-Opitz syndrome and Niemann-Pick disease type C. Smith-Lemli-Opitz syndrome results from a defect in the ability to manufacture cholesterol. (nih.gov)
Developmental Delay1
- Defects in cholesterol synthesis result in a wide variety of symptoms, from neonatal lethality to the relatively mild dysmorphic features and developmental delay found in individuals with Smith-Lemli-Opitz syndrome. (jci.org)
Malformation1
- CHARGE is a congenital disorder and an acronym for the constellation of medical problems that define this syndrome: coloboma of the eye, heart defects, atresia of the choanae, retardation of growth or development, genital hypoplasia, and ear malformation. (abainternational.org)
MeSH1
- Smith-Lemli-Opitz Syndrome" is a descriptor in the National Library of Medicine's controlled vocabulary thesaurus, MeSH (Medical Subject Headings) . (rush.edu)
Fetal1
- Fetal ultrasonography may reveal anomalies suggestive of Smith-Lemli-Opitz syndrome. (medscape.com)
Plasma cholesterol1
- The etiology of Smith-Lemli-Opitz syndrome was unknown until 1993, when Irons et al discovered that patients with Smith-Lemli-Opitz syndrome had low plasma cholesterol levels and accumulated sterol precursors such as 7DHC. (medscape.com)
Behavioral1
- The number of children diagnosed with CHARGE Syndrome is increasing, and several studies have reported that children with CHARGE syndrome exhibit multiple behavioral problems (e.g., repetitive behaviors, non-compliance, and social skills deficits) and educational problems. (abainternational.org)
Findings1
- A few individuals with findings of the 22q11.2 deletion syndrome have normal routine cytogenetic studies and no deletion by FISH, MLPA, CGH or microarray. (22q.org)
Proven2
- Currently, no treatment has proven effective long-term for patients with the syndrome. (medscape.com)
- As mentioned, no treatment has so far proven effective long-term for patients with Smith-Lemli-Opitz syndrome. (medscape.com)
Reveals1
- Modeling Smith-Lemli-Opitz syndrome with induced pluripotent stem cells reveals a causal role for Wnt/ß-catenin defects in neuronal cholesterol synthesis phenotypes. (harvard.edu)
Patients1
- The Special Care Clinic is home to a multidisciplinary team that focuses on providing care to patients with Ehlers-Danlos syndrome (EDS) . (childrenscolorado.org)
Cornelia1
- The third presenter will describe the treatment of severe aggression associated with Cornelia de Lange syndrome. (abainternational.org)
Multiple1
- The most severely affected individuals (those with the condition formerly referred to as Smith-Lemli-Opitz syndrome type II) have multiple congenital malformations and are often miscarried or stillborn or die in the first weeks of life. (medscape.com)
Dysplasia1
- Large polycystic kidneys with cystic dysplasia are a constant feature of Meckel syndrome. (orpha.net)
People2
- This graph shows the total number of publications written about "Smith-Lemli-Opitz Syndrome" by people in this website by year, and whether "Smith-Lemli-Opitz Syndrome" was a major or minor topic of these publications. (rush.edu)
- Below are the most recent publications written about "Smith-Lemli-Opitz Syndrome" by people in Profiles. (rush.edu)
Mental retardation1
- Another reason for children having an upturned nose, based on a medical condition is the Kaufman oculocerebrofacial syndrome which affects their facial features, mental retardation, and eye abnormalities amongst other aspects. (simplyaesthetic.co.uk)
Child2
- Child with Smith-Lemli-Opitz syndrome. (medscape.com)
- The next presenter will describe the treatment of severe problem behavior in a child with CHARGE syndrome. (abainternational.org)
Fatal1
- No treatment is currently available for Meckel syndrome which has a constantly fatal outcome. (orpha.net)
Patient1
- A Smith Lemli Opitz syndrome patient dia. (lokmanhekim.edu.tr)
Levels1
- a syndrome that is metabolically characterized by reduced serum cholesterol levels and elevated serum 7-dehydrocholesterol levels and phenotypically characterized by cognitive disability, facial dysmorphism, syndactyly of second and third toes, and holoprosencephaly in severe cases to minimal physical abnormalities and near-normal intelligence in mild cases. (nih.gov)
Study1
- In this study, principles of applied behavior analysis (ABA) were used to decrease self-injurious and disruptive behaviors for a 5-year old boy with CHARGE syndrome. (abainternational.org)
Group1
- The final presentation will summarize the assessment and treatment of self-injury and other problem behaviors in a group of children with Smith-Lemli-Opitz syndrome. (abainternational.org)