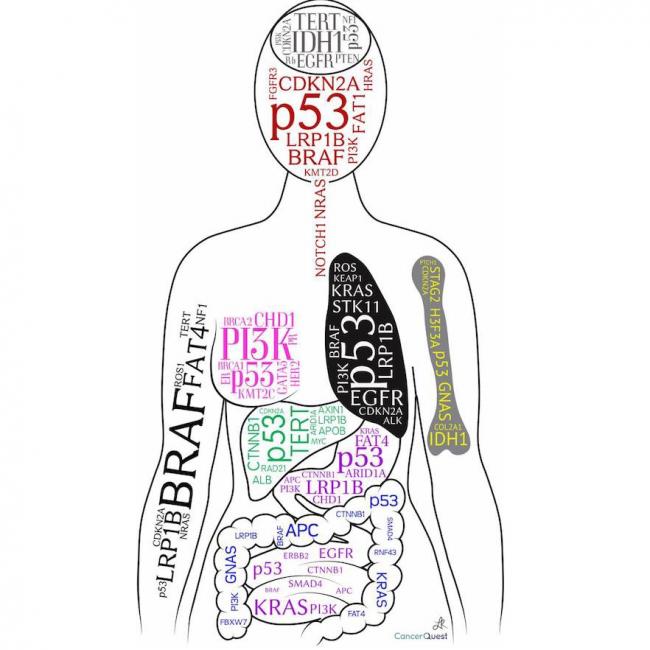
Sarcoma, Ewing
Sarcoma
Sarcoma, Synovial
Sarcoma, Kaposi
RNA-Binding Protein EWS
Proto-Oncogene Protein c-fli-1
Avian Sarcoma Viruses
Sarcoma, Experimental
Sarcoma 180
Sarcoma Viruses, Murine
Soft Tissue Neoplasms
Sarcoma, Avian
Oncogene Proteins, Fusion
Histiocytic Sarcoma
Sarcoma, Myeloid
Sarcoma, Endometrial Stromal
Sarcoma, Clear Cell
Neuroectodermal Tumors, Primitive, Peripheral
Neuroectodermal Tumors, Primitive
Sarcoma, Yoshida
Kirsten murine sarcoma virus
Osteosarcoma
Moloney murine sarcoma virus
Sarcoma Viruses, Feline
Rous sarcoma virus
Ifosfamide
Sarcoma, Small Cell
Rhabdomyosarcoma
Sarcoma, Alveolar Soft Part
Leiomyosarcoma
Herpesvirus 8, Human
Translocation, Genetic
Alpharetrovirus
Epidural Neoplasms
Chromosomes, Human, Pair 22
Cell Transformation, Viral
Harvey murine sarcoma virus
Cell Transformation, Neoplastic
Gammaretrovirus
Histiocytoma, Malignant Fibrous
Chondrosarcoma
Doxorubicin
Liposarcoma
Calmodulin-Binding Proteins
Gene Expression Regulation, Neoplastic
Nanodiamonds
Sarcoma 37
Combined Modality Therapy
Immunohistochemistry
Histiocytoma, Benign Fibrous
Reverse Transcriptase Polymerase Chain Reaction
Base Sequence
Abdominal Neoplasms
Prognosis
Keratin-17
Langerhans Cell Sarcoma
Vascular Neoplasms
Retroperitoneal Neoplasms
Chick Embryo
Neoplasms, Second Primary
Treatment Outcome
Dactinomycin
Receptor, IGF Type 1
Dendritic Cell Sarcoma, Follicular
Helper Viruses
Neoplasm Recurrence, Local
Antineoplastic Combined Chemotherapy Protocols
Chromosome Breakpoints
Muscle Neoplasms
Tumor Cells, Cultured
Neoplasm Transplantation
Transcription Factors
Avian leukosis virus
Mice, Nude
Etoposide
Hemangiosarcoma
Retroviridae
Retrospective Studies
Fibrosarcoma
Heterogeneous-Nuclear Ribonucleoproteins
Overexpression of p53 protein in primary Ewing's sarcoma of bone: relationship to tumour stage, response and prognosis. (1/874)
Biopsy tissues of 52 patients with Ewing's sarcoma of bone treated between 1983 and 1993 were examined immuno-histochemically to determine the significance of p53 protein in diagnosis and prognosis of Ewing's sarcoma. Mean age at diagnosis was 17 years (range 6-36) and minimum follow-up was 30 months. The tumours were located in the extremities and central bones in 35 and 17 patients respectively. Metastases were present in seven patients at diagnosis. Treatment consisted of chemotherapy, surgery and/or radiotherapy in all the patients. Overexpression of p53 protein was demonstrated in seven patients (14%). There was no relationship between expression of p53 and site of tumours. Patients who overexpressed p53 protein appeared to have more advanced diseases at diagnosis and poorer response to chemotherapy than those without p53 overexpression. The 5-year relapse-free survival and overall survival in patients without metastases at the time of diagnosis were 66% and 71%, respectively, in p53 protein-negative patients compared with 20% relapse-free and overall survival in those with p53 protein overexpression (P= 0.01). The poorer prognosis in p53 protein-positive patients was independent of site, local treatment or necrosis of the tumours (P < 0.05). Over-expression of p53 protein is an independent poor prognostic factor in Ewing's sarcoma of bone. (+info)Measurement of DNA cross-linking in patients on ifosfamide therapy using the single cell gel electrophoresis (comet) assay. (2/874)
The single cell gel electrophoresis comet assay has become established as a sensitive technique for measuring DNA strand breaks. The technique has been modified to allow the sensitive detection and quantitation of DNA interstrand cross-linking at the single cell level. Cells are irradiated immediately before analysis to deliver a fixed level of random strand breakage. After embedding of cells in agarose and lysis, the presence of cross-links retards the electrophoretic mobility of the alkaline denatured cellular DNA. Cross-links are, therefore, quantitated as the decrease in the comet tail moment compared with irradiated controls. Using this method, a linear response of cross-linking versus dose of chlorambucil over a wide dose range was demonstrated in human lymphocytes after drug treatment ex vivo. The method was also sensitive enough to determine cross-linking in clinical samples after chemotherapy. For example, crosslinking was observed in the lymphocytes of patients receiving ifosfamide (3 g/m2/day) as a continuous infusion for 3-5 days or as a 3-h infusion daily for 3 days. Cross-links were detected in all patients within 3 h, with no evidence of DNA single strand break formation. In patients receiving continuous infusion, a plateau of cross-linking was reached by 24 h. In the patients receiving ifosfamide over 3 h, a clear decrease in the peak level of cross-linking was observed before subsequent infusions. (+info)Differential transactivation by alternative EWS-FLI1 fusion proteins correlates with clinical heterogeneity in Ewing's sarcoma. (3/874)
The t(11;22)(q24;q12) translocation is present in up to 95% of cases of Ewing's sarcoma and results in the formation of an EWS-FLI1 fusion gene which encodes a chimeric transcription factor. The proximate role of EWS-FLI1 in the pathogenesis of Ewing's sarcoma is thought to involve the activation of as yet largely unknown target genes. Many alternative forms of EWS-FLI1 exist because of variations in the locations of the EWS and FLI1 genomic breakpoints. The most common form, designated "type 1," consists of the first seven exons of EWS joined to exons 6-9 of FLI1 and accounts for approximately 60% of cases. The "type 2" EWS-FLI1 fusion also includes FLI1 exon 5 and is present in another 25%. We and others have observed previously that the type 1 fusion is associated with a significantly better prognosis than the other fusion types. Because EWS-FLI1 is an aberrant transcription factor, we investigated whether these differences in clinical behavior may be correlated to functional differences by comparing transactivation by the type 1 EWS-FLI1 with other types in both heterologous cells (HeLa, NIH3T3) and homologous cells (Ewing's sarcoma cell lines). In a panel of seven Ewing's sarcoma cell lines, we found transactivation of a transiently transfected FLI1-responsive reporter construct to be significantly lower in cell lines with the type 1 fusion than in cell lines with the type 2 fusion (P = 0.003). Cotransfection of the same reporter construct with each of a series of seven EWS-FLI1 expression constructs (corresponding to the two major fusion types and five less common types) also showed that type 1 EWS-FLI1 was a significantly weaker transactivator than the type 2 product in both HeLa and NIH3T3 cells (P = 0.003, and P = 0.033, respectively). Electromobility shift assays showed equivalent binding of the type 1 and type 2 EWS-FLI1 to the consensus FLI1-responsive binding site, indicating that differences in transactivation were not due simply to differences in DNA binding affinity. The finding that the type 1 EWS-FLI1 fusion, associated with less aggressive clinical behavior, encodes a less active chimeric transcription factor may provide the basis for a molecular explanation of clinical heterogeneity in Ewing's sarcoma. (+info)Tumour volume as a predictor of necrosis after chemotherapy in Ewing's sarcoma. (4/874)
We studied the CT and MR scans, and the histology of 50 patients with primary Ewing's sarcoma of bone to determine the association between the change in tumour volume and necrosis after chemotherapy, and to ascertain their influence on prognosis. The mean age of the patients was 17 years. The limbs were involved in 40 and the axial bones in ten. The volume of the tumour at diagnosis varied from 31 to 1790 ml. There was a significant relationship between necrosis and the measured change in volume of the tumour after chemotherapy. Progression of the tumour despite chemotherapy was seen only in patients with necrosis of grades 4 to 6. Necrosis significantly influenced survival (p < 0.05), but the effect of change in volume was less significant. Change in volume of the tumour is a good predictor of necrosis induced by chemotherapy. Necrosis is a strong prognostic factor in Ewing's sarcoma. (+info)Tyrosine kinase Pyk2 mediates G-protein-coupled receptor regulation of the Ewing sarcoma RNA-binding protein EWS. (5/874)
Ewing family tumors result from the effects of chromosomal translocations that fuse the Ewing sarcoma (EWS) gene to various genes encoding transcription factors. The resulting chimeric EWS fusion proteins are transcriptional activators with transforming potential that have received much study. By contrast, the cellular function of somatic EWS remains obscure. EWS belongs to a family of RNA-binding proteins thought to play role in RNA synthesis or processing. Here, we show that EWS interacts with Pyk2, a protein tyrosine kinase implicated in a variety of signal transduction processes. G-protein-coupled receptor signaling and other stimuli of Pyk2 kinase activity significantly block the interaction between EWS and Pyk2. Furthermore, as assessed by sucrose gradient centrifugation, EWS partitions with dense ribosome-containing fractions in a manner that is enhanced by signaling from the G-protein-coupled m1 muscarinic acetylcholine receptor (mAChR). We conclude that extranuclear EWS is a previously unrecognized target of G-protein-coupled receptor regulation. (+info)Management of cancer in pregnancy: a case of Ewing's sarcoma of the pelvis in the third trimester. (6/874)
Ewing's sarcoma of the pelvic bones was diagnosed in a 21-year childbearing woman, raising major medical and ethical problems. The diagnostic and therapeutic approaches during the sixth month of gestation were tailored in order to cure the patient and avoid unnecessary toxicity to the fetus. Ancillary tests included ultrasound and MRI studies of the pelvis. Ifosfamide and adriamycin, premedicated by granisetron, were administered during gestation, and were found to be safe. Cesarean section was the preferred way of delivery since the tumor involved the pelvic bones. The outcome was a disease-free patient and a small healthy baby who is now two years of age. (+info)The EWS/TEC fusion protein encoded by the t(9;22) chromosomal translocation in human chondrosarcomas is a highly potent transcriptional activator. (7/874)
The EWS/TEC gene fusion generated by the t(9;22) chromosomal translocation found in extraskeletal myxoid chondrosarcomas encodes a fusion protein containing the amino-terminal domain of the EWS protein fused to the whole coding sequence of the orphan nuclear receptor TEC. We have compared the DNA-binding and transcriptional activation properties of various TEC isoforms and the corresponding EWS/TEC fusion proteins. Band-shift experiments show that the full-length TEC receptor can efficiently bind the NGFI-B Response Element (NBRE), whereas an isoform lacking the entire carboxyl-terminal domain of the receptor binds much less efficiently the NBRE. Addition of the amino-terminal domain of EWS to either isoforms does not alter significantly their DNA-binding properties to the NBRE. Co-transfection experiments of COS cells and human chondrocytes indicate that whereas TEC moderately activates transcription from a NBRE-containing promoter, the corresponding EWS/TEC fusion protein is a highly potent transcriptional activator of the same promoter, being approximately 270-fold more active than the native receptor. EWS/TEC may thus exert its oncogenic potential in chrondrosarcomas by activating the transcription of target genes involved in cell proliferation. (+info)Brain metastases in musculoskeletal sarcomas. (8/874)
BACKGROUND: In musculoskeletal sarcomas, brain metastases are rare, but severely affect quality of life. METHODS: All patients with musculoskeletal sarcomas who were treated at our institutions from 1975 to 1997 were reviewed for examples of brain metastasis. RESULTS: Of 480 sarcoma patients, 179 had distant metastases, including 20 patients with brain metastases (4.2%). Alveolar soft part sarcoma (3/4), extraskeletal Ewing's sarcoma (2/8), rhabdomyosarcoma (2/13) and bone Ewing's sarcoma (2/18) tended to metastasize to the brain. All 20 patients had distant or local relapses and 16 of the 20 patients had pulmonary metastases. Three patients underwent surgical treatment and two of them survived over 1 year. Mean survival after diagnosis of brain metastasis was 5.1 months. CONCLUSIONS: Patients with alveolar soft part sarcoma, Ewing's sarcoma, rhabdomyosarcoma and pulmonary metastases have a high risk of brain metastasis. (+info)Ewing sarcoma is a type of cancer that originates in bones or the soft tissues surrounding them, such as muscles and tendons. It primarily affects children and adolescents, although it can occur in adults as well. The disease is characterized by small, round tumor cells that typically grow quickly and are prone to metastasize (spread) to other parts of the body, most commonly the lungs, bones, and bone marrow.
Ewing sarcoma is caused by a genetic abnormality, specifically a chromosomal translocation that results in the fusion of two genes, EWSR1 and FLI1. This gene fusion leads to the formation of an abnormal protein that disrupts normal cell growth and division processes, ultimately resulting in cancer.
Symptoms of Ewing sarcoma can vary depending on the location and size of the tumor but may include pain or swelling in the affected area, fever, fatigue, and weight loss. Diagnosis typically involves imaging studies such as X-rays, CT scans, or MRI scans to locate the tumor, followed by a biopsy to confirm the presence of cancer cells. Treatment may involve surgery, radiation therapy, chemotherapy, or a combination of these approaches, depending on the stage and location of the disease.
Sarcoma is a type of cancer that develops from certain types of connective tissue (such as muscle, fat, fibrous tissue, blood vessels, or nerves) found throughout the body. It can occur in any part of the body, but it most commonly occurs in the arms, legs, chest, and abdomen.
Sarcomas are classified into two main groups: bone sarcomas and soft tissue sarcomas. Bone sarcomas develop in the bones, while soft tissue sarcomas develop in the soft tissues of the body, such as muscles, tendons, ligaments, fat, blood vessels, and nerves.
Sarcomas can be further classified into many subtypes based on their specific characteristics, such as the type of tissue they originate from, their genetic makeup, and their appearance under a microscope. The different subtypes of sarcoma have varying symptoms, prognoses, and treatment options.
Overall, sarcomas are relatively rare cancers, accounting for less than 1% of all cancer diagnoses in the United States each year. However, they can be aggressive and may require intensive treatment, such as surgery, radiation therapy, and chemotherapy.
Synovial sarcoma is a rare type of cancer that typically develops in the soft tissues surrounding the joints, such as the synovial membrane, which lines the joint capsules. Despite its name, synovial sarcoma does not necessarily arise from the synovium. It is called so due to its resemblance to this tissue under a microscope.
This form of sarcoma primarily affects young adults and can be found in various parts of the body, but it most commonly occurs in the extremities, particularly near the knees. Synovial sarcoma is characterized by specific genetic changes that result in the formation of fusion proteins, which contribute to uncontrolled cell growth and tumor development.
There are two main subtypes of synovial sarcoma: monophasic and biphasic. Monophasic synovial sarcoma is composed of either spindle-shaped (spaghetti-like) cells or epithelioid (roundish) cells, while biphasic synovial sarcoma contains both types of cells. A third subtype, called poorly differentiated synovial sarcoma, has a more aggressive behavior and is composed of small round cells that do not resemble the typical spindle or epithelioid cells.
Treatment for synovial sarcoma usually involves surgical removal of the tumor, often followed by radiation therapy and/or chemotherapy to reduce the risk of recurrence and metastasis. The prognosis varies depending on factors such as the size and location of the tumor, the patient's age, and the presence of metastases at diagnosis.
Kaposi sarcoma (KS) is a type of cancer that causes abnormal growths in the skin, lymph nodes, or other organs. It is caused by the Kaposi sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8 (HHV8). There are several forms of KS, including:
1. Classic KS: This form primarily affects older men of Mediterranean, Middle Eastern, or Ashkenazi Jewish descent. It tends to progress slowly and mainly involves the skin.
2. Endemic KS: Found in parts of Africa, this form predominantly affects children and young adults, regardless of their HIV status.
3. Immunosuppression-associated KS: This form is more aggressive and occurs in people with weakened immune systems due to organ transplantation or other causes.
4. Epidemic KS (AIDS-related KS): This is the most common form of KS, seen primarily in people with HIV/AIDS. The widespread use of antiretroviral therapy (ART) has significantly reduced its incidence.
KS lesions can appear as red, purple, or brown spots on the skin and may also affect internal organs such as the lungs, lymph nodes, or gastrointestinal tract. Symptoms vary depending on the location of the lesions but often include fever, fatigue, weight loss, and swelling in the legs or abdomen. Treatment options depend on the extent and severity of the disease and may involve local therapies (e.g., radiation, topical treatments), systemic therapies (e.g., chemotherapy, immunotherapy), or a combination of these approaches.
Ewing Sarcoma (EWS) RNA-Binding Protein, also known as EWSR1, is a protein that plays a role in gene expression by binding to RNA. It is a member of the FET family of proteins, which also includes FUS and TAF15. These proteins are involved in various cellular processes such as transcription, splicing, and translation.
Mutations in the EWSR1 gene have been associated with several types of cancer, most notably Ewing sarcoma, a rare tumor that typically affects children and adolescents. In Ewing sarcoma, a fusion protein is formed when EWSR1 combines with another protein, most commonly ETS translocation variant 1 (ETV1), FLI1, ERG or FEV. This fusion protein can lead to abnormal gene expression and tumor formation.
EWSR1 has also been found to be involved in other types of cancer such as acute myeloid leukemia, clear cell sarcoma, desmoplastic small round cell tumors and liposarcomas.
It's important to note that while EWSR1 is a RNA-binding protein, it can also bind to DNA in certain contexts, such as when it forms a fusion protein with an ETS transcription factor in Ewing sarcoma.
Proto-oncogene protein c-Fli-1 is a transcription factor that belongs to the ETS family and plays crucial roles in hematopoiesis, vascular development, and cell proliferation. The gene encoding this protein, called c-Fli-1, can be mutated or its expression can be dysregulated, leading to the formation of a proto-oncogene. When this happens, the protein can contribute to the development of various types of cancer, such as Ewing's sarcoma and acute myeloid leukemia. In these cases, the protein promotes cell growth and division, inhibits apoptosis (programmed cell death), and increases angiogenesis (the formation of new blood vessels). Overall, c-Fli-1 is an important regulator of normal cellular processes, but when its activity is deregulated, it can contribute to the development of cancer.
Avian sarcoma viruses (ASVs) are a group of retroviruses that primarily infect birds and cause various types of tumors, particularly sarcomas. These viruses contain an oncogene, which is a gene that has the ability to transform normal cells into cancerous ones. The oncogene in ASVs is often derived from cellular genes called proto-oncogenes, which are normally involved in regulating cell growth and division.
ASVs can be divided into two main types: non-defective and defective. Non-defective ASVs contain a complete set of viral genes that allow them to replicate independently, while defective ASVs lack some of the necessary viral genes and require assistance from other viruses to replicate.
One well-known example of an avian sarcoma virus is the Rous sarcoma virus (RSV), which was first discovered in chickens by Peyton Rous in 1910. RSV causes a highly malignant form of sarcoma in chickens and has been extensively studied as a model system for cancer research. The oncogene in RSV is called v-src, which is derived from the normal cellular gene c-src.
Avian sarcoma viruses have contributed significantly to our understanding of the molecular mechanisms underlying cancer development and have provided valuable insights into the role of oncogenes in tumorigenesis.
I'm sorry for any confusion, but "Sarcoma, Experimental" is not a recognized medical term or definition. Sarcomas are a type of cancer that develop in the body's connective tissues, such as bones, muscles, tendons, cartilage, and fat. There are many different types of sarcomas, classified based on the specific type of tissue they originate from.
Experimental, on the other hand, refers to something that is being tested or tried out for the first time, typically as part of a scientific experiment or clinical trial. In the context of cancer treatment, an experimental therapy might refer to a new drug, procedure, or device that is still being studied in clinical trials to determine its safety and effectiveness.
Therefore, "Sarcoma, Experimental" could potentially refer to a clinical trial or research study involving a new treatment for sarcoma, but it would not be a medical definition in and of itself. If you have any specific questions about sarcomas or experimental treatments, I would recommend consulting with a healthcare professional or medical researcher for more accurate information.
I'm sorry for any confusion, but "Sarcoma 180" is not a recognized medical term or an official classification of sarcomas in humans. It appears to be a term used primarily in research involving mice. Sarcoma 180 is a transplantable tumor that was first isolated from a mouse and has been used as a model for cancer research, particularly in studies involving immunotherapy and cancer treatment.
In general, sarcomas are cancers that develop from connective tissues such as bones, muscles, tendons, cartilages, nerves, and blood vessels. They can be further classified into various subtypes based on the specific type of tissue they originate from and their genetic characteristics. If you have any concerns about a specific medical condition or term, I would recommend consulting with a healthcare professional for accurate information.
Bone neoplasms are abnormal growths or tumors that develop in the bone. They can be benign (non-cancerous) or malignant (cancerous). Benign bone neoplasms do not spread to other parts of the body and are rarely a threat to life, although they may cause problems if they grow large enough to press on surrounding tissues or cause fractures. Malignant bone neoplasms, on the other hand, can invade and destroy nearby tissue and may spread (metastasize) to other parts of the body.
There are many different types of bone neoplasms, including:
1. Osteochondroma - a benign tumor that develops from cartilage and bone
2. Enchondroma - a benign tumor that forms in the cartilage that lines the inside of the bones
3. Chondrosarcoma - a malignant tumor that develops from cartilage
4. Osteosarcoma - a malignant tumor that develops from bone cells
5. Ewing sarcoma - a malignant tumor that develops in the bones or soft tissues around the bones
6. Giant cell tumor of bone - a benign or occasionally malignant tumor that develops from bone tissue
7. Fibrosarcoma - a malignant tumor that develops from fibrous tissue in the bone
The symptoms of bone neoplasms vary depending on the type, size, and location of the tumor. They may include pain, swelling, stiffness, fractures, or limited mobility. Treatment options depend on the type and stage of the tumor but may include surgery, radiation therapy, chemotherapy, or a combination of these treatments.
Sarcoma viruses, murine, are a group of RNA viruses that primarily affect mice and other rodents. They are classified as type C retroviruses, which means they contain an envelope, have reverse transcriptase enzyme activity, and replicate through a DNA intermediate.
The murine sarcoma viruses (MSVs) are associated with the development of various types of tumors in mice, particularly fibrosarcomas, which are malignant tumors that originate from fibroblasts, the cells that produce collagen and other fibers in connective tissue.
The MSVs are closely related to the murine leukemia viruses (MLVs), and together they form a complex called the murine leukemia virus-related viruses (MLVRVs). The MLVRVs can undergo recombination events, leading to the generation of new viral variants with altered biological properties.
The MSVs are important tools in cancer research because they can transform normal cells into tumor cells in vitro and in vivo. The study of these viruses has contributed significantly to our understanding of the molecular mechanisms underlying cancer development and progression.
Soft tissue neoplasms refer to abnormal growths or tumors that develop in the soft tissues of the body. Soft tissues include muscles, tendons, ligaments, fascia, nerves, blood vessels, fat, and synovial membranes (the thin layer of cells that line joints and tendons). Neoplasms can be benign (non-cancerous) or malignant (cancerous), and their behavior and potential for spread depend on the specific type of neoplasm.
Benign soft tissue neoplasms are typically slow-growing, well-circumscribed, and rarely spread to other parts of the body. They can often be removed surgically with a low risk of recurrence. Examples of benign soft tissue neoplasms include lipomas (fat tumors), schwannomas (nerve sheath tumors), and hemangiomas (blood vessel tumors).
Malignant soft tissue neoplasms, on the other hand, can grow rapidly, invade surrounding tissues, and may metastasize (spread) to distant parts of the body. They are often more difficult to treat than benign neoplasms and require a multidisciplinary approach, including surgery, radiation therapy, and chemotherapy. Examples of malignant soft tissue neoplasms include sarcomas, such as rhabdomyosarcoma (arising from skeletal muscle), leiomyosarcoma (arising from smooth muscle), and angiosarcoma (arising from blood vessels).
It is important to note that soft tissue neoplasms can occur in any part of the body, and their diagnosis and treatment require a thorough evaluation by a healthcare professional with expertise in this area.
I'm sorry for any confusion, but "Avian Sarcoma" is not a widely recognized medical term. The term "sarcoma" is used in pathology to describe a type of cancer that develops from certain types of connective tissue (such as bone, cartilage, muscle, or blood vessels).
On the other hand, "avian" refers to birds or related to birds. In medical literature, avian sarcomas are sometimes used to describe sarcomas that occur in birds. However, specific types of avian sarcomas would be defined by the type of cell from which they originate (like a fibrosarcoma, osteosarcoma, etc.).
If you're asking about a specific medical condition or context, could you please provide more details? I'm here to help!
An oncogene protein fusion is a result of a genetic alteration in which parts of two different genes combine to create a hybrid gene that can contribute to the development of cancer. This fusion can lead to the production of an abnormal protein that promotes uncontrolled cell growth and division, ultimately resulting in a malignant tumor. Oncogene protein fusions are often caused by chromosomal rearrangements such as translocations, inversions, or deletions and are commonly found in various types of cancer, including leukemia and sarcoma. These genetic alterations can serve as potential targets for cancer diagnosis and therapy.
Histiocytic sarcoma is a rare type of cancer that originates from histiocytes, which are cells that are part of the immune system and found in various tissues throughout the body. These cells normally function to help fight infection and remove foreign substances. In histiocytic sarcoma, there is an abnormal accumulation and proliferation of these cells, leading to the formation of tumors.
Histiocytic sarcoma can affect people of any age but is more commonly found in adults, with a slight male predominance. It can occur in various parts of the body, such as the lymph nodes, skin, soft tissues, and internal organs like the spleen, liver, and lungs. The exact cause of histiocytic sarcoma remains unknown, but it is not considered to be hereditary.
The symptoms of histiocytic sarcoma depend on the location and extent of the tumor(s). Common signs include swollen lymph nodes, fatigue, fever, weight loss, night sweats, and pain or discomfort in the affected area. Diagnosis typically involves a combination of imaging studies (like CT scans, PET scans, or MRI), biopsies, and laboratory tests to confirm the presence of histiocytic sarcoma and assess its extent.
Treatment for histiocytic sarcoma usually involves a multidisciplinary approach, including surgery, radiation therapy, and chemotherapy. The choice of treatment depends on several factors, such as the location and stage of the disease, the patient's overall health, and their personal preferences. Clinical trials may also be an option for some patients, allowing them to access new and experimental therapies.
Prognosis for histiocytic sarcoma is generally poor, with a five-year survival rate of approximately 15-30%. However, outcomes can vary significantly depending on individual factors, such as the patient's age, the extent of the disease at diagnosis, and the effectiveness of treatment. Continued research is necessary to improve our understanding of this rare cancer and develop more effective therapies for those affected.
A myeloid sarcoma is a rare type of cancer that can develop in various parts of the body. It is also known as a granulocytic sarcoma or chloroma.
Myeloid sarcomas occur when immature white blood cells, called myeloblasts, accumulate and form a tumor in an extramedullary site, which means outside of the bone marrow. These tumors can develop in various organs and tissues, such as the skin, soft tissue, bones, lymph nodes, or gastrointestinal tract.
Myeloid sarcomas are often associated with acute myeloid leukemia (AML), a type of blood cancer that affects the bone marrow's ability to produce healthy blood cells. However, they can also occur in individuals who have previously been treated for AML or other myeloid disorders, or rarely, in those without a known history of these conditions.
The diagnosis of myeloid sarcoma typically involves a biopsy of the affected tissue, followed by microscopic examination and immunohistochemical staining to confirm the presence of myeloblasts and other specific markers. Treatment options for myeloid sarcoma depend on several factors, including the patient's overall health, the extent and location of the disease, and whether it is associated with AML or another myeloid disorder. Treatment may include chemotherapy, radiation therapy, targeted therapy, or stem cell transplantation.
Endometrial stromal sarcoma is a rare type of cancer that arises from the connective tissue cells (stromal cells) of the endometrium, which is the inner lining of the uterus. This type of sarcoma is typically low-grade and slow-growing, but it can still metastasize or spread to other parts of the body.
Endometrial stromal sarcomas are usually diagnosed in postmenopausal women, although they can also occur in younger women. The most common symptom is abnormal vaginal bleeding, especially if it occurs after menopause. Other symptoms may include pelvic pain or a mass that can be felt during a physical examination.
The diagnosis of endometrial stromal sarcoma typically involves a combination of imaging studies, such as ultrasound, MRI, or CT scan, and a biopsy to confirm the presence of cancer cells. Treatment usually involves surgery to remove the uterus and surrounding tissues, followed by hormone therapy, radiation therapy, or chemotherapy, depending on the stage and grade of the tumor. Regular follow-up care is essential to monitor for recurrence and manage any long-term effects of treatment.
Sarcoma, clear cell, is a rare type of cancer that arises from certain types of connective tissue in the body. It is called "clear cell" because the cancer cells have a clear appearance when viewed under a microscope due to the presence of lipids or glycogen within the cytoplasm.
Clear cell sarcoma can occur in various parts of the body, but it most commonly affects the soft tissues of the extremities, such as the legs and arms. It is an aggressive cancer that tends to spread to other parts of the body, including the lungs, lymph nodes, and bones.
Clear cell sarcoma typically occurs in young adults, with a median age at diagnosis of around 30 years old. The exact cause of this type of sarcoma is not known, but it has been linked to genetic mutations involving the EWSR1 gene. Treatment for clear cell sarcoma usually involves surgery to remove the tumor, followed by radiation therapy and/or chemotherapy to kill any remaining cancer cells. Despite treatment, the prognosis for patients with clear cell sarcoma is generally poor, with a five-year survival rate of around 50%.
Neuroectodermal tumors, primitive, peripheral (PNET) are a group of rare and aggressive malignancies that primarily affect children and young adults. These tumors arise from the primitive neuroectodermal cells, which are the precursors to the nervous system. PNETs can occur in various locations throughout the body, but when they occur outside the central nervous system (CNS), they are referred to as peripheral PNETs (pPNETs).
Peripheral PNETs are similar to Ewing sarcoma, another type of small, round blue cell tumor that arises from primitive neuroectodermal cells. In fact, some researchers consider pPNETs and Ewing sarcomas to be part of the same disease spectrum, known as the Ewing family of tumors (EFT).
Peripheral PNETs can occur in any part of the body, but they most commonly affect the bones and soft tissues of the trunk, extremities, and head and neck region. The symptoms of pPNET depend on the location and size of the tumor, but they may include pain, swelling, decreased mobility, and systemic symptoms such as fever and weight loss.
The diagnosis of pPNET typically involves a combination of imaging studies (such as MRI or CT scans), biopsy, and molecular testing. The treatment usually involves a multimodal approach that includes surgery, chemotherapy, and radiation therapy. Despite aggressive treatment, the prognosis for patients with pPNET remains poor, with a five-year survival rate of approximately 30%.
Neuroectodermal tumors, primitive (PNETs) are a group of highly malignant and aggressive neoplasms that arise from neuroectodermal cells, which are the precursors to the nervous system during embryonic development. These tumors can occur anywhere in the body but are most commonly found in the central nervous system, particularly in the brain and spinal cord.
PNETs are characterized by small, round, blue cells that have a high degree of cellularity and mitotic activity. They are composed of undifferentiated or poorly differentiated cells that can differentiate along various neural lineages, including neuronal, glial, and epithelial. This feature makes their diagnosis challenging, as they can resemble other small round blue cell tumors, such as lymphomas, rhabdomyosarcomas, and Ewing sarcoma.
Immunohistochemical staining and molecular genetic testing are often required to confirm the diagnosis of PNETs. These tests typically reveal the expression of neural markers, such as NSE, Synaptophysin, and CD99, and the presence of specific chromosomal abnormalities, such as the EWS-FLI1 fusion gene in Ewing sarcoma.
PNETs are aggressive tumors with a poor prognosis, and their treatment typically involves a multimodal approach that includes surgery, radiation therapy, and chemotherapy. Despite these treatments, the five-year survival rate for patients with PNETs is less than 30%.
I'm sorry for the confusion, but "Sarcoma, Yoshida" is not a recognized medical term or a specific type of sarcoma in any major oncology reference or database. It appears that "Yoshida" might be referring to a person who described or studied a particular type of sarcoma. However, I cannot find any relevant information related to this exact term.
Sarcomas are cancers that develop from connective tissues such as bones, muscles, tendons, cartilages, nerves, and blood vessels. They can be categorized into two main groups: bone sarcomas and soft tissue sarcomas. There are many subtypes of sarcoma, each with its unique features, diagnostic criteria, and treatment approaches.
If you have more context or information about "Sarcoma, Yoshida," I would be happy to help you further research the topic. However, based on the available data, it is not possible to provide a medical definition for this term.
The Kirsten murine sarcoma virus (KiMSV) is a type of retrovirus that can cause tumors in mice. It was first discovered in 1968 by Charlotte Kirsten and her colleagues. KiMSV is a complex retrovirus, which means that it contains additional genes beyond the standard gag, pol, and env genes found in simple retroviruses.
In particular, KiMSV contains an oncogene called v-Ki-ras, which encodes a protein that can transform cells and lead to cancer. This oncogene is derived from the host cell's c-Ki-ras gene, which is involved in normal cell signaling pathways. When the viral oncogene is expressed in infected cells, it can cause uncontrolled cell growth and division, leading to the formation of tumors.
KiMSV primarily causes fibrosarcomas, a type of cancer that arises from connective tissue cells called fibroblasts. However, it has also been shown to induce other types of tumors in mice, including leukemias and lymphomas.
While KiMSV is not known to infect humans or cause disease in humans, the study of this virus and its oncogene have provided important insights into the mechanisms of cancer development and progression. The v-Ki-ras oncogene, for example, has been found to be mutated and activated in many human cancers, including lung, colon, and pancreatic cancers.
Osteosarcoma is defined as a type of cancerous tumor that arises from the cells that form bones (osteoblasts). It's the most common primary bone cancer, and it typically develops in the long bones of the body, such as the arms or legs, near the growth plates. Osteosarcoma can metastasize (spread) to other parts of the body, including the lungs, making it a highly malignant form of cancer. Symptoms may include bone pain, swelling, and fractures. Treatment usually involves a combination of surgery, chemotherapy, and/or radiation therapy.
Moloney murine sarcoma virus (Mo-MSV) is a type of retrovirus, specifically a sarcoma virus that infects mice. It was first discovered and isolated by John Moloney in 1960. Mo-MSV is a horizontally transmitted virus, meaning it is typically spread through the direct transfer of bodily fluids between infected and uninfected hosts.
Mo-MSV is closely related to Moloney leukemia virus (Mo-MLV), and both viruses are often found as co-infections in mice. Mo-MSV is associated with the development of sarcomas, which are malignant tumors that arise from connective tissues such as bone, cartilage, fat, muscle, or fibrous tissue.
The virus contains an RNA genome and integrates its genetic material into the host cell's DNA upon infection. Mo-MSV is capable of transforming cells by introducing oncogenes into the host cell's genome, which can lead to uncontrolled cell growth and ultimately result in cancer formation.
Mo-MSV has been extensively studied as a model system for retroviral infection and tumorigenesis, contributing significantly to our understanding of oncogene function and the molecular mechanisms underlying cancer development.
Sarcoma viruses in cats, also known as feline sarcoma viruses (FeSVs), are a group of retroviruses that can cause tumors and other diseases in felines. There are two main types of FeSVs: the feline leukemia virus (FeLV)-related sarcoma viruses and the independent feline sarcoma viruses.
The FeLV-related sarcoma viruses are formed when a cat is infected with FeLV, and the FeLV genome integrates into the host's DNA in such a way that it becomes rearranged and acquires new oncogenic properties. These rearranged FeLV proviruses can then cause various types of tumors, including fibrosarcomas, lymphosarcomas, and leukemias.
The independent feline sarcoma viruses, on the other hand, are not associated with FeLV infection. They contain their own unique oncogenes that can induce the formation of fibrosarcomas, a type of soft tissue cancer. These viruses are typically transmitted through direct contact with an infected cat or its saliva and can cause rapidly growing tumors at the site of inoculation.
It is important to note that not all cats infected with FeSVs will develop tumors, and other factors such as the cat's age, immune status, and genetic background may also play a role in the development of disease.
Rous sarcoma virus (RSV) is an avian retrovirus that was first discovered by Peyton Rous in 1910. It is the cause of a type of cancer called avian sarcoma, which affects birds, particularly chickens. The virus is transmitted through the spread of infected cells or cell-free filtrates and can induce tumors at the site of infection.
RSV contains an RNA genome that is reverse transcribed into DNA upon entry into the host cell. This DNA then integrates into the host's chromosomal DNA, leading to a persistent infection. The virus encodes several oncogenes, including src, which play a crucial role in the transformation of infected cells and the development of cancer.
RSV has been extensively studied as a model system for retroviral-induced tumorigenesis and has contributed significantly to our understanding of the molecular mechanisms underlying cancer development.
Ifosfamide is an alkylating agent, which is a type of chemotherapy medication. It works by interfering with the DNA of cancer cells, preventing them from dividing and growing. Ifosfamide is used to treat various types of cancers, such as testicular cancer, small cell lung cancer, ovarian cancer, cervical cancer, and certain types of sarcomas.
The medical definition of Ifosfamide is:
Ifosfamide is a synthetic antineoplastic agent, an oxazaphosphorine derivative, with the chemical formula C6H15Cl2N2O2P. It is used in the treatment of various malignancies, including germ cell tumors, sarcomas, lymphomas, and testicular cancer. The drug is administered intravenously and exerts its cytotoxic effects through the alkylation and cross-linking of DNA, leading to the inhibition of DNA replication and transcription. Ifosfamide can cause significant myelosuppression and has been associated with urotoxicity, neurotoxicity, and secondary malignancies. Therefore, it is essential to monitor patients closely during treatment and manage any adverse effects promptly.
Small cell sarcoma is a very rare and aggressive type of cancer that affects the connective tissues in the body, such as muscles, tendons, bones, cartilage, and fat. It is called "small cell" because the cancer cells are small and appear round or oval in shape, with scant cytoplasm and finely granular chromatin.
Small cell sarcoma typically occurs in adults between the ages of 40 and 70, and it can develop in any part of the body. However, it is most commonly found in the extremities, trunk, and retroperitoneum. The exact cause of small cell sarcoma is not known, but it is thought to be associated with genetic mutations that occur during a person's lifetime.
Small cell sarcoma can be difficult to diagnose because it often does not cause any symptoms until it has advanced to an aggressive stage. When symptoms do occur, they may include pain, swelling, or a lump in the affected area. Diagnosis typically involves a biopsy of the tumor tissue, followed by imaging tests such as CT scans, MRI scans, or PET scans to determine the extent of the cancer.
Treatment for small cell sarcoma usually involves surgery to remove the tumor, followed by radiation therapy and/or chemotherapy to kill any remaining cancer cells. However, because small cell sarcoma is so rare and aggressive, treatment options may be limited, and the prognosis is often poor. Clinical trials of new treatments are also an option for some patients.
Rhabdomyosarcoma is a type of cancer that develops in the body's soft tissues, specifically in the muscle cells. It is a rare and aggressive form of sarcoma, which is a broader category of cancers that affect the connective tissues such as muscles, tendons, cartilages, bones, blood vessels, and fatty tissues.
Rhabdomyosarcomas can occur in various parts of the body, including the head, neck, arms, legs, trunk, and genitourinary system. They are more common in children than adults, with most cases diagnosed before the age of 18. The exact cause of rhabdomyosarcoma is not known, but genetic factors and exposure to radiation or certain chemicals may increase the risk.
There are several subtypes of rhabdomyosarcoma, including embryonal, alveolar, pleomorphic, and spindle cell/sclerosing. The type and stage of the cancer determine the treatment options, which may include surgery, radiation therapy, chemotherapy, or a combination of these approaches. Early diagnosis and prompt treatment are crucial for improving the prognosis and long-term survival rates.
Alveolar Soft Part Sarcoma (ASPS) is a rare type of sarcoma, which is a cancer that develops in the body's connective or supportive tissues such as muscles, tendons, ligaments, cartilage, nerves, and blood vessels. ASPS typically arises in deep soft tissues, often in the legs or arms, but can also occur in other parts of the body like the head and neck region.
ASPS is called "alveolar" because the cancer cells sometimes form structures that look like the air sacs (alveoli) found in the lungs. The term "soft part" indicates that this type of sarcoma usually arises in the soft tissues of the body.
Histologically, ASPS is characterized by the presence of distinctive organoid nests or alveolar structures composed of large polygonal cells with eosinophilic cytoplasm and distinct cell borders. The nuclei are round to oval, with finely dispersed chromatin and prominent nucleoli. Immunohistochemically, ASPS cells typically express TFE3, a transcription factor that can be used in the diagnosis of this tumor type.
ASPS tends to grow slowly but can metastasize (spread) to other parts of the body, such as the lungs, brain, and bones. It primarily affects adolescents and young adults, with a slight female predominance. Treatment usually involves surgical resection, radiation therapy, and/or systemic treatment like targeted therapy or chemotherapy. The prognosis for ASPS is variable, depending on factors such as the tumor's size, location, and extent of metastasis at diagnosis.
Leiomyosarcoma is a type of cancer that arises from the smooth muscle cells, which are responsible for the involuntary contractions of various organs and blood vessels. It most commonly occurs in the uterus, soft tissues (such as muscles and fat), and the gastrointestinal tract.
Leiomyosarcomas can vary in their aggressiveness and may spread to other parts of the body (metastasize) through the bloodstream or lymphatic system. The prognosis for leiomyosarcoma depends on several factors, including the location and size of the tumor, the patient's age and overall health, and the extent of metastasis. Treatment typically involves surgical removal of the tumor, along with radiation therapy and/or chemotherapy to help prevent recurrence or spread of the cancer.
A cell line that is derived from tumor cells and has been adapted to grow in culture. These cell lines are often used in research to study the characteristics of cancer cells, including their growth patterns, genetic changes, and responses to various treatments. They can be established from many different types of tumors, such as carcinomas, sarcomas, and leukemias. Once established, these cell lines can be grown and maintained indefinitely in the laboratory, allowing researchers to conduct experiments and studies that would not be feasible using primary tumor cells. It is important to note that tumor cell lines may not always accurately represent the behavior of the original tumor, as they can undergo genetic changes during their time in culture.
Medical Definition of "Herpesvirus 8, Human" (HHV-8):
Human Herpesvirus 8 (HHV-8), also known as Kaposi's Sarcoma-associated Herpesvirus (KSHV), is a DNA virus from the family of Herpesviridae. It is the causative agent of several malignancies, including Kaposi's sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman's disease (MCD). HHV-8 is primarily transmitted through saliva, sexual contact, or organ transplantation. In immunocompromised individuals, such as those with HIV/AIDS, the risk of HHV-8-associated malignancies significantly increases. The virus establishes latency in infected cells and can periodically reactivate, causing inflammation and potentially leading to the development of cancer.
Translocation, genetic, refers to a type of chromosomal abnormality in which a segment of a chromosome is transferred from one chromosome to another, resulting in an altered genome. This can occur between two non-homologous chromosomes (non-reciprocal translocation) or between two homologous chromosomes (reciprocal translocation). Genetic translocations can lead to various clinical consequences, depending on the genes involved and the location of the translocation. Some translocations may result in no apparent effects, while others can cause developmental abnormalities, cancer, or other genetic disorders. In some cases, translocations can also increase the risk of having offspring with genetic conditions.
An alpharetrovirus is a type of retrovirus, which is a group of viruses that integrate their genetic material into the DNA of the host cell. Alpharetroviruses are characterized by their ability to cause persistent infections and are associated with various diseases in animals. One well-known example of an alpharetrovirus is the Rous sarcoma virus (RSV), which was the first retrovirus to be discovered and is known to cause cancer in chickens.
Alpharetroviruses have a complex structure, consisting of an outer envelope that contains glycoprotein spikes, and an inner core that contains the viral RNA genome and associated enzymes. The viral RNA genome contains three main genes: gag, pol, and env, which encode for the structural proteins, enzymes, and envelope proteins of the virus, respectively.
Alpharetroviruses are transmitted through various routes, including horizontal transmission (from host to host) and vertical transmission (from parent to offspring). They can cause a range of diseases, depending on the specific virus and the host species. In addition to RSV, other examples of alpharetroviruses include the avian leukosis virus, which causes tumors and immunosuppression in birds, and the Jaagsiekte sheep retrovirus, which causes a wasting disease in sheep.
It's worth noting that while alpharetroviruses are associated with diseases in animals, there are no known alpharetroviruses that infect humans. However, understanding the biology and behavior of these viruses in animal hosts can provide valuable insights into retroviral replication and pathogenesis, which may have implications for human health.
Epidural neoplasms refer to abnormal growths or tumors that develop in the epidural space, which is the area between the dura mater (the outermost protective covering of the spinal cord) and the vertebral column. These tumors can be either primary, originating directly from the cells in the epidural space, or secondary, resulting from the spread (metastasis) of cancerous cells from other parts of the body.
Epidural neoplasms can cause various symptoms due to the compression of the spinal cord and nerve roots. These symptoms may include localized back pain, radiating pain, sensory changes, motor weakness, and autonomic dysfunction. The diagnosis typically involves imaging studies such as MRI or CT scans, followed by a biopsy for histopathological examination to confirm the type and grade of the tumor. Treatment options depend on several factors, including the patient's overall health, the location and size of the tumor, and the type and extent of neurological deficits. Treatment may involve surgical resection, radiation therapy, chemotherapy, or a combination of these approaches.
Human chromosome pair 22 consists of two rod-shaped structures present in the nucleus of each cell in the human body. Each chromosome is made up of DNA tightly coiled around histone proteins, forming a complex structure called a chromatin.
Chromosome pair 22 is one of the 22 autosomal pairs of human chromosomes, meaning they are not sex chromosomes (X or Y). Chromosome 22 is the second smallest human chromosome, with each arm of the chromosome designated as p and q. The short arm is labeled "p," and the long arm is labeled "q."
Chromosome 22 contains several genes that are associated with various genetic disorders, including DiGeorge syndrome, velocardiofacial syndrome, and cat-eye syndrome, which result from deletions or duplications of specific regions on the chromosome. Additionally, chromosome 22 is the location of the NRXN1 gene, which has been associated with an increased risk for autism spectrum disorder (ASD) and schizophrenia when deleted or disrupted.
Understanding the genetic makeup of human chromosome pair 22 can provide valuable insights into human genetics, evolution, and disease susceptibility, as well as inform medical diagnoses, treatments, and research.
Cell transformation, viral refers to the process by which a virus causes normal cells to become cancerous or tumorigenic. This occurs when the genetic material of the virus integrates into the DNA of the host cell and alters its regulation, leading to uncontrolled cell growth and division. Some viruses known to cause cell transformation include human papillomavirus (HPV), hepatitis B virus (HBV), and certain types of herpesviruses.
Harvey murine sarcoma virus (HMSV) is a type of retrovirus, specifically a sarcoma virus that was first isolated from mice. It is named after J. Harvey, who discovered the virus in 1964. HMSV is closely related to Moloney murine leukemia virus (M-MuLV).
HMSV is a complex retrovirus, which contains several accessory genes that are not required for replication but contribute to viral pathogenesis and oncogenic transformation. The most well-known oncogene carried by HMSV is v-src, which encodes the pp60v-src protein tyrosine kinase. This oncogene was the first cellular oncogene (c-src) to be discovered, and it plays a crucial role in the transformation of cells and the development of sarcomas in infected mice.
HMSV infection typically occurs through the direct introduction of viral particles into susceptible tissues or by the transfer of infected cells. Once inside the host, HMSV integrates its genetic material into the host cell's DNA, leading to the expression of viral genes and the production of new virus particles. The activation of the v-src oncogene results in uncontrolled cell growth and division, ultimately leading to the formation of tumors.
In summary, Harvey murine sarcoma virus is a retrovirus that carries the v-src oncogene, causing uncontrolled cell growth and leading to the development of sarcomas in infected mice.
Neoplastic cell transformation is a process in which a normal cell undergoes genetic alterations that cause it to become cancerous or malignant. This process involves changes in the cell's DNA that result in uncontrolled cell growth and division, loss of contact inhibition, and the ability to invade surrounding tissues and metastasize (spread) to other parts of the body.
Neoplastic transformation can occur as a result of various factors, including genetic mutations, exposure to carcinogens, viral infections, chronic inflammation, and aging. These changes can lead to the activation of oncogenes or the inactivation of tumor suppressor genes, which regulate cell growth and division.
The transformation of normal cells into cancerous cells is a complex and multi-step process that involves multiple genetic and epigenetic alterations. It is characterized by several hallmarks, including sustained proliferative signaling, evasion of growth suppressors, resistance to cell death, enabling replicative immortality, induction of angiogenesis, activation of invasion and metastasis, reprogramming of energy metabolism, and evading immune destruction.
Neoplastic cell transformation is a fundamental concept in cancer biology and is critical for understanding the molecular mechanisms underlying cancer development and progression. It also has important implications for cancer diagnosis, prognosis, and treatment, as identifying the specific genetic alterations that underlie neoplastic transformation can help guide targeted therapies and personalized medicine approaches.
A gammaretrovirus is a type of retrovirus, which is a virus that contains RNA as its genetic material and uses the reverse transcriptase enzyme to produce DNA from its RNA genome. Gammaretroviruses are enveloped viruses, meaning they have a lipid membrane derived from the host cell. They are also classified as simple retroviruses because their genome only contains the genes gag, pol, and env.
Gammaretroviruses are known to cause diseases in animals, including leukemias and immunodeficiencies. One example of a gammaretrovirus is the feline leukemia virus (FeLV), which can cause a variety of symptoms in cats, including anemia, lymphoma, and immune suppression.
Gammaretroviruses have also been implicated in some human diseases, although they are not thought to be major causes of human disease. For example, the human T-cell leukemia virus type 1 (HTLV-1) is a retrovirus that is closely related to gammaretroviruses and can cause adult T-cell leukemia/lymphoma and tropical spastic paraparesis/ HTLV-associated myelopathy (TSP/HAM).
It's important to note that the classification of retroviruses has evolved over time, and some viruses that were once classified as gammaretroviruses are now considered to be part of other retrovirus genera.
Malignant fibrous histiocytoma (MFH) is not a specific type of histiocytoma; rather, it is a type of soft tissue sarcoma. Histiocytomas are benign tumors that arise from cells called histiocytes, which are part of the immune system. MFH, on the other hand, is a malignant (cancerous) tumor that can arise in various types of soft tissues, such as muscle, fat, tendons, and ligaments.
MFH was once thought to originate from histiocytes, but more recent research suggests that it may actually arise from undifferentiated mesenchymal cells, which are capable of developing into a variety of different cell types. MFH is the most common type of soft tissue sarcoma in adults over the age of 50 and typically presents as a painless mass in the extremities or retroperitoneum (the area in the back of the abdomen).
The tumor is characterized by the presence of fibroblastic and histiocytic-like cells, which can be quite pleomorphic (varied in shape and size) and may contain numerous mitotic figures (indicating rapid cell division). Treatment typically involves surgical excision, often followed by radiation therapy and/or chemotherapy. The prognosis for MFH depends on several factors, including the tumor's location, size, grade (degree of differentiation), and the patient's age and overall health.
Chondrosarcoma is a type of cancer that develops in the cartilaginous tissue, which is the flexible and smooth connective tissue found in various parts of the body such as the bones, ribs, and nose. It is characterized by the production of malignant cartilage cells that can invade surrounding tissues and spread to other parts of the body (metastasis).
Chondrosarcomas are typically slow-growing tumors but can be aggressive in some cases. They usually occur in adults over the age of 40, and men are more commonly affected than women. The most common sites for chondrosarcoma development include the bones of the pelvis, legs, and arms.
Treatment for chondrosarcoma typically involves surgical removal of the tumor, along with radiation therapy or chemotherapy in some cases. The prognosis for chondrosarcoma depends on several factors, including the size and location of the tumor, the grade of malignancy, and whether it has spread to other parts of the body.
Doxorubicin is a type of chemotherapy medication known as an anthracycline. It works by interfering with the DNA in cancer cells, which prevents them from growing and multiplying. Doxorubicin is used to treat a wide variety of cancers, including leukemia, lymphoma, breast cancer, lung cancer, ovarian cancer, and many others. It may be given alone or in combination with other chemotherapy drugs.
Doxorubicin is usually administered through a vein (intravenously) and can cause side effects such as nausea, vomiting, hair loss, mouth sores, and increased risk of infection. It can also cause damage to the heart muscle, which can lead to heart failure in some cases. For this reason, doctors may monitor patients' heart function closely while they are receiving doxorubicin treatment.
It is important for patients to discuss the potential risks and benefits of doxorubicin therapy with their healthcare provider before starting treatment.
Liposarcoma is a type of soft tissue sarcoma, which is a cancer that develops in the soft tissues of the body, such as fat, muscle, nerves, blood vessels, and fibrous tissues. Specifically, liposarcoma arises from fat cells (adipocytes) or their precursors.
There are several subtypes of liposarcoma, which differ in their appearance under the microscope, genetic features, and clinical behavior. These include well-differentiated, dedifferentiated, myxoid, round cell, and pleomorphic liposarcomas. The most common sites for liposarcoma are the thigh, retroperitoneum (the area behind the abdominal cavity), and the buttock.
Liposarcomas can grow slowly or rapidly, and they may spread to other parts of the body (metastasize) through the bloodstream or lymphatic system. Treatment typically involves surgical removal of the tumor, often followed by radiation therapy and/or chemotherapy. The prognosis for liposarcoma depends on several factors, including the type and grade of the tumor, its size and location, and whether it has spread to other parts of the body.
Calmodulin-binding proteins are a diverse group of proteins that have the ability to bind to calmodulin, a ubiquitous calcium-binding protein found in eukaryotic cells. Calmodulin plays a critical role in various cellular processes by regulating the activity of its target proteins in a calcium-dependent manner.
Calmodulin-binding proteins contain specific domains or motifs that enable them to interact with calmodulin. These domains can be classified into two main categories: IQ motifs and CaM motifs. The IQ motif is a short amino acid sequence that contains the consensus sequence IQXXXRGXXR, where X represents any amino acid. This motif binds to the C-lobe of calmodulin in a calcium-dependent manner. On the other hand, CaM motifs are longer sequences that can bind to both lobes of calmodulin with high affinity and in a calcium-dependent manner.
Calmodulin-binding proteins play crucial roles in various cellular functions, including signal transduction, gene regulation, cytoskeleton organization, and ion channel regulation. For example, calmodulin-binding proteins such as calcineurin and CaM kinases are involved in the regulation of immune responses, learning, and memory. Similarly, myosin regulatory light chains, which contain IQ motifs, play a critical role in muscle contraction by regulating the interaction between actin and myosin filaments.
In summary, calmodulin-binding proteins are a diverse group of proteins that interact with calmodulin to regulate various cellular processes. They contain specific domains or motifs that enable them to bind to calmodulin in a calcium-dependent manner, thereby modulating the activity of their target proteins.
Neoplastic gene expression regulation refers to the processes that control the production of proteins and other molecules from genes in neoplastic cells, or cells that are part of a tumor or cancer. In a normal cell, gene expression is tightly regulated to ensure that the right genes are turned on or off at the right time. However, in cancer cells, this regulation can be disrupted, leading to the overexpression or underexpression of certain genes.
Neoplastic gene expression regulation can be affected by a variety of factors, including genetic mutations, epigenetic changes, and signals from the tumor microenvironment. These changes can lead to the activation of oncogenes (genes that promote cancer growth and development) or the inactivation of tumor suppressor genes (genes that prevent cancer).
Understanding neoplastic gene expression regulation is important for developing new therapies for cancer, as targeting specific genes or pathways involved in this process can help to inhibit cancer growth and progression.
Nanodiamonds are a type of carbon nanoparticle with a diamond crystal structure. They have at least one dimension in the nanometer scale, typically ranging from 1 to 100 nanometers. These particles exhibit unique physical and chemical properties that make them valuable for various applications, including biomedical research and therapeutics.
In the context of medical definitions, nanodiamonds are often discussed as potential drug delivery vehicles or diagnostic agents due to their biocompatibility, high surface area-to-volume ratio, and ability to bind with different molecules. They can be functionalized with drugs, proteins, or other biomolecules, allowing for targeted drug release or imaging in specific cells or tissues.
However, it is essential to note that the use of nanodiamonds in medical applications is still under investigation and development. While they show promise as a novel platform for diagnostics and therapeutics, more research is needed to establish their safety and efficacy before widespread clinical adoption.
I'm sorry for any confusion, but "Sarcoma 37" is not a recognized medical term or classification for a specific type of sarcoma. Sarcomas are a diverse group of cancers that arise from various tissues such as bone, cartilage, fat, muscle, and connective tissue. They are typically classified based on the type of tissue they originate from, such as osteosarcoma (bone), chondrosarcoma (cartilage), liposarcoma (fat), rhabdomyosarcoma (muscle), or synovial sarcoma (synovial tissue).
The number "37" could potentially refer to a specific subtype within one of these classifications, or it might be part of a unique identifier for a particular patient's medical record. However, without more context, I cannot provide an accurate definition for "Sarcoma 37." If you have additional information or if you meant to ask about something else, please let me know and I will do my best to help.
Combined modality therapy (CMT) is a medical treatment approach that utilizes more than one method or type of therapy simultaneously or in close succession, with the goal of enhancing the overall effectiveness of the treatment. In the context of cancer care, CMT often refers to the combination of two or more primary treatment modalities, such as surgery, radiation therapy, and systemic therapies (chemotherapy, immunotherapy, targeted therapy, etc.).
The rationale behind using combined modality therapy is that each treatment method can target cancer cells in different ways, potentially increasing the likelihood of eliminating all cancer cells and reducing the risk of recurrence. The specific combination and sequence of treatments will depend on various factors, including the type and stage of cancer, patient's overall health, and individual preferences.
For example, a common CMT approach for locally advanced rectal cancer may involve preoperative (neoadjuvant) chemoradiation therapy, followed by surgery to remove the tumor, and then postoperative (adjuvant) chemotherapy. This combined approach allows for the reduction of the tumor size before surgery, increases the likelihood of complete tumor removal, and targets any remaining microscopic cancer cells with systemic chemotherapy.
It is essential to consult with a multidisciplinary team of healthcare professionals to determine the most appropriate CMT plan for each individual patient, considering both the potential benefits and risks associated with each treatment method.
Vincristine is an antineoplastic agent, specifically a vinca alkaloid. It is derived from the Madagascar periwinkle plant (Catharanthus roseus). Vincristine binds to tubulin, a protein found in microtubules, and inhibits their polymerization, which results in disruption of mitotic spindles leading to cell cycle arrest and apoptosis (programmed cell death). It is used in the treatment of various types of cancer including leukemias, lymphomas, and solid tumors. Common side effects include peripheral neuropathy, constipation, and alopecia.
Immunohistochemistry (IHC) is a technique used in pathology and laboratory medicine to identify specific proteins or antigens in tissue sections. It combines the principles of immunology and histology to detect the presence and location of these target molecules within cells and tissues. This technique utilizes antibodies that are specific to the protein or antigen of interest, which are then tagged with a detection system such as a chromogen or fluorophore. The stained tissue sections can be examined under a microscope, allowing for the visualization and analysis of the distribution and expression patterns of the target molecule in the context of the tissue architecture. Immunohistochemistry is widely used in diagnostic pathology to help identify various diseases, including cancer, infectious diseases, and immune-mediated disorders.
Benign fibrous histiocytoma (BFH) is a common benign tumor of the skin and superficial soft tissues. It primarily affects middle-aged adults and is more prevalent in men than women. The exact cause of BFH is unknown, but it's thought to arise from dermal fibroblasts or histiocytes.
Medical Definition: Benign Fibrous Histiocytoma (BFH) is a benign, slowly growing, solitary cutaneous or subcutaneous nodular tumor predominantly composed of a mixture of fibroblastic and histiocytic-like cells. The tumor typically presents as a well-circumscribed, firm, dome-shaped papule or nodule, ranging in size from a few millimeters to several centimeters. Histologically, BFH is characterized by the proliferation of spindle-shaped fibroblasts and histiocytes arranged in a storiform pattern, along with variable amounts of collagen deposition, multinucleated giant cells, and hemosiderin deposits. The lesion usually has a pushing border with no invasion into the surrounding tissues. BFH generally follows a benign clinical course, with local recurrence being uncommon following complete surgical excision.
Reverse Transcriptase Polymerase Chain Reaction (RT-PCR) is a laboratory technique used in molecular biology to amplify and detect specific DNA sequences. This technique is particularly useful for the detection and quantification of RNA viruses, as well as for the analysis of gene expression.
The process involves two main steps: reverse transcription and polymerase chain reaction (PCR). In the first step, reverse transcriptase enzyme is used to convert RNA into complementary DNA (cDNA) by reading the template provided by the RNA molecule. This cDNA then serves as a template for the PCR amplification step.
In the second step, the PCR reaction uses two primers that flank the target DNA sequence and a thermostable polymerase enzyme to repeatedly copy the targeted cDNA sequence. The reaction mixture is heated and cooled in cycles, allowing the primers to anneal to the template, and the polymerase to extend the new strand. This results in exponential amplification of the target DNA sequence, making it possible to detect even small amounts of RNA or cDNA.
RT-PCR is a sensitive and specific technique that has many applications in medical research and diagnostics, including the detection of viruses such as HIV, hepatitis C virus, and SARS-CoV-2 (the virus that causes COVID-19). It can also be used to study gene expression, identify genetic mutations, and diagnose genetic disorders.
A base sequence in the context of molecular biology refers to the specific order of nucleotides in a DNA or RNA molecule. In DNA, these nucleotides are adenine (A), guanine (G), cytosine (C), and thymine (T). In RNA, uracil (U) takes the place of thymine. The base sequence contains genetic information that is transcribed into RNA and ultimately translated into proteins. It is the exact order of these bases that determines the genetic code and thus the function of the DNA or RNA molecule.
Abdominal neoplasms refer to abnormal growths or tumors in the abdomen that can be benign (non-cancerous) or malignant (cancerous). These growths can occur in any of the organs within the abdominal cavity, including the stomach, small intestine, large intestine, liver, pancreas, spleen, and kidneys.
Abdominal neoplasms can cause various symptoms depending on their size, location, and type. Some common symptoms include abdominal pain or discomfort, bloating, changes in bowel habits, unexplained weight loss, fatigue, and fever. In some cases, abdominal neoplasms may not cause any symptoms until they have grown quite large or spread to other parts of the body.
The diagnosis of abdominal neoplasms typically involves a combination of physical exam, medical history, imaging studies such as CT scans or MRIs, and sometimes biopsy to confirm the type of tumor. Treatment options depend on the type, stage, and location of the neoplasm but may include surgery, radiation therapy, chemotherapy, targeted therapy, or a combination of these approaches.
Prognosis is a medical term that refers to the prediction of the likely outcome or course of a disease, including the chances of recovery or recurrence, based on the patient's symptoms, medical history, physical examination, and diagnostic tests. It is an important aspect of clinical decision-making and patient communication, as it helps doctors and patients make informed decisions about treatment options, set realistic expectations, and plan for future care.
Prognosis can be expressed in various ways, such as percentages, categories (e.g., good, fair, poor), or survival rates, depending on the nature of the disease and the available evidence. However, it is important to note that prognosis is not an exact science and may vary depending on individual factors, such as age, overall health status, and response to treatment. Therefore, it should be used as a guide rather than a definitive forecast.
Keratin-1
Langerhans cell sarcoma is a very rare and aggressive type of cancer that affects a specific group of cells called Langerhans cells, which are part of the immune system. These cells are normally found in the skin and mucous membranes, where they help to fight infection. In Langerhans cell sarcoma, these cells become malignant (cancerous) and can multiply and spread to other parts of the body.
Langerhans cell sarcoma is distinct from a more common type of cancer called Langerhans cell histiocytosis, which is not considered a true cancer but rather a disorder of the immune system. The exact cause of Langerhans cell sarcoma is not known, but it is thought to arise from genetic mutations that occur in Langerhans cells.
Symptoms of Langerhans cell sarcoma can vary depending on the location and extent of the cancer. Common symptoms may include skin rashes or lesions, fever, fatigue, weight loss, and swollen lymph nodes. Treatment for Langerhans cell sarcoma typically involves a combination of surgery, chemotherapy, and radiation therapy. However, because this is such a rare and aggressive cancer, treatment options may vary depending on the individual case.
Vascular neoplasms are a type of tumor that develops from cells that line the blood vessels or lymphatic vessels. These tumors can be benign (non-cancerous) or malignant (cancerous). Benign vascular neoplasms, such as hemangiomas and lymphangiomas, are usually harmless and may not require treatment unless they cause symptoms or complications. Malignant vascular neoplasms, on the other hand, are known as angiosarcomas and can be aggressive, spreading to other parts of the body and potentially causing serious health problems.
Angiosarcomas can develop in any part of the body but are most commonly found in the skin, particularly in areas exposed to radiation or chronic lymph edema. They can also occur in the breast, liver, spleen, and heart. Treatment for vascular neoplasms depends on the type, location, size, and stage of the tumor, as well as the patient's overall health. Treatment options may include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
Retroperitoneal neoplasms refer to abnormal growths or tumors that develop in the retroperitoneal space. This is the area located behind the peritoneum, which is the membrane that lines the abdominal cavity and covers the abdominal organs. The retroperitoneal space contains several vital structures such as the kidneys, adrenal glands, pancreas, aorta, and lymphatic vessels.
Retroperitoneal neoplasms can be benign or malignant (cancerous). Malignant retroperitoneal neoplasms are often aggressive and can invade surrounding tissues and organs, leading to various complications. Common types of retroperitoneal neoplasms include lymphomas, sarcomas, and metastatic tumors from other primary sites. Symptoms may vary depending on the size and location of the tumor but can include abdominal or back pain, weight loss, and swelling in the legs. Diagnosis typically involves imaging studies such as CT scans or MRI, followed by a biopsy to determine the type and grade of the tumor. Treatment options may include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
A chick embryo refers to the developing organism that arises from a fertilized chicken egg. It is often used as a model system in biological research, particularly during the stages of development when many of its organs and systems are forming and can be easily observed and manipulated. The study of chick embryos has contributed significantly to our understanding of various aspects of developmental biology, including gastrulation, neurulation, organogenesis, and pattern formation. Researchers may use various techniques to observe and manipulate the chick embryo, such as surgical alterations, cell labeling, and exposure to drugs or other agents.
A "second primary neoplasm" is a distinct, new cancer or malignancy that develops in a person who has already had a previous cancer. It is not a recurrence or metastasis of the original tumor, but rather an independent cancer that arises in a different location or organ system. The development of second primary neoplasms can be influenced by various factors such as genetic predisposition, environmental exposures, and previous treatments like chemotherapy or radiation therapy.
It is important to note that the definition of "second primary neoplasm" may vary slightly depending on the specific source or context. In general medical usage, it refers to a new, separate cancer; however, in some research or clinical settings, there might be more precise criteria for defining and diagnosing second primary neoplasms.
Treatment outcome is a term used to describe the result or effect of medical treatment on a patient's health status. It can be measured in various ways, such as through symptoms improvement, disease remission, reduced disability, improved quality of life, or survival rates. The treatment outcome helps healthcare providers evaluate the effectiveness of a particular treatment plan and make informed decisions about future care. It is also used in clinical research to compare the efficacy of different treatments and improve patient care.
Dactinomycin is an antineoplastic antibiotic, which means it is used to treat cancer. It is specifically used to treat certain types of testicular cancer, Wilms' tumor (a type of kidney cancer that occurs in children), and some gestational trophoblastic tumors (a type of tumor that can develop in the uterus after pregnancy). Dactinomycin works by interfering with the DNA in cancer cells, which prevents them from dividing and growing. It is often used in combination with other chemotherapy drugs as part of a treatment regimen.
Dactinomycin is administered intravenously (through an IV) and its use is usually limited to hospitals or specialized cancer treatment centers due to the need for careful monitoring during administration. Common side effects include nausea, vomiting, and hair loss. More serious side effects can include bone marrow suppression, which can lead to an increased risk of infection, and tissue damage at the site where the drug is injected. Dactinomycin can also cause severe allergic reactions in some people.
It's important to note that dactinomycin should only be used under the supervision of a qualified healthcare professional, as its use requires careful monitoring and management of potential side effects.
IGF-1R (Insulin-like Growth Factor 1 Receptor) is a transmembrane receptor tyrosine kinase that plays a crucial role in intracellular signaling pathways related to cell growth, differentiation, and survival. IGF-1R is primarily activated by its ligands, IGF-1 (Insulin-like Growth Factor 1) and IGF-2 (Insulin-like Growth Factor 2). Upon binding of the ligand, IGF-1R undergoes autophosphorylation and initiates a cascade of intracellular signaling events, primarily through the PI3K/AKT and RAS/MAPK pathways. These signaling cascades ultimately regulate various cellular processes such as glucose metabolism, protein synthesis, DNA replication, and cell cycle progression. Dysregulation of IGF-1R has been implicated in several diseases, including cancer, diabetes, and growth disorders.
Dendritic cell sarcoma, follicular is a very rare type of cancer that affects the dendritic cells, which are a type of immune cell found in the body. Specifically, this type of sarcoma arises from follicular dendritic cells, which are found in the lymph nodes and other lymphoid organs. These cells play an important role in helping the immune system recognize and respond to foreign invaders such as viruses and bacteria.
Dendritic cell sarcoma, follicular typically presents as a mass or enlargement of a lymph node or other lymphoid organ. It can also spread (metastasize) to other parts of the body. The symptoms of this type of cancer may vary depending on the location and extent of the tumor, but they can include swelling of lymph nodes, fever, night sweats, weight loss, and fatigue.
The exact cause of dendritic cell sarcoma, follicular is not known, but it is thought to arise from genetic mutations that occur in the cells over time. Treatment for this type of cancer typically involves a combination of surgery, radiation therapy, and chemotherapy, depending on the stage and location of the tumor.
It's important to note that medical definitions can be complex and technical, and they should not be used as a substitute for professional medical advice. If you have any concerns about your health or symptoms, please consult with a qualified healthcare provider.
Helper viruses, also known as "auxiliary" or "satellite" viruses, are defective viruses that depend on the assistance of a second virus, called a helper virus, to complete their replication cycle. They lack certain genes that are essential for replication, and therefore require the helper virus to provide these functions.
Helper viruses are often found in cases of dual infection, where both the helper virus and the dependent virus infect the same cell. The helper virus provides the necessary enzymes and proteins for the helper virus to replicate, package its genome into new virions, and bud off from the host cell.
One example of a helper virus is the hepatitis B virus (HBV), which can serve as a helper virus for hepatitis D virus (HDV) infection. HDV is a defective RNA virus that requires the HBV surface antigen to form an envelope around its nucleocapsid and be transmitted to other cells. In the absence of HBV, HDV cannot replicate or cause disease.
Understanding the role of helper viruses in viral infections is important for developing effective treatments and vaccines against viral diseases.
Local neoplasm recurrence is the return or regrowth of a tumor in the same location where it was originally removed or treated. This means that cancer cells have survived the initial treatment and started to grow again in the same area. It's essential to monitor and detect any local recurrence as early as possible, as it can affect the prognosis and may require additional treatment.
Antineoplastic combined chemotherapy protocols refer to a treatment plan for cancer that involves the use of more than one antineoplastic (chemotherapy) drug given in a specific sequence and schedule. The combination of drugs is used because they may work better together to destroy cancer cells compared to using a single agent alone. This approach can also help to reduce the likelihood of cancer cells becoming resistant to the treatment.
The choice of drugs, dose, duration, and frequency are determined by various factors such as the type and stage of cancer, patient's overall health, and potential side effects. Combination chemotherapy protocols can be used in various settings, including as a primary treatment, adjuvant therapy (given after surgery or radiation to kill any remaining cancer cells), neoadjuvant therapy (given before surgery or radiation to shrink the tumor), or palliative care (to alleviate symptoms and prolong survival).
It is important to note that while combined chemotherapy protocols can be effective in treating certain types of cancer, they can also cause significant side effects, including nausea, vomiting, hair loss, fatigue, and an increased risk of infection. Therefore, patients undergoing such treatment should be closely monitored and managed by a healthcare team experienced in administering chemotherapy.
A chromosome breakpoint is a specific location on a chromosome where a chromosomal rearrangement, such as a translocation or inversion, has occurred. A breakpoint is the point at which the chromosome has broken and then rejoined, often with another chromosome, resulting in a changed genetic sequence. These changes can have various consequences, including altered gene expression, loss of genetic material, or gain of new genetic material, which can lead to genetic disorders or predisposition to certain diseases. The identification and characterization of breakpoints are important for understanding the molecular basis of genomic rearrangements and their associated phenotypes.
A cell line is a culture of cells that are grown in a laboratory for use in research. These cells are usually taken from a single cell or group of cells, and they are able to divide and grow continuously in the lab. Cell lines can come from many different sources, including animals, plants, and humans. They are often used in scientific research to study cellular processes, disease mechanisms, and to test new drugs or treatments. Some common types of human cell lines include HeLa cells (which come from a cancer patient named Henrietta Lacks), HEK293 cells (which come from embryonic kidney cells), and HUVEC cells (which come from umbilical vein endothelial cells). It is important to note that cell lines are not the same as primary cells, which are cells that are taken directly from a living organism and have not been grown in the lab.
Muscle neoplasms are abnormal growths or tumors that develop in the muscle tissue. They can be benign (non-cancerous) or malignant (cancerous). Benign muscle neoplasms are typically slow-growing and do not spread to other parts of the body, while malignant muscle neoplasms, also known as soft tissue sarcomas, can grow quickly, invade nearby tissues, and metastasize (spread) to distant parts of the body.
Soft tissue sarcomas can arise from any of the muscles in the body, including the skeletal muscles (voluntary muscles that attach to bones and help with movement), smooth muscles (involuntary muscles found in the walls of blood vessels, digestive tract, and other organs), or cardiac muscle (the specialized muscle found in the heart).
There are many different types of soft tissue sarcomas, each with its own set of characteristics and prognosis. Treatment for muscle neoplasms typically involves a combination of surgery, radiation therapy, and chemotherapy, depending on the type, size, location, and stage of the tumor.
'Tumor cells, cultured' refers to the process of removing cancerous cells from a tumor and growing them in controlled laboratory conditions. This is typically done by isolating the tumor cells from a patient's tissue sample, then placing them in a nutrient-rich environment that promotes their growth and multiplication.
The resulting cultured tumor cells can be used for various research purposes, including the study of cancer biology, drug development, and toxicity testing. They provide a valuable tool for researchers to better understand the behavior and characteristics of cancer cells outside of the human body, which can lead to the development of more effective cancer treatments.
It is important to note that cultured tumor cells may not always behave exactly the same way as they do in the human body, so findings from cell culture studies must be validated through further research, such as animal models or clinical trials.
Neoplasm transplantation is not a recognized or established medical procedure in the field of oncology. The term "neoplasm" refers to an abnormal growth of cells, which can be benign or malignant (cancerous). "Transplantation" typically refers to the surgical transfer of living cells, tissues, or organs from one part of the body to another or between individuals.
The concept of neoplasm transplantation may imply the transfer of cancerous cells or tissues from a donor to a recipient, which is not a standard practice due to ethical considerations and the potential harm it could cause to the recipient. In some rare instances, researchers might use laboratory animals to study the transmission and growth of human cancer cells, but this is done for scientific research purposes only and under strict regulatory guidelines.
In summary, there is no medical definition for 'Neoplasm Transplantation' as it does not represent a standard or ethical medical practice.
The Musculoskeletal System is a complex system composed of the bones, joints, muscles, tendons, ligaments, and associated tissues that work together to provide form, support, stability, and movement to the body. It serves various functions including:
1. Protection: The musculoskeletal system protects vital organs by encasing them in bones, such as the ribcage protecting the lungs and heart, and the skull protecting the brain.
2. Support and Movement: Muscles and bones work together to enable movement and maintain posture. Muscles contract to pull on bones, causing joint motion and producing movements like walking, running, or jumping.
3. Storage: Bones act as a reservoir for essential minerals like calcium and phosphorus, which can be released into the bloodstream when needed.
4. Hematopoiesis: Within the bone marrow, hematopoietic cells produce blood cells, including red blood cells, white blood cells, and platelets.
5. Endocrine Function: Bone tissue is also an endocrine organ, producing hormones like osteocalcin and FGF23 that regulate various physiological processes, such as energy metabolism and mineral homeostasis.
Dysfunctions or injuries in the musculoskeletal system can result in conditions like arthritis, fractures, muscle strains, tendonitis, and other painful or debilitating ailments that impact an individual's quality of life and mobility.
Transcription factors are proteins that play a crucial role in regulating gene expression by controlling the transcription of DNA to messenger RNA (mRNA). They function by binding to specific DNA sequences, known as response elements, located in the promoter region or enhancer regions of target genes. This binding can either activate or repress the initiation of transcription, depending on the properties and interactions of the particular transcription factor. Transcription factors often act as part of a complex network of regulatory proteins that determine the precise spatiotemporal patterns of gene expression during development, differentiation, and homeostasis in an organism.
Avian leukosis virus (ALV) is a type of retrovirus that primarily affects chickens and other birds. It is responsible for a group of diseases known as avian leukosis, which includes various types of tumors and immunosuppressive conditions. The virus is transmitted horizontally through the shedder's dander, feathers, and vertical transmission through infected eggs.
There are several subgroups of ALV (A, B, C, D, E, and J), each with different host ranges and pathogenicity. Some strains can cause rapid death in young chickens, while others may take years to develop clinical signs. The most common form of the disease is neoplastic, characterized by the development of various types of tumors such as lymphomas, myelomas, and sarcomas.
Avian leukosis virus infection can have significant economic impacts on the poultry industry due to decreased growth rates, increased mortality, and condemnation of infected birds at processing. Control measures include eradication programs, biosecurity practices, vaccination, and breeding for genetic resistance.
"Nude mice" is a term used in the field of laboratory research to describe a strain of mice that have been genetically engineered to lack a functional immune system. Specifically, nude mice lack a thymus gland and have a mutation in the FOXN1 gene, which results in a failure to develop a mature T-cell population. This means that they are unable to mount an effective immune response against foreign substances or organisms.
The name "nude" refers to the fact that these mice also have a lack of functional hair follicles, resulting in a hairless or partially hairless phenotype. This feature is actually a secondary consequence of the same genetic mutation that causes their immune deficiency.
Nude mice are commonly used in research because their weakened immune system makes them an ideal host for transplanted tumors, tissues, and cells from other species, including humans. This allows researchers to study the behavior of these foreign substances in a living organism without the complication of an immune response. However, it's important to note that because nude mice lack a functional immune system, they must be kept in sterile conditions and are more susceptible to infection than normal mice.
Etoposide is a chemotherapy medication used to treat various types of cancer, including lung cancer, testicular cancer, and certain types of leukemia. It works by inhibiting the activity of an enzyme called topoisomerase II, which is involved in DNA replication and transcription. By doing so, etoposide can interfere with the growth and multiplication of cancer cells.
Etoposide is often administered intravenously in a hospital or clinic setting, although it may also be given orally in some cases. The medication can cause a range of side effects, including nausea, vomiting, hair loss, and an increased risk of infection. It can also have more serious side effects, such as bone marrow suppression, which can lead to anemia, bleeding, and a weakened immune system.
Like all chemotherapy drugs, etoposide is not without risks and should only be used under the close supervision of a qualified healthcare provider. It is important for patients to discuss the potential benefits and risks of this medication with their doctor before starting treatment.
Fungal antigens are substances found on or produced by fungi that can stimulate an immune response in a host organism. They can be proteins, polysaccharides, or other molecules that are recognized as foreign by the host's immune system. Fungal antigens can be used in diagnostic tests to identify fungal infections, and they can also be targets of immune responses during fungal infections. In some cases, fungal antigens may contribute to the pathogenesis of fungal diseases by inducing inflammatory or allergic reactions. Examples of fungal antigens include the cell wall components of Candida albicans and the extracellular polysaccharide galactomannan produced by Aspergillus fumigatus.
Hemangiosarcoma is a type of cancer that arises from the cells that line the blood vessels (endothelial cells). It most commonly affects middle-aged to older dogs, but it can also occur in cats and other animals, as well as rarely in humans.
This cancer can develop in various parts of the body, including the skin, heart, spleen, liver, and lungs. Hemangiosarcomas of the skin tend to be more benign and have a better prognosis than those that arise internally.
Hemangiosarcomas are highly invasive and often metastasize (spread) to other organs, making them difficult to treat. The exact cause of hemangiosarcoma is not known, but exposure to certain chemicals, radiation, and viruses may increase the risk of developing this cancer. Treatment options typically include surgery, chemotherapy, and/or radiation therapy, depending on the location and stage of the tumor.
Antineoplastic agents are a class of drugs used to treat malignant neoplasms or cancer. These agents work by inhibiting the growth and proliferation of cancer cells, either by killing them or preventing their division and replication. Antineoplastic agents can be classified based on their mechanism of action, such as alkylating agents, antimetabolites, topoisomerase inhibitors, mitotic inhibitors, and targeted therapy agents.
Alkylating agents work by adding alkyl groups to DNA, which can cause cross-linking of DNA strands and ultimately lead to cell death. Antimetabolites interfere with the metabolic processes necessary for DNA synthesis and replication, while topoisomerase inhibitors prevent the relaxation of supercoiled DNA during replication. Mitotic inhibitors disrupt the normal functioning of the mitotic spindle, which is essential for cell division. Targeted therapy agents are designed to target specific molecular abnormalities in cancer cells, such as mutated oncogenes or dysregulated signaling pathways.
It's important to note that antineoplastic agents can also affect normal cells and tissues, leading to various side effects such as nausea, vomiting, hair loss, and myelosuppression (suppression of bone marrow function). Therefore, the use of these drugs requires careful monitoring and management of their potential adverse effects.
Retroviridae is a family of viruses that includes human immunodeficiency virus (HIV) and other viruses that primarily use RNA as their genetic material. The name "retrovirus" comes from the fact that these viruses reverse transcribe their RNA genome into DNA, which then becomes integrated into the host cell's genome. This is a unique characteristic of retroviruses, as most other viruses use DNA as their genetic material.
Retroviruses can cause a variety of diseases in animals and humans, including cancer, neurological disorders, and immunodeficiency syndromes like AIDS. They have a lipid membrane envelope that contains glycoprotein spikes, which allow them to attach to and enter host cells. Once inside the host cell, the viral RNA is reverse transcribed into DNA by the enzyme reverse transcriptase, which is then integrated into the host genome by the enzyme integrase.
Retroviruses can remain dormant in the host genome for extended periods of time, and may be reactivated under certain conditions to produce new viral particles. This ability to integrate into the host genome has also made retroviruses useful tools in molecular biology, where they are used as vectors for gene therapy and other genetic manipulations.
Retrospective studies, also known as retrospective research or looking back studies, are a type of observational study that examines data from the past to draw conclusions about possible causal relationships between risk factors and outcomes. In these studies, researchers analyze existing records, medical charts, or previously collected data to test a hypothesis or answer a specific research question.
Retrospective studies can be useful for generating hypotheses and identifying trends, but they have limitations compared to prospective studies, which follow participants forward in time from exposure to outcome. Retrospective studies are subject to biases such as recall bias, selection bias, and information bias, which can affect the validity of the results. Therefore, retrospective studies should be interpreted with caution and used primarily to generate hypotheses for further testing in prospective studies.
Fibrosarcoma is a type of soft tissue cancer that develops in the fibrous (or connective) tissue found throughout the body, including tendons, ligaments, and muscles. It is characterized by the malignant proliferation of fibroblasts, which are the cells responsible for producing collagen, a structural protein found in connective tissue.
The tumor typically presents as a painless, firm mass that grows slowly over time. Fibrosarcomas can occur at any age but are more common in adults between 30 and 60 years old. The exact cause of fibrosarcoma is not well understood, but it has been linked to radiation exposure, certain chemicals, and genetic factors.
There are several subtypes of fibrosarcoma, including adult-type fibrosarcoma, infantile fibrosarcoma, and dedifferentiated fibrosarcoma. Treatment usually involves surgical removal of the tumor, often followed by radiation therapy and/or chemotherapy to reduce the risk of recurrence. The prognosis for patients with fibrosarcoma depends on several factors, including the size and location of the tumor, the patient's age and overall health, and the presence or absence of metastasis (spread of cancer to other parts of the body).
Heterogeneous Nuclear Ribonucleoproteins (hnRNPs) are a type of nuclear protein complex associated with nascent RNA transcripts in the nucleus of eukaryotic cells. They play crucial roles in various aspects of RNA metabolism, including processing, transport, stability, and translation.
The term "heterogeneous" refers to the diverse range of proteins that make up these complexes, while "nuclear" indicates their location within the nucleus. The hnRNPs are composed of a core protein component and associated RNA molecules, primarily heterogeneous nuclear RNAs (hnRNAs) or pre-messenger RNAs (pre-mRNAs).
There are over 20 different hnRNP proteins identified so far, each with distinct functions and structures. Some of the well-known hnRNPs include hnRNP A1, hnRNP C, and hnRNP U. These proteins contain several domains that facilitate RNA binding, protein-protein interactions, and post-translational modifications.
The primary function of hnRNPs is to regulate gene expression at the post-transcriptional level by interacting with RNA molecules. They participate in splicing, 3' end processing, export, localization, stability, and translation of mRNAs. Dysregulation of hnRNP function has been implicated in various human diseases, including neurological disorders and cancer.
 Ewing sarcoma
Ewing sarcoma
Extraskeletal Ewing sarcoma
Bone sarcoma
John H. Healey
Small-blue-round-cell tumor
Askin's tumor
FET protein family
Mary Beckerle
VPS13B
List of OMIM disorder codes
Codman triangle
List of vaginal tumors
Lourdes Medical Bureau
NOV (gene)
James Ewing (pathologist)
Vilen Prokofyev
Septic arthritis
RNA-binding protein EWS
Diaphysis
Ewing family of tumors
Neuroectodermal neoplasm
Deaths in January 2023
ANGAU General Hospital
Periodic acid-Schiff stain
FLI1
Deaths in July 2020
STYXL1
ERG (gene)
BMI1
Mir-708 microRNA precursor family
S. P. Beebe
Ewing sarcoma - Wikipedia
 Ewing sarcoma: MedlinePlus Genetics
Ewing sarcoma: MedlinePlus Genetics
 Ewing Sarcoma: Practice Essentials, Etiology, Epidemiology
Ewing Sarcoma: Practice Essentials, Etiology, Epidemiology
 Ewing Sarcoma
Ewing Sarcoma
 Ewing's Sarcoma : Wheeless' Textbook of Orthopaedics
Ewing's Sarcoma : Wheeless' Textbook of Orthopaedics
 Ewing Sarcoma Clinical Trials | Memorial Sloan Kettering Cancer Center
Ewing Sarcoma Clinical Trials | Memorial Sloan Kettering Cancer Center
 Better Imaging Method for Ewing Sarcoma Response Assessment
Better Imaging Method for Ewing Sarcoma Response Assessment
 Ewing's sarcoma overview - wikidoc
Ewing's sarcoma overview - wikidoc
 Ewing Sarcoma: Diagnosis, Treatment, Research and Support
Ewing Sarcoma: Diagnosis, Treatment, Research and Support
 Ewing's sarcoma
Ewing's sarcoma
Ewing sarcoma
Ewing's Sarcoma Research Grant - Sloan-Kettering Cancer Center
 Ewing Sarcoma Treatment | Children's Hospital Pittsburgh
Ewing Sarcoma Treatment | Children's Hospital Pittsburgh
Information for "Ewing's sarcoma laboratory tests" - wikidoc
Virtual Pediatric Hospital: Paediapaedia: Ewing Sarcoma, Chest Wall (Askin's Tumor)
 Fundraiser for Diane Dean by Shana Baker : Caleb is fighting Ewing's Sarcoma - Share
Fundraiser for Diane Dean by Shana Baker : Caleb is fighting Ewing's Sarcoma - Share
 Jody Hirsch Fund for Ewing's Sarcoma Research - University of Florida Advancement
Jody Hirsch Fund for Ewing's Sarcoma Research - University of Florida Advancement
 Chemotherapy for osteosarcoma and Ewing's sarcoma | Lund University Publications
Chemotherapy for osteosarcoma and Ewing's sarcoma | Lund University Publications
Ewing Sarcoma: Practice Essentials, Etiology, Epidemiology
 FDA Grants Orphan Drug Designation for Nantcell's Ganitumab in Ewing Sarcoma | 2017-04-14 | FDANews
FDA Grants Orphan Drug Designation for Nantcell's Ganitumab in Ewing Sarcoma | 2017-04-14 | FDANews
Endothelins may modulate invasion and proliferation of Ewing's sarcoma and neuroblastoma | Clinical Science | Portland Press
 Ewing sarcoma - femur | Radiology Case | Radiopaedia.org
Ewing sarcoma - femur | Radiology Case | Radiopaedia.org
 NKX2.2 (Neuroendocrine and Ewing's Sarcoma Marker) Monoclonal Antibody (NX2/294) (4821-MSM1-P1ABX)
...
NKX2.2 (Neuroendocrine and Ewing's Sarcoma Marker) Monoclonal Antibody (NX2/294) (4821-MSM1-P1ABX)
...
 Signature-based small molecule screening identifies cytosine arabinoside as an EWS/FLI modulator in Ewing sarcoma. | Broad...
Signature-based small molecule screening identifies cytosine arabinoside as an EWS/FLI modulator in Ewing sarcoma. | Broad...
 Phase II study of sequential gemcitabine followed by docetaxel for recurrent Ewing sarcoma, osteosarcoma, or unresectable or...
Phase II study of sequential gemcitabine followed by docetaxel for recurrent Ewing sarcoma, osteosarcoma, or unresectable or...
What Type of Cancer Is Ewing Sarcoma? Causes, Symptoms, Survival Rate - Hdkino.org
 Peripheral neuroepithelioma crude neuroectodermal growth and atypical Ewing's sarcoma. | Abstract
Peripheral neuroepithelioma crude neuroectodermal growth and atypical Ewing's sarcoma. | Abstract
 A live single-cell reporter assay links intratumor heterogeneity to metastatic proclivity in Ewing sarcoma | FUJIFILM...
A live single-cell reporter assay links intratumor heterogeneity to metastatic proclivity in Ewing sarcoma | FUJIFILM...
 "CD99 triggering induces methuosis of Ewing sarcoma cells through IGF-1" by Maria Cristina Manara, Mario Terracciano et al.
"CD99 triggering induces methuosis of Ewing sarcoma cells through IGF-1" by Maria Cristina Manara, Mario Terracciano et al.
 Ewing's Sarcoma - Pathology - Orthobullets
Ewing's Sarcoma - Pathology - Orthobullets
Ewing's52
- Definitive diagnosis was determined with a biopsy, which revealed the small, blue round cells of Ewing's sarcoma (Figure 4). (medscape.com)
- Ewing's sarcoma was first identified by Dr. Ewing in the 1920s. (medscape.com)
- The classic radiographic appearance of Ewing's sarcoma is onionskin periostitis and is often used as a preliminary means of differentiating these tumors. (medscape.com)
- The spiculated or 'sunburst' type of periosteal reaction can also be seen with Ewing's sarcoma, but is more often seen in osteosarcoma. (medscape.com)
- The recommended treatment of Ewing's sarcoma is surgical resection or radiation ablation of the primary tumor, followed by multidrug chemo therapy for possible distant metastasis. (medscape.com)
- The 'onionskin' periostitis is classic for Ewing's sarcoma. (medscape.com)
- Impact of EWS-ETS fusion type on disease progression in Ewing's sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. (medscape.com)
- Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. (medscape.com)
- Oncogenic partnerships: EWS-FLI1 protein interactions initiate key pathways of Ewing's sarcoma. (medscape.com)
- A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing's sarcoma. (medscape.com)
- Prognostic factors in Ewing's tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. (medscape.com)
- I was curious if anyone is aware of the clinical trial for TK216 for Ewing's Sarcoma. (cancer.org)
- Ewing's sarcoma, now often referred to as Ewing's Family of Tumors (EFST), is a small blue round cell sarcoma of bone, but it also can occur exclusively in the soft tissues. (curesarcoma.org)
- Ewing's sarcoma or peripheral primitive neuroectodermal tumor. (knowcancer.com)
- Eighteen patients with poor risk Ewing's sarcoma (including 11 patients with metastatic disease at presentation) received consolidation therapy of busulphan and melphalan with autologous stem cell rescue. (nih.gov)
- We conclude that high-dose busulphan/melphalan is well-tolerated and should be evaluated for efficacy in a larger series of patients with high risk Ewing's sarcoma. (nih.gov)
- Megachemotherapy followed by autologous stem cell transplantation in children with Ewing's sarcoma. (nih.gov)
- Ewing's sarcoma is a rare bone cancer that primarily affects individuals under the age of 30. (nih.gov)
- Ewing's sarcoma is treated with cytotoxic drug combinations, and there is a need for new treatment options. (nih.gov)
- Ewing's sarcoma / Peripheral Primitive Neuroectodermal Tumors (PNET) of bone is a type of cancer usually found in children and young adults. (heartdiseasepreventioninstitute.org)
- Ewing's sarcoma can also arise in soft tissue (extra-skeletal). (heartdiseasepreventioninstitute.org)
- Ewing's sarcoma is a radiosensitive tumour (Infante-Cossio et al. (journalcra.com)
- 2005). Multimodality therapy consisting of an initial biopsy, aggressive combination of surgery, chemotherapy and localized radiotherapy is the treatment of choice for Ewing's sarcoma of the head and neck region and may result in long-term survival (Vikas Prasad et al. (journalcra.com)
- The poster presentation will take place at the American Society of Clinical Oncology (ASCO) Annual Meeting: Illumination and Innovation and is entitled Pilot Trial of Vigil Immunotherapy in Ewing's Sarcoma. (marycrowley.org)
- Ewing's sarcoma is a somewhat a rare type of tumor, which usually develops in children and teenagers but can also occur in young adults [1] . (guidelineshealth.com)
- Ewing's sarcoma most commonly occurs in the area of the pelvis, chest and the legs affecting the hip bone, ribs or shoulder blades and the middle of long bones, respectively. (guidelineshealth.com)
- However, fractures of the bone without a known etiology or repeated fractures and marked bone pain are classic symptoms of Ewing's sarcoma. (guidelineshealth.com)
- As Ewing's sarcoma continues to grow within your body, swelling or lumps formation will be seen, which will be soft and tender to touch. (guidelineshealth.com)
- As with most type of cancers, weight loss will be experienced along with Ewing's sarcoma, which may be pronounced in some children but not so in others. (guidelineshealth.com)
- It is important to note that Ewing's sarcoma is comparatively rare and there is a high chance that your child will be affected with some other mild form of infection of fever, which you will get to know upon your doctor's visit. (guidelineshealth.com)
- In case, you notice these early signs and symptoms of Ewing's sarcoma, then we recommend you to consult your healthcare team asap. (guidelineshealth.com)
- Ewing's sarcoma is a rare type of cancer that arises in the bones or surrounding soft tissues, causing symptoms such as pain or constant pain in a region of the body with bone, excessive tiredness or the appearance of a fracture without an apparent cause. (thelightlifeblog.com)
- Depending on when it is identified, Ewing's sarcoma can be cured, however, it is usually necessary to do high doses of chemotherapy or radiation to completely eliminate the cancer. (thelightlifeblog.com)
- In the early stages, Ewing's sarcoma does not usually cause symptoms, however, as the disease progresses, some signs and symptoms may appear that are not very specific, and Ewing's sarcoma may be confused with other bone diseases. (thelightlifeblog.com)
- The specific cause of Ewing's sarcoma is not yet known, however the disease does not appear to be hereditary and, therefore, there is no risk of passing from parents to children, even if there are other cases in the family. (thelightlifeblog.com)
- Initially, Ewing's sarcoma can be quite difficult to identify, as the symptoms are similar to more common situations such as sprains or ligament ruptures. (thelightlifeblog.com)
- Thus, in order to confirm the diagnosis of Ewing's sarcoma, the doctor, in addition to evaluating the symptoms, indicates the performance of imaging exams with the objective of identifying bone alterations and suggestive of a tumor, such as tomography, X-ray and magnetic resonance. (thelightlifeblog.com)
- Treatment for Ewing's sarcoma may vary depending on the size of the tumor. (thelightlifeblog.com)
- Surgery for Ewing's sarcoma consists of removing the affected part of the bone and surrounding tissues, but in the case of larger tumors, removal of a limb may be necessary.Then, chemo or radiotherapy sessions may be recommended again to ensure the elimination of cancer cells and decrease the risk of metastasis. (thelightlifeblog.com)
- Localized Ewing's sarcoma affects only the bone in which it developed and the tissues next to the bone, such as muscle and tendon. (hoapb.com)
- The following is a general overview of treatment for localized Ewing's sarcoma. (hoapb.com)
- The multi-modality approach to treatment for Ewing's sarcoma requires that patients be treated by a multi-disciplinary team consisting of the primary care physician, an orthopedic surgeon experienced in bone tumors, a pathologist, radiation oncologists, pediatric oncologists, rehabilitation specialists, pediatric nurse specialists, social workers, and others. (hoapb.com)
- An experienced team is best found in specialty cancer centers that treat many patients with Ewing's sarcoma. (hoapb.com)
- The primary cooperative group evaluating Ewing's sarcoma treatment in the U.S. is the Children's Cancer Study Group. (hoapb.com)
- Effective treatment of localized Ewing's sarcoma requires both local and systemic therapy. (hoapb.com)
- Most patients diagnosed with localized Ewing's sarcoma actually have micrometastases that are undetectable by current procedures. (hoapb.com)
- The presence of micrometastases may cause Ewing's sarcoma recurrence following local treatment with surgery alone, radiation alone or surgery plus radiation. (hoapb.com)
- Thus, systemic therapy is nearly always given to patients with Ewing's sarcoma to treat undetectable micrometastases. (hoapb.com)
- The ultimate goal of surgery for localized Ewing's sarcoma is to remove the cancer without amputation. (hoapb.com)
- Even with the advent of chemotherapy as systemic treatment and radiation as local treatment, surgery is still an important component of treatment for Ewing's sarcoma. (hoapb.com)
- The main improvement in the treatment of localized Ewing's sarcoma over the past 30 years has been the advent of combination chemotherapy. (hoapb.com)
- The most common approach for the treatment of localized Ewing's sarcoma is to remove as much tumor as possible surgically, deliver local radiation to eradicate microscopic tumor not removed by surgery and to administer systemic combination chemotherapy to eradicate micrometastases. (hoapb.com)
Tumors19
- In 1920, James Ewing discerned that these tumors are a distinct type of cancer. (wikipedia.org)
- Extraosseous (or extraskeletal) Ewing sarcoma describes tumors in the soft tissues around bones, such as cartilage. (medlineplus.gov)
- Analysis of prognostic factors in ewing sarcoma family of tumors: review of St. Jude Children's Research Hospital studies. (medscape.com)
- A pilot study of low-dose anti-angiogenic chemotherapy in combination with standard multiagent chemotherapy for patients with newly diagnosed metastatic Ewing sarcoma family of tumors: A Children's Oncology Group (COG) Phase II study NCT00061893. (medscape.com)
- Here you can find out all about the Ewing family of tumors, including risk factors, symptoms, how they are found, and how they are treated. (cancer.org)
- For instance, genetic sequencing is helping researchers gather data on mutations associated with Ewing sarcoma, while other research has revealed new insights into the role of a certain protein, called EWS-FLI1, which is seen in most of these tumors. (stbaldricks.org)
- Ewing sarcoma has also been called peripheral primitive neuroectodermal tumor, Askin tumor (Ewing sarcoma of the chest wall), extraosseous Ewing sarcoma (Ewing sarcoma in tissue other than bone), and Ewing sarcoma family of tumors . (vicc.org)
- Proton therapy is used to treat Ewing sarcoma, and many other types of cancer, because it delivers radiation directly to the tumors, minimizing damage to surrounding healthy tissue. (chop.edu)
- Ewing sarcoma tumors include Ewing sarcoma, Askin tumor, and peripheral primitive neuroectodermal tumors. (medscape.com)
- The most common combination- EWS exon 7 fused to FLI1 exon 6 (type 1 translocation)-occurs in approximately 50-64% of tumors of Ewing sarcomas. (medscape.com)
- DNA methylation and gene expression profiling of ewing sarcoma primary tumors reveal genes that are potential targets of epigenetic inactivation. (nih.gov)
- Progress in the treatment of Ewing's sarcoma has improved survival from about 10%, before the introduction of chemotherapy, to about 75% today for patients with localized tumors. (go.jp)
- Ewing Sarcoma tumors can occur in any bone in the body but most often occur in the long bones of the legs or arms, as well as the ribs, spine, and pelvis. (dfeetcancer.org)
- The 2 most common malignant bone tumors in children are osteosarcoma and Ewing sarcoma. (medscape.com)
- Ewing sarcoma accounts for approximately 5% of biopsy-analyzed bone tumors and approximately 33% of primary bone tumors. (medscape.com)
- Because the clinical symptoms of Ewing sarcoma are nonspecific and because they frequently mimic osteomyelitis or other malignant tumors, such as leukemia, an initial conventional radiographic and/or magnetic resonance imaging (MRI) examination is performed. (medscape.com)
- The three most common forms of primary bone cancers are osteosarcoma, Ewing sarcoma family of tumors, and chondrosarcoma. (healthline.com)
- Ewing sarcoma family of tumors (ESFTs) strikes adolescents and young adults, but these tumors can sometimes affect children as young as 5 years old. (healthline.com)
- It acts as a valuable marker for Ewing sarcoma, with a sensitivity of 93% and a specificity of 89%, and aids in the differential diagnosis of small round cell tumors. (neobiotechnologies.com)
Chemotherapy16
- These patients also have to deal with tremendous side effects due to high-dose chemotherapy and the debilitating surgeries that are often part of treatment for Ewing sarcoma. (stbaldricks.org)
- Typically, chemotherapy is the first-line treatment for Ewing sarcoma. (healthline.com)
- Although surgery, chemotherapy, and radiation therapy are the most commonly used treatments for Ewing sarcoma, they are not the latest treatments available. (healthline.com)
- Before chemotherapy was combined with surgery and radiation therapy, the long-term survival rate for Ewing sarcoma was less than 10% . (healthline.com)
- A clinical trial to investigate the early prediction of toxicity following chemotherapy in Ewing sarcoma using blood biomarkers and correlation with age-dependent pharmacokinetic variation started in 2014. (bcrt.org.uk)
- The first arm will enroll up to 30 patients with Ewing sarcoma, a rare and deadly pediatric bone cancer, and will investigate seclidemstat in combination with topotecan and cyclophosphamide, a commonly used second- and third- line chemotherapy regimen. (foxchase.org)
- As in the overwhelming majority of pediatric cancers, it usually remains unmutated in Ewing sarcoma, a pediatric bone cancer whose standard treatment includes tough rounds of chemotherapy. (childrenshospital.org)
- In pre-clinical studies, she says, the stapled-peptide agent was synergistic with standard chemotherapy for Ewing sarcoma. (childrenshospital.org)
- The treatment for Ewing Sarcoma typically consists of chemotherapy combined with surgery and/or radiation. (dfeetcancer.org)
- Surgery and/or radiation is often combined with Ewing Sarcoma chemotherapy. (dfeetcancer.org)
- Standard care for Ewing Sarcoma patients revolves around VDC/IE chemotherapy and surgery/radiation. (dfeetcancer.org)
- Oncologists are left to try alternate chemotherapy agents in hopes to slowdown or stop the rapidly dividing Ewing Sarcoma cells. (dfeetcancer.org)
- Finally, the recurrent Ewing Sarcoma patient is faced with months and sometimes years of additional chemotherapy, surgeries, medical care, and a much lower survival rate. (dfeetcancer.org)
- High-dose ifosfamide has shown superior survival benefit over three other chemotherapy regimens for patients with recurring or refractory primary Ewing sarcoma (RR-ES) in the practice-changing rEECur trial . (medscape.com)
- adult and pediatric patients with Ewing sarcoma, as part of a multi-phase, combination chemotherapy regimen. (nih.gov)
- Ewing Sarcoma: The recommended dose is 1250 mcg/m 2 intravenously once every 3 weeks for 51 weeks, as part of a multi-agent combination chemotherapy regimen. (nih.gov)
Osteosarcoma10
- Ewing sarcoma accounts for about 1.5 percent of all childhood cancers, and it is the second most common type of bone tumor in children (the most common type of bone cancer is called osteosarcoma). (medlineplus.gov)
- Ewing sarcoma - the second most common bone cancer after osteosarcoma - often originates in the long, large bones of the body, including the hip, thigh, shin, chest, and arm bones. (stbaldricks.org)
- Nutritional status of children and young adults with Ewing sarcoma or osteosarcoma at diagnosis and during multimodality therapy. (nih.gov)
- Osteosarcoma, chondrosarcoma and Ewing sarcoma: Clinical aspects, biomarker discovery and liquid biopsy. (nih.gov)
- Background/Aim: Ewing sarcoma can arise in patients after osteosarcoma or vice versa. (iiarjournals.org)
- Patients and Methods: The database of the Cooperative Osteosarcoma Study Group (1980-09/2022) was searched for all patients with an osteosarcoma (including undifferentiated pleomorphic sarcoma of the bone) who also suffered from Ewing sarcoma (incl. (iiarjournals.org)
- Conclusion: Secondary osteosarcoma arising after Ewing sarcoma is almost exclusively associated with radiation. (iiarjournals.org)
- The combination of Ewing sarcoma with a later osteosarcoma is a feared complication in both pediatric and adult oncology. (iiarjournals.org)
- Secondary Ewing sarcoma, arising after an osteosarcoma, is also a possibility, but clearly represents the much less common order of events ( 4 - 10 ). (iiarjournals.org)
- Osteosarcoma is also sometimes known as osteogenic sarcoma. (healthline.com)
Round cell sarcoma7
- It is a type of small round cell sarcoma. (wikipedia.org)
- Undifferentiated round cell sarcoma may also occur in the bone or soft tissue. (vicc.org)
- Undifferentiated round cell sarcoma usually occurs in the bones or the muscles that are attached to bones and that help the body move. (vicc.org)
- Undifferentiated round cell sarcoma with BCOR-CCNB3 rearrangements. (vicc.org)
- In this type of round cell sarcoma, the BCOR gene is joined to the CCNB3 gene. (vicc.org)
- To diagnose round cell sarcoma, the tumor cells are checked for this gene change. (vicc.org)
- Undifferentiated round cell sarcoma with CIC-DUX4 rearrangements. (vicc.org)
Primitive neuroectodermal tumours3
- Impact of FDG PET for staging of Ewing sarcomas and primitive neuroectodermal tumours. (medscape.com)
- Ewing sarcoma (ES)/primitive neuroectodermal tumours (PNET, together defined as Ewing sarcoma [ES]) is a primary neoplasm of the skeletal system, first described by James Ewing in 1921 (Gupta et al. (journalcra.com)
- Chronic osteomyelitis , extra-skeletal Ewing sarcoma , malignant small cell tumour and soft tissue -based primitive neuroectodermal tumours were excluded. (bvsalud.org)
Cause of Ewing sarcoma2
- The cause of Ewing sarcoma is unknown, most cases appearing to occur randomly. (wikipedia.org)
- The cause of Ewing sarcoma in children is still unknown. (childrensoncologygroup.org)
Signs and symptoms of Ewing2
- What are the signs and symptoms of Ewing sarcoma? (stbaldricks.org)
- Signs and symptoms of Ewing sarcoma include swelling and pain near the tumor. (vicc.org)
Diagnosis8
- Because Ewing sarcomas are rare, they are often not considered in a differential diagnosis until biopsy reveals a neoplasm known as a small round blue cell tumor. (medscape.com)
- Ewing sarcoma should be considered in the differential diagnosis if a patient aged 10-30 years has a soft tissue or bony mass that causes the physician to consider the presence of a neoplasm. (medscape.com)
- Utilisation of fluorescent multiplex PCR and laser-induced capillary electrophoresis for the diagnosis of Ewing family of tumours in formalin-fixed paraffin-embedded tissues. (medscape.com)
- Approximately 1 in 4 children with Ewing sarcoma will have metastatic disease at the time of first diagnosis. (stbaldricks.org)
- This article provides information about the diagnosis, staging and treatment of this pediatric Ewing sarcoma which is a cancer of the connective tissues, including bone, muscle and cartilage. (oncolink.org)
- Cancer abdominal en ninos sintomas - Cancer abdominal en ninos sintomas, Cura varicelor 1 etapa Posts navigation Top news Tratament home pentru masaj prostatitis [ 6, 7] Diagnosis of prostatitis is made with histopathological examination of the biopsy specimens retrieved because of suspect prostate cancer. (comunicaliber.ro)
- Molecular techniques and new imaging modalities are affecting the diagnosis and classification of patients with Ewing's sarcoma. (go.jp)
- No single morphologic or functional imaging method provides findings for a specific diagnosis of Ewing sarcoma, but the results do contribute to tumor staging. (medscape.com)
PNET1
- [ 4 ] An association exists between Ewing sarcoma and primitive peripheral neuroectodermal tumor (PNET). (medscape.com)
EWSR14
- a reciprocal translocation between chromosomes 11 and 22, t(11,22), which fuses the Ewing Sarcoma Breakpoint Region 1 (EWSR1) gene of chromosome 22 (which encodes the EWS protein) to the Friend Leukemia Virus Integration 1 (FLI1) gene (which encodes Friend Leukemia Integration 1 transcription factor (FLI1), a member of the ETS transcription factor family) of chromosome 11. (wikipedia.org)
- The most common mutation that causes Ewing sarcoma involves two genes, the EWSR1 gene on chromosome 22 and the FLI1 gene on chromosome 11 . (medlineplus.gov)
- The EWSR1/FLI1 fusion gene occurs in approximately 85 percent of Ewing sarcomas. (medlineplus.gov)
- Translocation of EWSR1 (Ewing sarcoma breakpoint region 1) with an ETS (E26 transformation-specific) transcription factor gene occurs in more than 95% of Ewing sarcomas. (medscape.com)
Young adults6
- Ewing sarcoma occurs most often in teenagers and young adults and represents 2% of childhood cancers. (wikipedia.org)
- Ewing sarcomas most often occur in children and young adults. (medlineplus.gov)
- Doctors will be able to quickly see evidence of tumor spread in 25% of children and young adults with Ewing sarcoma when they are diagnosed. (cancer.net)
- Ewing sarcoma is most common in adolescents and young adults (teens through mid-20s). (vicc.org)
- Ewing's sarcoma of the bone is a rare, highly aggressive tumor that typically affects children and young adults. (go.jp)
- Ewing sarcoma is a very rare cancer of the bone and soft tissue that mainly affects children and young adults, particularly in the second decade of life, explained lead author Martin McCabe, MD, clinical senior lecturer in pediatric, teenage, and young adult cancer at the University of Manchester, UK. (medscape.com)
Malignant4
- Extraskeletal Ewing sarcoma is a rare, poorly differentiated, highly malignant, soft tissue tumor, derived from neuroectoderm, that is morphologically indistinguishable from skeletal Ewing sarcoma but is located in extraosseous locations, with the most common being: chest wall, paravertebral region, abdominopelvic area (with predilection for the retroperitoneal space), gluteal region and lower extremities. (mendelian.co)
- Ewing Sarcoma (ES) is the second most common primary malignant bone tumor in children and adolescents. (oncotarget.com)
- Ewing sarcoma is a highly malignant primary bone tumor that is derived from red bone marrow. (medscape.com)
- Ewing sarcoma is a highly malignant tumour predominantly found in children . (bvsalud.org)
Symptoms4
- Jason Yustein, M.D., Ph.D. , a St. Baldrick's Foundation Board Member and Scientific Program Committee Member, at Emory University School of Medicine, Georgia, explains Ewing sarcoma symptoms, treatment options, and research opportunities. (stbaldricks.org)
- These and other signs and symptoms may be caused by Ewing sarcoma or by other conditions . (vicc.org)
- See Clinical Presentation for more specific information on the signs, symptoms, patient history, and physical examination for Ewing sarcoma. (medscape.com)
- Síntomas del sarcoma vaccin papillomavirus effet In best anti inflammatory drugs for prostatitis article, we look at the causes and symptoms of chronic prostatitis. (comunicaliber.ro)
Cancers6
- Unfortunately, the survival outcome for Ewing sarcoma patients is less than most other types of pediatric cancers. (stbaldricks.org)
- To now be working with a cancer research center of the caliber of Fox Chase Cancer Center further affirms the potential of seclidemstat to have a meaningful impact on the treatment of Ewing sarcoma and other cancers. (foxchase.org)
- While the pathway is intact in most pediatric cancers, research finds that drugs targeting the pathway can curb tumor cell proliferation in Ewing sarcoma. (childrenshospital.org)
- Their collaboration has produced a new clinical trial for patients with Ewing sarcoma and selected other childhood cancers. (childrenshospital.org)
- Most research cell lines for Ewing sarcoma and many other cancers possess a mutated TP53 gene, likely because these cells tend to grow better in the laboratory. (childrenshospital.org)
- The NIH invites applications to use whole genome sequencing at an NHGRI-supported sequencing center to investigate the genetic etiology of structural birth defects, and to further elucidate the genetic contribution to childhood cancers and the genomic contributions to treatment failure for childhood sarcomas. (nih.gov)
Treatment16
- Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children's Oncology Group. (medscape.com)
- Treatment for Ewing sarcoma usually involves surgery, which helps remove the tumor from your body. (healthline.com)
- Although it's not used in every case, surgery is a common treatment for Ewing sarcoma. (healthline.com)
- Recurrent Ewing sarcoma is a tumor that has come back after treatment. (cancer.net)
- See Treatment and Medication for more specific information regarding pharmacologic and other therapies for Ewing sarcoma. (medscape.com)
- How could this project improve treatment options for Ewing sarcoma patients? (bcrt.org.uk)
- Planet Ayurveda provides best cancer sarcoma pelvico herbal remedies for ayurvedic treatment of Prostatitis. (comunicaliber.ro)
- Salarius believes this treatment combination and its use as a second- and third-line therapy could greatly expand the addressable patient population for seclidemstat and improve outcomes by allowing physicians to use seclidemstat earlier in the Ewing sarcoma continuum of care. (foxchase.org)
- Given the fact that Ewing Sarcoma's family had to struggle to pay for his medication and often expensive hospital stays, his mom started thinking about the dilemma of quitting her job in order to take care of her children or keeping her job in order to pay for his expensive treatment. (kennyslaught.com)
- Introduction Foods for Ewing Sarcoma should be personalized for each individual and also must adapt when cancer treatment or tumor genetic change. (addon.life)
- How can I find treatment of Ewing Sarcoma? (marham.pk)
- Amputation, which is the removal of part or all of the limb, is also seen in Ewing Sarcoma treatment. (dfeetcancer.org)
- However, if this frontline treatment in unsuccessful and the cancer returns, called recurrent Ewing Sarcoma, there is no agreed upon treatment protocol. (dfeetcancer.org)
- Gralow explained in an interview that treatment of Ewing sarcoma differs from one cancer center to another. (medscape.com)
- During Sarcoma Awareness Month , we recognize the importance of not only raising awareness of this rare cancer type, but also of accelerating our understanding of disease mechanisms driving sarcomas in the pursuit of innovative treatment methods. (arimagenomics.com)
- Through awareness and rigorous scientific research, we can champion early detection, better treatment, and improved patient outcomes for those battling sarcoma. (arimagenomics.com)
Askin1
- There are several types of Ewing sarcoma, including Ewing sarcoma of bone, extraosseous Ewing sarcoma, peripheral primitive neuroectodermal tumor (pPNET), and Askin tumor. (medlineplus.gov)
Extraosseous1
- Extraskeletal Ewing Sarcoma Is also known as extraosseous ewing sarcoma, extraskeletal ewing tumor, eoe, extraosseous ewing tumor. (mendelian.co)
Clinical2
- There are always new treatments in clinical trials for Ewing sarcoma. (healthline.com)
- This, added to clinical data showing drug activity across Ewing and other sarcomas, support the further exploration of seclidemstat in these high unmet need patient populations. (foxchase.org)
Children's Oncol1
- Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: A report from the Children's Oncology Group. (medscape.com)
Gene4
- A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. (medscape.com)
- More recently, NKX2.2 protein was identified as a target of EWS-FLI-1, the fusion protein specific to Ewing sarcoma, and was shown to be differentially upregulated in Ewing sarcoma on the basis of array-based gene expression analysis. (neobiotechnologies.com)
- Genetically, Ewing sarcomas are characterized by chromosomal translocations between one gene and a transcription factor, resulting in dysregulation of thousands of downstream targets and oncogenesis. (arimagenomics.com)
- Research led by Stephen Lessnick and Emily Theisen at Nationwide Children's Hospital used Arima Hi-C to precisely define the global changes in chromatin structure associated with Ewing sarcoma and further link these structural changes to alterations in gene expression. (arimagenomics.com)
Recurrence1
- Sharma et al concluded that 18 F-FDG PET/CT demonstrated high accuracy in diagnosing the recurrence of primary skeletal Ewing sarcoma. (medscape.com)
Occur1
- Sarcomas can occur in various parts of the body, including bone and connective tissues, and affect both pediatric and adult populations. (arimagenomics.com)
Bone tumor1
- Approximately 87 percent of Ewing sarcomas are Ewing sarcoma of bone, which is a bone tumor that usually occurs in the thigh bones (femurs), pelvis, ribs, or shoulder blades. (medlineplus.gov)
Child's doctor1
- To diagnose Ewing sarcoma, your child's doctor will do an exam, take a detailed medical history and order some tests. (uwhealth.org)
Patients12
- Is there a need for dedicated bone imaging in addition to 18F-FDG PET/CT imaging in pediatric sarcoma patients? (medscape.com)
- Role of bone marrow biopsy for staging new patients with Ewing sarcoma: A systematic review. (medscape.com)
- Patients with Ewing sarcoma in the distal extremities have the best prognosis. (cancer.net)
- Patients with Ewing sarcoma in the proximal extremities have an intermediate prognosis, followed by patients with Ewing sarcoma in central or pelvic sites. (cancer.net)
- Establish pharmacokinetic profiles for vincristine, ifosfamide, doxorubicin, etoposide and cyclophosphamide in Ewing sarcoma patients and to compare the results between children, adolescents and adults. (bcrt.org.uk)
- Investigate potential relationships between pharmacokinetics, pharmacogenetics and biomarkers of toxicity in adolescent Ewing sarcoma patients, as compared to children and adults. (bcrt.org.uk)
- The third patient arm will investigate seclidemstat as a single agent in up to 15 patients with select sarcomas that share a biology similar to Ewing sarcoma, also referred to as FET-rearranged or Ewing-related sarcomas. (foxchase.org)
- In data released at ASCO 2021, a subset of patients with advanced FET-rearranged sarcomas treated with single-agent seclidemstat resulted in stable disease (SD) and prolonged time to progression (TTP) which Salarius believes suggests disease control, a clinically relevant endpoint for soft tissue sarcomas. (foxchase.org)
- In this report, we present two consecutive cases of patients with Ewing's Sarcoma, diagnosed, and treated at our institute. (go.jp)
- The findings from the rEECur trial "could help physicians talk with patients and their families about the likelihood of response, survival, and toxicity for each regimen available for relapsed Ewing sarcoma based on objective, randomized data," she commented in an ASCO press release. (medscape.com)
- A total of 182 radiographs from our Sarcoma Centre (118 healthy, 44 Ewing, 20 osteomyelitis ) from 58 different paediatric (≤18 years) patients were collected. (bvsalud.org)
- As a "jumpstart" for the larger Precision Medicine initiative, expected to begin in FY16, there is a special opportunity to propose cohorts of pediatric sarcoma patients in which the cancer has failed to respond to therapy. (nih.gov)
Pelvis1
- The swelling is most likely to be visible if the sarcoma is located on a bone near the surface of the body, but when it occurs in other places deeper in the body, like on the pelvis, it may not be visible. (wikipedia.org)
Soft-tissue5
- Ewing sarcoma is a type of pediatric cancer that forms in bone or soft tissue. (wikipedia.org)
- Tests that examine the bone and soft tissue are used to diagnose and stage Ewing sarcoma. (vicc.org)
- Ewing sarcoma is a cancer that arises from bone or the soft tissue around bone. (uwhealth.org)
- Ewing Sarcoma, named after James R. Ewing, who described the first cases in 1921, is an aggressive cancer that affects bones or nearby soft tissue. (dfeetcancer.org)
- Doxorubicin is also used alone and in combination with other medications to treat certain types of thyroid cancer and certain types of soft tissue or bone sarcomas (cancer that forms in muscles and bones). (nih.gov)
Occurs3
- Ewing sarcoma is a cancerous tumor that occurs in bones or soft tissues, such as cartilage or nerves. (medlineplus.gov)
- Ewing sarcoma (ES) is a pediatric bone cancer that occurs in children and adolescents. (nih.gov)
- But in this case, the researchers wanted to analyze Ewing sarcoma cell lines with normal, "wild-type" TP53 , since those are more representative of the disease when it occurs in children and adolescents. (childrenshospital.org)
Rhabdomyosarcoma1
- Recently, Arima technology has been used to study the mechanisms of several sarcomas, including rhabdomyosarcoma and Ewing sarcoma, as well as Kaposi's sarcoma-associated herpesvirus known to cause Kaposi sarcoma. (arimagenomics.com)
20211
- Salarius believes data released during ASCO 2021 demonstrated synergy in an Ewing sarcoma cell line when seclidemstat was used in combination with topotecan and cyclophosphamide. (foxchase.org)
Translocation4
- Bailly RA, Bosselut R, Zucman J, Cormier F, Delattre O, Roussel M, Thomas G, Ghysdael J. DNA-binding and transcriptional activation properties of the EWS-FLI-1 fusion protein resulting from the t(11;22) translocation in Ewing sarcoma. (medlineplus.gov)
- In the early 1980s, Ewing sarcoma and the peripheral primitive neuroectodermal tumor were found to contain the same reciprocal translocation between chromosomes 11 and 22, t(11;22). (medscape.com)
- Some argue that without a translocation, the tumor does not belong to Ewing sarcoma). (medscape.com)
- The most common translocation seen in about 85% of all Ewing tumor is the t(11;22) translocation. (medscape.com)
Pediatric cancer1
- Ewing sarcoma is a highly aggressive pediatric cancer. (arimagenomics.com)
Primary1
- Kaposi sarcoma-associated herpesvirus (KSHV) is a human herpesviruses that is known to cause several diseases, including Kaposi sarcoma, primary effusion lymphoma, and AIDS-related multicentric Castleman disease. (arimagenomics.com)
James Ewing1
- It was first described by James Ewing in 1921. (medscape.com)
Childhood2
- Ewing sarcoma is more common in males (1.6 male:1 female) and usually presents in childhood or early adulthood, with a peak between 10 and 20 years of age. (wikipedia.org)
- Ewing sarcoma is a type of childhood cancer that is most frequently found in children and adolescents between the ages of 10 and 20 years old. (stbaldricks.org)
Bones3
- Ewing sarcoma is a type of cancer that starts in bones and soft tissues. (healthline.com)
- Procedures that make pictures of the bones and soft tissues and nearby areas help diagnose Ewing sarcoma and show how far the cancer has spread. (vicc.org)
- The most frequent sites of metastases in Ewing's sarcoma include lungs, bones and bone marrow. (go.jp)
Chromatin1
- Using Arima technology to explore genome-wide 3D chromatin conformation data, Dr. Lessnick's team was able to fill significant knowledge gaps in Ewing sarcoma. (arimagenomics.com)
Radiograph3
- Radiograph of an 11-year-old boy with a large Ewing sarcoma in the right pelvic area. (medscape.com)
- Radiograph of Ewing sarcoma of the os naviculare, a rare location for the tumor. (medscape.com)
- The purpose of this study was to develop an artificial intelligence algorithm , which can determine imaging features in a common radiograph to distinguish osteomyelitis from Ewing sarcoma . (bvsalud.org)
Spine1
- At the age of seven, I was diagnosed with Ewing sarcoma on my spine which is a type of bone cancer. (aspire.org.uk)
Paraffin-embedded1
- Formalin-fixed, paraffin-embedded human Ewing's Sarcoma stained with NKX2.2 Rabbit Recombinant Monoclonal Antibody (NX2/1422R). (neobiotechnologies.com)