Neuroectodermal Tumors, Primitive
Neuroectodermal Tumors
Neuroectodermal Tumors, Primitive, Peripheral
Neuroectodermal Tumor, Melanotic
Sarcoma, Ewing
Medulloblastoma
Cerebellar Neoplasms
Supratentorial Neoplasms
Brain Neoplasms
Proto-Oncogene Protein c-fli-1
RNA-Binding Protein EWS
Pinealoma
Chromosomes, Human, Pair 22
Ependymoma
Spinal Cord Neoplasms
Sarcoma, Small Cell
Neuroblastoma
Rhabdoid Tumor
Tumor Markers, Biological
Neoplasms, Germ Cell and Embryonal
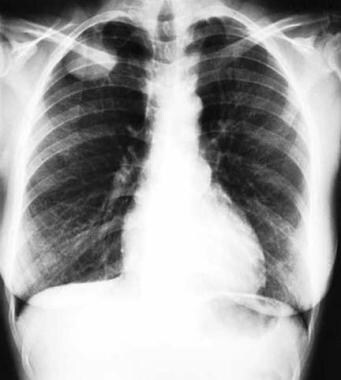
Thoracic Neoplasms
Central Nervous System Neoplasms
Neural Plate
Chromosomes, Human, Pair 11
Oncogene Proteins, Fusion
Astrocytoma
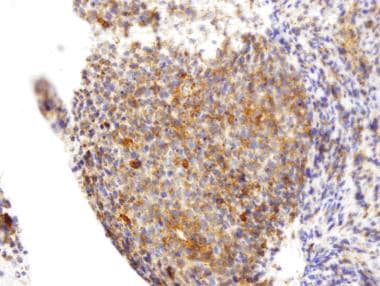
Immunohistochemistry
Ifosfamide
Fatal Outcome
Tomography, X-Ray Computed
Translocation, Genetic
Ganglioneuroma
Paraparesis
Thoracic Wall
Teratoma
Neoplasms, Nerve Tissue
Peripheral Nervous System Neoplasms
Tumor Burden
Tumor Necrosis Factor-alpha
Etoposide
Pineal Gland
Antigens, Neoplasm
Combined Modality Therapy
Chromosomes, Human, Pair 17
Rhabdomyosarcoma
Neurofilament Proteins
Glioma
Abdominal Neoplasms
Magnetic Resonance Imaging
Retinoblastoma
Tumor Cells, Cultured
Thoracic Surgical Procedures
Cranial Irradiation
Cyclophosphamide
Gangliosides
Phosphopyruvate Hydratase
Soft Tissue Neoplasms
Synaptophysin
Genes, myc
In Situ Hybridization, Fluorescence
Antineoplastic Combined Chemotherapy Protocols
Reverse Transcriptase Polymerase Chain Reaction
Treatment Outcome
Thiotepa
Neoplasm Proteins
Cell Transformation, Neoplastic
Cell Division
Transcription Factors
Neoplasm Recurrence, Local
Polymerase Chain Reaction
RNA, Messenger
Wilms Tumor
Cell Differentiation
Immunoenzyme Techniques
Gene Amplification
Gene Expression Regulation, Neoplastic
Neoplasms, Experimental
Genes, Tumor Suppressor
Molecular Sequence Data
Base Sequence
Biopsy, Needle
Disease Progression
Doxorubicin
Carcinoid Tumor
Gene Rearrangement
Survival Analysis
Neoplasms
Biopsy
DNA-Binding Proteins
Prognosis
Tumor Suppressor Protein p53
Dactinomycin
Disease-Free Survival
Survival Rate
Glial Fibrillary Acidic Protein
Neuroendocrine Tumors
Chromosome Aberrations
Tumor Suppressor Proteins
Proto-Oncogene Proteins
Tumor Microenvironment
Blotting, Western
Neoplasm Metastasis
Phenotype
Adults with Ewing's sarcoma/primitive neuroectodermal tumor: adverse effect of older age and primary extraosseous disease on outcome. (1/57)
OBJECTIVE: To assess outcome and prognostic factors for survival of adults with Ewing's sarcoma/primitive neuroectodermal tumor (PNET). BACKGROUND: Ewing's sarcoma/PNET is a disease of childhood rarely seen in adults. Accordingly, there is a relative paucity of published literature pertaining to outcome for adults with this disease. METHODS: Between 1979 and 1996, 37 patients with newly diagnosed Ewing's sarcoma/PNET were evaluated and treated at the Adult Sarcoma Program at Dana-Farber Cancer Institute and Brigham & Women's Hospital. Twenty-six patients had localized disease at presentation and 11 had metastatic disease. All but two patients received multiagent chemotherapy. Local treatment consisted of surgery (7 patients), surgery and radiation therapy (19), radiation therapy (6), or no local treatment (5). Median follow-up for living patients was 100 months (range 8 to 199). RESULTS: The 5-year survival rate for the group overall was 37%+/-9%. The 5-year local control rate was 85%+/-7%. Significant favorable predictors for survival on univariate analysis included localized disease at presentation, primary origin in bone, primary size <8 cm, and a favorable objective response to chemotherapy. Patients with localized disease had a 5-year survival rate of 49%+/-11% compared with 0% for those with metastatic disease at presentation. Multivariate analysis showed three significant independent predictors for death: metastatic disease at presentation, primary origin in extraosseous tissue versus bone, and age 26 years or older. CONCLUSION: Adult patients with Ewing's sarcoma/PNET at highest risk for death are those who are older than 26 years and have metastatic disease or an extraosseous primary tumor. The development of novel therapies should target these high-risk groups. (+info)Chromosomal imbalances and DNA amplifications in SV40 large T antigen-induced primitive neuroectodermal tumor cell lines of the rat. (2/57)
Comparative genomic in situ hybridization analysis of four cell lines derived from SV40 large T antigen-induced primitive neuroectodermal tumors of the rat revealed non-recurrent chromosomal copy number changes and DNA amplifications at chromosomal bands 2q34, 4q43qter and 15q12qter in cell lines TZ102, TZ103 and TZ107, respectively. Semi-quantitative PCR and western blot analysis demonstrated amplification and over-expression of the rat N-ras proto-oncogene in TZ102. Furthermore, all cell lines displayed aneuploid cell populations and variable chromosome numbers as assessed by flow cytometry and cytogenetics. These findings suggest that DNA amplification as well as genomic instability may contribute to the pathogenesis of SV40 large T antigen-induced primitive neuroectodermal tumors of the rat. (+info)Stem cell factor is not essential for cell survival and proliferation of soft tissue sarcoma of neuroectodermal origin. (3/57)
BACKGROUND AND OBJECTIVE: Stem cell factor (SCF), and its receptor (c-kit) play key roles in the expansion and differentiation of hematopoietic progenitor cells, in melanoblasts and primordial germ cells, making it possible that SCF and c-kit are involved in neoplastic processes deriving from these cells. C-kit has been described to be expressed at different levels in neuroblastoma and in soft tissue sarcoma of neuroectodermal origin, and seems to be required for survival processes. In this study we investigate how c-kit expression is regulated and whether a SCF autocrine loop is essential for survival of sarcoma cell lines. DESIGN AND METHODS: C-kit modulation and internalization was evaluated incubating cells with rhSCF. Cell differentiation and proliferation experiments were performed to test whether c-kit expression is related to cell cycle progression or to differentiation processes. Cell cultures were treated with neutralizing antibody and antisense oligonucleotides in order to assess the possible significance of the SCF autocrine loop. RESULTS: In vitro SCF stimulation induces c-kit down-regulation; this phenomenon could be connected with receptor internalization, and new protein synthesis is necessary for its re-expression. The cell proliferation arrest in G0/G1 does not modify c-kit expression while down-regulation of c-kit was demonstrated after cells had been treated with differentiating agents. SCF neutralization does not influence either the S phase or apoptosis in sarcoma cell lines. INTERPRETATION AND CONCLUSIONS: In sarcoma cell lines, c-kit is regulated by differentiation processes; moreover our results suggest that c-kit activity, but probably not the SCF autocrine loop, is essential for survival of these cell lines. (+info)Synovial sarcoma, histologically mimicking primitive neuroectodermal tumor/Ewing's sarcoma at distant sites. (4/57)
We report a case of synovial sarcoma (SS) showing unusual histology at distant sites. A 47-year-old man was aware of a tumor on the sole of his left foot. After preoperative chemotherapy with a diagnosis of SS, wide excision was performed. During postoperative chemotherapy, multiple tumorous lesions developed in the bone (including the whole spine) and both lungs. The patient died 1 year later. Histologically, the excised tumor of the foot showed a biphasic cellular pattern typical of SS, whereas at autopsy the bone and lung lesions were composed only of undifferentiated small round cells with cytoplasmic fibrillar processes. Homer-Wright rosettes were also observed. Immunohistochemically, 80% of the bone and lung tumor cells expressed MIC2 protein homogeneously. To clarify whether the bone and lung round cell tumors were metastatic lesions or second malignancies, especially primary primitive neuroectodermal tumor (PNET)/Ewing's sarcoma (ES), we performed reverse transcription-polymerase chain reaction (RT-PCR) analysis of tumor type-specific fusion gene transcripts. The SYT/SSX fusion transcript was identified in both the foot and lung lesions, whereas the EWS/FLI1 transcript was not detected in either lesion. Therefore, we concluded that the multiple bone and lung tumors were poorly differentiated metastatic tumors, which arose from the SS of the foot. We also conclude that the identification of chimeric fusion transcripts can be successfully applied to poorly differentiated sarcomas and will help in the differential diagnosis of tumors that cannot be distinguished by conventional morphological examinations. Also, it should be remembered that cytoplasmic staining for MIC2 protein may occur in sarcomas other than PNET/ES. (+info)Targeted oncogenesis reveals a distinct tissue-specific utilization of alternative promoters of the human mineralocorticoid receptor gene in transgenic mice. (5/57)
The human mineralocorticoid receptor (hMR) is a nuclear receptor mediating aldosterone action, whose expression is driven by two alternative promoters, P1 and P2, flanking the two first 5'-untranslated exons. In vivo characterization of hMR regulatory regions was performed by targeted oncogenesis in mice using P1 or P2 directing expression of the large T antigen of SV40 (TAg). While transgenic P1.TAg founders rapidly developed lethal hibernomas from brown fat, cerebral primitive neuroectodermal tumors and facial leiomyosarcomas occurred in P2.TAg mice. Quantitative analyses of mouse MR (mMR) and transgene expression indicate that P1 promoter was transcriptionally active in all MR-expressing tissues, directing strong TAg expression in testis and salivary glands, moderate in lung, brain, uterus, liver, and heart but, unlike mMR, rather low in colon and kidney. Importantly, the renal transgene expression colocalized with mMR in the distal nephron. In contrast, P2 promoter was approximately 10 times less potent than P1, with no activity in the brain and colon. Several immortalized cell lines were established from both neoplastic and normal tissues of transgenic mice. These cells exhibited differentiated characteristics and maintained MR expression, thus providing useful models for further studies exploring the widespread expression and functions of MR. Our results demonstrate that hMR gene expression in vivo is controlled by complex regulatory mechanisms involving distinct tissue-specific utilization of alternative promoters. (+info)A splice variant of the neuron-restrictive silencer factor repressor is expressed in small cell lung cancer: a potential role in derepression of neuroendocrine genes and a useful clinical marker. (6/57)
The neuron-restrictive silencer factor [NRSF (RE-1 silencing transcription factor/X box repressor)] is a transcriptional silencer, which we have previously implicated in deregulation of the vasopressin promoter in small cell lung cancer (SCLC). Here we describe a novel splice variant of the NRSF transcript, which is highly expressed in SCLCs. The variant was detected in both established cell lines and primary SCLC cultures as well as in some primitive neuroectodermal tumor biopsies. It was present at very low levels in human brain tissue, non-SCLC tumors, and normal bronchial epithelium. This human splice variant, which is massively overexpressed in SCLCs, incorporates a 50-bp insert between exons 5 and 6, introducing a stop codon and predicting translation of a truncated NRSF isoform. We propose that the encoded isoform may antagonize repression of the vasopressin promoter and other "neuronal" genes with neuron-restrictive silencer elements in SCLCs. Thus, up-regulated expression of this NRSF isoform may be a key early factor in defining the neuroendocrine phenotype of these tumors. The NRSF splice variant represents a specific clinical marker that could prove useful in detection of the majority of SCLCs. (+info)Aloe-emodin is a new type of anticancer agent with selective activity against neuroectodermal tumors. (7/57)
Here we report that aloe-emodin (AE), a hydroxyanthraquinone present in Aloe vera leaves, has a specific in vitro and in vivo antineuroectodermal tumor activity. The growth of human neuroectodermal tumors is inhibited in mice with severe combined immunodeficiency without any appreciable toxic effects on the animals. The compound does not inhibit the proliferation of normal fibroblasts nor that of hemopoietic progenitor cells. The cytotoxicity mechanism consists of the induction of apoptosis, whereas the selectivity against neuroectodermal tumor cells is founded on a specific energy-dependent pathway of drug incorporation. Taking into account its unique cytotoxicity profile and mode of action, AE might represent a conceptually new lead antitumor drug. (+info)Presence of new alternative exons in human and mouse Fli-1 genes. (8/57)
The mouse Fli-1 proto-oncogene is activated by proviral integration of four murine leukemia retroviruses and its human counterpart is translocated (11,22) in Ewing tumors. We have identified two alternative exons 1 by RACE analysis from a human neuroectodermal tumor. Exons 1a and 1b are located respectively 1.3 and 2.5 kb upstream from the published exon 1. Translation of these alternative messengers is predicted to generate very similar proteins. The sequence upstream from exon 1b showed functional promoter activity. Exon 1b was not conserved in the mouse but was detected in every analyzed human cell, whereas exon 1a was present only in a subset of them and also in various mouse cell lines. These results suggest that both mouse and human Fli-1 gene expression might be under the control of several independent promoter regions. (+info)Neuroectodermal tumors, primitive (PNETs) are a group of highly malignant and aggressive neoplasms that arise from neuroectodermal cells, which are the precursors to the nervous system during embryonic development. These tumors can occur anywhere in the body but are most commonly found in the central nervous system, particularly in the brain and spinal cord.
PNETs are characterized by small, round, blue cells that have a high degree of cellularity and mitotic activity. They are composed of undifferentiated or poorly differentiated cells that can differentiate along various neural lineages, including neuronal, glial, and epithelial. This feature makes their diagnosis challenging, as they can resemble other small round blue cell tumors, such as lymphomas, rhabdomyosarcomas, and Ewing sarcoma.
Immunohistochemical staining and molecular genetic testing are often required to confirm the diagnosis of PNETs. These tests typically reveal the expression of neural markers, such as NSE, Synaptophysin, and CD99, and the presence of specific chromosomal abnormalities, such as the EWS-FLI1 fusion gene in Ewing sarcoma.
PNETs are aggressive tumors with a poor prognosis, and their treatment typically involves a multimodal approach that includes surgery, radiation therapy, and chemotherapy. Despite these treatments, the five-year survival rate for patients with PNETs is less than 30%.
Neuroectodermal tumors (NETs) are a diverse group of neoplasms that arise from the embryonic cells of the neural crest, which is a part of the ectoderm that gives rise to various tissues such as peripheral nerves, nerve sheath, adrenal medulla, and melanocytes. These tumors can occur in both children and adults, and they can be benign or malignant.
The term "neuroectodermal tumor" encompasses a wide range of tumors, including:
1. Neuroblastoma: This is the most common extracranial solid tumor in children, which arises from the sympathetic nervous system. It typically affects children under the age of 5 and can occur anywhere along the sympathetic chain, but it most commonly occurs in the abdomen.
2. Ganglioneuroblastoma: This is a rare tumor that arises from the same cells as neuroblastoma, but it tends to have a more favorable prognosis. It can occur at any age, but it is most common in children under 10 years old.
3. Pheochromocytoma and Paraganglioma: These are rare tumors that arise from the chromaffin cells of the adrenal gland or other sympathetic ganglia. They can produce excessive amounts of catecholamines, leading to hypertension and other symptoms.
4. Medulloblastoma: This is a malignant brain tumor that arises from the cerebellum. It is the most common malignant brain tumor in children.
5. Malignant peripheral nerve sheath tumors (MPNSTs): These are rare tumors that arise from the cells that surround and protect nerves. They can occur sporadically or in association with neurofibromatosis type 1.
6. Merkel cell carcinoma: This is a rare and aggressive skin cancer that arises from the Merkel cells, which are located in the epidermis and function as touch receptors.
The diagnosis of NETs typically involves imaging studies such as CT or MRI scans, as well as biopsy and histopathological examination. Treatment may include surgery, radiation therapy, chemotherapy, or targeted therapy depending on the type and stage of the tumor.
Neuroectodermal tumors, primitive, peripheral (PNET) are a group of rare and aggressive malignancies that primarily affect children and young adults. These tumors arise from the primitive neuroectodermal cells, which are the precursors to the nervous system. PNETs can occur in various locations throughout the body, but when they occur outside the central nervous system (CNS), they are referred to as peripheral PNETs (pPNETs).
Peripheral PNETs are similar to Ewing sarcoma, another type of small, round blue cell tumor that arises from primitive neuroectodermal cells. In fact, some researchers consider pPNETs and Ewing sarcomas to be part of the same disease spectrum, known as the Ewing family of tumors (EFT).
Peripheral PNETs can occur in any part of the body, but they most commonly affect the bones and soft tissues of the trunk, extremities, and head and neck region. The symptoms of pPNET depend on the location and size of the tumor, but they may include pain, swelling, decreased mobility, and systemic symptoms such as fever and weight loss.
The diagnosis of pPNET typically involves a combination of imaging studies (such as MRI or CT scans), biopsy, and molecular testing. The treatment usually involves a multimodal approach that includes surgery, chemotherapy, and radiation therapy. Despite aggressive treatment, the prognosis for patients with pPNET remains poor, with a five-year survival rate of approximately 30%.
A neuroectodermal tumor, melanotic, also known as a melanotic neuroectodermal tumor of infancy or MNTI, is a rare, typically benign but locally aggressive tumor that originates from the neural crest cells, a type of stem cell that gives rise to various tissues in the body, including nerve cells and pigment-producing cells called melanocytes.
MNTIs are usually found in the head and neck region of infants under one year of age, although they can occur in older children and adults as well. These tumors are characterized by the presence of melanin, a dark pigment that gives them a black or brown color.
MNTIs typically grow rapidly and can cause symptoms such as swelling, pain, or difficulty breathing if they are located in the head or neck region. Treatment usually involves surgical removal of the tumor, and the prognosis is generally good, especially if the tumor is completely removed. However, there is a risk of recurrence, so close follow-up with a healthcare provider is necessary.
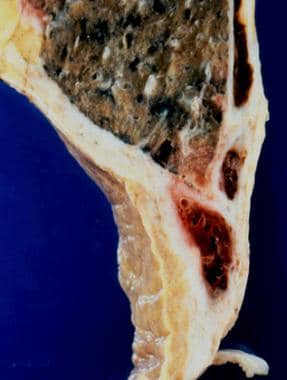
Ewing sarcoma is a type of cancer that originates in bones or the soft tissues surrounding them, such as muscles and tendons. It primarily affects children and adolescents, although it can occur in adults as well. The disease is characterized by small, round tumor cells that typically grow quickly and are prone to metastasize (spread) to other parts of the body, most commonly the lungs, bones, and bone marrow.
Ewing sarcoma is caused by a genetic abnormality, specifically a chromosomal translocation that results in the fusion of two genes, EWSR1 and FLI1. This gene fusion leads to the formation of an abnormal protein that disrupts normal cell growth and division processes, ultimately resulting in cancer.
Symptoms of Ewing sarcoma can vary depending on the location and size of the tumor but may include pain or swelling in the affected area, fever, fatigue, and weight loss. Diagnosis typically involves imaging studies such as X-rays, CT scans, or MRI scans to locate the tumor, followed by a biopsy to confirm the presence of cancer cells. Treatment may involve surgery, radiation therapy, chemotherapy, or a combination of these approaches, depending on the stage and location of the disease.
Medulloblastoma is a type of malignant brain tumor that originates in the cerebellum, which is the part of the brain located at the back of the skull and controls coordination and balance. It is one of the most common types of pediatric brain tumors, although it can also occur in adults.
Medulloblastomas are typically made up of small, round cancer cells that grow quickly and can spread to other parts of the central nervous system, such as the spinal cord. They are usually treated with a combination of surgery, radiation therapy, and chemotherapy. The exact cause of medulloblastoma is not known, but it is thought to be related to genetic mutations or abnormalities that occur during development.
Cerebellar neoplasms refer to abnormal growths or tumors that develop in the cerebellum, which is the part of the brain responsible for coordinating muscle movements and maintaining balance. These tumors can be benign (non-cancerous) or malignant (cancerous), and they can arise from various types of cells within the cerebellum.
The most common type of cerebellar neoplasm is a medulloblastoma, which arises from primitive nerve cells in the cerebellum. Other types of cerebellar neoplasms include astrocytomas, ependymomas, and brain stem gliomas. Symptoms of cerebellar neoplasms may include headaches, vomiting, unsteady gait, coordination problems, and visual disturbances. Treatment options depend on the type, size, and location of the tumor, as well as the patient's overall health and age. Treatment may involve surgery, radiation therapy, chemotherapy, or a combination of these approaches.
Supratentorial neoplasms refer to tumors that originate in the region of the brain located above the tentorium cerebelli, which is a dual layer of dura mater (the protective outer covering of the brain) that separates the cerebrum from the cerebellum. This area includes the cerebral hemispheres, basal ganglia, thalamus, hypothalamus, and pineal gland. Supratentorial neoplasms can be benign or malignant and may arise from various cell types such as neurons, glial cells, meninges, or blood vessels. They can cause a variety of neurological symptoms depending on their size, location, and rate of growth.
Brain neoplasms, also known as brain tumors, are abnormal growths of cells within the brain. These growths can be benign (non-cancerous) or malignant (cancerous). Benign brain tumors typically grow slowly and do not spread to other parts of the body. However, they can still cause serious problems if they press on sensitive areas of the brain. Malignant brain tumors, on the other hand, are cancerous and can grow quickly, invading surrounding brain tissue and spreading to other parts of the brain or spinal cord.
Brain neoplasms can arise from various types of cells within the brain, including glial cells (which provide support and insulation for nerve cells), neurons (nerve cells that transmit signals in the brain), and meninges (the membranes that cover the brain and spinal cord). They can also result from the spread of cancer cells from other parts of the body, known as metastatic brain tumors.
Symptoms of brain neoplasms may vary depending on their size, location, and growth rate. Common symptoms include headaches, seizures, weakness or paralysis in the limbs, difficulty with balance and coordination, changes in speech or vision, confusion, memory loss, and changes in behavior or personality.
Treatment for brain neoplasms depends on several factors, including the type, size, location, and grade of the tumor, as well as the patient's age and overall health. Treatment options may include surgery, radiation therapy, chemotherapy, targeted therapy, or a combination of these approaches. Regular follow-up care is essential to monitor for recurrence and manage any long-term effects of treatment.
Proto-oncogene protein c-Fli-1 is a transcription factor that belongs to the ETS family and plays crucial roles in hematopoiesis, vascular development, and cell proliferation. The gene encoding this protein, called c-Fli-1, can be mutated or its expression can be dysregulated, leading to the formation of a proto-oncogene. When this happens, the protein can contribute to the development of various types of cancer, such as Ewing's sarcoma and acute myeloid leukemia. In these cases, the protein promotes cell growth and division, inhibits apoptosis (programmed cell death), and increases angiogenesis (the formation of new blood vessels). Overall, c-Fli-1 is an important regulator of normal cellular processes, but when its activity is deregulated, it can contribute to the development of cancer.
Ewing Sarcoma (EWS) RNA-Binding Protein, also known as EWSR1, is a protein that plays a role in gene expression by binding to RNA. It is a member of the FET family of proteins, which also includes FUS and TAF15. These proteins are involved in various cellular processes such as transcription, splicing, and translation.
Mutations in the EWSR1 gene have been associated with several types of cancer, most notably Ewing sarcoma, a rare tumor that typically affects children and adolescents. In Ewing sarcoma, a fusion protein is formed when EWSR1 combines with another protein, most commonly ETS translocation variant 1 (ETV1), FLI1, ERG or FEV. This fusion protein can lead to abnormal gene expression and tumor formation.
EWSR1 has also been found to be involved in other types of cancer such as acute myeloid leukemia, clear cell sarcoma, desmoplastic small round cell tumors and liposarcomas.
It's important to note that while EWSR1 is a RNA-binding protein, it can also bind to DNA in certain contexts, such as when it forms a fusion protein with an ETS transcription factor in Ewing sarcoma.
A pinealoma is a rare type of brain tumor that originates in the pineal gland, a small endocrine gland located in the center of the brain. The pineal gland is responsible for producing melatonin, a hormone that helps regulate sleep-wake cycles. Pinealomas can be benign or malignant, with malignant pinealomas being more aggressive and likely to spread to other parts of the body.
Pinealomas are typically classified as either pineocytomas or pineoblastomas, depending on their appearance under a microscope. Pineocytomas are slow-growing and less aggressive, while pineoblastomas are fast-growing and more likely to spread. Symptoms of pinealomas can include headaches, nausea, vomiting, vision problems, and hormonal imbalances.
Treatment for pinealomas typically involves surgery to remove as much of the tumor as possible, followed by radiation therapy and/or chemotherapy to kill any remaining cancer cells. The prognosis for pinealomas varies depending on the type and stage of the tumor, as well as the patient's age and overall health.
Human chromosome pair 22 consists of two rod-shaped structures present in the nucleus of each cell in the human body. Each chromosome is made up of DNA tightly coiled around histone proteins, forming a complex structure called a chromatin.
Chromosome pair 22 is one of the 22 autosomal pairs of human chromosomes, meaning they are not sex chromosomes (X or Y). Chromosome 22 is the second smallest human chromosome, with each arm of the chromosome designated as p and q. The short arm is labeled "p," and the long arm is labeled "q."
Chromosome 22 contains several genes that are associated with various genetic disorders, including DiGeorge syndrome, velocardiofacial syndrome, and cat-eye syndrome, which result from deletions or duplications of specific regions on the chromosome. Additionally, chromosome 22 is the location of the NRXN1 gene, which has been associated with an increased risk for autism spectrum disorder (ASD) and schizophrenia when deleted or disrupted.
Understanding the genetic makeup of human chromosome pair 22 can provide valuable insights into human genetics, evolution, and disease susceptibility, as well as inform medical diagnoses, treatments, and research.
Ependymoma is a type of brain or spinal cord tumor that develops from the ependymal cells that line the ventricles (fluid-filled spaces) in the brain, or the central canal of the spinal cord. These tumors can be benign or malignant, and they can cause various symptoms depending on their location and size.
Ependymomas are relatively rare, accounting for about 2-3% of all primary brain and central nervous system tumors. They most commonly occur in children and young adults, but they can also affect older individuals. Treatment typically involves surgical removal of the tumor, followed by radiation therapy or chemotherapy, depending on the grade and location of the tumor. The prognosis for ependymomas varies widely, with some patients experiencing long-term survival and others having more aggressive tumors that are difficult to treat.
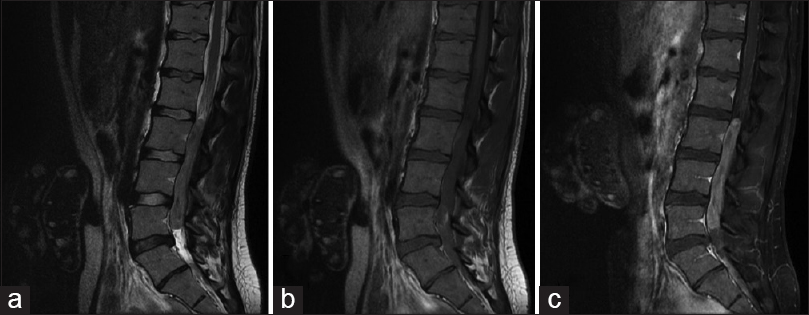
Spinal cord neoplasms refer to abnormal growths or tumors within the spinal cord. These can be benign (non-cancerous) or malignant (cancerous). They originate from the cells within the spinal cord itself (primary tumors), or they may spread to the spinal cord from other parts of the body (metastatic tumors). Spinal cord neoplasms can cause various symptoms depending on their location and size, including back pain, neurological deficits, and even paralysis. Treatment options include surgery, radiation therapy, and chemotherapy.
Small cell sarcoma is a very rare and aggressive type of cancer that affects the connective tissues in the body, such as muscles, tendons, bones, cartilage, and fat. It is called "small cell" because the cancer cells are small and appear round or oval in shape, with scant cytoplasm and finely granular chromatin.
Small cell sarcoma typically occurs in adults between the ages of 40 and 70, and it can develop in any part of the body. However, it is most commonly found in the extremities, trunk, and retroperitoneum. The exact cause of small cell sarcoma is not known, but it is thought to be associated with genetic mutations that occur during a person's lifetime.
Small cell sarcoma can be difficult to diagnose because it often does not cause any symptoms until it has advanced to an aggressive stage. When symptoms do occur, they may include pain, swelling, or a lump in the affected area. Diagnosis typically involves a biopsy of the tumor tissue, followed by imaging tests such as CT scans, MRI scans, or PET scans to determine the extent of the cancer.
Treatment for small cell sarcoma usually involves surgery to remove the tumor, followed by radiation therapy and/or chemotherapy to kill any remaining cancer cells. However, because small cell sarcoma is so rare and aggressive, treatment options may be limited, and the prognosis is often poor. Clinical trials of new treatments are also an option for some patients.
Bone neoplasms are abnormal growths or tumors that develop in the bone. They can be benign (non-cancerous) or malignant (cancerous). Benign bone neoplasms do not spread to other parts of the body and are rarely a threat to life, although they may cause problems if they grow large enough to press on surrounding tissues or cause fractures. Malignant bone neoplasms, on the other hand, can invade and destroy nearby tissue and may spread (metastasize) to other parts of the body.
There are many different types of bone neoplasms, including:
1. Osteochondroma - a benign tumor that develops from cartilage and bone
2. Enchondroma - a benign tumor that forms in the cartilage that lines the inside of the bones
3. Chondrosarcoma - a malignant tumor that develops from cartilage
4. Osteosarcoma - a malignant tumor that develops from bone cells
5. Ewing sarcoma - a malignant tumor that develops in the bones or soft tissues around the bones
6. Giant cell tumor of bone - a benign or occasionally malignant tumor that develops from bone tissue
7. Fibrosarcoma - a malignant tumor that develops from fibrous tissue in the bone
The symptoms of bone neoplasms vary depending on the type, size, and location of the tumor. They may include pain, swelling, stiffness, fractures, or limited mobility. Treatment options depend on the type and stage of the tumor but may include surgery, radiation therapy, chemotherapy, or a combination of these treatments.
Neuroblastoma is defined as a type of cancer that develops from immature nerve cells found in the fetal or early postnatal period, called neuroblasts. It typically occurs in infants and young children, with around 90% of cases diagnosed before age five. The tumors often originate in the adrenal glands but can also arise in the neck, chest, abdomen, or spine. Neuroblastoma is characterized by its ability to spread (metastasize) to other parts of the body, including bones, bone marrow, lymph nodes, and skin. The severity and prognosis of neuroblastoma can vary widely, depending on factors such as the patient's age at diagnosis, stage of the disease, and specific genetic features of the tumor.
A rhabdoid tumor is a rare and aggressive type of cancer that typically develops in the kidneys of children, but can also occur in other areas of the body such as the brain, soft tissues, and lungs. These tumors are characterized by the presence of cells with a unique appearance, known as rhabdoid cells, which have large nuclei, prominent nucleoli, and eosinophilic inclusions.
Rhabdoid tumors can occur in both children and adults, but they are most commonly found in children under the age of 3. They are often resistant to conventional cancer treatments such as chemotherapy and radiation therapy, making them difficult to treat. The prognosis for patients with rhabdoid tumors is generally poor, with a high rate of recurrence and metastasis.
The exact cause of rhabdoid tumors is not known, but they are associated with mutations in the SMARCB1 or SMARCA4 genes, which are involved in regulating gene expression and maintaining genomic stability. These genetic changes can occur spontaneously or may be inherited from a parent.
Treatment for rhabdoid tumors typically involves a combination of surgery, chemotherapy, and radiation therapy. In some cases, stem cell transplantation or targeted therapies may also be used. Despite aggressive treatment, the prognosis for patients with rhabdoid tumors remains poor, with a five-year survival rate of less than 20%.
Maxillary neoplasms refer to abnormal growths or tumors in the maxilla, which is the upper jaw bone. These growths can be benign (non-cancerous) or malignant (cancerous). Benign neoplasms are slow-growing and do not spread to other parts of the body, while malignant neoplasms can invade surrounding tissues and spread to distant sites.
Maxillary neoplasms can cause various symptoms such as swelling, pain, numbness, loose teeth, or difficulty in chewing or swallowing. They may also cause nasal congestion, nosebleeds, or visual changes if they affect the eye or orbit. The diagnosis of maxillary neoplasms usually involves a combination of clinical examination, imaging studies such as CT or MRI scans, and biopsy to determine the type and extent of the tumor.
Treatment options for maxillary neoplasms depend on several factors, including the type, size, location, and stage of the tumor, as well as the patient's overall health and preferences. Treatment may include surgery, radiation therapy, chemotherapy, or a combination of these modalities. Regular follow-up care is essential to monitor for recurrence or metastasis and ensure optimal outcomes.
Tumor markers are substances that can be found in the body and their presence can indicate the presence of certain types of cancer or other conditions. Biological tumor markers refer to those substances that are produced by cancer cells or by other cells in response to cancer or certain benign (non-cancerous) conditions. These markers can be found in various bodily fluids such as blood, urine, or tissue samples.
Examples of biological tumor markers include:
1. Proteins: Some tumor markers are proteins that are produced by cancer cells or by other cells in response to the presence of cancer. For example, prostate-specific antigen (PSA) is a protein produced by normal prostate cells and in higher amounts by prostate cancer cells.
2. Genetic material: Tumor markers can also include genetic material such as DNA, RNA, or microRNA that are shed by cancer cells into bodily fluids. For example, circulating tumor DNA (ctDNA) is genetic material from cancer cells that can be found in the bloodstream.
3. Metabolites: Tumor markers can also include metabolic products produced by cancer cells or by other cells in response to cancer. For example, lactate dehydrogenase (LDH) is an enzyme that is released into the bloodstream when cancer cells break down glucose for energy.
It's important to note that tumor markers are not specific to cancer and can be elevated in non-cancerous conditions as well. Therefore, they should not be used alone to diagnose cancer but rather as a tool in conjunction with other diagnostic tests and clinical evaluations.
Neoplasms, germ cell and embryonal are types of tumors that originate from the abnormal growth of cells. Here's a brief medical definition for each:
1. Neoplasms: Neoplasms refer to abnormal tissue growths or masses, which can be benign (non-cancerous) or malignant (cancerous). They result from uncontrolled cell division and may invade surrounding tissues or spread to other parts of the body through a process called metastasis.
2. Germ Cell Tumors: These are rare tumors that develop from the germ cells, which give rise to sperm and eggs in the reproductive organs (ovaries and testes). They can be benign or malignant and may occur in both children and adults. Germ cell tumors can also arise outside of the reproductive organs, a condition known as extragonadal germ cell tumors.
3. Embryonal Tumors: These are a type of malignant neoplasm that primarily affects infants and young children. They develop from embryonic cells, which are immature cells present during fetal development. Embryonal tumors can occur in various organs, including the brain (medulloblastomas), nervous system (primitive neuroectodermal tumors or PNETs), and other areas like the kidneys and liver.
It is essential to note that these conditions require professional medical evaluation and treatment by healthcare professionals with expertise in oncology and related fields.
Thoracic neoplasms refer to abnormal growths or tumors that develop in the thorax, which is the area of the body that includes the chest and lungs. These neoplasms can be benign (non-cancerous) or malignant (cancerous). Malignant thoracic neoplasms are often referred to as lung cancer, but they can also include other types of cancer such as mesothelioma, thymoma, and esophageal cancer.
Thoracic neoplasms can cause various symptoms depending on their location and size. Common symptoms include coughing, chest pain, shortness of breath, hoarseness, and difficulty swallowing. Treatment options for thoracic neoplasms depend on the type, stage, and location of the tumor, as well as the patient's overall health. Treatment may include surgery, radiation therapy, chemotherapy, targeted therapy, or a combination of these approaches.
Central nervous system (CNS) neoplasms refer to a group of abnormal growths or tumors that develop within the brain or spinal cord. These tumors can be benign or malignant, and their growth can compress or disrupt the normal functioning of surrounding brain or spinal cord tissue.
Benign CNS neoplasms are slow-growing and rarely spread to other parts of the body. However, they can still cause significant problems if they grow large enough to put pressure on vital structures within the brain or spinal cord. Malignant CNS neoplasms, on the other hand, are aggressive tumors that can invade and destroy surrounding tissue. They may also spread to other parts of the CNS or, rarely, to other organs in the body.
CNS neoplasms can arise from various types of cells within the brain or spinal cord, including nerve cells, glial cells (which provide support and insulation for nerve cells), and supportive tissues such as blood vessels. The specific type of CNS neoplasm is often used to help guide treatment decisions and determine prognosis.
Symptoms of CNS neoplasms can vary widely depending on the location and size of the tumor, but may include headaches, seizures, weakness or paralysis, vision or hearing changes, balance problems, memory loss, and changes in behavior or personality. Treatment options for CNS neoplasms may include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
The neural plate is a structure formed during the embryonic development of vertebrates. It is a thickened plate of ectodermal cells located on the dorsal surface of the developing embryo. The neural plate gives rise to the central nervous system, including the brain and spinal cord.
The process of neural plate formation begins with the specification of ectodermal cells into neural fated cells, a process that is regulated by various signaling molecules. Once specified, these cells undergo morphological changes, resulting in the thickening of the ectoderm to form the neural plate.
The neural plate then undergoes a series of folding movements, leading to the formation of the neural tube, which eventually develops into the brain and spinal cord. The edges of the neural plate, known as the neural folds, come together and fuse, forming a closed tube. Failure of the neural folds to fuse properly can result in neural tube defects, such as spina bifida.
Overall, the neural plate is a critical structure in the development of the nervous system in vertebrates, and its formation and subsequent development are tightly regulated by various genetic and environmental factors.
Human chromosome pair 11 consists of two rod-shaped structures present in the nucleus of each cell in the human body. Each member of the pair is a single chromosome, and together they contain the genetic material that is inherited from both parents. They are located on the eleventh position in the standard karyotype, which is a visual representation of the 23 pairs of human chromosomes.
Chromosome 11 is one of the largest human chromosomes and contains an estimated 135 million base pairs. It contains approximately 1,400 genes that provide instructions for making proteins, as well as many non-coding RNA molecules that play a role in regulating gene expression.
Chromosome 11 is known to contain several important genes and genetic regions associated with various human diseases and conditions. For example, it contains the Wilms' tumor 1 (WT1) gene, which is associated with kidney cancer in children, and the neurofibromatosis type 1 (NF1) gene, which is associated with a genetic disorder that causes benign tumors to grow on nerves throughout the body. Additionally, chromosome 11 contains the region where the ABO blood group genes are located, which determine a person's blood type.
It's worth noting that human chromosomes come in pairs because they contain two copies of each gene, one inherited from the mother and one from the father. This redundancy allows for genetic diversity and provides a backup copy of essential genes, ensuring their proper function and maintaining the stability of the genome.
An oncogene protein fusion is a result of a genetic alteration in which parts of two different genes combine to create a hybrid gene that can contribute to the development of cancer. This fusion can lead to the production of an abnormal protein that promotes uncontrolled cell growth and division, ultimately resulting in a malignant tumor. Oncogene protein fusions are often caused by chromosomal rearrangements such as translocations, inversions, or deletions and are commonly found in various types of cancer, including leukemia and sarcoma. These genetic alterations can serve as potential targets for cancer diagnosis and therapy.
Astrocytoma is a type of brain tumor that arises from astrocytes, which are star-shaped glial cells in the brain. These tumors can occur in various parts of the brain and can have different grades of malignancy, ranging from low-grade (I or II) to high-grade (III or IV). Low-grade astrocytomas tend to grow slowly and may not cause any symptoms for a long time, while high-grade astrocytomas are more aggressive and can grow quickly, causing neurological problems.
Symptoms of astrocytoma depend on the location and size of the tumor but may include headaches, seizures, weakness or numbness in the limbs, difficulty speaking or swallowing, changes in vision or behavior, and memory loss. Treatment options for astrocytomas include surgery, radiation therapy, chemotherapy, or a combination of these approaches. The prognosis for astrocytoma varies widely depending on the grade and location of the tumor, as well as the age and overall health of the patient.
Immunohistochemistry (IHC) is a technique used in pathology and laboratory medicine to identify specific proteins or antigens in tissue sections. It combines the principles of immunology and histology to detect the presence and location of these target molecules within cells and tissues. This technique utilizes antibodies that are specific to the protein or antigen of interest, which are then tagged with a detection system such as a chromogen or fluorophore. The stained tissue sections can be examined under a microscope, allowing for the visualization and analysis of the distribution and expression patterns of the target molecule in the context of the tissue architecture. Immunohistochemistry is widely used in diagnostic pathology to help identify various diseases, including cancer, infectious diseases, and immune-mediated disorders.
Orbital neoplasms refer to abnormal growths or tumors that develop in the orbit, which is the bony cavity that contains the eyeball, muscles, nerves, fat, and blood vessels. These neoplasms can be benign (non-cancerous) or malignant (cancerous), and they can arise from various types of cells within the orbit.
Orbital neoplasms can cause a variety of symptoms depending on their size, location, and rate of growth. Common symptoms include protrusion or displacement of the eyeball, double vision, limited eye movement, pain, swelling, and numbness in the face. In some cases, orbital neoplasms may not cause any noticeable symptoms, especially if they are small and slow-growing.
There are many different types of orbital neoplasms, including:
1. Optic nerve glioma: a rare tumor that arises from the optic nerve's supportive tissue.
2. Orbital meningioma: a tumor that originates from the membranes covering the brain and extends into the orbit.
3. Lacrimal gland tumors: benign or malignant growths that develop in the lacrimal gland, which produces tears.
4. Orbital lymphangioma: a non-cancerous tumor that arises from the lymphatic vessels in the orbit.
5. Rhabdomyosarcoma: a malignant tumor that develops from the skeletal muscle cells in the orbit.
6. Metastatic tumors: cancerous growths that spread to the orbit from other parts of the body, such as the breast, lung, or prostate.
The diagnosis and treatment of orbital neoplasms depend on several factors, including the type, size, location, and extent of the tumor. Imaging tests, such as CT scans and MRI, are often used to visualize the tumor and determine its extent. A biopsy may also be performed to confirm the diagnosis and determine the tumor's type and grade. Treatment options include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
Vincristine is an antineoplastic agent, specifically a vinca alkaloid. It is derived from the Madagascar periwinkle plant (Catharanthus roseus). Vincristine binds to tubulin, a protein found in microtubules, and inhibits their polymerization, which results in disruption of mitotic spindles leading to cell cycle arrest and apoptosis (programmed cell death). It is used in the treatment of various types of cancer including leukemias, lymphomas, and solid tumors. Common side effects include peripheral neuropathy, constipation, and alopecia.
Ifosfamide is an alkylating agent, which is a type of chemotherapy medication. It works by interfering with the DNA of cancer cells, preventing them from dividing and growing. Ifosfamide is used to treat various types of cancers, such as testicular cancer, small cell lung cancer, ovarian cancer, cervical cancer, and certain types of sarcomas.
The medical definition of Ifosfamide is:
Ifosfamide is a synthetic antineoplastic agent, an oxazaphosphorine derivative, with the chemical formula C6H15Cl2N2O2P. It is used in the treatment of various malignancies, including germ cell tumors, sarcomas, lymphomas, and testicular cancer. The drug is administered intravenously and exerts its cytotoxic effects through the alkylation and cross-linking of DNA, leading to the inhibition of DNA replication and transcription. Ifosfamide can cause significant myelosuppression and has been associated with urotoxicity, neurotoxicity, and secondary malignancies. Therefore, it is essential to monitor patients closely during treatment and manage any adverse effects promptly.
A fatal outcome is a term used in medical context to describe a situation where a disease, injury, or illness results in the death of an individual. It is the most severe and unfortunate possible outcome of any medical condition, and is often used as a measure of the severity and prognosis of various diseases and injuries. In clinical trials and research, fatal outcome may be used as an endpoint to evaluate the effectiveness and safety of different treatments or interventions.
X-ray computed tomography (CT or CAT scan) is a medical imaging method that uses computer-processed combinations of many X-ray images taken from different angles to produce cross-sectional (tomographic) images (virtual "slices") of the body. These cross-sectional images can then be used to display detailed internal views of organs, bones, and soft tissues in the body.
The term "computed tomography" is used instead of "CT scan" or "CAT scan" because the machines take a series of X-ray measurements from different angles around the body and then use a computer to process these data to create detailed images of internal structures within the body.
CT scanning is a noninvasive, painless medical test that helps physicians diagnose and treat medical conditions. CT imaging provides detailed information about many types of tissue including lung, bone, soft tissue and blood vessels. CT examinations can be performed on every part of the body for a variety of reasons including diagnosis, surgical planning, and monitoring of therapeutic responses.
In computed tomography (CT), an X-ray source and detector rotate around the patient, measuring the X-ray attenuation at many different angles. A computer uses this data to construct a cross-sectional image by the process of reconstruction. This technique is called "tomography". The term "computed" refers to the use of a computer to reconstruct the images.
CT has become an important tool in medical imaging and diagnosis, allowing radiologists and other physicians to view detailed internal images of the body. It can help identify many different medical conditions including cancer, heart disease, lung nodules, liver tumors, and internal injuries from trauma. CT is also commonly used for guiding biopsies and other minimally invasive procedures.
In summary, X-ray computed tomography (CT or CAT scan) is a medical imaging technique that uses computer-processed combinations of many X-ray images taken from different angles to produce cross-sectional images of the body. It provides detailed internal views of organs, bones, and soft tissues in the body, allowing physicians to diagnose and treat medical conditions.
A cell line that is derived from tumor cells and has been adapted to grow in culture. These cell lines are often used in research to study the characteristics of cancer cells, including their growth patterns, genetic changes, and responses to various treatments. They can be established from many different types of tumors, such as carcinomas, sarcomas, and leukemias. Once established, these cell lines can be grown and maintained indefinitely in the laboratory, allowing researchers to conduct experiments and studies that would not be feasible using primary tumor cells. It is important to note that tumor cell lines may not always accurately represent the behavior of the original tumor, as they can undergo genetic changes during their time in culture.
Translocation, genetic, refers to a type of chromosomal abnormality in which a segment of a chromosome is transferred from one chromosome to another, resulting in an altered genome. This can occur between two non-homologous chromosomes (non-reciprocal translocation) or between two homologous chromosomes (reciprocal translocation). Genetic translocations can lead to various clinical consequences, depending on the genes involved and the location of the translocation. Some translocations may result in no apparent effects, while others can cause developmental abnormalities, cancer, or other genetic disorders. In some cases, translocations can also increase the risk of having offspring with genetic conditions.
A ganglioneuroma is a type of benign (noncancerous) tumor that arises from the nerve cells called ganglia in the autonomic nervous system. These tumors typically develop in the abdomen or chest and are most commonly found in children and adolescents, although they can occur at any age.
Ganglioneuromas are composed of mature nerve cells (ganglion cells) and supporting tissue called stroma. They tend to grow slowly and usually do not cause any symptoms unless they become very large or press on nearby organs. In some cases, ganglioneuromas may produce hormones that can cause symptoms such as diarrhea, flushing, or heart palpitations.
While ganglioneuromas are generally benign, there is a small risk that they may become malignant (cancerous) and develop into a type of tumor called a ganglioneuroblastoma or neuroblastoma. For this reason, it is important to monitor these tumors closely and remove them if they grow too large or cause symptoms.
Treatment for ganglioneuromas typically involves surgical removal of the tumor. In some cases, radiation therapy or chemotherapy may also be recommended, particularly if there is a risk of malignant transformation.
Skull neoplasms refer to abnormal growths or tumors that develop within the skull. These growths can be benign (non-cancerous) or malignant (cancerous). They can originate from various types of cells, such as bone cells, nerve cells, or soft tissues. Skull neoplasms can cause various symptoms depending on their size and location, including headaches, seizures, vision problems, hearing loss, and neurological deficits. Treatment options include surgery, radiation therapy, and chemotherapy. It is important to note that a neoplasm in the skull can also refer to metastatic cancer, which has spread from another part of the body to the skull.
Paraparesis is a medical term that refers to a mild to moderate form of paralysis affecting the lower limbs, specifically the legs. It is characterized by partial loss of strength and mobility, which may result in difficulty walking or maintaining balance. Paraparesis can be caused by various conditions such as spinal cord injuries, multiple sclerosis, spina bifida, or other neurological disorders affecting the spinal cord.
The term "para" means "two," and "paresis" comes from the Greek word "paresis," which means "loosening" or "relaxation." Therefore, paraparesis implies weakness or partial paralysis in two lower extremities. It is important to note that while paraparesis can impact a person's ability to walk and perform daily activities, it does not necessarily lead to complete loss of movement or sensation in the affected limbs.
Proper diagnosis and management of the underlying cause are crucial for improving symptoms and preventing further progression of paraparesis. Treatment options may include physical therapy, medications, assistive devices, or surgical interventions depending on the specific condition causing the paraparesis.
The thoracic wall refers to the anatomical structure that surrounds and protects the chest cavity or thorax, which contains the lungs, heart, and other vital organs. It is composed of several components:
1. Skeletal framework: This includes the 12 pairs of ribs, the sternum (breastbone) in the front, and the thoracic vertebrae in the back. The upper seven pairs of ribs are directly attached to the sternum in the front through costal cartilages. The lower five pairs of ribs are not directly connected to the sternum but are joined to the ribs above them.
2. Muscles: The thoracic wall contains several muscles, including the intercostal muscles (located between the ribs), the scalene muscles (at the side and back of the neck), and the serratus anterior muscle (on the sides of the chest). These muscles help in breathing by expanding and contracting the ribcage.
3. Soft tissues: The thoracic wall also contains various soft tissues, such as fascia, nerves, blood vessels, and fat. These structures support the functioning of the thoracic organs and contribute to the overall stability and protection of the chest cavity.
The primary function of the thoracic wall is to protect the vital organs within the chest cavity while allowing for adequate movement during respiration. Additionally, it provides a stable base for the attachment of various muscles involved in upper limb movement and posture.
A teratoma is a type of germ cell tumor, which is a broad category of tumors that originate from the reproductive cells. A teratoma contains developed tissues from all three embryonic germ layers: ectoderm, mesoderm, and endoderm. This means that a teratoma can contain various types of tissue such as hair, teeth, bone, and even more complex organs like eyes, thyroid, or neural tissue.
Teratomas are usually benign (non-cancerous), but they can sometimes be malignant (cancerous) and can spread to other parts of the body. They can occur anywhere in the body, but they're most commonly found in the ovaries and testicles. When found in these areas, they are typically removed surgically.
Teratomas can also occur in other locations such as the sacrum, coccyx (tailbone), mediastinum (the area between the lungs), and pineal gland (a small gland in the brain). These types of teratomas can be more complex to treat due to their location and potential to cause damage to nearby structures.
Neoplasms of nerve tissue are abnormal growths or tumors that originate in the nervous system, including the brain, spinal cord, and peripheral nerves. These neoplasms can be benign or malignant (cancerous) and can cause a variety of symptoms depending on their location and size.
Benign nerve tissue neoplasms are typically slow-growing and do not spread to other parts of the body. Examples include schwannomas, neurofibromas, and meningiomas. These tumors arise from the supporting cells of the nervous system, such as Schwann cells, which produce the myelin sheath that insulates nerve fibers.
Malignant nerve tissue neoplasms, on the other hand, are cancerous and can invade nearby tissues and spread to other parts of the body. These tumors are less common than benign neoplasms and can be difficult to treat. Examples include glioblastoma multiforme, a highly aggressive brain cancer, and malignant peripheral nerve sheath tumors, which arise from the cells that surround peripheral nerves.
Symptoms of nerve tissue neoplasms can vary widely depending on their location and size. Some common symptoms include headaches, seizures, weakness or numbness in the limbs, difficulty with coordination or balance, and changes in vision, hearing, or speech. Treatment options for nerve tissue neoplasms may include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
Peripheral nervous system (PNS) neoplasms refer to tumors that originate in the peripheral nerves, which are the nerves outside the brain and spinal cord. These tumors can be benign or malignant (cancerous). Benign tumors, such as schwannomas and neurofibromas, grow slowly and do not spread to other parts of the body. Malignant tumors, such as malignant peripheral nerve sheath tumors (MPNSTs), can invade nearby tissues and may metastasize (spread) to other organs.
PNS neoplasms can cause various symptoms depending on their location and size. Common symptoms include pain, weakness, numbness, or tingling in the affected area. In some cases, PNS neoplasms may not cause any symptoms until they become quite large. Treatment options for PNS neoplasms depend on several factors, including the type, size, and location of the tumor, as well as the patient's overall health. Treatment options may include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
Tumor burden is a term used to describe the total amount of cancer in the body. It can refer to the number of tumors, the size of the tumors, or the amount of cancer cells in the body. In research and clinical trials, tumor burden is often measured to assess the effectiveness of treatments or to monitor disease progression. High tumor burden can cause various symptoms and complications, depending on the type and location of the cancer. It can also affect a person's prognosis and treatment options.
Tumor Necrosis Factor-alpha (TNF-α) is a cytokine, a type of small signaling protein involved in immune response and inflammation. It is primarily produced by activated macrophages, although other cell types such as T-cells, natural killer cells, and mast cells can also produce it.
TNF-α plays a crucial role in the body's defense against infection and tissue injury by mediating inflammatory responses, activating immune cells, and inducing apoptosis (programmed cell death) in certain types of cells. It does this by binding to its receptors, TNFR1 and TNFR2, which are found on the surface of many cell types.
In addition to its role in the immune response, TNF-α has been implicated in the pathogenesis of several diseases, including autoimmune disorders such as rheumatoid arthritis, inflammatory bowel disease, and psoriasis, as well as cancer, where it can promote tumor growth and metastasis.
Therapeutic agents that target TNF-α, such as infliximab, adalimumab, and etanercept, have been developed to treat these conditions. However, these drugs can also increase the risk of infections and other side effects, so their use must be carefully monitored.
Etoposide is a chemotherapy medication used to treat various types of cancer, including lung cancer, testicular cancer, and certain types of leukemia. It works by inhibiting the activity of an enzyme called topoisomerase II, which is involved in DNA replication and transcription. By doing so, etoposide can interfere with the growth and multiplication of cancer cells.
Etoposide is often administered intravenously in a hospital or clinic setting, although it may also be given orally in some cases. The medication can cause a range of side effects, including nausea, vomiting, hair loss, and an increased risk of infection. It can also have more serious side effects, such as bone marrow suppression, which can lead to anemia, bleeding, and a weakened immune system.
Like all chemotherapy drugs, etoposide is not without risks and should only be used under the close supervision of a qualified healthcare provider. It is important for patients to discuss the potential benefits and risks of this medication with their doctor before starting treatment.
The pineal gland, also known as the epiphysis cerebri, is a small endocrine gland located in the brain. It is shaped like a pinecone, hence its name, and is situated near the center of the brain, between the two hemispheres, attached to the third ventricle. The primary function of the pineal gland is to produce melatonin, a hormone that helps regulate sleep-wake cycles and circadian rhythms in response to light and darkness. Additionally, it plays a role in the onset of puberty and has been suggested to have other functions related to cognition, mood, and reproduction, although these are not as well understood.
Neoplasm antigens, also known as tumor antigens, are substances that are produced by cancer cells (neoplasms) and can stimulate an immune response. These antigens can be proteins, carbohydrates, or other molecules that are either unique to the cancer cells or are overexpressed or mutated versions of normal cellular proteins.
Neoplasm antigens can be classified into two main categories: tumor-specific antigens (TSAs) and tumor-associated antigens (TAAs). TSAs are unique to cancer cells and are not expressed by normal cells, while TAAs are present at low levels in normal cells but are overexpressed or altered in cancer cells.
TSAs can be further divided into viral antigens and mutated antigens. Viral antigens are produced when cancer is caused by a virus, such as human papillomavirus (HPV) in cervical cancer. Mutated antigens are the result of genetic mutations that occur during cancer development and are unique to each patient's tumor.
Neoplasm antigens play an important role in the immune response against cancer. They can be recognized by the immune system, leading to the activation of immune cells such as T cells and natural killer (NK) cells, which can then attack and destroy cancer cells. However, cancer cells often develop mechanisms to evade the immune response, allowing them to continue growing and spreading.
Understanding neoplasm antigens is important for the development of cancer immunotherapies, which aim to enhance the body's natural immune response against cancer. These therapies include checkpoint inhibitors, which block proteins that inhibit T cell activation, and therapeutic vaccines, which stimulate an immune response against specific tumor antigens.
Combined modality therapy (CMT) is a medical treatment approach that utilizes more than one method or type of therapy simultaneously or in close succession, with the goal of enhancing the overall effectiveness of the treatment. In the context of cancer care, CMT often refers to the combination of two or more primary treatment modalities, such as surgery, radiation therapy, and systemic therapies (chemotherapy, immunotherapy, targeted therapy, etc.).
The rationale behind using combined modality therapy is that each treatment method can target cancer cells in different ways, potentially increasing the likelihood of eliminating all cancer cells and reducing the risk of recurrence. The specific combination and sequence of treatments will depend on various factors, including the type and stage of cancer, patient's overall health, and individual preferences.
For example, a common CMT approach for locally advanced rectal cancer may involve preoperative (neoadjuvant) chemoradiation therapy, followed by surgery to remove the tumor, and then postoperative (adjuvant) chemotherapy. This combined approach allows for the reduction of the tumor size before surgery, increases the likelihood of complete tumor removal, and targets any remaining microscopic cancer cells with systemic chemotherapy.
It is essential to consult with a multidisciplinary team of healthcare professionals to determine the most appropriate CMT plan for each individual patient, considering both the potential benefits and risks associated with each treatment method.
Human chromosome pair 17 consists of two rod-shaped structures present in the nucleus of each human cell. Each chromosome is made up of DNA tightly coiled around histone proteins, forming a complex called chromatin. Chromosomes carry genetic information in the form of genes, which are segments of DNA that contain instructions for the development and function of an organism.
Human cells typically have 23 pairs of chromosomes, for a total of 46 chromosomes. Pair 17 is one of the autosomal pairs, meaning it is not a sex chromosome (X or Y). Chromosome 17 is a medium-sized chromosome and contains an estimated 800 million base pairs of DNA. It contains approximately 1,500 genes that provide instructions for making proteins and regulating various cellular processes.
Chromosome 17 is associated with several genetic disorders, including inherited cancer syndromes such as Li-Fraumeni syndrome and hereditary nonpolyposis colorectal cancer (HNPCC). Mutations in genes located on chromosome 17 can increase the risk of developing various types of cancer, including breast, ovarian, colon, and pancreatic cancer.
Rhabdomyosarcoma is a type of cancer that develops in the body's soft tissues, specifically in the muscle cells. It is a rare and aggressive form of sarcoma, which is a broader category of cancers that affect the connective tissues such as muscles, tendons, cartilages, bones, blood vessels, and fatty tissues.
Rhabdomyosarcomas can occur in various parts of the body, including the head, neck, arms, legs, trunk, and genitourinary system. They are more common in children than adults, with most cases diagnosed before the age of 18. The exact cause of rhabdomyosarcoma is not known, but genetic factors and exposure to radiation or certain chemicals may increase the risk.
There are several subtypes of rhabdomyosarcoma, including embryonal, alveolar, pleomorphic, and spindle cell/sclerosing. The type and stage of the cancer determine the treatment options, which may include surgery, radiation therapy, chemotherapy, or a combination of these approaches. Early diagnosis and prompt treatment are crucial for improving the prognosis and long-term survival rates.
Neurofilament proteins (NFs) are type IV intermediate filament proteins that are specific to neurons. They are the major structural components of the neuronal cytoskeleton and play crucial roles in maintaining the structural integrity, stability, and diameter of axons. Neurofilaments are composed of three subunits: light (NFL), medium (NFM), and heavy (NFH) neurofilament proteins, which differ in their molecular weights. These subunits assemble into heteropolymers to form the neurofilament core, while the C-terminal tails of NFH and NFM extend outward from the core, interacting with other cellular components and participating in various neuronal functions. Increased levels of neurofilament proteins, particularly NFL, in cerebrospinal fluid (CSF) and blood are considered biomarkers for axonal damage and neurodegeneration in several neurological disorders, such as Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS).
A glioma is a type of tumor that originates from the glial cells in the brain. Glial cells are non-neuronal cells that provide support and protection for nerve cells (neurons) within the central nervous system, including providing nutrients, maintaining homeostasis, and insulating neurons.
Gliomas can be classified into several types based on the specific type of glial cell from which they originate. The most common types include:
1. Astrocytoma: Arises from astrocytes, a type of star-shaped glial cells that provide structural support to neurons.
2. Oligodendroglioma: Develops from oligodendrocytes, which produce the myelin sheath that insulates nerve fibers.
3. Ependymoma: Originate from ependymal cells, which line the ventricles (fluid-filled spaces) in the brain and spinal cord.
4. Glioblastoma multiforme (GBM): A highly aggressive and malignant type of astrocytoma that tends to spread quickly within the brain.
Gliomas can be further classified based on their grade, which indicates how aggressive and fast-growing they are. Lower-grade gliomas tend to grow more slowly and may be less aggressive, while higher-grade gliomas are more likely to be aggressive and rapidly growing.
Symptoms of gliomas depend on the location and size of the tumor but can include headaches, seizures, cognitive changes, and neurological deficits such as weakness or paralysis in certain parts of the body. Treatment options for gliomas may include surgery, radiation therapy, chemotherapy, or a combination of these approaches.
Abdominal neoplasms refer to abnormal growths or tumors in the abdomen that can be benign (non-cancerous) or malignant (cancerous). These growths can occur in any of the organs within the abdominal cavity, including the stomach, small intestine, large intestine, liver, pancreas, spleen, and kidneys.
Abdominal neoplasms can cause various symptoms depending on their size, location, and type. Some common symptoms include abdominal pain or discomfort, bloating, changes in bowel habits, unexplained weight loss, fatigue, and fever. In some cases, abdominal neoplasms may not cause any symptoms until they have grown quite large or spread to other parts of the body.
The diagnosis of abdominal neoplasms typically involves a combination of physical exam, medical history, imaging studies such as CT scans or MRIs, and sometimes biopsy to confirm the type of tumor. Treatment options depend on the type, stage, and location of the neoplasm but may include surgery, radiation therapy, chemotherapy, targeted therapy, or a combination of these approaches.
Medical Definition:
Magnetic Resonance Imaging (MRI) is a non-invasive diagnostic imaging technique that uses a strong magnetic field and radio waves to create detailed cross-sectional or three-dimensional images of the internal structures of the body. The patient lies within a large, cylindrical magnet, and the scanner detects changes in the direction of the magnetic field caused by protons in the body. These changes are then converted into detailed images that help medical professionals to diagnose and monitor various medical conditions, such as tumors, injuries, or diseases affecting the brain, spinal cord, heart, blood vessels, joints, and other internal organs. MRI does not use radiation like computed tomography (CT) scans.
Retinoblastoma is a rare type of eye cancer that primarily affects young children, typically developing in the retina (the light-sensitive tissue at the back of the eye) before the age of 5. This malignancy originates from immature retinal cells called retinoblasts and can occur in one or both eyes (bilateral or unilateral).
There are two main types of Retinoblastoma: heritable and non-heritable. The heritable form is caused by a genetic mutation that can be inherited from a parent or may occur spontaneously during embryonic development. This type often affects both eyes and has an increased risk of developing other cancers. Non-heritable Retinoblastoma, on the other hand, occurs due to somatic mutations (acquired during life) that affect only the retinal cells in one eye.
Symptoms of Retinoblastoma may include a white pupil or glow in photographs, crossed eyes, strabismus (misalignment of the eyes), poor vision, redness, or swelling in the eye. Treatment options depend on various factors such as the stage and location of the tumor(s), patient's age, and overall health. These treatments may include chemotherapy, radiation therapy, laser therapy, cryotherapy (freezing), thermotherapy (heating), or enucleation (removal of the affected eye) in advanced cases.
Early detection and prompt treatment are crucial for improving the prognosis and preserving vision in children with Retinoblastoma. Regular eye examinations by a pediatric ophthalmologist or oncologist are recommended to monitor any changes and ensure timely intervention if necessary.
'Tumor cells, cultured' refers to the process of removing cancerous cells from a tumor and growing them in controlled laboratory conditions. This is typically done by isolating the tumor cells from a patient's tissue sample, then placing them in a nutrient-rich environment that promotes their growth and multiplication.
The resulting cultured tumor cells can be used for various research purposes, including the study of cancer biology, drug development, and toxicity testing. They provide a valuable tool for researchers to better understand the behavior and characteristics of cancer cells outside of the human body, which can lead to the development of more effective cancer treatments.
It is important to note that cultured tumor cells may not always behave exactly the same way as they do in the human body, so findings from cell culture studies must be validated through further research, such as animal models or clinical trials.
Thoracic surgical procedures refer to the operations that are performed on the thorax, which is the part of the body that lies between the neck and the abdomen and includes the chest cage, lungs, heart, great blood vessels, esophagus, diaphragm, and other organs in the chest cavity. These surgical procedures can be either open or minimally invasive (using small incisions and specialized instruments) and are performed to diagnose, treat, or manage various medical conditions affecting the thoracic organs, such as:
1. Lung cancer: Thoracic surgeons perform lung resections (lobectomy, segmentectomy, wedge resection) to remove cancerous lung tissue. They may also perform mediastinal lymph node dissection to assess the spread of the disease.
2. Esophageal surgery: Surgeries like esophagectomy are performed to treat esophageal cancer or other conditions affecting the esophagus, such as severe GERD (gastroesophageal reflux disease).
3. Chest wall surgery: This includes procedures to repair or replace damaged ribs, sternum, or chest wall muscles and treat conditions like pectus excavatum or tumors in the chest wall.
4. Heart surgery: Thoracic surgeons collaborate with cardiac surgeons to perform surgeries on the heart, such as coronary artery bypass grafting (CABG), valve repair/replacement, and procedures for treating aneurysms or dissections of the aorta.
5. Diaphragm surgery: Procedures like diaphragm plication are performed to treat paralysis or weakness of the diaphragm that can lead to respiratory insufficiency.
6. Mediastinal surgery: This involves operating on the mediastinum, the area between the lungs, to remove tumors, cysts, or other abnormal growths.
7. Pleural surgery: Procedures like pleurodesis or decortication are performed to manage conditions affecting the pleura (the membrane surrounding the lungs), such as pleural effusions, pneumothorax, or empyema.
8. Lung surgery: Thoracic surgeons perform procedures on the lungs, including lobectomy, segmentectomy, or pneumonectomy to treat lung cancer, benign tumors, or other lung diseases.
9. Tracheal surgery: This includes procedures to repair or reconstruct damaged trachea or remove tumors and growths in the airway.
10. Esophageal surgery: Collaborating with general surgeons, thoracic surgeons perform esophagectomy and other procedures to treat esophageal cancer, benign tumors, or other conditions affecting the esophagus.
Cranial irradiation is a medical treatment that involves the use of radiation therapy to target the brain. It is often used to treat various conditions affecting the brain, such as brain tumors, leukemia, and certain neurological disorders. The radiation is directed at the skull and can be focused on specific areas of the brain or delivered more broadly, depending on the nature and location of the condition being treated.
The goal of cranial irradiation may be to destroy cancer cells, reduce the size of tumors, prevent the spread of cancer, or provide symptomatic relief for patients with advanced disease. However, it is important to note that cranial irradiation can have side effects, including hair loss, fatigue, memory problems, and cognitive changes, among others. These side effects can vary in severity and duration depending on the individual patient and the specific treatment regimen.
The term "DNA, neoplasm" is not a standard medical term or concept. DNA refers to deoxyribonucleic acid, which is the genetic material present in the cells of living organisms. A neoplasm, on the other hand, is a tumor or growth of abnormal tissue that can be benign (non-cancerous) or malignant (cancerous).
In some contexts, "DNA, neoplasm" may refer to genetic alterations found in cancer cells. These genetic changes can include mutations, amplifications, deletions, or rearrangements of DNA sequences that contribute to the development and progression of cancer. Identifying these genetic abnormalities can help doctors diagnose and treat certain types of cancer more effectively.
However, it's important to note that "DNA, neoplasm" is not a term that would typically be used in medical reports or research papers without further clarification. If you have any specific questions about DNA changes in cancer cells or neoplasms, I would recommend consulting with a healthcare professional or conducting further research on the topic.
Cyclophosphamide is an alkylating agent, which is a type of chemotherapy medication. It works by interfering with the DNA of cancer cells, preventing them from dividing and growing. This helps to stop the spread of cancer in the body. Cyclophosphamide is used to treat various types of cancer, including lymphoma, leukemia, multiple myeloma, and breast cancer. It can be given orally as a tablet or intravenously as an injection.
Cyclophosphamide can also have immunosuppressive effects, which means it can suppress the activity of the immune system. This makes it useful in treating certain autoimmune diseases, such as rheumatoid arthritis and lupus. However, this immunosuppression can also increase the risk of infections and other side effects.
Like all chemotherapy medications, cyclophosphamide can cause a range of side effects, including nausea, vomiting, hair loss, fatigue, and increased susceptibility to infections. It is important for patients receiving cyclophosphamide to be closely monitored by their healthcare team to manage these side effects and ensure the medication is working effectively.
Gangliosides are a type of complex lipid molecule known as sialic acid-containing glycosphingolipids. They are predominantly found in the outer leaflet of the cell membrane, particularly in the nervous system. Gangliosides play crucial roles in various biological processes, including cell recognition, signal transduction, and cell adhesion. They are especially abundant in the ganglia (nerve cell clusters) of the peripheral and central nervous systems, hence their name.
Gangliosides consist of a hydrophobic ceramide portion and a hydrophilic oligosaccharide chain that contains one or more sialic acid residues. The composition and structure of these oligosaccharide chains can vary significantly among different gangliosides, leading to the classification of various subtypes, such as GM1, GD1a, GD1b, GT1b, and GQ1b.
Abnormalities in ganglioside metabolism or expression have been implicated in several neurological disorders, including Parkinson's disease, Alzheimer's disease, and various lysosomal storage diseases like Tay-Sachs and Gaucher's diseases. Additionally, certain bacterial toxins, such as botulinum neurotoxin and tetanus toxin, target gangliosides to gain entry into neuronal cells, causing their toxic effects.
Phosphopyruvate Hydratase is an enzyme also known as Enolase. It plays a crucial role in the glycolytic pathway, which is a series of reactions that occur in the cell to break down glucose into pyruvate, producing ATP and NADH as energy-rich intermediates.
Specifically, Phosphopyruvate Hydratase catalyzes the conversion of 2-phospho-D-glycerate (2-PG) to phosphoenolpyruvate (PEP), which is the second to last step in the glycolytic pathway. This reaction includes the removal of a water molecule from 2-PG, resulting in the formation of PEP and the release of a molecule of water.
The enzyme requires magnesium ions as a cofactor for its activity, and it is inhibited by fluoride ions. Deficiency or dysfunction of Phosphopyruvate Hydratase can lead to various metabolic disorders, including some forms of muscular dystrophy and neurodegenerative diseases.
Soft tissue neoplasms refer to abnormal growths or tumors that develop in the soft tissues of the body. Soft tissues include muscles, tendons, ligaments, fascia, nerves, blood vessels, fat, and synovial membranes (the thin layer of cells that line joints and tendons). Neoplasms can be benign (non-cancerous) or malignant (cancerous), and their behavior and potential for spread depend on the specific type of neoplasm.
Benign soft tissue neoplasms are typically slow-growing, well-circumscribed, and rarely spread to other parts of the body. They can often be removed surgically with a low risk of recurrence. Examples of benign soft tissue neoplasms include lipomas (fat tumors), schwannomas (nerve sheath tumors), and hemangiomas (blood vessel tumors).
Malignant soft tissue neoplasms, on the other hand, can grow rapidly, invade surrounding tissues, and may metastasize (spread) to distant parts of the body. They are often more difficult to treat than benign neoplasms and require a multidisciplinary approach, including surgery, radiation therapy, and chemotherapy. Examples of malignant soft tissue neoplasms include sarcomas, such as rhabdomyosarcoma (arising from skeletal muscle), leiomyosarcoma (arising from smooth muscle), and angiosarcoma (arising from blood vessels).
It is important to note that soft tissue neoplasms can occur in any part of the body, and their diagnosis and treatment require a thorough evaluation by a healthcare professional with expertise in this area.
Lomustine is a medical term for a specific antineoplastic agent, which is a type of medication used to treat cancer. It's a nitrosourea compound that is classified as an alkylating agent, meaning it works by preventing the reproduction of cancer cells. Lomustine is used in the treatment of various types of cancer, including brain tumors, Hodgkin's lymphoma, and non-Hodgkin's lymphoma. It's usually administered orally in the form of a capsule. As with any medication, it can have side effects, which can include nausea, vomiting, and lowered blood cell counts.
Synaptophysin is a protein found in the presynaptic vesicles of neurons, which are involved in the release of neurotransmitters during synaptic transmission. It is often used as a marker for neuronal differentiation and is widely expressed in neuroendocrine cells and tumors. Synaptophysin plays a role in the regulation of neurotransmitter release and has been implicated in various neurological disorders, including Alzheimer's disease and synaptic dysfunction-related conditions.
I'm sorry for any confusion, but "Genes, myc" is not a recognized medical term or abbreviation. It seems like there might be a misunderstanding or a missing word in the request. "Myc" could refer to the Myc family of transcription factors that are involved in cell growth and division, and are often deregulated in cancer. However, without more context, it's difficult to provide an accurate definition. If you could provide more information or clarify your question, I would be happy to help further!
In situ hybridization, fluorescence (FISH) is a type of molecular cytogenetic technique used to detect and localize the presence or absence of specific DNA sequences on chromosomes through the use of fluorescent probes. This technique allows for the direct visualization of genetic material at a cellular level, making it possible to identify chromosomal abnormalities such as deletions, duplications, translocations, and other rearrangements.
The process involves denaturing the DNA in the sample to separate the double-stranded molecules into single strands, then adding fluorescently labeled probes that are complementary to the target DNA sequence. The probe hybridizes to the complementary sequence in the sample, and the location of the probe is detected by fluorescence microscopy.
FISH has a wide range of applications in both clinical and research settings, including prenatal diagnosis, cancer diagnosis and monitoring, and the study of gene expression and regulation. It is a powerful tool for identifying genetic abnormalities and understanding their role in human disease.
Antineoplastic combined chemotherapy protocols refer to a treatment plan for cancer that involves the use of more than one antineoplastic (chemotherapy) drug given in a specific sequence and schedule. The combination of drugs is used because they may work better together to destroy cancer cells compared to using a single agent alone. This approach can also help to reduce the likelihood of cancer cells becoming resistant to the treatment.
The choice of drugs, dose, duration, and frequency are determined by various factors such as the type and stage of cancer, patient's overall health, and potential side effects. Combination chemotherapy protocols can be used in various settings, including as a primary treatment, adjuvant therapy (given after surgery or radiation to kill any remaining cancer cells), neoadjuvant therapy (given before surgery or radiation to shrink the tumor), or palliative care (to alleviate symptoms and prolong survival).
It is important to note that while combined chemotherapy protocols can be effective in treating certain types of cancer, they can also cause significant side effects, including nausea, vomiting, hair loss, fatigue, and an increased risk of infection. Therefore, patients undergoing such treatment should be closely monitored and managed by a healthcare team experienced in administering chemotherapy.
Reverse Transcriptase Polymerase Chain Reaction (RT-PCR) is a laboratory technique used in molecular biology to amplify and detect specific DNA sequences. This technique is particularly useful for the detection and quantification of RNA viruses, as well as for the analysis of gene expression.
The process involves two main steps: reverse transcription and polymerase chain reaction (PCR). In the first step, reverse transcriptase enzyme is used to convert RNA into complementary DNA (cDNA) by reading the template provided by the RNA molecule. This cDNA then serves as a template for the PCR amplification step.
In the second step, the PCR reaction uses two primers that flank the target DNA sequence and a thermostable polymerase enzyme to repeatedly copy the targeted cDNA sequence. The reaction mixture is heated and cooled in cycles, allowing the primers to anneal to the template, and the polymerase to extend the new strand. This results in exponential amplification of the target DNA sequence, making it possible to detect even small amounts of RNA or cDNA.
RT-PCR is a sensitive and specific technique that has many applications in medical research and diagnostics, including the detection of viruses such as HIV, hepatitis C virus, and SARS-CoV-2 (the virus that causes COVID-19). It can also be used to study gene expression, identify genetic mutations, and diagnose genetic disorders.
Treatment outcome is a term used to describe the result or effect of medical treatment on a patient's health status. It can be measured in various ways, such as through symptoms improvement, disease remission, reduced disability, improved quality of life, or survival rates. The treatment outcome helps healthcare providers evaluate the effectiveness of a particular treatment plan and make informed decisions about future care. It is also used in clinical research to compare the efficacy of different treatments and improve patient care.
Thiotepa is an antineoplastic (cancer-fighting) drug. It belongs to a class of medications called alkylating agents, which work by interfering with the DNA of cancer cells, preventing them from dividing and growing. Thiotepa is used in the treatment of various types of cancers, including breast, ovarian, and bladder cancer.
It may be administered intravenously (into a vein), intravesically (into the bladder), or intrathecally (into the spinal cord). The specific dosage and duration of treatment will depend on the type and stage of cancer being treated, as well as the patient's overall health status.
Like all chemotherapy drugs, thiotepa can have significant side effects, including nausea, vomiting, hair loss, and a weakened immune system. It is important for patients to discuss these potential risks with their healthcare provider before starting treatment.
A neoplasm is a tumor or growth that is formed by an abnormal and excessive proliferation of cells, which can be benign or malignant. Neoplasm proteins are therefore any proteins that are expressed or produced in these neoplastic cells. These proteins can play various roles in the development, progression, and maintenance of neoplasms.
Some neoplasm proteins may contribute to the uncontrolled cell growth and division seen in cancer, such as oncogenic proteins that promote cell cycle progression or inhibit apoptosis (programmed cell death). Others may help the neoplastic cells evade the immune system, allowing them to proliferate undetected. Still others may be involved in angiogenesis, the formation of new blood vessels that supply the tumor with nutrients and oxygen.
Neoplasm proteins can also serve as biomarkers for cancer diagnosis, prognosis, or treatment response. For example, the presence or level of certain neoplasm proteins in biological samples such as blood or tissue may indicate the presence of a specific type of cancer, help predict the likelihood of cancer recurrence, or suggest whether a particular therapy will be effective.
Overall, understanding the roles and behaviors of neoplasm proteins can provide valuable insights into the biology of cancer and inform the development of new diagnostic and therapeutic strategies.
Neoplastic cell transformation is a process in which a normal cell undergoes genetic alterations that cause it to become cancerous or malignant. This process involves changes in the cell's DNA that result in uncontrolled cell growth and division, loss of contact inhibition, and the ability to invade surrounding tissues and metastasize (spread) to other parts of the body.
Neoplastic transformation can occur as a result of various factors, including genetic mutations, exposure to carcinogens, viral infections, chronic inflammation, and aging. These changes can lead to the activation of oncogenes or the inactivation of tumor suppressor genes, which regulate cell growth and division.
The transformation of normal cells into cancerous cells is a complex and multi-step process that involves multiple genetic and epigenetic alterations. It is characterized by several hallmarks, including sustained proliferative signaling, evasion of growth suppressors, resistance to cell death, enabling replicative immortality, induction of angiogenesis, activation of invasion and metastasis, reprogramming of energy metabolism, and evading immune destruction.
Neoplastic cell transformation is a fundamental concept in cancer biology and is critical for understanding the molecular mechanisms underlying cancer development and progression. It also has important implications for cancer diagnosis, prognosis, and treatment, as identifying the specific genetic alterations that underlie neoplastic transformation can help guide targeted therapies and personalized medicine approaches.
Cell division is the process by which a single eukaryotic cell (a cell with a true nucleus) divides into two identical daughter cells. This complex process involves several stages, including replication of DNA, separation of chromosomes, and division of the cytoplasm. There are two main types of cell division: mitosis and meiosis.
Mitosis is the type of cell division that results in two genetically identical daughter cells. It is a fundamental process for growth, development, and tissue repair in multicellular organisms. The stages of mitosis include prophase, prometaphase, metaphase, anaphase, and telophase, followed by cytokinesis, which divides the cytoplasm.
Meiosis, on the other hand, is a type of cell division that occurs in the gonads (ovaries and testes) during the production of gametes (sex cells). Meiosis results in four genetically unique daughter cells, each with half the number of chromosomes as the parent cell. This process is essential for sexual reproduction and genetic diversity. The stages of meiosis include meiosis I and meiosis II, which are further divided into prophase, prometaphase, metaphase, anaphase, and telophase.
In summary, cell division is the process by which a single cell divides into two daughter cells, either through mitosis or meiosis. This process is critical for growth, development, tissue repair, and sexual reproduction in multicellular organisms.
Transcription factors are proteins that play a crucial role in regulating gene expression by controlling the transcription of DNA to messenger RNA (mRNA). They function by binding to specific DNA sequences, known as response elements, located in the promoter region or enhancer regions of target genes. This binding can either activate or repress the initiation of transcription, depending on the properties and interactions of the particular transcription factor. Transcription factors often act as part of a complex network of regulatory proteins that determine the precise spatiotemporal patterns of gene expression during development, differentiation, and homeostasis in an organism.
Local neoplasm recurrence is the return or regrowth of a tumor in the same location where it was originally removed or treated. This means that cancer cells have survived the initial treatment and started to grow again in the same area. It's essential to monitor and detect any local recurrence as early as possible, as it can affect the prognosis and may require additional treatment.
Ectoderm is the outermost of the three primary germ layers in a developing embryo, along with the endoderm and mesoderm. The ectoderm gives rise to the outer covering of the body, including the skin, hair, nails, glands, and the nervous system, which includes the brain, spinal cord, and peripheral nerves. It also forms the lining of the mouth, anus, nose, and ears. Essentially, the ectoderm is responsible for producing all the epidermal structures and the neural crest cells that contribute to various derivatives such as melanocytes, adrenal medulla, smooth muscle, and peripheral nervous system components.
Polymerase Chain Reaction (PCR) is a laboratory technique used to amplify specific regions of DNA. It enables the production of thousands to millions of copies of a particular DNA sequence in a rapid and efficient manner, making it an essential tool in various fields such as molecular biology, medical diagnostics, forensic science, and research.
The PCR process involves repeated cycles of heating and cooling to separate the DNA strands, allow primers (short sequences of single-stranded DNA) to attach to the target regions, and extend these primers using an enzyme called Taq polymerase, resulting in the exponential amplification of the desired DNA segment.
In a medical context, PCR is often used for detecting and quantifying specific pathogens (viruses, bacteria, fungi, or parasites) in clinical samples, identifying genetic mutations or polymorphisms associated with diseases, monitoring disease progression, and evaluating treatment effectiveness.
Messenger RNA (mRNA) is a type of RNA (ribonucleic acid) that carries genetic information copied from DNA in the form of a series of three-base code "words," each of which specifies a particular amino acid. This information is used by the cell's machinery to construct proteins, a process known as translation. After being transcribed from DNA, mRNA travels out of the nucleus to the ribosomes in the cytoplasm where protein synthesis occurs. Once the protein has been synthesized, the mRNA may be degraded and recycled. Post-transcriptional modifications can also occur to mRNA, such as alternative splicing and addition of a 5' cap and a poly(A) tail, which can affect its stability, localization, and translation efficiency.
Wilms tumor, also known as nephroblastoma, is a type of kidney cancer that primarily affects children. It occurs in the cells of the developing kidneys and is named after Dr. Max Wilms, who first described this type of tumor in 1899. Wilms tumor typically develops before the age of 5, with most cases occurring in children under the age of 3.
The medical definition of Wilms tumor is:
A malignant, embryonal kidney tumor originating from the metanephric blastema, which is a mass of undifferentiated cells in the developing kidney. Wilms tumor is characterized by its rapid growth and potential for spread (metastasis) to other parts of the body, particularly the lungs and liver. The tumor usually presents as a large, firm, and irregular mass in the abdomen, and it may be associated with various symptoms such as abdominal pain, swelling, or blood in the urine.
Wilms tumor is typically treated with a combination of surgery, chemotherapy, and radiation therapy. The prognosis for children with Wilms tumor has improved significantly over the past few decades due to advances in treatment methods and early detection.
Monoclonal antibodies are a type of antibody that are identical because they are produced by a single clone of cells. They are laboratory-produced molecules that act like human antibodies in the immune system. They can be designed to attach to specific proteins found on the surface of cancer cells, making them useful for targeting and treating cancer. Monoclonal antibodies can also be used as a therapy for other diseases, such as autoimmune disorders and inflammatory conditions.
Monoclonal antibodies are produced by fusing a single type of immune cell, called a B cell, with a tumor cell to create a hybrid cell, or hybridoma. This hybrid cell is then able to replicate indefinitely, producing a large number of identical copies of the original antibody. These antibodies can be further modified and engineered to enhance their ability to bind to specific targets, increase their stability, and improve their effectiveness as therapeutic agents.
Monoclonal antibodies have several mechanisms of action in cancer therapy. They can directly kill cancer cells by binding to them and triggering an immune response. They can also block the signals that promote cancer growth and survival. Additionally, monoclonal antibodies can be used to deliver drugs or radiation directly to cancer cells, increasing the effectiveness of these treatments while minimizing their side effects on healthy tissues.
Monoclonal antibodies have become an important tool in modern medicine, with several approved for use in cancer therapy and other diseases. They are continuing to be studied and developed as a promising approach to treating a wide range of medical conditions.
Cell differentiation is the process by which a less specialized cell, or stem cell, becomes a more specialized cell type with specific functions and structures. This process involves changes in gene expression, which are regulated by various intracellular signaling pathways and transcription factors. Differentiation results in the development of distinct cell types that make up tissues and organs in multicellular organisms. It is a crucial aspect of embryonic development, tissue repair, and maintenance of homeostasis in the body.
Immunoenzyme techniques are a group of laboratory methods used in immunology and clinical chemistry that combine the specificity of antibody-antigen reactions with the sensitivity and amplification capabilities of enzyme reactions. These techniques are primarily used for the detection, quantitation, or identification of various analytes (such as proteins, hormones, drugs, viruses, or bacteria) in biological samples.
In immunoenzyme techniques, an enzyme is linked to an antibody or antigen, creating a conjugate. This conjugate then interacts with the target analyte in the sample, forming an immune complex. The presence and amount of this immune complex can be visualized or measured by detecting the enzymatic activity associated with it.
There are several types of immunoenzyme techniques, including:
1. Enzyme-linked Immunosorbent Assay (ELISA): A widely used method for detecting and quantifying various analytes in a sample. In ELISA, an enzyme is attached to either the capture antibody or the detection antibody. After the immune complex formation, a substrate is added that reacts with the enzyme, producing a colored product that can be measured spectrophotometrically.
2. Immunoblotting (Western blot): A method used for detecting specific proteins in a complex mixture, such as a protein extract from cells or tissues. In this technique, proteins are separated by gel electrophoresis and transferred to a membrane, where they are probed with an enzyme-conjugated antibody directed against the target protein.
3. Immunohistochemistry (IHC): A method used for detecting specific antigens in tissue sections or cells. In IHC, an enzyme-conjugated primary or secondary antibody is applied to the sample, and the presence of the antigen is visualized using a chromogenic substrate that produces a colored product at the site of the antigen-antibody interaction.
4. Immunofluorescence (IF): A method used for detecting specific antigens in cells or tissues by employing fluorophore-conjugated antibodies. The presence of the antigen is visualized using a fluorescence microscope.
5. Enzyme-linked immunosorbent assay (ELISA): A method used for detecting and quantifying specific antigens or antibodies in liquid samples, such as serum or culture supernatants. In ELISA, an enzyme-conjugated detection antibody is added after the immune complex formation, and a substrate is added that reacts with the enzyme to produce a colored product that can be measured spectrophotometrically.
These techniques are widely used in research and diagnostic laboratories for various applications, including protein characterization, disease diagnosis, and monitoring treatment responses.
Gene amplification is a process in molecular biology where a specific gene or set of genes are copied multiple times, leading to an increased number of copies of that gene within the genome. This can occur naturally in cells as a response to various stimuli, such as stress or exposure to certain chemicals, but it can also be induced artificially through laboratory techniques for research purposes.
In cancer biology, gene amplification is often associated with tumor development and progression, where the amplified genes can contribute to increased cell growth, survival, and drug resistance. For example, the overamplification of the HER2/neu gene in breast cancer has been linked to more aggressive tumors and poorer patient outcomes.
In diagnostic and research settings, gene amplification techniques like polymerase chain reaction (PCR) are commonly used to detect and analyze specific genes or genetic sequences of interest. These methods allow researchers to quickly and efficiently generate many copies of a particular DNA sequence, facilitating downstream analysis and detection of low-abundance targets.
Neoplastic gene expression regulation refers to the processes that control the production of proteins and other molecules from genes in neoplastic cells, or cells that are part of a tumor or cancer. In a normal cell, gene expression is tightly regulated to ensure that the right genes are turned on or off at the right time. However, in cancer cells, this regulation can be disrupted, leading to the overexpression or underexpression of certain genes.
Neoplastic gene expression regulation can be affected by a variety of factors, including genetic mutations, epigenetic changes, and signals from the tumor microenvironment. These changes can lead to the activation of oncogenes (genes that promote cancer growth and development) or the inactivation of tumor suppressor genes (genes that prevent cancer).
Understanding neoplastic gene expression regulation is important for developing new therapies for cancer, as targeting specific genes or pathways involved in this process can help to inhibit cancer growth and progression.
Experimental neoplasms refer to abnormal growths or tumors that are induced and studied in a controlled laboratory setting, typically in animals or cell cultures. These studies are conducted to understand the fundamental mechanisms of cancer development, progression, and potential treatment strategies. By manipulating various factors such as genetic mutations, environmental exposures, and pharmacological interventions, researchers can gain valuable insights into the complex processes underlying neoplasm formation and identify novel targets for cancer therapy. It is important to note that experimental neoplasms may not always accurately represent human cancers, and further research is needed to translate these findings into clinically relevant applications.
Tumor suppressor genes are a type of gene that helps to regulate and prevent cells from growing and dividing too rapidly or in an uncontrolled manner. They play a critical role in preventing the formation of tumors and cancer. When functioning properly, tumor suppressor genes help to repair damaged DNA, control the cell cycle, and trigger programmed cell death (apoptosis) when necessary. However, when these genes are mutated or altered, they can lose their ability to function correctly, leading to uncontrolled cell growth and the development of tumors. Examples of tumor suppressor genes include TP53, BRCA1, and BRCA2.
Molecular sequence data refers to the specific arrangement of molecules, most commonly nucleotides in DNA or RNA, or amino acids in proteins, that make up a biological macromolecule. This data is generated through laboratory techniques such as sequencing, and provides information about the exact order of the constituent molecules. This data is crucial in various fields of biology, including genetics, evolution, and molecular biology, allowing for comparisons between different organisms, identification of genetic variations, and studies of gene function and regulation.
A base sequence in the context of molecular biology refers to the specific order of nucleotides in a DNA or RNA molecule. In DNA, these nucleotides are adenine (A), guanine (G), cytosine (C), and thymine (T). In RNA, uracil (U) takes the place of thymine. The base sequence contains genetic information that is transcribed into RNA and ultimately translated into proteins. It is the exact order of these bases that determines the genetic code and thus the function of the DNA or RNA molecule.
A needle biopsy is a medical procedure in which a thin, hollow needle is used to remove a small sample of tissue from a suspicious or abnormal area of the body. The tissue sample is then examined under a microscope to check for cancer cells or other abnormalities. Needle biopsies are often used to diagnose lumps or masses that can be felt through the skin, but they can also be guided by imaging techniques such as ultrasound, CT scan, or MRI to reach areas that cannot be felt. There are several types of needle biopsy procedures, including fine-needle aspiration (FNA) and core needle biopsy. FNA uses a thin needle and gentle suction to remove fluid and cells from the area, while core needle biopsy uses a larger needle to remove a small piece of tissue. The type of needle biopsy used depends on the location and size of the abnormal area, as well as the reason for the procedure.
Disease progression is the worsening or advancement of a medical condition over time. It refers to the natural course of a disease, including its development, the severity of symptoms and complications, and the impact on the patient's overall health and quality of life. Understanding disease progression is important for developing appropriate treatment plans, monitoring response to therapy, and predicting outcomes.
The rate of disease progression can vary widely depending on the type of medical condition, individual patient factors, and the effectiveness of treatment. Some diseases may progress rapidly over a short period of time, while others may progress more slowly over many years. In some cases, disease progression may be slowed or even halted with appropriate medical interventions, while in other cases, the progression may be inevitable and irreversible.
In clinical practice, healthcare providers closely monitor disease progression through regular assessments, imaging studies, and laboratory tests. This information is used to guide treatment decisions and adjust care plans as needed to optimize patient outcomes and improve quality of life.
Doxorubicin is a type of chemotherapy medication known as an anthracycline. It works by interfering with the DNA in cancer cells, which prevents them from growing and multiplying. Doxorubicin is used to treat a wide variety of cancers, including leukemia, lymphoma, breast cancer, lung cancer, ovarian cancer, and many others. It may be given alone or in combination with other chemotherapy drugs.
Doxorubicin is usually administered through a vein (intravenously) and can cause side effects such as nausea, vomiting, hair loss, mouth sores, and increased risk of infection. It can also cause damage to the heart muscle, which can lead to heart failure in some cases. For this reason, doctors may monitor patients' heart function closely while they are receiving doxorubicin treatment.
It is important for patients to discuss the potential risks and benefits of doxorubicin therapy with their healthcare provider before starting treatment.
A carcinoid tumor is a type of slow-growing neuroendocrine tumor that usually originates in the digestive tract, particularly in the small intestine. These tumors can also arise in other areas such as the lungs, appendix, and rarely in other organs. Carcinoid tumors develop from cells of the diffuse endocrine system (also known as the neuroendocrine system) that are capable of producing hormones or biologically active amines.
Carcinoid tumors can produce and release various hormones and bioactive substances, such as serotonin, histamine, bradykinins, prostaglandins, and tachykinins, which can lead to a variety of symptoms. The most common syndrome associated with carcinoid tumors is the carcinoid syndrome, characterized by flushing, diarrhea, abdominal cramping, and wheezing or difficulty breathing.
Carcinoid tumors are typically classified as functional or nonfunctional based on whether they produce and secrete hormones that cause symptoms. Functional carcinoid tumors account for approximately 30% of cases and can lead to the development of carcinoid syndrome, while nonfunctional tumors do not produce significant amounts of hormones and are often asymptomatic until they grow large enough to cause local or distant complications.
Treatment options for carcinoid tumors depend on the location, size, and extent of the tumor, as well as whether it is functional or nonfunctional. Treatment may include surgery, medications (such as somatostatin analogs, chemotherapy, or targeted therapies), and radiation therapy. Regular follow-up with imaging studies and biochemical tests is essential to monitor for recurrence and assess treatment response.
"Gene rearrangement" is a process that involves the alteration of the order, orientation, or copy number of genes or gene segments within an organism's genome. This natural mechanism plays a crucial role in generating diversity and specificity in the immune system, particularly in vertebrates.
In the context of the immune system, gene rearrangement occurs during the development of B-cells and T-cells, which are responsible for adaptive immunity. The process involves breaking and rejoining DNA segments that encode antigen recognition sites, resulting in a unique combination of gene segments and creating a vast array of possible antigen receptors.
There are two main types of gene rearrangement:
1. V(D)J recombination: This process occurs in both B-cells and T-cells. It involves the recombination of variable (V), diversity (D), and joining (J) gene segments to form a functional antigen receptor gene. In humans, there are multiple copies of V, D, and J segments for each antigen receptor gene, allowing for a vast number of possible combinations.
2. Class switch recombination: This process occurs only in mature B-cells after antigen exposure. It involves the replacement of the constant (C) region of the immunoglobulin heavy chain gene with another C region, resulting in the production of different isotypes of antibodies (IgG, IgA, or IgE) that have distinct effector functions while maintaining the same antigen specificity.
These processes contribute to the generation of a diverse repertoire of antigen receptors, allowing the immune system to recognize and respond effectively to a wide range of pathogens.
Survival analysis is a branch of statistics that deals with the analysis of time to event data. It is used to estimate the time it takes for a certain event of interest to occur, such as death, disease recurrence, or treatment failure. The event of interest is called the "failure" event, and survival analysis estimates the probability of not experiencing the failure event until a certain point in time, also known as the "survival" probability.
Survival analysis can provide important information about the effectiveness of treatments, the prognosis of patients, and the identification of risk factors associated with the event of interest. It can handle censored data, which is common in medical research where some participants may drop out or be lost to follow-up before the event of interest occurs.
Survival analysis typically involves estimating the survival function, which describes the probability of surviving beyond a certain time point, as well as hazard functions, which describe the instantaneous rate of failure at a given time point. Other important concepts in survival analysis include median survival times, restricted mean survival times, and various statistical tests to compare survival curves between groups.
Neoplasms are abnormal growths of cells or tissues in the body that serve no physiological function. They can be benign (non-cancerous) or malignant (cancerous). Benign neoplasms are typically slow growing and do not spread to other parts of the body, while malignant neoplasms are aggressive, invasive, and can metastasize to distant sites.
Neoplasms occur when there is a dysregulation in the normal process of cell division and differentiation, leading to uncontrolled growth and accumulation of cells. This can result from genetic mutations or other factors such as viral infections, environmental exposures, or hormonal imbalances.
Neoplasms can develop in any organ or tissue of the body and can cause various symptoms depending on their size, location, and type. Treatment options for neoplasms include surgery, radiation therapy, chemotherapy, immunotherapy, and targeted therapy, among others.
A biopsy is a medical procedure in which a small sample of tissue is taken from the body to be examined under a microscope for the presence of disease. This can help doctors diagnose and monitor various medical conditions, such as cancer, infections, or autoimmune disorders. The type of biopsy performed will depend on the location and nature of the suspected condition. Some common types of biopsies include:
1. Incisional biopsy: In this procedure, a surgeon removes a piece of tissue from an abnormal area using a scalpel or other surgical instrument. This type of biopsy is often used when the lesion is too large to be removed entirely during the initial biopsy.
2. Excisional biopsy: An excisional biopsy involves removing the entire abnormal area, along with a margin of healthy tissue surrounding it. This technique is typically employed for smaller lesions or when cancer is suspected.
3. Needle biopsy: A needle biopsy uses a thin, hollow needle to extract cells or fluid from the body. There are two main types of needle biopsies: fine-needle aspiration (FNA) and core needle biopsy. FNA extracts loose cells, while a core needle biopsy removes a small piece of tissue.
4. Punch biopsy: In a punch biopsy, a round, sharp tool is used to remove a small cylindrical sample of skin tissue. This type of biopsy is often used for evaluating rashes or other skin abnormalities.
5. Shave biopsy: During a shave biopsy, a thin slice of tissue is removed from the surface of the skin using a sharp razor-like instrument. This technique is typically used for superficial lesions or growths on the skin.
After the biopsy sample has been collected, it is sent to a laboratory where a pathologist will examine the tissue under a microscope and provide a diagnosis based on their findings. The results of the biopsy can help guide further treatment decisions and determine the best course of action for managing the patient's condition.
DNA-binding proteins are a type of protein that have the ability to bind to DNA (deoxyribonucleic acid), the genetic material of organisms. These proteins play crucial roles in various biological processes, such as regulation of gene expression, DNA replication, repair and recombination.
The binding of DNA-binding proteins to specific DNA sequences is mediated by non-covalent interactions, including electrostatic, hydrogen bonding, and van der Waals forces. The specificity of binding is determined by the recognition of particular nucleotide sequences or structural features of the DNA molecule.
DNA-binding proteins can be classified into several categories based on their structure and function, such as transcription factors, histones, and restriction enzymes. Transcription factors are a major class of DNA-binding proteins that regulate gene expression by binding to specific DNA sequences in the promoter region of genes and recruiting other proteins to modulate transcription. Histones are DNA-binding proteins that package DNA into nucleosomes, the basic unit of chromatin structure. Restriction enzymes are DNA-binding proteins that recognize and cleave specific DNA sequences, and are widely used in molecular biology research and biotechnology applications.
Prognosis is a medical term that refers to the prediction of the likely outcome or course of a disease, including the chances of recovery or recurrence, based on the patient's symptoms, medical history, physical examination, and diagnostic tests. It is an important aspect of clinical decision-making and patient communication, as it helps doctors and patients make informed decisions about treatment options, set realistic expectations, and plan for future care.
Prognosis can be expressed in various ways, such as percentages, categories (e.g., good, fair, poor), or survival rates, depending on the nature of the disease and the available evidence. However, it is important to note that prognosis is not an exact science and may vary depending on individual factors, such as age, overall health status, and response to treatment. Therefore, it should be used as a guide rather than a definitive forecast.
Tumor suppressor protein p53, also known as p53 or tumor protein p53, is a nuclear phosphoprotein that plays a crucial role in preventing cancer development and maintaining genomic stability. It does so by regulating the cell cycle and acting as a transcription factor for various genes involved in apoptosis (programmed cell death), DNA repair, and cell senescence (permanent cell growth arrest).
In response to cellular stress, such as DNA damage or oncogene activation, p53 becomes activated and accumulates in the nucleus. Activated p53 can then bind to specific DNA sequences and promote the transcription of target genes that help prevent the proliferation of potentially cancerous cells. These targets include genes involved in cell cycle arrest (e.g., CDKN1A/p21), apoptosis (e.g., BAX, PUMA), and DNA repair (e.g., GADD45).
Mutations in the TP53 gene, which encodes p53, are among the most common genetic alterations found in human cancers. These mutations often lead to a loss or reduction of p53's tumor suppressive functions, allowing cancer cells to proliferate uncontrollably and evade apoptosis. As a result, p53 has been referred to as "the guardian of the genome" due to its essential role in preventing tumorigenesis.
Dactinomycin is an antineoplastic antibiotic, which means it is used to treat cancer. It is specifically used to treat certain types of testicular cancer, Wilms' tumor (a type of kidney cancer that occurs in children), and some gestational trophoblastic tumors (a type of tumor that can develop in the uterus after pregnancy). Dactinomycin works by interfering with the DNA in cancer cells, which prevents them from dividing and growing. It is often used in combination with other chemotherapy drugs as part of a treatment regimen.
Dactinomycin is administered intravenously (through an IV) and its use is usually limited to hospitals or specialized cancer treatment centers due to the need for careful monitoring during administration. Common side effects include nausea, vomiting, and hair loss. More serious side effects can include bone marrow suppression, which can lead to an increased risk of infection, and tissue damage at the site where the drug is injected. Dactinomycin can also cause severe allergic reactions in some people.
It's important to note that dactinomycin should only be used under the supervision of a qualified healthcare professional, as its use requires careful monitoring and management of potential side effects.
Disease-free survival (DFS) is a term used in medical research and clinical practice, particularly in the field of oncology. It refers to the length of time after primary treatment for a cancer during which no evidence of the disease can be found. This means that the patient shows no signs or symptoms of the cancer, and any imaging studies or other tests do not reveal any tumors or other indications of the disease.
DFS is often used as an important endpoint in clinical trials to evaluate the effectiveness of different treatments for cancer. By measuring the length of time until the cancer recurs or a new cancer develops, researchers can get a better sense of how well a particular treatment is working and whether it is improving patient outcomes.
It's important to note that DFS is not the same as overall survival (OS), which refers to the length of time from primary treatment until death from any cause. While DFS can provide valuable information about the effectiveness of cancer treatments, it does not necessarily reflect the impact of those treatments on patients' overall survival.
A cell line is a culture of cells that are grown in a laboratory for use in research. These cells are usually taken from a single cell or group of cells, and they are able to divide and grow continuously in the lab. Cell lines can come from many different sources, including animals, plants, and humans. They are often used in scientific research to study cellular processes, disease mechanisms, and to test new drugs or treatments. Some common types of human cell lines include HeLa cells (which come from a cancer patient named Henrietta Lacks), HEK293 cells (which come from embryonic kidney cells), and HUVEC cells (which come from umbilical vein endothelial cells). It is important to note that cell lines are not the same as primary cells, which are cells that are taken directly from a living organism and have not been grown in the lab.
Medical survival rate is a statistical measure used to determine the percentage of patients who are still alive for a specific period of time after their diagnosis or treatment for a certain condition or disease. It is often expressed as a five-year survival rate, which refers to the proportion of people who are alive five years after their diagnosis. Survival rates can be affected by many factors, including the stage of the disease at diagnosis, the patient's age and overall health, the effectiveness of treatment, and other health conditions that the patient may have. It is important to note that survival rates are statistical estimates and do not necessarily predict an individual patient's prognosis.
Glial Fibrillary Acidic Protein (GFAP) is a type of intermediate filament protein that is primarily found in astrocytes, which are a type of star-shaped glial cells in the central nervous system (CNS). These proteins play an essential role in maintaining the structural integrity and stability of astrocytes. They also participate in various cellular processes such as responding to injury, providing support to neurons, and regulating the extracellular environment.
GFAP is often used as a marker for astrocytic activation or reactivity, which can occur in response to CNS injuries, neuroinflammation, or neurodegenerative diseases. Elevated GFAP levels in cerebrospinal fluid (CSF) or blood can indicate astrocyte damage or dysfunction and are associated with several neurological conditions, including traumatic brain injury, stroke, multiple sclerosis, Alzheimer's disease, and Alexander's disease.
Karyotyping is a medical laboratory test used to study the chromosomes in a cell. It involves obtaining a sample of cells from a patient, usually from blood or bone marrow, and then staining the chromosomes so they can be easily seen under a microscope. The chromosomes are then arranged in pairs based on their size, shape, and other features to create a karyotype. This visual representation allows for the identification and analysis of any chromosomal abnormalities, such as extra or missing chromosomes, or structural changes like translocations or inversions. These abnormalities can provide important information about genetic disorders, diseases, and developmental problems.
Neuroendocrine tumors (NETs) are a diverse group of neoplasms that arise from cells of the neuroendocrine system, which is composed of dispersed neuroendocrine cells throughout the body, often in close association with nerves and blood vessels. These cells have the ability to produce and secrete hormones or hormone-like substances in response to various stimuli. NETs can occur in a variety of organs, including the lungs, pancreas, small intestine, colon, rectum, stomach, and thyroid gland, as well as in some less common sites such as the thymus, adrenal glands, and nervous system.
NETs can be functional or nonfunctional, depending on whether they produce and secrete hormones or hormone-like substances that cause specific symptoms related to hormonal excess. Functional NETs may give rise to a variety of clinical syndromes, such as carcinoid syndrome, Zollinger-Ellison syndrome, pancreatic neuroendocrine tumor syndrome (also known as Verner-Morrison or WDHA syndrome), and others. Nonfunctional NETs are more likely to present with symptoms related to the size and location of the tumor, such as abdominal pain, intestinal obstruction, or bleeding.
The diagnosis of NETs typically involves a combination of imaging studies, biochemical tests (e.g., measurement of serum hormone levels), and histopathological examination of tissue samples obtained through biopsy or surgical resection. Treatment options depend on the type, location, stage, and grade of the tumor, as well as the presence or absence of functional symptoms. They may include surgery, radiation therapy, chemotherapy, targeted therapy, and/or peptide receptor radionuclide therapy (PRRT).
Chromosome aberrations refer to structural and numerical changes in the chromosomes that can occur spontaneously or as a result of exposure to mutagenic agents. These changes can affect the genetic material encoded in the chromosomes, leading to various consequences such as developmental abnormalities, cancer, or infertility.
Structural aberrations include deletions, duplications, inversions, translocations, and rings, which result from breaks and rearrangements of chromosome segments. Numerical aberrations involve changes in the number of chromosomes, such as aneuploidy (extra or missing chromosomes) or polyploidy (multiples of a complete set of chromosomes).
Chromosome aberrations can be detected and analyzed using various cytogenetic techniques, including karyotyping, fluorescence in situ hybridization (FISH), and comparative genomic hybridization (CGH). These methods allow for the identification and characterization of chromosomal changes at the molecular level, providing valuable information for genetic counseling, diagnosis, and research.
A chromosome deletion is a type of genetic abnormality that occurs when a portion of a chromosome is missing or deleted. Chromosomes are thread-like structures located in the nucleus of cells that contain our genetic material, which is organized into genes.
Chromosome deletions can occur spontaneously during the formation of reproductive cells (eggs or sperm) or can be inherited from a parent. They can affect any chromosome and can vary in size, from a small segment to a large portion of the chromosome.
The severity of the symptoms associated with a chromosome deletion depends on the size and location of the deleted segment. In some cases, the deletion may be so small that it does not cause any noticeable symptoms. However, larger deletions can lead to developmental delays, intellectual disabilities, physical abnormalities, and various medical conditions.
Chromosome deletions are typically detected through a genetic test called karyotyping, which involves analyzing the number and structure of an individual's chromosomes. Other more precise tests, such as fluorescence in situ hybridization (FISH) or chromosomal microarray analysis (CMA), may also be used to confirm the diagnosis and identify the specific location and size of the deletion.
Tumor suppressor proteins are a type of regulatory protein that helps control the cell cycle and prevent cells from dividing and growing in an uncontrolled manner. They work to inhibit tumor growth by preventing the formation of tumors or slowing down their progression. These proteins can repair damaged DNA, regulate gene expression, and initiate programmed cell death (apoptosis) if the damage is too severe for repair.
Mutations in tumor suppressor genes, which provide the code for these proteins, can lead to a decrease or loss of function in the resulting protein. This can result in uncontrolled cell growth and division, leading to the formation of tumors and cancer. Examples of tumor suppressor proteins include p53, Rb (retinoblastoma), and BRCA1/2.
In the field of medicine, "time factors" refer to the duration of symptoms or time elapsed since the onset of a medical condition, which can have significant implications for diagnosis and treatment. Understanding time factors is crucial in determining the progression of a disease, evaluating the effectiveness of treatments, and making critical decisions regarding patient care.
For example, in stroke management, "time is brain," meaning that rapid intervention within a specific time frame (usually within 4.5 hours) is essential to administering tissue plasminogen activator (tPA), a clot-busting drug that can minimize brain damage and improve patient outcomes. Similarly, in trauma care, the "golden hour" concept emphasizes the importance of providing definitive care within the first 60 minutes after injury to increase survival rates and reduce morbidity.
Time factors also play a role in monitoring the progression of chronic conditions like diabetes or heart disease, where regular follow-ups and assessments help determine appropriate treatment adjustments and prevent complications. In infectious diseases, time factors are crucial for initiating antibiotic therapy and identifying potential outbreaks to control their spread.
Overall, "time factors" encompass the significance of recognizing and acting promptly in various medical scenarios to optimize patient outcomes and provide effective care.
Proto-oncogene proteins are normal cellular proteins that play crucial roles in various cellular processes, such as signal transduction, cell cycle regulation, and apoptosis (programmed cell death). They are involved in the regulation of cell growth, differentiation, and survival under physiological conditions.
When proto-oncogene proteins undergo mutations or aberrations in their expression levels, they can transform into oncogenic forms, leading to uncontrolled cell growth and division. These altered proteins are then referred to as oncogene products or oncoproteins. Oncogenic mutations can occur due to various factors, including genetic predisposition, environmental exposures, and aging.
Examples of proto-oncogene proteins include:
1. Ras proteins: Involved in signal transduction pathways that regulate cell growth and differentiation. Activating mutations in Ras genes are found in various human cancers.
2. Myc proteins: Regulate gene expression related to cell cycle progression, apoptosis, and metabolism. Overexpression of Myc proteins is associated with several types of cancer.
3. EGFR (Epidermal Growth Factor Receptor): A transmembrane receptor tyrosine kinase that regulates cell proliferation, survival, and differentiation. Mutations or overexpression of EGFR are linked to various malignancies, such as lung cancer and glioblastoma.
4. Src family kinases: Intracellular tyrosine kinases that regulate signal transduction pathways involved in cell proliferation, survival, and migration. Dysregulation of Src family kinases is implicated in several types of cancer.
5. Abl kinases: Cytoplasmic tyrosine kinases that regulate various cellular processes, including cell growth, differentiation, and stress responses. Aberrant activation of Abl kinases, as seen in chronic myelogenous leukemia (CML), leads to uncontrolled cell proliferation.
Understanding the roles of proto-oncogene proteins and their dysregulation in cancer development is essential for developing targeted cancer therapies that aim to inhibit or modulate these aberrant signaling pathways.
The tumor microenvironment (TME) is a complex and dynamic setting that consists of various cellular and non-cellular components, which interact with each other and contribute to the growth, progression, and dissemination of cancer. The TME includes:
1. Cancer cells: These are the malignant cells that grow uncontrollably, invade surrounding tissues, and can spread to distant organs.
2. Stromal cells: These are non-cancerous cells present within the tumor, including fibroblasts, immune cells, adipocytes, and endothelial cells. They play a crucial role in supporting the growth of cancer cells by providing structural and nutritional support, modulating the immune response, and promoting angiogenesis (the formation of new blood vessels).
3. Extracellular matrix (ECM): This is the non-cellular component of the TME, consisting of a network of proteins, glycoproteins, and polysaccharides that provide structural support and regulate cell behavior. The ECM can be remodeled by both cancer and stromal cells, leading to changes in tissue stiffness, architecture, and signaling pathways.
4. Soluble factors: These include various cytokines, chemokines, growth factors, and metabolites that are secreted by both cancer and stromal cells. They can act as signaling molecules, influencing cell behavior, survival, proliferation, and migration.
5. Blood vessels: The formation of new blood vessels (angiogenesis) within the TME is essential for providing nutrients and oxygen to support the growth of cancer cells. The vasculature in the TME is often disorganized, leading to hypoxic (low oxygen) regions and altered drug delivery.
6. Immune cells: The TME contains various immune cell populations, such as tumor-associated macrophages (TAMs), dendritic cells, natural killer (NK) cells, and different subsets of T lymphocytes. These cells can either promote or inhibit the growth and progression of cancer, depending on their phenotype and activation status.
7. Niche: A specific microenvironment within the TME that supports the survival and function of cancer stem cells (CSCs) or tumor-initiating cells. The niche is often characterized by unique cellular components, signaling molecules, and physical properties that contribute to the maintenance and propagation of CSCs.
Understanding the complex interactions between these various components in the TME can provide valuable insights into cancer biology and help inform the development of novel therapeutic strategies.
Western blotting is a laboratory technique used in molecular biology to detect and quantify specific proteins in a mixture of many different proteins. This technique is commonly used to confirm the expression of a protein of interest, determine its size, and investigate its post-translational modifications. The name "Western" blotting distinguishes this technique from Southern blotting (for DNA) and Northern blotting (for RNA).
The Western blotting procedure involves several steps:
1. Protein extraction: The sample containing the proteins of interest is first extracted, often by breaking open cells or tissues and using a buffer to extract the proteins.
2. Separation of proteins by electrophoresis: The extracted proteins are then separated based on their size by loading them onto a polyacrylamide gel and running an electric current through the gel (a process called sodium dodecyl sulfate-polyacrylamide gel electrophoresis or SDS-PAGE). This separates the proteins according to their molecular weight, with smaller proteins migrating faster than larger ones.
3. Transfer of proteins to a membrane: After separation, the proteins are transferred from the gel onto a nitrocellulose or polyvinylidene fluoride (PVDF) membrane using an electric current in a process called blotting. This creates a replica of the protein pattern on the gel but now immobilized on the membrane for further analysis.
4. Blocking: The membrane is then blocked with a blocking agent, such as non-fat dry milk or bovine serum albumin (BSA), to prevent non-specific binding of antibodies in subsequent steps.
5. Primary antibody incubation: A primary antibody that specifically recognizes the protein of interest is added and allowed to bind to its target protein on the membrane. This step may be performed at room temperature or 4°C overnight, depending on the antibody's properties.
6. Washing: The membrane is washed with a buffer to remove unbound primary antibodies.
7. Secondary antibody incubation: A secondary antibody that recognizes the primary antibody (often coupled to an enzyme or fluorophore) is added and allowed to bind to the primary antibody. This step may involve using a horseradish peroxidase (HRP)-conjugated or alkaline phosphatase (AP)-conjugated secondary antibody, depending on the detection method used later.
8. Washing: The membrane is washed again to remove unbound secondary antibodies.
9. Detection: A detection reagent is added to visualize the protein of interest by detecting the signal generated from the enzyme-conjugated or fluorophore-conjugated secondary antibody. This can be done using chemiluminescent, colorimetric, or fluorescent methods.
10. Analysis: The resulting image is analyzed to determine the presence and quantity of the protein of interest in the sample.
Western blotting is a powerful technique for identifying and quantifying specific proteins within complex mixtures. It can be used to study protein expression, post-translational modifications, protein-protein interactions, and more. However, it requires careful optimization and validation to ensure accurate and reproducible results.
Neoplasm metastasis is the spread of cancer cells from the primary site (where the original or primary tumor formed) to other places in the body. This happens when cancer cells break away from the original (primary) tumor and enter the bloodstream or lymphatic system. The cancer cells can then travel to other parts of the body and form new tumors, called secondary tumors or metastases.
Metastasis is a key feature of malignant neoplasms (cancers), and it is one of the main ways that cancer can cause harm in the body. The metastatic tumors may continue to grow and may cause damage to the organs and tissues where they are located. They can also release additional cancer cells into the bloodstream or lymphatic system, leading to further spread of the cancer.
The metastatic tumors are named based on the location where they are found, as well as the type of primary cancer. For example, if a patient has a primary lung cancer that has metastasized to the liver, the metastatic tumor would be called a liver metastasis from lung cancer.
It is important to note that the presence of metastases can significantly affect a person's prognosis and treatment options. In general, metastatic cancer is more difficult to treat than cancer that has not spread beyond its original site. However, there are many factors that can influence a person's prognosis and response to treatment, so it is important for each individual to discuss their specific situation with their healthcare team.
A phenotype is the physical or biochemical expression of an organism's genes, or the observable traits and characteristics resulting from the interaction of its genetic constitution (genotype) with environmental factors. These characteristics can include appearance, development, behavior, and resistance to disease, among others. Phenotypes can vary widely, even among individuals with identical genotypes, due to differences in environmental influences, gene expression, and genetic interactions.
Antineoplastic agents are a class of drugs used to treat malignant neoplasms or cancer. These agents work by inhibiting the growth and proliferation of cancer cells, either by killing them or preventing their division and replication. Antineoplastic agents can be classified based on their mechanism of action, such as alkylating agents, antimetabolites, topoisomerase inhibitors, mitotic inhibitors, and targeted therapy agents.
Alkylating agents work by adding alkyl groups to DNA, which can cause cross-linking of DNA strands and ultimately lead to cell death. Antimetabolites interfere with the metabolic processes necessary for DNA synthesis and replication, while topoisomerase inhibitors prevent the relaxation of supercoiled DNA during replication. Mitotic inhibitors disrupt the normal functioning of the mitotic spindle, which is essential for cell division. Targeted therapy agents are designed to target specific molecular abnormalities in cancer cells, such as mutated oncogenes or dysregulated signaling pathways.
It's important to note that antineoplastic agents can also affect normal cells and tissues, leading to various side effects such as nausea, vomiting, hair loss, and myelosuppression (suppression of bone marrow function). Therefore, the use of these drugs requires careful monitoring and management of their potential adverse effects.
 Primitive neuroectodermal tumor
Primitive neuroectodermal tumor
Gastrointestinal neuroectodermal tumor
Melanotic neuroectodermal tumor of infancy
Central nervous system primitive neuroectodermal tumor
Choroid plexus papilloma
Canine brain tumors
Diktyoma
Tropomyosin receptor kinase C
Oral pigmentation
Neuroectodermal neoplasm
Pericardial effusion
Medulloblastoma
Pineoblastoma
MIR517C
MicroRNA 517c
FET protein family
Chlorotoxin
Palisade (pathology)
Congenital malformations of the dermatoglyphs
Congenital erosive and vesicular dermatosis
Pediatric ependymoma
Otmar Wiestler
Model organism
Animal disease model
SV40
Merkel cell polyomavirus
Atypical teratoid rhabdoid tumor
ERG (gene)
Aleksey Abrikosov
Betulinic acid
Primitive neuroectodermal tumor - Wikipedia
 Primitive Neuroectodermal Tumors: Background, Epidemiology, Clinical Features
Primitive Neuroectodermal Tumors: Background, Epidemiology, Clinical Features
 Thyroid dysfunction in patients with childhood-onset medulloblastoma or primitive neuroectodermal tumor
Thyroid dysfunction in patients with childhood-onset medulloblastoma or primitive neuroectodermal tumor
Neuroectodermal Tumors, Primitive, Peripheral | Profiles RNS
Melanotic Neuroectodermal Tumor of Infancy Differential Diagnoses
 Brain and Nervous System Cancers (for Parents) - East Tenneesee Children's
Brain and Nervous System Cancers (for Parents) - East Tenneesee Children's
Pathology of Nonmesothelial Cancers of the Pleura: Definition, Etiology, Epidemiology
Gigantiform melanotic neuroectodermal tumor of infancy<...
 Primary vaginal Ewing's sarcoma or primitive neuroectodermal tumor in a 17-year-old woman: a case report | Journal of Medical...
Primary vaginal Ewing's sarcoma or primitive neuroectodermal tumor in a 17-year-old woman: a case report | Journal of Medical...
Primary Vulvar Ewing Sarcoma/Primitive Neuroectodermal Tumor: A Report of 2 Cases and Review of the Literature | International...
 Dental strategy with immediate palatal obturator in a patient with melanotic neuroectodermal tumor of infancy: case report
Dental strategy with immediate palatal obturator in a patient with melanotic neuroectodermal tumor of infancy: case report
 Ellen M. Basu, MD, PhD - MSK Pediatric Hematologist-Oncologist
Ellen M. Basu, MD, PhD - MSK Pediatric Hematologist-Oncologist
 Esthesioneuroblastoma
Esthesioneuroblastoma
 Antineoplastons (PDQ®) - NCI
Antineoplastons (PDQ®) - NCI
Recurrence of melanotic neuroectodermal tumor of infancy after 2 months of complete enucleation in a 3 month old child: a case...
A germline mutation of CDKN2A and a novel RPLP1-C19MC fusion detected in a rare melanotic neuroectodermal tumor of infancy: a...
Neuroendoscopic biopsy of ventricular tumors: a multicentric experience in: Neurosurgical Focus Volume 30 Issue 4 (2011)...
Neuroendoscopic biopsy of ventricular tumors: a multicentric experience in: Neurosurgical Focus Volume 30 Issue 4 (2011)...
 Current Oncology | Free Full-Text | Localized Colonic Small-Cell Carcinoma with Pathological Complete Response after...
Current Oncology | Free Full-Text | Localized Colonic Small-Cell Carcinoma with Pathological Complete Response after...
 Sutter Medical Center Advanced Gamma Knife Treatment | Sutter Health
Sutter Medical Center Advanced Gamma Knife Treatment | Sutter Health
 United States Cancer Statistics: Public Information Data 1999 - 2002 Archive
United States Cancer Statistics: Public Information Data 1999 - 2002 Archive
Plus it
 Brain Tumor Treatment: Neuro-Oncology Expertise | UVA Health
Brain Tumor Treatment: Neuro-Oncology Expertise | UVA Health
 Brain Tumors | Boston Children's Hospital
Brain Tumors | Boston Children's Hospital
 Edward C. McCarron, MD| Surgical Oncology | MedStar Health
Edward C. McCarron, MD| Surgical Oncology | MedStar Health
 Malignant Carcinoid Syndrome | Harvard Catalyst Profiles | Harvard Catalyst
Malignant Carcinoid Syndrome | Harvard Catalyst Profiles | Harvard Catalyst
 Volume 185 Issue 4 | Dermatology | Karger Publishers
Volume 185 Issue 4 | Dermatology | Karger Publishers
 Advanced Search Results - Public Health Image Library(PHIL)
Advanced Search Results - Public Health Image Library(PHIL)
 Down-Regulation of miR-101 in Endothelial Cells Promotes Blood Vessel Formation through Reduced Repression of EZH2 | PLOS ONE
Down-Regulation of miR-101 in Endothelial Cells Promotes Blood Vessel Formation through Reduced Repression of EZH2 | PLOS ONE
 Ewing Sarcoma: Diagnosis, Treatment, Research and Support
Ewing Sarcoma: Diagnosis, Treatment, Research and Support
PNET10
- PNET belongs to the Ewing family of tumors. (wikipedia.org)
- citation needed] The peripheral PNET (pPNET) is now thought to be virtually identical to Ewing sarcoma: "Current evidence indicates that both Ewing's sarcoma and PNET have a similar neural phenotype and, because they share an identical chromosome translocation, they should be viewed as the same tumor, differing only in their degree of neural differentiation. (wikipedia.org)
- After the diagnosis of a CNS PNET is confirmed, management includes neoadjuvant chemotherapy and radiation (to reduce tumor size burden), complete surgical resection with confirmed negative margins, and/or additional adjuvant post-surgical chemotherapy. (wikipedia.org)
- We investigated the clinical characteristics of patients who developed thyroid dysfunction and evaluated the risk factors for hypothyroidism following radiotherapy and chemotherapy in pediatric patients with medulloblastoma or primitive neuroectodermal tumor (PNET). (e-apem.org)
- Medulloblastoma and primitive neuroectodermal tumor (PNET) account for approximately 15%, and 3% of pediatric malignant tumors of the central nervous system (CNS), respectively [ 1 ]. (e-apem.org)
- Ewing sarcoma/primitive neuroectodermal tumor (ES/PNET) family of tumor is a very aggressive malignant round cell tumor characterized by translocations involving EWS-FLI1 genes. (bmj.com)
- Ewing sarcoma/PNET should be considered in the differential diagnosis of any undifferentiated tumors involving the lower gynecologic tract and all axillary tests including molecular tests should be performed for correct diagnosis because prolonged survival is possible for this dreadful disease after complete surgical resection, followed by adjuvant therapy. (bmj.com)
- Pathologists have long known that Ewing sarcoma looks very similar to an even rarer soft tissue tumor called primitive neuroectodermal tumor ( PNET ). (sarcomahelp.org)
- Background: There is evidence that exposure to chlorinated solvents may be associated with childhood medulloblastoma and primitive neuroectodermal tumor (M/PNET) risk. (cdc.gov)
- Medulloblastoma to be considered a type of Primitive Neuroectodermal Tumors (PNET), but that term isn't used anymore. (cincinnatichildrens.org)
Primitive neuroectodermal26
- Primitive neuroectodermal tumor is a malignant (cancerous) neural crest tumor. (wikipedia.org)
- Medulloblastoma Ependymoma Ewing family of tumors "primitive neuroectodermal tumor" at Dorland's Medical Dictionary Smoll, N. R. (2012). (wikipedia.org)
- Primitive neuroectodermal tumors (PNETs) are a group of highly malignant tumors composed of small round cells of neuroectodermal origin that affect soft tissue and bone. (medscape.com)
- Primitive neuroectodermal tumors (PNETs) exhibit great diversity in their clinical manifestations and pathologic similarities with other small round cell tumors. (medscape.com)
- Peripheral primitive neuroectodermal tumors (pPNETS) are the focus of this review. (medscape.com)
- Photomicrograph shows characteristic cytoplasmic CD-99 staining in peripheral primitive neuroectodermal tumor. (medscape.com)
- peripheral primitive neuroectodermal tumors (pPNETs) and Ewing family of tumors (EFTs) are often referred to interchangeably in the literature. (medscape.com)
- [ 3 ] Generally, Ewing family of tumors (EFTs) and peripheral primitive neuroectodermal tumors (pPNETs) represent different manifestations of the same tumor and have similar genetic alterations. (medscape.com)
- Ewing sarcoma, however, is more common in bone, while peripheral primitive neuroectodermal tumors (pPNETs) are more common in soft tissues. (medscape.com)
- Based on molecular cytogenetic analysis, both EFTs and peripheral primitive neuroectodermal tumors (pPNETs) are known to share the same reciprocal translocations, most commonly between chromosomes 11 and 22. (medscape.com)
- Peripheral primitive neuroectodermal tumors (pPNETs) often exhibit aggressive clinical behavior, with worse outcomes than other small, round cell tumors. (medscape.com)
- The incidence of peripheral primitive neuroectodermal tumors (pPNETs) is likely underreported in the literature because recent diagnostic advances have allowed these tumors to be distinguished from other small, poorly differentiated, round cell tumors. (medscape.com)
- Although peripheral primitive neuroectodermal tumors (pPNETs) are exceedingly rare, the annual incidence of tumors from the larger Ewing family of tumors (EFTs) from birth to age 20 years is 2.9 per million population. (medscape.com)
- In most large series published to date, peripheral primitive neuroectodermal tumors (pPNETs) usually present in the second decade of life, with a slight male preponderance. (medscape.com)
- Most peripheral primitive neuroectodermal tumors (pPNETs) manifest in the thoracopulmonary region (Askin tumor), pelvis, abdomen, and extremities. (medscape.com)
- Cole M, Parajuli S, Laske D, Goldstein L, Morrison T, Mukherjee A, Tumelty K, Tetzlaff E, von Mehren M, Inniss S. Peripheral primitive neuroectodermal tumor of the dura in a 51-year-old woman following intensive treatment for breast cancer. (jefferson.edu)
- Sacral intraspinal extradural primitive neuroectodermal tumor. (jefferson.edu)
- The histologic appearance of MNTI is usually that of a small, dark, cell neoplasm suggestive of neuroblastoma, rhabdomyosarcoma, Ewing sarcoma, lymphoma, desmoplastic small round cell tumor, and peripheral primitive neuroectodermal tumor. (medscape.com)
- Primitive neuroectodermal tumor of the cervix uteri: a case report -- changing concepts in therapy. (nih.gov)
- Treatment of a supratentorial primitive neuroectodermal tumor using magnetic resonance-guided laser-induced thermal therapy. (medtronic.com)
- There are several types of Ewing sarcoma, including Ewing sarcoma of bone, extraosseous Ewing sarcoma, peripheral primitive neuroectodermal tumor (pPNET), and Askin tumor. (medlineplus.gov)
- Primitive Neuroectodermal Tumor in Infancy - An Unusual Clinical Presentation. (pediatriconcall.com)
- Examination of the surgical specimen revealed findings characteristic for Askin Tumor - malignant small round cell tumor, consistent with primitive neuroectodermal tumor, 95% necrotic. (virtualpediatrichospital.org)
- Ewing sarcomas are histologically similar and identical to peripheral primitive neuroectodermal tumors. (drrathresearch.org)
- Ewing's sarcoma is the common name for primitive neuroectodermal tumor . (wikidoc.org)
- Medulloblastomas are primitive neuroectodermal tumors that commonly manifest as a posterior fossa mass and obstructive hydrocephalus. (msdmanuals.com)
Medulloblastoma8
- Medulloblastoma is a primitive cerebellar tumor of neuroectodermal origin. (oncolink.org)
- 1 - 3 Intracranial brain tumors, most commonly medulloblastoma [ Figure 2 ], can metastasize to the peritoneal cavity via ventriculo-peritoneal shunts used to divert excess cerebrospinal fluid. (cytojournal.com)
- Alexander was two years old when he was diagnosed with medulloblastoma, the most common pediatric brain tumor. (ouralexander.org)
- Medulloblastoma is defined by the World Health Organization (WHO) as "an embryonal neuroepithelial tumor arising in the cerebellum or dorsal brainstem, presenting mainly in childhood and consisting of densely packed small round undifferentiated cells with mild to moderate nuclear pleomorphism and high mitotic count. (medscape.com)
- Medulloblastoma is the most common malignant central nervous system (CNS) tumor of childhood, with an annual incidence of about 0.5-0.8/100,000 in children younger than 19 years. (medscape.com)
- Dissemination of medulloblastoma within cerebrospinal fluid (CSF) pathways is a defining pathobiologic characteristic of this tumor, and about 30% of patients will have CSF metastasis at presentation. (medscape.com)
- Medulloblastoma Medulloblastomas are invasive and rapidly growing childhood central nervous system tumors that develop in the posterior fossa (containing the brain stem and cerebellum). (msdmanuals.com)
- Currently, the neoplasms classified as ET are the medulloblastoma (MB), embryonal tumors with multilayered rosettes (ETMR), medulloepithelioma (ME), CNS neuroblastoma (NB), CNS ganglioneuroblastoma (GNB), atypical teratoid/rhabdoid tumors (AT/RT), and CNS embryonal tumors with rhabdoid features. (bvsalud.org)
Melanotic neuroectodermal9
- Consider clinical, radiographic, laboratory, and histologic findings when establishing a proper differential diagnosis for melanotic neuroectodermal tumor of infancy (MNTI). (medscape.com)
- Neville B, Damm D, Allen C. Melanotic Neuroectodermal Tumor of Infancy. (medscape.com)
- Melanotic neuroectodermal tumor of infancy--a neoplasm of neural crese origin. (medscape.com)
- Cutler LS, Chaudhry AP, Topazian R. Melanotic neuroectodermal tumor of infancy: an ultrastructural study, literature review, and reevaluation. (medscape.com)
- Melanotic neuroectodermal tumor of infancy is a rare neoplasm of possibly neural crest origin, and it predominantly occurs in the premaxillas of infants less than 12 months old. (smu.ac.za)
- The aim of this paper is to report a dental strategy with a differentiated immediate palatal obturator in a case of melanotic neuroectodermal tumor of infancy, held at the National Cancer Institute José Alencar Gomes da Silva, by performing a literature review and addressing the multidisciplinary treatment as a guarantee of integral care for the patient. (bvsalud.org)
- Melanotic neuroectodermal tumour of infancy (MNTI), first reported by Krompecher in 1918, is an uncommon pigmented tumour affecting predominantly the craniofacial bones of the newborn infants 1. (journalcra.com)
- A germline mutation of CDKN2A and a novel RPLP1-C19MC fusion detected in a rare melanotic neuroectodermal tumor of infancy: a case report. (ox.ac.uk)
- BACKGROUND: Melanotic neuroectodermal tumor of infancy (MNTI) is exceptionally rare and occurs predominantly in the head and neck (92.8 % cases). (ox.ac.uk)
Infancy1
- CONCLUSIONS: In the absence of somatic copy number variations or mutations, the fully transformed phenotype of the MNTI may have arisen in infancy because of the combined effects of a germline CDKN2A mutation, tumor promoting somatic fusion genes and epigenetic deregulation. (ox.ac.uk)
Small round cell tumors1
- Ovarian desmoplastic small round cell tumors: Prognosis is poor! (scirp.org)
Ewing9
- Further advances in immunohistochemical analyses have helped further distinguish PNETs and Ewing family of tumors (EFTs) from other small, round, poorly differentiated tumors, including rhabdomyosarcoma, neuroblastoma, and lymphoma. (medscape.com)
- These tumors occur primarily in children and adolescents and share a number of characteristics with EWING SARCOMA. (jefferson.edu)
- The tumor which bears his name is generally referred to as Ewing's sarcoma when spoken and either Ewing's sarcoma or Ewing sarcoma when written. (sarcomahelp.org)
- Ewing sarcoma is classified into various types depending on the site of development of the malignant tumor . (hdkino.org)
- Ewing sarcoma is a cancerous tumor that occurs in bones or soft tissues, such as cartilage or nerves. (medlineplus.gov)
- These types of Ewing sarcoma can be distinguished from one another by the tissue in which the tumor develops. (medlineplus.gov)
- Approximately 87 percent of Ewing sarcomas are Ewing sarcoma of bone, which is a bone tumor that usually occurs in the thigh bones (femurs), pelvis, ribs, or shoulder blades. (medlineplus.gov)
- Extraosseous (or extraskeletal) Ewing sarcoma describes tumors in the soft tissues around bones, such as cartilage. (medlineplus.gov)
- Ewing sarcoma accounts for about 1.5 percent of all childhood cancers, and it is the second most common type of bone tumor in children (the most common type of bone cancer is called osteosarcoma). (medlineplus.gov)
Askin3
- If it is discovered in the chest, it is called an Askin tumor. (hdkino.org)
- A type of pPNET found in the chest is called Askin tumor. (medlineplus.gov)
- Askin tumor, osteosarcoma, osteomyelitis. (virtualpediatrichospital.org)
Neoplasms8
- While most tumors metastatic to the serous membranes are of epithelial origin, cytologists should be aware that non-epithelial neoplasms can also cause malignant effusions including sarcomas, melanomas, germ cell tumors, and, more rarely, brain tumors. (cytojournal.com)
- Diagnosing non-epithelial malignancies in effusion specimens based entirely upon their cytomorphologic features is difficult because these neoplasms often exhibit considerable morphological overlap and their cytomorphology can differ from the original tumor. (cytojournal.com)
- Common non-epithelial neoplasms that may cause malignant effusions include malignant melanoma, sarcomas, and other neoplasms including germ cell tumors [ Figure 1 ]. (cytojournal.com)
- Benign musculoskeletal neoplasms are one hundred times more common than malignant soft tissue tumors. (drrathresearch.org)
- Studies reliant on molecular detection of tumor-associated virus in isolation, however extensive, are inconclusive because association between PyVs and naturally occurring neoplasms varies and because PyV infections are highly prevalent, yet tumor formation is rare ( 3 , 4 , 12 - 14 ). (cdc.gov)
- Pancreatic neuroendocrine neoplasms (pNENs) are relatively rare epithelial malignancies originating from pancreatic neuroendocrine cells, pathologically classified into well-differentiated pancreatic neuroendocrine tumors (pNETs) and poorly-differentiated pancreatic neuroendocrine carcinoma (pNECs). (bvsalud.org)
- Well-differentiated nonfunctional pancreatic neuroendocrine tumors are often indolent neoplasms without lymph node (LN) metastasis at diagnosis. (bvsalud.org)
- All these tumors are classified as malignant-grade IV neoplasms, and the prognosis of patients with these neoplasms is very poor. (bvsalud.org)
Metastasis2
- The American Joint Committee on Cancer (AJCC) tumor-node-metastasis (TNM) classification and the International Federation of Gynecology and Obstetrics (FIGO) staging system for germ cell tumors are listed below (see Tables 1 and 2). (medscape.com)
- Pretreatment factors that influence outcome of the Ewing's sarcoma are site of the tumor, size of the tumor, serum LDH levels, and site of metastasis. (wikidoc.org)
Group of highly malignant tumors1
- Embryonal tumors (ETs) of the central nervous system (CNS) comprise a large heterogeneous group of highly malignant tumors that predominantly affect children and adolescents. (bvsalud.org)
PNETs4
- The PNETs were histologically indistinguishable from the human counterparts and have been used to identify new genes involved in human brain tumor carcinogenesis. (wikipedia.org)
- Tumors that demonstrate neural differentiation by light microscopy, immunohistochemistry, or electron microscopy have been traditionally labeled PNETs, and those that are undifferentiated by these analyses have been diagnosed as Ewing's sarcoma. (wikipedia.org)
- Stout first described PNETs in 1918, and these tumors were thought to arise directly from nerves. (medscape.com)
- PNETs are a group of tumors that can happen anywhere in the brain. (kidshealth.org)
Embryonal tumors with multilayered rosettes1
- Multiple chromosomal translocations were identified by RNA-Seq, and fusion genes included RPLP1-C19MC, potentially deregulating the C19MC cluster, an imprinted locus containing microRNA genes reactivated by gene fusion in embryonal tumors with multilayered rosettes. (ox.ac.uk)
Neuroblastoma2
- The differential diagnosis of a malignant effusion is accordingly broad, especially for the small round blue cell tumors that includes not only mesenchymal tumors, but also non-mesenchymal tumors, such as neuroblastoma and Wilms tumor. (cytojournal.com)
- Other rare non-epithelial malignancies that may be encountered in effusion cytology include neuroblastoma, Wilms tumor, and metastatic brain tumors. (cytojournal.com)
Family of tumors2
- This has made classifying this family of tumors challenging and controversial. (medscape.com)
- What is Ewing's Sarcoma Family of Tumors? (sarcomahelp.org)
Carcinoma1
- Germ cell tumors of the brain include germinoma, teratoma, embryonal carcinoma and yolk-sac tumors. (cincinnatichildrens.org)
Germ cell1
- The most common type of germ cell tumor of the brain is a germinoma. (indiasurgerytour.com)
Cancer11
- The notion of controlling tumor growth through a naturally occurring biochemical mechanism in the body that directs cancer cells into normal channels of differentiation is one of the theoretical foundations of antineoplaston therapy. (cancer.gov)
- Brain tumors account for only 1-2% of all cancer. (uvahealth.com)
- Not all brain tumors are cancer. (uvahealth.com)
- Swedish Research Counsil (R.J.A.N.), National Cancer Institute (NCI) CA069246 and CA86355 (X.O.B.), and the American Brain Tumor Association (T.W.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. (plos.org)
- VEGF, produced in large amounts by cancer cells during tumor growth, interacts with its receptors VEGFR1 and VEGFR2 thereby causing endothelial cell survival, proliferation, and sprouting [7] . (plos.org)
- In clinical practice, she concentrates on germ cell tumors, while her epidemiologic research primarily focuses on colorectal cancer screening and prevention. (dana-farber.org)
- Atypical teratoid rhabdoid tumor (ATRT) is a rare and aggressive type of cancer that can appear in the brain or spinal cord. (cincinnatichildrens.org)
- When cancer cells spread to the brain from another organ (such as the lung or breast), is called as a secondary tumor or metastatic tumor. (indiasurgerytour.com)
- Burzynski, a MD Ph.D. has a twenty-year track record of curing or controlling the re-growth of malignant brain tumors in children and adults with an innovative cancer therapy. (ouralexander.org)
- Our results suggest that a micronutrient mixture containing vitamin C, EGCG, Lysine, Proline and other natural components is effective in inhibiting cancer cells proliferation, secretion of MMPs and cancer invasion through Matrigel in vitro and in inhibiting the tumor growth of grafted saracoma cell lines in nude mice. (drrathresearch.org)
- Ewing's sarcoma is a small round-cell tumor in which cancer cells are found in the bone or in soft tissue . (wikidoc.org)
Benign10
- Benign brain tumors, including pituitary tumors, still require experienced care. (uvahealth.com)
- Learn about your options for benign brain tumor treatment . (uvahealth.com)
- Although childhood brain tumors can be benign (non-cancerous) or malignant (cancerous), both types can be life-threatening. (childrenshospital.org)
- The term "benign" refers to a condition, tumor, or growth that is not cancerous. (indiasurgerytour.com)
- Benign tumors usually grow slowly. (indiasurgerytour.com)
- If a benign tumor is big enough, its size and weight can press on nearby blood vessels, nerves, or organs, or otherwise cause problems. (indiasurgerytour.com)
- While some benign brain tumors may pose a health risk, including risk of disability and death, most are usually successfully treated with techniques such as surgery. (indiasurgerytour.com)
- Benign tumor occurring in the 8th cranial nerve (the acoustic nerve) between the pons and the cerebellum. (indiasurgerytour.com)
- This is a benign, congenital tumor. (indiasurgerytour.com)
- Benign tumor arising from the meninges, the membranes covering the brain and spinal cord. (indiasurgerytour.com)
Prognosis3
- It is a high-grade and poorly differentiated neuroendocrine tumor, usually diagnosed in advanced stages, with a poor prognosis and few therapeutic options in that setting. (mdpi.com)
- Nevertheless, children with brain tumors generally have a better prognosis than adults with a similar condition. (childrenshospital.org)
- After administration of preoperative chemotherapy , patients with minimal or no residual viable tumor have a significantly better prognosis than do patients with larger amounts of viable tumor. (wikidoc.org)
Suggest that these tumors2
- Immunohistochemical and cytogenetic studies suggest that these tumors all have a common origin. (medscape.com)
- While the origin of these tumors is still not definitively known, the two theories with the most support suggest that these tumors arise from a primitive cell derived either from an embryologic tissue called the neural crest, or from resident cells in the body (called mesenchymal stem cells) that have a capability to become one of a variety of tissue types. (sarcomahelp.org)
Lymphoma1
- 1 The most frequent primary tumor types of anterior mediastinal masses are thymic tumors and lymphoma, with approximate proportions of 35% and 25% among mediastinal lesions, respectively. (allenpress.com)
Malignant brain tumors1
- Raccoon polyomavirus may contribute to the development of malignant brain tumors of raccoons. (cdc.gov)
Children and adolescents3
- [ 9 ] Of note, these tumors are rare in African American children and children of Asian descent, with most worldwide cases occurring in white and Hispanic children and adolescents. (medscape.com)
- About 2,200 children and adolescents in the United States are diagnosed with a brain tumor each year. (childrenshospital.org)
- Most children and adolescents who develop brain tumors survive into adulthood. (childrenshospital.org)
Ewing's4
- Subsequently, these two tumors have been grouped into a class of cancers entitled Ewing's Sarcoma Family of Tumor (ESFT), all of which demonstrate this translocation. (sarcomahelp.org)
- Tumors in the Ewing's family of sarcomas are made of primitive cells, which are cells that haven't yet decided what type of cell they are. (sarcomahelp.org)
- Ewing's sarcoma is the second most common malignant bone tumor in patients younger than 20 years. (wikidoc.org)
- Common physical examination findings of Ewing's sarcoma are fever, localized swelling, and tenderness at the site of the tumor. (wikidoc.org)
Metastatic brain1
- Your primary and metastatic brain tumor patients may benefit from Visualase™ MRI-guided laser ablation. (medtronic.com)
Diagnosis4
- The tumor will then need to be biopsied to confirm the diagnosis. (wikipedia.org)
- A brain tumor diagnosis brings on lots of questions. (uvahealth.com)
- Brain Tumor Diagnosis? (uvahealth.com)
- Most veterinary diagnostic laboratories receive large numbers of raccoon ( Procyon lotor ) carcasses for diagnosis, yet tumors of any type are rarely reported ( 15 - 17 ). (cdc.gov)
Pediatric8
- They account for 4-17% of all pediatric soft tissue tumors. (medscape.com)
- and (3) treatment of germ cell tumors in pediatric patients. (dana-farber.org)
- In clinical care, Dr. Frazier is the national expert on germ cell tumors in pediatric patients and oversees the care of these patients referred to DFCI. (dana-farber.org)
- In addition, she is cochair of two national protocols that opened in 1999 for the treatment of low-risk and high-risk pediatric germ cell tumors and chair of the COG Germ Cell subcommittee in COG Rare Tumors. (dana-farber.org)
- Evaluation of prevalence and outcomes of serial tyrosine kinase inhibitor use in pediatric patients with advanced solid tumors. (dana-farber.org)
- Correction to: Radiotherapy for Atypical Teratoid/Rhabdoid Tumor (ATRT) on the Pediatric Proton/Photon Consortium Registry (PPCR). (mayo.edu)
- Radiotherapy for Atypical Teratoid/Rhabdoid Tumor (ATRT) on the Pediatric Proton/Photon Consortium Registry (PPCR). (mayo.edu)
- Back in Los Angeles, we scrambled for other options but we were unable to find any other viable non-toxic therapy that had any record of success with pediatric brain tumors. (ouralexander.org)
Immunohistochemical1
- Malignant gastrointestinal neuroectodermal tumor: clinicopathologic, immunohistochemical, ultrastructural, and molecular analysis of 16 cases with a reappraisal of clear cell sarcoma-like tumors of the gastrointestinal tract. (jefferson.edu)
Aggressive2
- With fast-growing tumors (or less aggressive ones that have reached a large size), vision problems become apparent more quickly. (kidshealth.org)
- A group of highly aggressive tumors that affect soft tissues and bone. (hdkino.org)
Cancerous2
- Cancerous brain tumors are further classified as either primary or secondary tumors. (indiasurgerytour.com)
- Although primary brain tumors often shed cancerous cells to other sites in the central nervous system (the brain or spine), they rarely spread to other parts of the body. (indiasurgerytour.com)
Carcinoid3
- A symptom complex associated with CARCINOID TUMOR and characterized by attacks of severe flushing of the skin, diarrheal watery stools, bronchoconstriction, sudden drops in blood pressure, edema, and ascites. (harvard.edu)
- The carcinoid tumors are usually located in the gastrointestinal tract and metastasize to the liver. (harvard.edu)
- Retrospective review of serotonergic medication tolerability in patients with neuroendocrine tumors with biochemically proven carcinoid syndrome. (harvard.edu)
Glioma3
- A tumor that develops in any area of the brain stem is called a brain stem glioma . (kidshealth.org)
- A tumor that develops along this pathway is called an optic pathway glioma. (kidshealth.org)
- The most common type of brain tumor is glioma. (cincinnatichildrens.org)
Meningioma1
- Meningeal tumors , such as meningioma, which arise from the membranes (meninges) surrounding the brain and spinal cord. (cincinnatichildrens.org)
Neural crest1
- Esthesioneuroblastoma is an uncommon tumor of neural crest origin arising in the nasal cavity. (nih.gov)
Ovarian1
- Management of ovarian germ cell tumors. (medscape.com)
Neuronal3
- Using gene transfer of SV40 large T-antigen in neuronal precursor cells of rats, a brain tumor model was established. (wikipedia.org)
- Mixed neuronal-glial tumors , which have abnormal neuron cells and abnormal glial cells. (cincinnatichildrens.org)
- The cells of this neoplasm resemble those of other neuronal tumors, and hence, immunochemistry markers have been utilized, such as smooth muscle actin, epithelial membrane antigen, vimentin, and lately antibodies for INI1. (bvsalud.org)
Primary9
- Exhaustive genomic, transcriptomic, epigenetic and pathological characterization was performed on the excised primary tumor and a derived cell line. (ox.ac.uk)
- These lesions are accessible through endoscopic/endobronchial ultrasound-guided or computed tomography-guided fine-needle aspiration cytology and represent a wide range of primary and metastatic tumors. (allenpress.com)
- Tumors metastatic to mediastinal lymph nodes represent the most common mediastinal lesions and must be differentiated from primary lesions. (allenpress.com)
- Primary tumors start in the brain, whereas secondary tumors spread to the brain from another site such as the breast or lung. (indiasurgerytour.com)
- The term "acoustic neuroma" is actually a misnomer since it this a primary intracranial tumor of the myelin forming cells called "Schwann cells" (schwannoma). (indiasurgerytour.com)
- Meningiomas represent approximately 20% of all primary brain tumors and occur most commonly in middle-aged women. (indiasurgerytour.com)
- Tumors that begin in brain tissue are known as primary brain tumors. (indiasurgerytour.com)
- Secondary tumors in the brain are far more common than primary brain tumors. (indiasurgerytour.com)
- The cause of primary brain tumors is unknown. (indiasurgerytour.com)
Genes2
- The model was used to confirm p53 as one of the genes involved in human medulloblastomas, but since only about 10% of the human tumors showed mutations in that gene, the model can be used to identify the other binding partners of SV40 Large T- antigen, other than p53. (wikipedia.org)
- Translocations that fuse the EWSR1 gene with other genes that are related to the FLI1 gene can also cause these types of tumors, although these alternative translocations are relatively uncommon. (medlineplus.gov)
Chemotherapy2
- We can then tell which brain tumors will respond best to chemotherapy, gene therapy, or other therapies. (uvahealth.com)
- Brain tumors are commonly treated with surgery and/or other therapies including chemotherapy and radiation . (childrenshospital.org)
Anlage3
- Retinal anlage" tumors. (medscape.com)
- The pigmented ameloblastomas or "retinal anlage" tumors. (medscape.com)
- It has also been suggested that the tumor arises from the retinal anlage by a pinching-off process of neuroepithelium during the formation of embryonic eye7. (journalcra.com)
Brain38
- There are many different types of brain and nervous system cancers, and doctors categorize them based on where the tumors are, the type of cells involved, and how quickly they grow. (kidshealth.org)
- Ependymomas are tumors that develop in the brain cells that make cerebrospinal fluid. (kidshealth.org)
- Tumors in the back of the brain are more common. (kidshealth.org)
- All of these tumors can metastasize (spread) through the cerebrospinal fluid that surrounds the brain and the spinal cord. (kidshealth.org)
- The Leksell Gamma Knife® is a sophisticated instrument that pinpoints and delivers precise beams of radiation to tumors and other brain abnormalities. (sutterhealth.org)
- This surgery-free process is exceptionally effective at reducing or eliminating brain tumors, blood vessel abnormalities, functional disorders and other conditions, offering a noninvasive alternative to open brain surgery that eliminates the risk of surgical complications. (sutterhealth.org)
- When you choose UVA Health for brain tumor treatment, you get expert care from a deeply experienced team that understands your challenges. (uvahealth.com)
- We don't just treat your brain tumor. (uvahealth.com)
- You'll have access to clinical trials and a team of neuro-oncologists who focus only on brain tumors. (uvahealth.com)
- But our neuro-oncologists only see patients with brain tumors. (uvahealth.com)
- What is a brain tumor? (childrenshospital.org)
- Brain tumors are relatively rare in children, occurring in only five of every 100,000 children. (childrenshospital.org)
- However, as scientists continue to learn more about the specific genetic mutations that occur in childhood brain tumors, they are starting to develop targeted treatments (precision medicine) that can be used in brain tumor treatment. (childrenshospital.org)
- If your child is diagnosed with a brain tumor, you will learn there are many different brain tumor types and classifications based upon the tumor's cell structure, composition, rate of growth, location, and other characteristics. (childrenshospital.org)
- The name and classification of the tumor may change as your doctor gains information about your child's brain tumor or if the tumor changes over time. (childrenshospital.org)
- The types of brain tumors most common in children are not the same as those most common in adults. (childrenshospital.org)
- Childhood brain tumors frequently appear in different locations and behave differently than brain tumors in adults. (childrenshospital.org)
- Learn more about the specific types of brain tumors. (childrenshospital.org)
- Choroid plexus tumors arise in the tissue located in the spaces of the brain called ventricles. (childrenshospital.org)
- Between 10 and 20 percent of brain tumors that occur within the first year of life are choroid plexus tumors. (childrenshospital.org)
- What are the symptoms of brain tumors? (childrenshospital.org)
- Each child may experience symptoms of a brain tumor differently, and symptoms vary depending on the size and location of the tumor - both in the brain and elsewhere in the central nervous system. (childrenshospital.org)
- Patients come from around the world come to Cincinnati Children's for expert treatment for brain tumors in children or young adults. (cincinnatichildrens.org)
- Choroid plexus tumors , which arise from cells lining the ventricles of the brain. (cincinnatichildrens.org)
- Tumors in the brain are categorized according to several factors, including where they're located, the type of cells involved, and how quickly they're growing. (indiasurgerytour.com)
- The tumor occurs in the lowest part of the brain. (indiasurgerytour.com)
- It is the most common brain tumor in children. (indiasurgerytour.com)
- Most germ cell tumors that arise in the brain occur in people younger than 30. (indiasurgerytour.com)
- Some inherited conditions increase the risk of brain tumors, including neurofibromatosis, Von Hippel-Lindau syndrome, Li-Fraumeni syndrome, and Turcot's syndrome. (indiasurgerytour.com)
- The other hypothesis is that HER-specific CAR T cells safely can be administered through an indwelling CNS catheter to allow the T cells to directly interact with the tumor cells for each patient enrolled on the study safely can be delivered directly into the brain via indwelling catheter. (fredhutch.org)
- Now FDA-cleared to ablate epileptic foci and tumors in the brain. (medtronic.com)
- FDA-cleared to ablate tumors in the brain, this minimally invasive surgical option may ultimately help patients to a speedier recovery for the continuation of their treatment. (medtronic.com)
- He explained that the FDA controlled his protocols and it required that Alexander have the tumor return in his brain after using chemo and or radiation. (ouralexander.org)
- We explained that our son had suffered through a total of sixteen hours of brain surgery to be tumor free. (ouralexander.org)
- [ 4 ] Adult cases are unusual, accounting for less than 1% of brain tumors. (medscape.com)
- High-grade brain tumors, consistently located in the frontal lobes and olfactory tracts, were detected in 10 raccoons during March 2010-May 2012 in California and Oregon, suggesting an emerging, infectious origin. (cdc.gov)
- JCV, for example, induces brain tumors when intracerebrally inoculated in experimental animals ( 7 - 11 ). (cdc.gov)
- In northern California and southern Oregon, we diagnosed 10 cases of olfactory tract/frontal lobe brain tumors in free-ranging raccoons during March 2010-May 2012. (cdc.gov)
Occur2
- Subependymal giant cell tumors may occur in children and adults who have a condition called tuberous sclerosis . (cincinnatichildrens.org)
- Tumors may occur at any age, but many specific tumors have a particular age group in which they are most common. (indiasurgerytour.com)
Glial3
- Summary: Astrocytoma, or pituicytoma, of the posterior pituitary is a relatively rare entity consisting of poorly characterized glial tumor cells. (ajnr.org)
- Gliomas also can be named according to the type of glial cells involved or the location of the tumor. (childrenshospital.org)
- These tumors begin from "glial" cells, which support functions of the nervous system. (cincinnatichildrens.org)
Epithelial3
- The same age differential is noted with respect to the more common odontogenic tumors (eg, ameloblastoma, odontoma, adenomatoid odontogenic tumor, calcifying epithelial odontogenic tumor, ameloblastic fibroma, odontogenic myxoma, odontogenic fibroma). (medscape.com)
- According to Krompecher, this tumor derives from epithelial nests evolved at the time of embryonic fusion of the facial processes. (journalcra.com)
- Most tumors metastatic to the serous membranes are of epithelial origin. (cytojournal.com)
Pituitary2
- Sometimes, a tumor that's pressing on the pituitary gland can cause growth problems. (kidshealth.org)
- A review of the literature reveals only a few reports of this tumor, and there has been scanty discussion of the imaging findings of posterior pituitary astrocytomas compared with lesions of the anterior pituitary gland. (ajnr.org)
Bone1
- Osteosarcoma accounts for about 60% of malignant bone tumors between the ages of 10 to 20. (drrathresearch.org)
Rare tumor1
- It is a rare tumor, usually occurring in children and young adults under 25 years of age. (wikipedia.org)