Rhabdomyosarcoma
Rhabdomyosarcoma, Embryonal
Rhabdomyosarcoma, Alveolar
Muscle Neoplasms
PAX7 Transcription Factor
Paired Box Transcription Factors
Soft Tissue Neoplasms
Sarcoma
Mesenchymoma
Sarcoma, Ewing
Chromosomes, Human, Pair 13
Forkhead Transcription Factors
Oncogene Proteins, Fusion
Desmin
Chromosomes, Human, Pair 2
Combined Modality Therapy
Translocation, Genetic
Sertoli-Leydig Cell Tumor
Myogenin
MyoD Protein
Thoracic Neoplasms
Tumor Cells, Cultured
Gene Expression Regulation, Neoplastic
Dactinomycin
Gene Fusion
Enterovirus A, Human
Pulmonary Blastoma
Prenatal sonographic features of embryonal rhabdomyosarcoma. (1/128)
We describe a case of fetal rhabdomyosarcoma detected during the third trimester of pregnancy by prenatal sonography. At 33 weeks' gestation, sonography performed because of suspected polyhydramnios showed a solid mass of 120 x 54 mm arising from the anterior wall of the fetal thoracic cage. Another mass within the left maxillary area which originated from the left orbital floor was also detected. In the abdomen, there were multiple round masses in and around the liver. As the previous scan at 28 weeks had appeared normal, the multiple masses which became visible and enlarged rapidly in different locations led us to believe that there was fetal cancer. The most likely diagnosis was rhabdomyosarcoma (which was later confirmed), because it is the most prevalent soft-tissue tumor in children and may develop within or outside muscle anywhere in the body and at any age. Two other reported cases which were detected by prenatal ultrasound examination are also discussed. (+info)Differentiated embryonal rhabdomyosarcoma in a cow. (2/128)
An embryonal rhabdomyosarcoma was found in the pleura of a 2-year-old Holstein cow after first delivery. The most predominant cells in the tumor were relatively small in size, but considerable numbers of more differentiated cells of larger sizes mingled with the small cells. The most differentiated cells were characterized by multinucleation, abundant cytoplasm containing cross-striated fibrils, intense immunoreactivity for desmin, and weak or negative reactivity for vimentin. Such cells, lacking mitotic activity and displaying weak or no reactivity for proliferating cell nuclear antigen, were considered to be malignant counterparts of myotubes or muscle fibers. This neoplasm seems to follow normal skeletal muscle embryogenesis, and to be capable of differentiation into the final stage of muscle development. (+info)Conservative treatment followed by chemotherapy with doxorubicin and ifosfamide for cervical sarcoma botryoides in young females. (3/128)
Sarcoma botryoides of the cervix is an extremely rare tumour and seems to be associated with a better prognosis than its vaginal counterpart. Recent studies have suggested that it is possible to limit surgery to local excision in stage I cases. We report three cases of young subjects treated successfully with polypectomy or diathermy loop excision followed by adjuvant chemotherapy. One patient had a local recurrence which was treated with further local excision. All subjects remain alive without evidence of recurrence and with normal menstrual function 36, 38 and 38 months following initial diagnosis. A conservative surgical approach to early cervical sarcoma botryoides is possible. The efficacy of adjuvant chemotherapy and the regimen of choice still need to be investigated. (+info)The zinc finger protein GLI induces cellular sensitivity to the mTOR inhibitor rapamycin. (4/128)
The protein synthetic machinery is activated by diverse genetic alterations during tumor progression in vivo and represents an attractive target for cancer therapy. We show that rapamycin inhibits the induction of transformed foci in vitro by GLI, a transcription factor that functions in the sonic hedgehog-patched pathway in tumors. In control cells, which were nontransformed epithelioid RK3E cells and derivative c-MYC- or RAS-transformed sister cell lines, rapamycin inhibits mTOR and mTOR-dependent activities but increases global protein synthesis, perhaps by activating a feedback mechanism. In GLI-transformed cells, rapamycin inhibits global protein synthesis and turnover and prevents cellular proliferation. In contrast to its effects on protein synthesis, rapamycin affects bromodeoxyuridine incorporation and cell cycle occupancy of GLI cells and control cells to a similar extent. Rare, variant GLI cells isolated by selection in rapamycin are also drug-resistant for protein metabolism and for cell cycle progression through G1. Our results indicate that sensitivity to rapamycin can be induced by a specific oncogene and that inhibition of global protein metabolism is linked to the rapamycin-sensitive phenotype. (+info)Perinatal management of a neonate with airway obstruction caused by rhabdomyosarcoma of the tongue. (5/128)
Intra-oral masses in neonates can seriously compromise the airway, potentially causing hypoxia and death if not recognized and managed appropriately. We report a case in which an intra-oral mass was diagnosed on antenatal ultrasound scan. Preparation for delivery involved a multidisciplinary team approach, with a strategy for management at delivery. The child was delivered by elective Caesarean section and had a patent airway. A tracheostomy was performed immediately after delivery. The infant underwent a debulking procedure 3 weeks after birth. A histological diagnosis of embryonal rhabdomyosarcoma was made and a course of chemotherapy commenced. The child had a partial response to treatment with considerable shrinkage of the tongue mass. We discuss the management options in neonates with intra-oral masses to provide an adequate airway and maintain fetal oxygenation. The differential diagnosis of fetal oral masses is reviewed. (+info)Benefit of intensified therapy for patients with local or regional embryonal rhabdomyosarcoma: results from the Intergroup Rhabdomyosarcoma Study IV. (6/128)
PURPOSE: To compare failure-free survival (FFS) and survival for patients with local or regional embryonal rhabdomyosarcoma treated on the Intergroup Rhabdomyosarcoma Study (IRS)-IV with that of comparable patients treated on IRS-III. PATIENTS AND METHODS: Patients were retrospectively classified as low- or intermediate-risk. Low-risk patients were defined as those with primary tumors at favorable sites, completely resected or microscopic residual, or orbit/eyelid primaries with gross residual disease and tumors less than 5 cm at unfavorable sites but completely resected. Intermediate-risk patients were all other patients with local or regional tumors. RESULTS: Three-year FFS improved from 72% on IRS-III to 78% on IRS-IV for patients with intermediate-risk embryonal rhabdomyosarcoma (P =.02). Subset analysis revealed two groups that benefited most from IRS-IV therapy. FFS at 3 years for patients with resectable node-positive or unresectable (group III) embryonal rhabdomyosarcoma arising at certain favorable sites (head and neck [not orbit/eyelid or parameningeal] and genitourinary [not bladder or prostate]) improved from 72% on IRS-III to 92% on IRS-IV (P =.01). Similarly, 3-year FFS for patients with completely resected tumor or with only microscopic disease remaining (group I or II) at unfavorable sites improved from 71% on IRS-III to 86% on IRS-IV (P =.04). Only patients with unresectable embryonal rhabdomyosarcoma (group III) at unfavorable sites had no improvement in outcome on IRS-IV (3-year FFS for IRS-III and IRS-IV, 72% and 75%, respectively; P =.31). CONCLUSION: IRS-IV therapy benefited certain subgroups of patients with intermediate-risk embryonal rhabdomyosarcoma. A doubling of the intensity of cyclophosphamide (or ifosfamide equivalent) dosing per cycle between IRS-III and IRS-IV is thought to be a key contributing factor for this improvement. (+info)Myogenin is a specific marker for rhabdomyosarcoma: an immunohistochemical study in paraffin-embedded tissues. (7/128)
Myogenin belongs to a group of myogenic regulatory proteins whose expression determines commitment and differentiation of primitive mesenchymal cells into skeletal muscle. The expression of myogenin has been demonstrated to be extremely specific for rhabdomyoblastic differentiation, which makes it a useful marker in the differential diagnosis of rhabdomyosarcomas (RMS) from other malignant small round cell tumors of childhood. Commercially available antibodies capable of detecting myogenin in routinely processed formalin-fixed paraffin-embedded (FFPE) tissue are now available. In this study, we evaluated myogenin expression using the monoclonal myf-4 antibody (Novocastra Labs) on FFPE in a large number of pediatric tumors in order to define the clinical utility of this marker. A total of 119 tumors were studied. These included 48 alveolar RMS (ARMS), 20 embryonal RMS (ERMS), one spindle cell RMS, 16 Ewing's sarcomas (ES), six nephroblastomas, two ectomesenchymomas, seven precursor hematopoietic neoplasms, five olfactory neuroblastomas, three neuroblastomas, six desmoplastic small round cell tumors, and five rhabdoid tumors. Distinct nuclear staining for myogenin was noted in all 69 RMS. Notably, the number of positive tumor cells differed between the ARMS and ERMS. In ARMS, the majority of tumor cells (75 to 100%) were positive, in contrast to ERMS, in which the positivity ranged from rare + to 25% in all but three tumors. Additionally, myogenin positivity was seen in two of two ectomesenchymomas and in two nephroblastomas with myogenous differentiation. All other tumors were clearly negative. Our results indicate that staining for myogenin is an extremely reliable and specific marker for rhabdomyoblastic differentiation. It gives consistent and easily interpretable results in routinely fixed tissues. (+info)High-dose thiotepa and hematopoietic stem cell transplantation in pediatric malignant mesenchymal tumors: a phase II study. (8/128)
The prognosis of metastatic malignant mesenchymal tumors (MMT) remains poor. Given the chemosensitivity of these neoplasms, a phase II study of high-dose thiotepa (HDT) was performed to evaluate the efficacy of this drug in this particular subset of pediatric tumors. Between 1986 and 1998, 18 patients, previously treated with conventional therapy for metastatic or refractory MMT, entered the study. Thiotepa was administered at a daily dose of 300 mg/m2 for 3 consecutive days. Hematopoietic stem cell rescue, consisting of bone marrow transplantation or peripheral stem cell transplantation, was performed 2 days after completion of HDT. A response exceeding 50% was observed in 6/18 patients (response rate 33%). Toxicity was severe but never led to death. HDT used at a dose of 900 mg/m2 yields measurable anti-tumor activity in previously treated patients. The next step in these particularly poor prognosis metastatic MMT will be to investigate HDT combined with other drugs, known to be efficient at high doses. (+info)Rhabdomyosarcoma is a type of cancer that develops in the body's soft tissues, specifically in the muscle cells. It is a rare and aggressive form of sarcoma, which is a broader category of cancers that affect the connective tissues such as muscles, tendons, cartilages, bones, blood vessels, and fatty tissues.
Rhabdomyosarcomas can occur in various parts of the body, including the head, neck, arms, legs, trunk, and genitourinary system. They are more common in children than adults, with most cases diagnosed before the age of 18. The exact cause of rhabdomyosarcoma is not known, but genetic factors and exposure to radiation or certain chemicals may increase the risk.
There are several subtypes of rhabdomyosarcoma, including embryonal, alveolar, pleomorphic, and spindle cell/sclerosing. The type and stage of the cancer determine the treatment options, which may include surgery, radiation therapy, chemotherapy, or a combination of these approaches. Early diagnosis and prompt treatment are crucial for improving the prognosis and long-term survival rates.
Rhabdomyosarcoma, embryonal is a type of soft tissue sarcoma, which is a cancer that develops in the body's connective tissues, such as muscles, tendons, ligaments, and cartilage. Specifically, embryonal rhabdomyosarcoma is a subtype of rhabdomyosarcoma that arises from cells that are in the process of becoming muscle cells. This type of cancer typically affects children, with most cases diagnosed before the age of 10.
Embryonal rhabdomyosarcoma can develop in various parts of the body, including the head and neck, genitourinary tract (reproductive and urinary organs), and extremities. The tumors are often aggressive and fast-growing, but they can be treated with a combination of surgery, radiation therapy, and chemotherapy.
The medical definition of embryonal rhabdomyosarcoma is: "A malignant neoplasm composed of small, round to avoid cells with hyperchromatic nuclei and scant cytoplasm, often arranged in a loose, fascicular pattern. It arises from primitive muscle cells and typically affects children and adolescents. The tumor can develop in various parts of the body, including the head and neck, genitourinary tract, and extremities."
Alveolar Rhabdomyosarcoma (ARMS) is a type of soft tissue sarcoma, which is a rare cancer that affects the muscles and connective tissues. ARMS is characterized by the presence of specific genetic alterations involving the PAX3 or PAX7 genes, which are fused with the FOXO1 gene. These genetic changes lead to the formation of abnormal proteins that promote uncontrolled cell growth and division, resulting in the development of tumors.
ARMS typically affects children and adolescents, although it can occur in adults as well. The most common sites for ARMS include the extremities, trunk, head, and neck. The alveolar subtype is named for its histological resemblance to lung tissue, with tumors forming small, thin-walled cavities or spaces that look like the air sacs (alveoli) in the lungs.
ARMS tends to be more aggressive than other types of rhabdomyosarcoma and has a higher risk of metastasis (spreading to other parts of the body). Treatment usually involves a combination of surgery, radiation therapy, and chemotherapy. The prognosis for ARMS depends on several factors, including the patient's age, the size and location of the tumor, and the extent of spread at the time of diagnosis.
Muscle neoplasms are abnormal growths or tumors that develop in the muscle tissue. They can be benign (non-cancerous) or malignant (cancerous). Benign muscle neoplasms are typically slow-growing and do not spread to other parts of the body, while malignant muscle neoplasms, also known as soft tissue sarcomas, can grow quickly, invade nearby tissues, and metastasize (spread) to distant parts of the body.
Soft tissue sarcomas can arise from any of the muscles in the body, including the skeletal muscles (voluntary muscles that attach to bones and help with movement), smooth muscles (involuntary muscles found in the walls of blood vessels, digestive tract, and other organs), or cardiac muscle (the specialized muscle found in the heart).
There are many different types of soft tissue sarcomas, each with its own set of characteristics and prognosis. Treatment for muscle neoplasms typically involves a combination of surgery, radiation therapy, and chemotherapy, depending on the type, size, location, and stage of the tumor.
PAX7 is a transcription factor that belongs to the PAX (paired box) family of proteins, which are characterized by the presence of a paired domain that binds to DNA. Specifically, PAX7 contains two DNA-binding domains: a paired domain and a homeodomain.
PAX7 is primarily expressed in satellite cells, which are muscle stem cells responsible for postnatal muscle growth, maintenance, and regeneration. PAX7 plays a critical role in the self-renewal and survival of satellite cells, and its expression is required for their activation and differentiation into mature muscle fibers.
As a transcription factor, PAX7 binds to specific DNA sequences in the regulatory regions of target genes and regulates their expression. This regulation can either activate or repress gene transcription, depending on the context and other factors that interact with PAX7.
PAX7 has been implicated in various muscle-related diseases, including muscular dystrophies and muscle wasting disorders. Its expression is often downregulated in these conditions, leading to a decrease in satellite cell function and muscle regeneration capacity. Therefore, understanding the role of PAX7 in muscle biology and disease has important implications for developing new therapies for muscle-related diseases.
Paired box (PAX) transcription factors are a group of proteins that regulate gene expression during embryonic development and in some adult tissues. They are characterized by the presence of a paired box domain, a conserved DNA-binding motif that recognizes specific DNA sequences. PAX proteins play crucial roles in various developmental processes, such as the formation of the nervous system, eyes, and pancreas. Dysregulation of PAX genes has been implicated in several human diseases, including cancer.
Soft tissue neoplasms refer to abnormal growths or tumors that develop in the soft tissues of the body. Soft tissues include muscles, tendons, ligaments, fascia, nerves, blood vessels, fat, and synovial membranes (the thin layer of cells that line joints and tendons). Neoplasms can be benign (non-cancerous) or malignant (cancerous), and their behavior and potential for spread depend on the specific type of neoplasm.
Benign soft tissue neoplasms are typically slow-growing, well-circumscribed, and rarely spread to other parts of the body. They can often be removed surgically with a low risk of recurrence. Examples of benign soft tissue neoplasms include lipomas (fat tumors), schwannomas (nerve sheath tumors), and hemangiomas (blood vessel tumors).
Malignant soft tissue neoplasms, on the other hand, can grow rapidly, invade surrounding tissues, and may metastasize (spread) to distant parts of the body. They are often more difficult to treat than benign neoplasms and require a multidisciplinary approach, including surgery, radiation therapy, and chemotherapy. Examples of malignant soft tissue neoplasms include sarcomas, such as rhabdomyosarcoma (arising from skeletal muscle), leiomyosarcoma (arising from smooth muscle), and angiosarcoma (arising from blood vessels).
It is important to note that soft tissue neoplasms can occur in any part of the body, and their diagnosis and treatment require a thorough evaluation by a healthcare professional with expertise in this area.
Orbital neoplasms refer to abnormal growths or tumors that develop in the orbit, which is the bony cavity that contains the eyeball, muscles, nerves, fat, and blood vessels. These neoplasms can be benign (non-cancerous) or malignant (cancerous), and they can arise from various types of cells within the orbit.
Orbital neoplasms can cause a variety of symptoms depending on their size, location, and rate of growth. Common symptoms include protrusion or displacement of the eyeball, double vision, limited eye movement, pain, swelling, and numbness in the face. In some cases, orbital neoplasms may not cause any noticeable symptoms, especially if they are small and slow-growing.
There are many different types of orbital neoplasms, including:
1. Optic nerve glioma: a rare tumor that arises from the optic nerve's supportive tissue.
2. Orbital meningioma: a tumor that originates from the membranes covering the brain and extends into the orbit.
3. Lacrimal gland tumors: benign or malignant growths that develop in the lacrimal gland, which produces tears.
4. Orbital lymphangioma: a non-cancerous tumor that arises from the lymphatic vessels in the orbit.
5. Rhabdomyosarcoma: a malignant tumor that develops from the skeletal muscle cells in the orbit.
6. Metastatic tumors: cancerous growths that spread to the orbit from other parts of the body, such as the breast, lung, or prostate.
The diagnosis and treatment of orbital neoplasms depend on several factors, including the type, size, location, and extent of the tumor. Imaging tests, such as CT scans and MRI, are often used to visualize the tumor and determine its extent. A biopsy may also be performed to confirm the diagnosis and determine the tumor's type and grade. Treatment options include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
Urogenital neoplasms refer to abnormal growths or tumors that occur in the urinary and genital organs. These can include various types of cancer, such as bladder cancer, kidney cancer, prostate cancer, testicular cancer, cervical cancer, ovarian cancer, and others. Some urogenital neoplasms may be benign (non-cancerous), while others are malignant (cancerous) and can spread to other parts of the body.
The term "urogenital" refers to the combined urinary and genital systems in the human body. The urinary system includes the kidneys, ureters, bladder, and urethra, which are responsible for filtering waste from the blood and eliminating it as urine. The genital system includes the reproductive organs such as the ovaries, fallopian tubes, uterus, vagina, prostate gland, testicles, and penis.
Urogenital neoplasms can cause various symptoms depending on their location and size. Common symptoms include blood in urine, pain during urination, difficulty urinating, abnormal discharge, lumps or swelling in the genital area, and unexplained weight loss. If you experience any of these symptoms, it is important to consult a healthcare professional for further evaluation and treatment.
Sarcoma is a type of cancer that develops from certain types of connective tissue (such as muscle, fat, fibrous tissue, blood vessels, or nerves) found throughout the body. It can occur in any part of the body, but it most commonly occurs in the arms, legs, chest, and abdomen.
Sarcomas are classified into two main groups: bone sarcomas and soft tissue sarcomas. Bone sarcomas develop in the bones, while soft tissue sarcomas develop in the soft tissues of the body, such as muscles, tendons, ligaments, fat, blood vessels, and nerves.
Sarcomas can be further classified into many subtypes based on their specific characteristics, such as the type of tissue they originate from, their genetic makeup, and their appearance under a microscope. The different subtypes of sarcoma have varying symptoms, prognoses, and treatment options.
Overall, sarcomas are relatively rare cancers, accounting for less than 1% of all cancer diagnoses in the United States each year. However, they can be aggressive and may require intensive treatment, such as surgery, radiation therapy, and chemotherapy.
Mesenchymoma is a very rare type of tumor that contains a mixture of different types of mesenchymal tissues, such as muscle, fat, bone, cartilage, or fibrous tissue. It typically occurs in children and young adults, and can be found in various parts of the body, including the head, neck, retroperitoneum (the area behind the abdominal cavity), and the limbs.
Mesenchymomas are usually slow-growing and may not cause any symptoms until they reach a large size. Treatment typically involves surgical removal of the tumor, but radiation therapy or chemotherapy may also be used in some cases. The prognosis for mesenchymoma depends on several factors, including the location and size of the tumor, the patient's age and overall health, and the specific types of tissue that are present in the tumor.
Ewing sarcoma is a type of cancer that originates in bones or the soft tissues surrounding them, such as muscles and tendons. It primarily affects children and adolescents, although it can occur in adults as well. The disease is characterized by small, round tumor cells that typically grow quickly and are prone to metastasize (spread) to other parts of the body, most commonly the lungs, bones, and bone marrow.
Ewing sarcoma is caused by a genetic abnormality, specifically a chromosomal translocation that results in the fusion of two genes, EWSR1 and FLI1. This gene fusion leads to the formation of an abnormal protein that disrupts normal cell growth and division processes, ultimately resulting in cancer.
Symptoms of Ewing sarcoma can vary depending on the location and size of the tumor but may include pain or swelling in the affected area, fever, fatigue, and weight loss. Diagnosis typically involves imaging studies such as X-rays, CT scans, or MRI scans to locate the tumor, followed by a biopsy to confirm the presence of cancer cells. Treatment may involve surgery, radiation therapy, chemotherapy, or a combination of these approaches, depending on the stage and location of the disease.
Human chromosome pair 13 consists of two rod-shaped structures present in the nucleus of each cell in the human body. Each chromosome is made up of DNA tightly coiled around histone proteins, forming a complex structure called a chromatin.
Chromosomes carry genetic information in the form of genes, which are sequences of DNA that code for specific traits and functions. Human cells typically have 23 pairs of chromosomes, for a total of 46 chromosomes. Chromosome pair 13 is one of the autosomal pairs, meaning it is not a sex chromosome (X or Y).
Chromosome pair 13 contains several important genes that are associated with various genetic disorders, such as cri-du-chat syndrome and Phelan-McDermid syndrome. Cri-du-chat syndrome is caused by a deletion of the short arm of chromosome 13 (13p), resulting in distinctive cat-like crying sounds in infants, developmental delays, and intellectual disabilities. Phelan-McDermid syndrome is caused by a deletion or mutation of the terminal end of the long arm of chromosome 13 (13q), leading to developmental delays, intellectual disability, absent or delayed speech, and autistic behaviors.
It's important to note that while some genetic disorders are associated with specific chromosomal abnormalities, many factors can contribute to the development and expression of these conditions, including environmental influences and interactions between multiple genes.
Forkhead transcription factors (FOX) are a family of proteins that play crucial roles in the regulation of gene expression through the process of binding to specific DNA sequences, thereby controlling various biological processes such as cell growth, differentiation, and apoptosis. These proteins are characterized by a conserved DNA-binding domain, known as the forkhead box or FOX domain, which adopts a winged helix structure that recognizes and binds to the consensus sequence 5'-(G/A)(T/C)AA(C/A)A-3'.
The FOX family is further divided into subfamilies based on the structure of their DNA-binding domains, with each subfamily having distinct functions. For example, FOXP proteins are involved in brain development and function, while FOXO proteins play a key role in regulating cellular responses to stress and metabolism. Dysregulation of forkhead transcription factors has been implicated in various diseases, including cancer, diabetes, and neurodegenerative disorders.
An oncogene protein fusion is a result of a genetic alteration in which parts of two different genes combine to create a hybrid gene that can contribute to the development of cancer. This fusion can lead to the production of an abnormal protein that promotes uncontrolled cell growth and division, ultimately resulting in a malignant tumor. Oncogene protein fusions are often caused by chromosomal rearrangements such as translocations, inversions, or deletions and are commonly found in various types of cancer, including leukemia and sarcoma. These genetic alterations can serve as potential targets for cancer diagnosis and therapy.
Desmin is a type of intermediate filament protein that is primarily found in the cardiac and skeletal muscle cells, as well as in some types of smooth muscle cells. It is an important component of the cytoskeleton, which provides structural support to the cell and helps maintain its shape. Desmin plays a crucial role in maintaining the integrity of the sarcomere, which is the basic contractile unit of the muscle fiber. Mutations in the desmin gene can lead to various forms of muscular dystrophy and other inherited muscle disorders.
Human chromosome pair 2 consists of two rod-shaped structures present in the nucleus of each cell of the human body. Each member of the pair contains thousands of genes and other genetic material, encoded in the form of DNA molecules. Chromosomes are the physical carriers of inheritance, and human cells typically contain 23 pairs of chromosomes for a total of 46 chromosomes.
Chromosome pair 2 is one of the autosomal pairs, meaning that it is not a sex chromosome (X or Y). Each member of chromosome pair 2 is approximately 247 million base pairs in length and contains an estimated 1,000-1,300 genes. These genes play crucial roles in various biological processes, including development, metabolism, and response to environmental stimuli.
Abnormalities in chromosome pair 2 can lead to genetic disorders, such as cat-eye syndrome (CES), which is characterized by iris abnormalities, anal atresia, hearing loss, and intellectual disability. This disorder arises from the presence of an extra copy of a small region on chromosome 2, resulting in partial trisomy of this region. Other genetic conditions associated with chromosome pair 2 include proximal 2q13.3 microdeletion syndrome and Potocki-Lupski syndrome (PTLS).
Vaginal neoplasms refer to abnormal growths or tumors in the vagina. These growths can be benign (non-cancerous) or malignant (cancerous). The two main types of vaginal neoplasms are:
1. Vaginal intraepithelial neoplasia (VAIN): This is a condition where the cells on the inner lining of the vagina become abnormal but have not invaded deeper tissues. VAIN can be low-grade or high-grade, depending on the severity of the cell changes.
2. Vaginal cancer: This is a malignant tumor that arises from the cells in the vagina. The two main types of vaginal cancer are squamous cell carcinoma and adenocarcinoma. Squamous cell carcinoma is the most common type, accounting for about 85% of all cases.
Risk factors for vaginal neoplasms include human papillomavirus (HPV) infection, smoking, older age, history of cervical cancer or precancerous changes, and exposure to diethylstilbestrol (DES) in utero. Treatment options depend on the type, stage, and location of the neoplasm but may include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
Combined modality therapy (CMT) is a medical treatment approach that utilizes more than one method or type of therapy simultaneously or in close succession, with the goal of enhancing the overall effectiveness of the treatment. In the context of cancer care, CMT often refers to the combination of two or more primary treatment modalities, such as surgery, radiation therapy, and systemic therapies (chemotherapy, immunotherapy, targeted therapy, etc.).
The rationale behind using combined modality therapy is that each treatment method can target cancer cells in different ways, potentially increasing the likelihood of eliminating all cancer cells and reducing the risk of recurrence. The specific combination and sequence of treatments will depend on various factors, including the type and stage of cancer, patient's overall health, and individual preferences.
For example, a common CMT approach for locally advanced rectal cancer may involve preoperative (neoadjuvant) chemoradiation therapy, followed by surgery to remove the tumor, and then postoperative (adjuvant) chemotherapy. This combined approach allows for the reduction of the tumor size before surgery, increases the likelihood of complete tumor removal, and targets any remaining microscopic cancer cells with systemic chemotherapy.
It is essential to consult with a multidisciplinary team of healthcare professionals to determine the most appropriate CMT plan for each individual patient, considering both the potential benefits and risks associated with each treatment method.
Translocation, genetic, refers to a type of chromosomal abnormality in which a segment of a chromosome is transferred from one chromosome to another, resulting in an altered genome. This can occur between two non-homologous chromosomes (non-reciprocal translocation) or between two homologous chromosomes (reciprocal translocation). Genetic translocations can lead to various clinical consequences, depending on the genes involved and the location of the translocation. Some translocations may result in no apparent effects, while others can cause developmental abnormalities, cancer, or other genetic disorders. In some cases, translocations can also increase the risk of having offspring with genetic conditions.
A Sertoli-Leydig cell tumor is a rare type of sex cord-stromal tumor that develops in the ovaries. These tumors arise from the cells that produce hormones and help to form and maintain the ovarian tissue. Sertoli-Leydig cell tumors can occur in people of any age but are most commonly found in women between the ages of 20 and 40.
These tumors can be functional, meaning they produce hormones, or nonfunctional. Functional Sertoli-Leydig cell tumors may cause symptoms related to the production of male hormones (androgens), such as excess facial hair, a deepened voice, and irregular menstrual periods. Nonfunctional tumors typically do not cause any specific symptoms and are often found during routine pelvic examinations or imaging studies performed for other reasons.
Sertoli-Leydig cell tumors are usually slow-growing and can vary in size. Most of these tumors are benign (not cancerous), but some can be malignant (cancerous) and may spread to other parts of the body. Treatment typically involves surgical removal of the tumor, and additional therapies such as chemotherapy or radiation therapy may be recommended depending on the stage and grade of the tumor. Regular follow-up care is essential to monitor for any recurrence of the tumor.
Myogenin is defined as a protein that belongs to the family of myogenic regulatory factors (MRFs). These proteins play crucial roles in the development, growth, and repair of skeletal muscle cells. Myogenin is specifically involved in the differentiation and fusion of myoblasts to form multinucleated myotubes, which are essential for the formation of mature skeletal muscle fibers. It functions as a transcription factor that binds to specific DNA sequences, thereby regulating the expression of genes required for muscle cell differentiation. Myogenin also plays a role in maintaining muscle homeostasis and may contribute to muscle regeneration following injury or disease.
MyoD protein is a member of the family of muscle regulatory factors (MRFs) that play crucial roles in the development and regulation of skeletal muscle. MyoD is a transcription factor, which means it binds to specific DNA sequences and helps control the transcription of nearby genes into messenger RNA (mRNA).
MyoD protein is encoded by the MYOD1 gene and is primarily expressed in skeletal muscle cells, where it functions as a master regulator of muscle differentiation. During myogenesis, MyoD is activated and initiates the expression of various genes involved in muscle-specific functions, such as contractile proteins and ion channels.
MyoD protein can also induce cell cycle arrest and promote the differentiation of non-muscle cells into muscle cells, a process known as transdifferentiation. This property has been explored in regenerative medicine for potential therapeutic applications.
In summary, MyoD protein is a key regulator of skeletal muscle development, differentiation, and maintenance, and it plays essential roles in the regulation of gene expression during myogenesis.
Vincristine is an antineoplastic agent, specifically a vinca alkaloid. It is derived from the Madagascar periwinkle plant (Catharanthus roseus). Vincristine binds to tubulin, a protein found in microtubules, and inhibits their polymerization, which results in disruption of mitotic spindles leading to cell cycle arrest and apoptosis (programmed cell death). It is used in the treatment of various types of cancer including leukemias, lymphomas, and solid tumors. Common side effects include peripheral neuropathy, constipation, and alopecia.
A cell line that is derived from tumor cells and has been adapted to grow in culture. These cell lines are often used in research to study the characteristics of cancer cells, including their growth patterns, genetic changes, and responses to various treatments. They can be established from many different types of tumors, such as carcinomas, sarcomas, and leukemias. Once established, these cell lines can be grown and maintained indefinitely in the laboratory, allowing researchers to conduct experiments and studies that would not be feasible using primary tumor cells. It is important to note that tumor cell lines may not always accurately represent the behavior of the original tumor, as they can undergo genetic changes during their time in culture.
Thoracic neoplasms refer to abnormal growths or tumors that develop in the thorax, which is the area of the body that includes the chest and lungs. These neoplasms can be benign (non-cancerous) or malignant (cancerous). Malignant thoracic neoplasms are often referred to as lung cancer, but they can also include other types of cancer such as mesothelioma, thymoma, and esophageal cancer.
Thoracic neoplasms can cause various symptoms depending on their location and size. Common symptoms include coughing, chest pain, shortness of breath, hoarseness, and difficulty swallowing. Treatment options for thoracic neoplasms depend on the type, stage, and location of the tumor, as well as the patient's overall health. Treatment may include surgery, radiation therapy, chemotherapy, targeted therapy, or a combination of these approaches.
'Tumor cells, cultured' refers to the process of removing cancerous cells from a tumor and growing them in controlled laboratory conditions. This is typically done by isolating the tumor cells from a patient's tissue sample, then placing them in a nutrient-rich environment that promotes their growth and multiplication.
The resulting cultured tumor cells can be used for various research purposes, including the study of cancer biology, drug development, and toxicity testing. They provide a valuable tool for researchers to better understand the behavior and characteristics of cancer cells outside of the human body, which can lead to the development of more effective cancer treatments.
It is important to note that cultured tumor cells may not always behave exactly the same way as they do in the human body, so findings from cell culture studies must be validated through further research, such as animal models or clinical trials.
Pelvic neoplasms refer to abnormal growths or tumors located in the pelvic region. These growths can be benign (non-cancerous) or malignant (cancerous). They can originate from various tissues within the pelvis, including the reproductive organs (such as ovaries, uterus, cervix, vagina, and vulva in women; and prostate, testicles, and penis in men), the urinary system (kidneys, ureters, bladder, and urethra), the gastrointestinal tract (colon, rectum, and anus), as well as the muscles, nerves, blood vessels, and other connective tissues.
Malignant pelvic neoplasms can invade surrounding tissues and spread to distant parts of the body (metastasize). The symptoms of pelvic neoplasms may vary depending on their location, size, and type but often include abdominal or pelvic pain, bloating, changes in bowel or bladder habits, unusual vaginal bleeding or discharge, and unintentional weight loss. Early detection and prompt treatment are crucial for improving the prognosis of malignant pelvic neoplasms.
Neoplastic gene expression regulation refers to the processes that control the production of proteins and other molecules from genes in neoplastic cells, or cells that are part of a tumor or cancer. In a normal cell, gene expression is tightly regulated to ensure that the right genes are turned on or off at the right time. However, in cancer cells, this regulation can be disrupted, leading to the overexpression or underexpression of certain genes.
Neoplastic gene expression regulation can be affected by a variety of factors, including genetic mutations, epigenetic changes, and signals from the tumor microenvironment. These changes can lead to the activation of oncogenes (genes that promote cancer growth and development) or the inactivation of tumor suppressor genes (genes that prevent cancer).
Understanding neoplastic gene expression regulation is important for developing new therapies for cancer, as targeting specific genes or pathways involved in this process can help to inhibit cancer growth and progression.
Dactinomycin is an antineoplastic antibiotic, which means it is used to treat cancer. It is specifically used to treat certain types of testicular cancer, Wilms' tumor (a type of kidney cancer that occurs in children), and some gestational trophoblastic tumors (a type of tumor that can develop in the uterus after pregnancy). Dactinomycin works by interfering with the DNA in cancer cells, which prevents them from dividing and growing. It is often used in combination with other chemotherapy drugs as part of a treatment regimen.
Dactinomycin is administered intravenously (through an IV) and its use is usually limited to hospitals or specialized cancer treatment centers due to the need for careful monitoring during administration. Common side effects include nausea, vomiting, and hair loss. More serious side effects can include bone marrow suppression, which can lead to an increased risk of infection, and tissue damage at the site where the drug is injected. Dactinomycin can also cause severe allergic reactions in some people.
It's important to note that dactinomycin should only be used under the supervision of a qualified healthcare professional, as its use requires careful monitoring and management of potential side effects.
A gene fusion, also known as a chromosomal translocation or fusion gene, is an abnormal genetic event where parts of two different genes combine to create a single, hybrid gene. This can occur due to various mechanisms such as chromosomal rearrangements, deletions, or inversions, leading to the formation of a chimeric gene with new and often altered functions.
Gene fusions can result in the production of abnormal fusion proteins that may contribute to cancer development and progression by promoting cell growth, inhibiting apoptosis (programmed cell death), or activating oncogenic signaling pathways. In some cases, gene fusions are specific to certain types of cancer and serve as valuable diagnostic markers and therapeutic targets for personalized medicine.
Enterovirus A, Human is a type of enterovirus that infects humans. Enteroviruses are small, single-stranded RNA viruses that belong to the Picornaviridae family. There are over 100 different types of enteroviruses, and they are divided into several species, including Enterovirus A, B, C, D, and Rhinovirus.
Enterovirus A includes several important human pathogens, such as polioviruses (which have been largely eradicated thanks to vaccination efforts), coxsackieviruses, echoviruses, and enterovirus 71. These viruses are typically transmitted through the fecal-oral route or respiratory droplets and can cause a range of illnesses, from mild symptoms like fever, rash, and sore throat to more severe diseases such as meningitis, encephalitis, myocarditis, and paralysis.
Poliovirus, which is the most well-known member of Enterovirus A, was responsible for causing poliomyelitis, a highly infectious disease that can lead to irreversible paralysis. However, due to widespread vaccination programs, wild poliovirus transmission has been eliminated in many parts of the world, and only a few countries still report cases of polio caused by vaccine-derived viruses.
Coxsackieviruses and echoviruses can cause various symptoms, including fever, rash, mouth sores, muscle aches, and respiratory illnesses. In some cases, they can also lead to more severe diseases such as meningitis or myocarditis. Enterovirus 71 is a significant pathogen that can cause hand, foot, and mouth disease, which is a common childhood illness characterized by fever, sore throat, and rash on the hands, feet, and mouth. In rare cases, enterovirus 71 can also lead to severe neurological complications such as encephalitis and polio-like paralysis.
Prevention measures for enterovirus A infections include good hygiene practices, such as washing hands frequently, avoiding close contact with sick individuals, and practicing safe food handling. Vaccination is available for poliovirus and can help prevent the spread of vaccine-derived viruses. No vaccines are currently available for other enterovirus A infections, but research is ongoing to develop effective vaccines against these viruses.
Pulmonary blastoma is a rare and aggressive type of lung cancer that primarily affects adults, but it can also occur in children. It's characterized by the rapid growth of primitive, undifferentiated cells that form tumors in the lungs. There are two main types of pulmonary blastomas:
1. Pleuropulmonary blastoma (PPB): This type is more common in children and adolescents. PPB can be further divided into three subtypes based on the age at diagnosis and the extent of tumor spread: Type I, Type II, and Type III. Types II and III are more aggressive and have a higher risk of metastasis compared to Type I.
2. Lung sarcomatoid carcinoma with pulmonary blastomatous components (LSC-PBC): This type is primarily found in adults and is considered a variant of lung sarcomatoid carcinoma, which is an aggressive subtype of non-small cell lung cancer. LSC-PBC contains both epithelial and mesenchymal elements, with the latter showing blastomatous features.
Both types of pulmonary blastomas have a poor prognosis due to their rapid growth and high likelihood of metastasis. Treatment typically involves surgical resection, chemotherapy, and radiation therapy. However, given the rarity of this condition, treatment options may vary depending on individual cases and access to specialized care.
Nephroblastoma overexpressed protein, also known as NOV or CCN3, is a member of the CCN family of proteins that are involved in cell growth, differentiation, and migration. It was originally identified as being highly expressed in nephroblastoma (also known as Wilms' tumor), a type of kidney cancer that typically affects children. NOV has been found to play a role in various biological processes, including angiogenesis, cell adhesion, and apoptosis. It can act as both a positive and negative regulator of cell growth and differentiation, depending on the context. Abnormal expression of NOV has been implicated in several types of cancer, including nephroblastoma, breast cancer, and prostate cancer.
 Embryonal rhabdomyosarcoma
Embryonal rhabdomyosarcoma
Sarcoma botryoides
Rhabdomyoblast
Santosh G. Honavar
PAX7
Enterovirus 71
Enterovirus
RMST (gene)
Rhabdomyosarcoma
Spindle cell rhabdomyosarcoma
TSPAN31
Alveolar rhabdomyosarcoma
Fibroblast growth factor receptor 1
Splenogonadal fusion
P53
Margaret Ransone Murray
Malignant ectomesenchymoma
Mir-1 microRNA precursor family
Cancer epigenetics
PAX3
Vulvar tumors
List of MeSH codes (C04)
List of diseases (R)
Small-blue-round-cell tumor
Beckwith-Wiedemann syndrome
Hedgehog pathway inhibitor
ERMS
International Classification of Diseases for Oncology
Index of oncology articles
Sarcoma
Intergroup Rhabdomyosarcoma Study Group
Embryonal rhabdomyosarcoma - Wikipedia
 EHMT2 epigenetically suppresses Wnt signaling and is a potential target in embryonal rhabdomyosarcoma | eLife
EHMT2 epigenetically suppresses Wnt signaling and is a potential target in embryonal rhabdomyosarcoma | eLife
 MicroRNA-101 is repressed by EZH2 and its restoration inhibits tumorigenic features in embryonal rhabdomyosarcoma
MicroRNA-101 is repressed by EZH2 and its restoration inhibits tumorigenic features in embryonal rhabdomyosarcoma
 Embryonal rhabdomyosarcoma - Libre Pathology
Embryonal rhabdomyosarcoma - Libre Pathology
 Embryonal Rhabdomyosarcoma of the Conjunctiva: A Clinicopathologic and Immunohistochemical Study - Fingerprint
- Experts...
Embryonal Rhabdomyosarcoma of the Conjunctiva: A Clinicopathologic and Immunohistochemical Study - Fingerprint
- Experts...
 Embryonal rhabdomyosarcoma (Concept Id: C0206656)
- MedGen - NCBI
Embryonal rhabdomyosarcoma (Concept Id: C0206656)
- MedGen - NCBI
 Vaginal Cancer: Overview, Risk Factors, Pathogenesis
Vaginal Cancer: Overview, Risk Factors, Pathogenesis
Orbit embryonal rhabdomyosarcoma (Concept Id: C1335127)
- MedGen - NCBI
Focally anaplastic embryonal rhabdomyosarcoma
 Leptomeningeal Relapse Of Embryonal Rhabdomyosarcoma After 15 Years - Irish Medical Journal
Leptomeningeal Relapse Of Embryonal Rhabdomyosarcoma After 15 Years - Irish Medical Journal
 Hedgehog pathway activity in pediatric embryonal rhabdomyosarcoma and undifferentiated sarcoma:a report from the Children's...
Hedgehog pathway activity in pediatric embryonal rhabdomyosarcoma and undifferentiated sarcoma:a report from the Children's...
The Hippo Transducer YAP1 Transforms Activated Satellite Cells and Is a Potent Effector of Embryonal Rhabdomyosarcoma Formation...
 Rhabdomyosarcoma: MedlinePlus Medical Encyclopedia
Rhabdomyosarcoma: MedlinePlus Medical Encyclopedia
 Is Rhabdomyosarcoma Soft Tissue Sarcoma? 7 Symptoms, 4 Stages
Is Rhabdomyosarcoma Soft Tissue Sarcoma? 7 Symptoms, 4 Stages
 The NOTCH1/SNAIL1/MEF2C Pathway Regulates Growth and Self-Renewal in Embryonal Rhabdomyosarcoma.
The NOTCH1/SNAIL1/MEF2C Pathway Regulates Growth and Self-Renewal in Embryonal Rhabdomyosarcoma.
Clinicopathologic along with survival fits of embryonal rhabdomyosarcoma driven simply by RAS/RAF mutations. | Thrombin...
 articles • APPLIED RADIOLOGY
articles • APPLIED RADIOLOGY
 Vol. 21 No. 2 (2015)
| Bangladesh Journal of Otorhinolaryngology
Vol. 21 No. 2 (2015)
| Bangladesh Journal of Otorhinolaryngology
 J. Javier Gorgoso-Varela - Search Results - PubMed
J. Javier Gorgoso-Varela - Search Results - PubMed
 The botanical drug PBI-05204, a supercritical CO2 extract of Nerium oleander, sensitizes alveolar and embryonal...
The botanical drug PBI-05204, a supercritical CO2 extract of Nerium oleander, sensitizes alveolar and embryonal...
 Rhabdomyosarcoma : Wheeless' Textbook of Orthopaedics
Rhabdomyosarcoma : Wheeless' Textbook of Orthopaedics
Table A1 - Molecular Epidemiology of Eastern Equine Encephalitis Virus, New York - Volume 14, Number 3-March 2008 - Emerging...
 Pediatric Rhabdomyosarcoma - Conditions and Treatments | Children's National Hospital
Pediatric Rhabdomyosarcoma - Conditions and Treatments | Children's National Hospital
 Block 7 Reproduction Creighton Univ W Xpl Pt 4 - ProProfs Quiz
Block 7 Reproduction Creighton Univ W Xpl Pt 4 - ProProfs Quiz
 Abhinav Adhikari, Ph.D. | Harvard Catalyst Profiles | Harvard Catalyst
Abhinav Adhikari, Ph.D. | Harvard Catalyst Profiles | Harvard Catalyst
 United States Cancer Statistics: Public Information Data 1999 - 2002 Archive
United States Cancer Statistics: Public Information Data 1999 - 2002 Archive
 Rhabdomyosarcoma
Rhabdomyosarcoma
 miRNA Expression Profiling for Well-Differentiated and De-Differentiated Liposarcoma
miRNA Expression Profiling for Well-Differentiated and De-Differentiated Liposarcoma
 Diagnostic Immunohistochemistry - Vulva and Vagina Tumor
Diagnostic Immunohistochemistry - Vulva and Vagina Tumor
 ALSF Childhood Cancer Research Grants | Alex's Lemonade Stand Foundation for Childhood Cancer
ALSF Childhood Cancer Research Grants | Alex's Lemonade Stand Foundation for Childhood Cancer
ERMS10
- ERMS accounts for 60% to 70% of rhabdomyosarcoma, the other being alveolar rhabdomyosarcoma (ARMS), also known as PAX-fusion positive or fusion-positive rhabdomyosarcoma. (wikipedia.org)
- These two subtypes of rhabdomyosarcoma, ERMS and ARMS, also are caused by different genetic mutation pathways. (wikipedia.org)
- When examining embryonal rhabdomyosarcoma tumors vs. alveolar rhabdomyosarcoma tumors, a 2013 study had discovered that there were more rates of mutation in ERMS tumors. (wikipedia.org)
- Wnt signaling is downregulated in embryonal rhabdomyosarcoma (ERMS) and contributes to the block of differentiation. (elifesciences.org)
- Embryonal rhabdomyosarcoma (ERMS) accounts for the majority (~60%) of all RMS cases. (elifesciences.org)
- We previously reported that EZH2 inhibition induces cell cycle arrest followed by myogenic differentiation of RMS cells of the embryonal subtype (eRMS). (figshare.com)
- Here we report that YAP1 activity is elevated in human embryonal rhabdomyosarcoma (ERMS). (elsevierpure.com)
- However, mechanisms regulating TPC self-renewal are largely unknown, especially in embryonal rhabdomyosarcoma (ERMS)-a common pediatric cancer of muscle. (duke.edu)
- Embryonal rhabdomyosarcoma (ERMS) is the most common type. (childrensoncologygroup.org)
- Her research allowed for developing an easy PCR-based assay to phenotype embryonal (ERMS) and alveolar (ARMS) rhabdomyosarcoma based on the methylation of imprinted regions within DLK1-MEG3 locus. (louisville.edu)
Tumors14
- Embryonal rhabdomyosarcoma is also known as PAX-fusion negative or fusion-negative rhabdomyosarcoma, as tumors of this subtype are unified by their lack of a PAX3-FOXO1 fusion oncogene (or other PAX fusions seen in alveolar rhabdomyosarcoma). (wikipedia.org)
- These types of tumors are classified as embryonal rhabdomyosarcoma "because of their remarkable resemblance to developing embryonic and fetal skeletal muscle. (wikipedia.org)
- The study had use whole genome sequencing to sequence the DNA from 16 RMS tumors and found that RAS pathway mutations tend to be more associated with intermediate and high-risk embryonal Rhabdomyosarcoma. (wikipedia.org)
- The spectrum of tumors found in Gorlin Syndrome includes basal cell carcinoma, medulloblastoma, and rarely, rhabdomyosarcoma (RMS). (uea.ac.uk)
- Rhabdomyosarcoma is a type of sarcoma made up of tumors that arise from muscle tissue and spread throughout the body. (medicinenet.com)
- Children with genitourinary tract cancers may manifest with a painless scrotal lump (paratesticular tumors), a projecting grape-like mass in the vagina ("botryoid" rhabdomyosarcoma), blood in the urine (bladder tumors), or frequent urination , often with a burning sensation or hesitation. (medicinenet.com)
- Rhabdomyosarcoma belongs to a group of tumors known as soft-tissue sarcomas and is the most common cancer in this group. (merckmanuals.com)
- The p53 gene was examined in primary or metastatic tumors from six patients with rhabdomyosarcoma (RMS) and in five RMS cell lines by screening methods including single-strand conformation polymorphism analysis, the RNase protection assay, sequencing of complementary DNA subclones, and Southern blotting. (nih.gov)
- Six original tumors were of embryonal histology, four alveolar, and one mixed. (nih.gov)
- p53 mutations were identified in four of the six tumors or cell lines derived from tumors with embryonal histology and in one of the four with alveolar histology. (nih.gov)
- Rhabdomyosarcoma is a member of the group of "small round blue cell" tumors, and must be distinguished from morphologically similar pediatric tumors. (umich.edu)
- Testing can aid in distinguishing alveolar rhabdomyosarcoma from other similar tumors. (umich.edu)
- Approximately 20% of tumors diagnosed as alveolar rhabdomyosarcoma based on histologic grounds have been found to be negative for a PAX/FOXO1 translocation or fusion transcript. (umich.edu)
- These guidelines do not pertain to the management of gastrointestinal stroma tumors (GISTs), rhabdomyosarcoma, Ewing sarcoma, desmoplastic round cell tumors, and primitive neuroectodermal tumors. (medscape.com)
Skeletal muscle6
- Embryonal rhabdomyosarcoma (EMRS) is a rare histological form of cancer in the connective tissue wherein the mesenchymally-derived malignant cells resemble the primitive developing skeletal muscle of the embryo. (wikipedia.org)
- Abstract Background Rhabdomyosarcoma (RMS) is a pediatric soft tissue sarcoma arising from myogenic precursors that have lost their capability to differentiate into skeletal muscle. (figshare.com)
- Rhabdomyosarcoma is a rare type of cancer that starts in the cells that develop into skeletal muscle cells. (childrensnational.org)
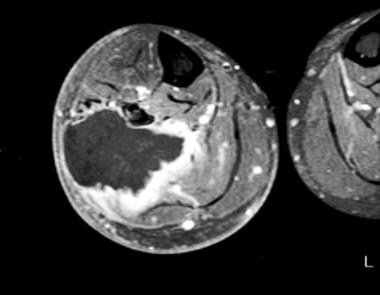
- Rhabdomyosarcoma (RMS) is a malignant tumor with skeletal muscle cell morphology. (radiopaedia.org)
- Rhabdomyosarcomas are thought not to arise from skeletal muscle, but rather to differentiate into a tumor that resembles skeletal muscle 7 . (radiopaedia.org)
- Rhabdomyosarcoma is a cancer arising from embryonal mesenchymal cells that have potential to differentiate into skeletal muscle cells. (merckmanuals.com)
Sarcoma2
- Is Rhabdomyosarcoma Soft Tissue Sarcoma? (medicinenet.com)
- Treatment of rhabdomyosarcoma (RMS), the most common a soft tissue sarcoma in childhood, provides intensive multimodal therapy, with radiotherapy (RT) playing a critical role for local tumor control. (saludintegral.hn)
Self-Renewal in Embryonal Rhabdomyosarcoma1
- Scholars@Duke publication: The NOTCH1/SNAIL1/MEF2C Pathway Regulates Growth and Self-Renewal in Embryonal Rhabdomyosarcoma. (duke.edu)
Cause of rhabdomyosarcoma is unknown1
- The cause of rhabdomyosarcoma is unknown. (medlineplus.gov)
Botryoid2
- Embryonal rhabdomyosarcoma can be further divided into three subcategories: the botryoid, spindle cell, and not-otherwise-specified (NOS). (wikipedia.org)
- The Horn-Enterline classification uses morphologic characteristics to divide rhabdomyosarcoma into the embryonal, alveolar, botryoid, and pleomorphic subtypes. (wikipedia.org)
Exact cause of rhabdomyosarcoma1
- The exact cause of rhabdomyosarcoma is not known. (childrensnational.org)
Alveolar rhabdomyosarcomas3
- The paternally imprinted DLK1-GTL2 locus is differentially methylated in embryonal and alveolar rhabdomyosarcomas. (louisville.edu)
- The t(2;13)(q35;q14) translocation joins the PAX3 and FOXO1 (FKHR) genes in approximately 60% of alveolar rhabdomyosarcomas, while the less common t(1;13)(p36;q14) joins PAX7 with FOXO1 in approximately 20% of cases. (umich.edu)
- This assay is expected to be positive in approximately 80% of all alveolar rhabdomyosarcomas. (umich.edu)
Morphologic1
- A poorly circumscribed morphologic variant of rhabdomyosarcoma. (nih.gov)
Patients with rhabdomyosarcoma1
- Five additional Costello syndrome patients with rhabdomyosarcoma: proposal for a tumor screening protocol. (atlasgeneticsoncology.org)
Head and neck5
- Pediatric head and neck rhabdomyosarcoma: An analysis of treatment and survival in the United States (1975-2016). (nih.gov)
- Clinicopathological analysis of head and neck rhabdomyosarcoma: A series of 10 cases and literature review. (nih.gov)
- Rhabdomyosarcoma of the head and neck in children: review and update. (nih.gov)
- Long term survival in rhabdomyosarcoma of head and neck. (minervamedica.it)
- Embryonal occurs in the head and neck, or the genital or urinary organs. (curesarcoma.org)
Spindle cell2
- As a result, the World Health Organization currently takes into account both molecular genetics and morphology to classify rhabdomyosarcoma into the embryonal, alveolar, spindle cell/sclerosing, and pleomorphic subtypes. (wikipedia.org)
- Histologically, embryonal rhabdomyosarcoma commonly presents as alternating loose and dense patches of cells, including round cell and spindle cell components. (wikipedia.org)
Occur3
- Cancers that occur most frequently in affected individuals include a cancer of muscle tissue called rhabdomyosarcoma, a form of kidney cancer known as Wilms tumor, and a cancer of the blood-forming tissue known as leukemia. (nih.gov)
- Rhabdomyosarcoma can occur in many places in the body. (medlineplus.gov)
- Two recurrent and distinctive chromosomal translocations occur in alveolar rhabdomyosarcoma. (umich.edu)
Pediatric Rhabdomyosarcoma1
- Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: An analysis of 2,600 patients. (merckmanuals.com)
Survival2
- Clinicopathologic along with survival fits of embryonal rhabdomyosarcoma driven simply by RAS/RAF mutations. (thrombininhibitors.com)
- Dr. Schneider's work led to the identification of changes in genomic imprinting of DLK1-MEG3 locus in rhabdomyosarcoma and acute myeloid leukemia (AML) and pointed to their role in diagnosis and patient survival prediction. (louisville.edu)
Diagnosis2
- A Review of the Role of Cytogenetics in the Diagnosis of Orbital Rhabdomyosarcoma. (nih.gov)
- Up to 20% of patients with rhabdomyosarcomas have metastases at the time of diagnosis 7 . (radiopaedia.org)
Symptoms4
- Call your provider if your child has symptoms of rhabdomyosarcoma. (medlineplus.gov)
- The symptoms of rhabdomyosarcoma (RMS) might differ greatly depending on where the tumor develops. (medicinenet.com)
- What are the symptoms of rhabdomyosarcoma in children? (childrensnational.org)
- The symptoms of rhabdomyosarcoma are a lot like those of other, more common, health conditions. (childrensnational.org)
Mesenchymal1
- Rhabdomyosarcoma (RMS) is a soft tissue tumor derived from mesenchymal tissue with myogenic differentiation and associated with the embryogenesis of striated muscle. (biomedcentral.com)
ARMS1
- Alveolar rhabdomyosarcoma (ARMS) makes up about 25-40% of RMS. (childrensoncologygroup.org)
Malignant1
- Rhabdomyosarcoma is a cancerous (malignant) tumor of the muscles that are attached to the bones. (medlineplus.gov)
Wilms1
- Wilms Tumor Wilms tumor is an embryonal cancer of the kidney composed of blastemal, stromal, and epithelial elements. (merckmanuals.com)
Childhood2
- Childhood rhabdomyosarcoma treatment (PDQ) health professional version. (medlineplus.gov)
- Rhabdomyosarcomas are the most common soft tissue tumor in children and account for 5-8% of childhood cancers 6,7 , and 19% of all pediatric soft tissue sarcomas 7 . (radiopaedia.org)
Extremities1
- When present in the extremities in children, embryonal rhabdomyosarcomas may cause bowing of the adjacent long bones. (radiopaedia.org)
Epidemiology1
- 3. Linardic CM, Wexler LH: Rhabdomyosarcoma: Epidemiology and Genetic Susceptibilty. (merckmanuals.com)
Congenital1
- A case of congenital rhabdomyosarcoma. (nih.gov)
Necrosis1
- Embryonal rhabdomyosarcomas tend to be more homogeneous, whereas alveolar and pleomorphic rhabdomyosarcomas frequently have areas of necrosis 6 . (radiopaedia.org)
Embryonic1
- Rhabdomyosarcoma, mostly of the embryonic subtype is the tumor the most frequently encountered in CS. (atlasgeneticsoncology.org)
Chemotherapy2
- Many different chemotherapy drugs are active against rhabdomyosarcoma. (medlineplus.gov)
- Intergroup Rhabdomyosarcoma Study-4 risk stratification was used, with treatment based on a multimodality-regimen with chemotherapy (Vincristine/Ifosfamide/Etoposide and Vincristine/Actinomycin-D/Cyclophosphamide) and appropriate local therapy. (ecancer.org)
Sarcomas1
- Lymph node involvement is not characteristic of most sarcomas except rhabdomyosarcoma and synovial, clear cell, vascular, and epithelioid sarcomas. (medscape.com)
Children9
- Embryonal rhabdomyosarcoma is the most common in young children but there has been report of a second age peak in adolescence years. (wikipedia.org)
- Most children with rhabdomyosarcoma do not have any known risk factors. (medlineplus.gov)
- With intensive treatment, most children with rhabdomyosarcoma are able to survive long-term. (medlineplus.gov)
- What is rhabdomyosarcoma in children? (childrensnational.org)
- Which children are at risk for rhabdomyosarcoma? (childrensnational.org)
- How is rhabdomyosarcoma in children diagnosed? (childrensnational.org)
- Two thirds of rhabdomyosarcomas are diagnosed in children 7 years of age. (merckmanuals.com)
- Lizzy was diagnosed with Stage Four Embryonal rhabdomyosarcoma, a rare form of cancer found in children. (kxxv.com)
- children born with high birth weights or larger than expected when born have increased risk of embryonal rhabdomyosarcoma. (curesarcoma.org)
Common3
- Embryonal rhabdomyosarcoma is the more common of the two major sub-types of rhabdomyosarcoma. (wikipedia.org)
- It is the most common type of rhabdomyosarcoma. (medicinenet.com)
- Embryonal is the most common type. (childrensnational.org)
Mutations2
- Embryonal rhabdomyosarcoma results from copy number alterations as well as mutations in the RAS pathway. (wikipedia.org)
- Preclinical studies have demonstrated that resistance to FGFR inhibitors can be acquired through mutations in the FGFR gatekeeper residue, as clinically observed for FGFR4 in embryonal rhabdomyosarcoma and neuroendocrine breast carcinomas. (rcsb.org)
Radiation1
- Although the specific risk factors for rhabdomyosarcoma are unknown, there is an increased risk of rhabdomyosarcoma due to radiation exposure or the use of certain recreational drugs by the mother during pregnancy and certain genetic conditions. (medicinenet.com)
Type2
- Treatment depends on the site and type of rhabdomyosarcoma. (medlineplus.gov)
- Rhabdomyosarcoma is a type of cancer. (childrensnational.org)
Variants1
- P3F and P7F were detected in 25/49 (51%) and 14/85 (16.5%) of alveolar and embryonal variants, respectively. (ecancer.org)
Disease1
- Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease. (nih.gov)