Floxuridine
Infusions, Intra-Arterial
Hepatic Artery
Prodrugs
Esters
Leucovorin
Colorectal Neoplasms
Camptothecin
Fluorouracil
Colorectal liver metastasis thymidylate synthase staining correlates with response to hepatic arterial floxuridine. (1/548)
We assessed whether intensity of colorectal liver metastasis staining with the thymidylate synthase (TS) antibody TS106 predicted response to hepatic arterial infusion (HAI) of floxuridine chemotherapy. Liver metastasis biopsies were taken during laparotomy for hepatic arterial cannulation and stained using the TS106 monoclonal antibody. Staining intensity was designated at histological examination by two independent assessors as either "high" or "low." Patients were treated by HAI, and liver metastasis response was assessed by comparison of computed tomography scan tumor volume before and after 4 months of treatment. A significant correlation (Fisher's exact test, P = 0.01) was noted between partial response to HAI and TS106 staining intensity in patients with colorectal liver metastases. Seventy-five percent of patients with evidence of a partial response had low TS staining compared with 29% of nonresponders. There was a significant difference (Fisher's exact test, P = 0.01) in the proportion of low (9 of 16) compared with high (3 of 20) TS staining tumors in which a partial response occurred. There was no significant difference (logrank test, P = 0.4) in survival from hepatic cannulation and HAI treatment of high (median, 322 days; interquartile range, 236-411) compared with low (median, 335 days; interquartile range, 301-547) TS staining patients. This study demonstrates an inverse correlation between TS immunohistochemical staining intensity in colorectal liver metastases and response to HAI. The results suggest that a prospective assessment of TS staining intensity in colorectal liver metastases would be useful to determine whether this method can be used to define patients who will benefit from HAI chemotherapy. (+info)Suppression of replication of multidrug-resistant HIV type 1 variants by combinations of thymidylate synthase inhibitors with zidovudine or stavudine. (2/548)
The replication of recombinant multidrug-resistant HIV-1 clones modeled on clinically derived resistant HIV-1 strains from patients receiving long-term combination therapy with zidovudine (AZT) plus 2',3'-dideoxycytidine was found to regain sensitivity to AZT and stavudine (D4T) as a consequence of a pharmacologically induced decrease in de novo dTMP synthesis. The host-cell system used was phytohemagglutinin-stimulated peripheral blood mononuclear cells; dTMP and dTTP depletion were induced by single exposures to a low level of the thymidylate synthase inhibitor 5-fluorouracil (5-FU) or its deoxynucleoside, 2'-deoxy-5-fluorouridine. The host-cell response to the latter was biphasic: a very rapid decrease in the rate of de novo dTMP formation and, consequently, in intracellular dTTP pools, followed by slower recovery in both indices over 3 to 24 h. With the additional presence of AZT or D4T, however, replication of the multidrug-resistant HIV-1 strains remained inhibited, indicating dependence of HIV DNA chain termination by AZT-5'-monophosphate or 2',3'-didehydro-2', 3'-dideoxythymidine-5'-monophosphate in these resistant strains on simultaneous inhibition of host-cell de novo synthesis of thymidine nucleotides. No effect on viability of control (uninfected) phytohemagglutinin-stimulated/peripheral blood mononuclear cells was noted on 6-day exposures to 5-FU or 2'-deoxy-5-fluorouridine alone or in combination with AZT or D4T, even at drug levels severalfold higher than those used in the viral inhibition studies. These studies may provide useful information for the potential clinical use of AZT/5-FU or D4T/5-FU combinations for the prevention or reversal of multidrug resistance associated with long-term dideoxynucleoside combination therapy. (+info)Ligand-mediated induction of thymidylate synthase occurs by enzyme stabilization. Implications for autoregulation of translation. (3/548)
Thymidylate synthase (TS) is indispensable in the de novo synthesis of dTMP. As such, it has been an important target at which anti-neoplastic drugs are directed. The fluoropyrimidines 5-fluorouracil and 5-fluoro-2'-deoxyuridine are cytotoxic as a consequence of inhibition of TS by the metabolite 5-fluoro-2'-deoxyuridine 5'-monophosphate (FdUMP). This inhibition occurs through formation of a stable ternary complex among the enzyme, the nucleotide analog, and the co-substrate N5, N10-methylenetetrahydrofolate. Numerous studies have shown that cellular concentrations of TS undergo about a 2-4-fold induction following treatment with TS inhibitors. An extensive body of in vitro studies has led to the proposal that this induction occurs because of relief of the translational repression brought on by the binding of TS to its own mRNA. In the current study, we have tested several predictions of this autoregulatory translation model. In contrast to expectations, we find that fluoropyrimidines do not cause a change in the extent of ribosome binding to TS mRNA. Furthermore, mutations within the mRNA that abolish its ability to bind TS have no effect on the induction. Finally, enzyme turnover measurements show that the induction is associated with an increase in the stability of the TS polypeptide. Our results, in total, indicate that enzyme stabilization, rather than translational derepression, is the primary mechanism of TS induction by fluoropyrimidines and call into question the general applicability of the autoregulatory translation model. (+info)Pharmacokinetics and bioavailability of oral 5'-deoxy-5-fluorouridine in cancer patients. (4/548)
AIMS: Oral administration of 5-fluorouracil (FUra), an important cytotoxic agent, is limited by a wide variation in bioavailability. 5'-deoxy-5-fluorouridine (dFUrd), a masked form of FUra, has shown promise clinically when given intravenously or orally as a solution or tablet. This study investigates the efficacy of an oral capsule formulation of dFUrd in generating continuous systemic levels of this compound in cancer patients. METHODS: Six patients with advanced intestinal or ovarian malignancies were given three cycles of dFUrd, days 1-5, at intervals of 4 weeks. The doses of dFUrd were 600 mg m-2 three times daily, 800 mg m-2 three times daily, and 1000 mg m-2 three times daily, on cycles one, two and three, respectively (total dose 36 g m-2 ). The initial dose in each cycle was given as a slow intravenous injection over 10 min, and the remainder orally. Plasma and urine levels of dFUrd and two of its metabolites, FUra and 5,6-dihydro-5-fluorouracil (FUraH2 ), were monitored in six patients at each dose level. RESULTS: All six patients completed the study, receiving three different doses over a 3 month period, following which one had achieved a partial response, one had stable disease, and four had developed progressive disease. Side-effects were negligible, and only two instances of transient diarrhoea WHO grade 1 were seen. Total body clearance (CLtot) of intravenous dFUrd decreased with increasing dose; 2.7, 2.0 and 1.3 l min-1 m-2, following doses of 600, 800 and 1000 mg m-2, respectively. The mean elimination half-life of intravenous dFUrd increased with the dose from 15 to 22 min. Oral dFUrd was rapidly absorbed with a lag time of less than 20 min. The mean elimination half-life (t1/2, z ) of oral dFUrd was 32-45 min in the dose range 600-1000 mg m-2. The AUC of FUra and FUraH2 increased overproportionally with increasing intravenous doses of dFUrd. The mean systemic bioavailability of oral dFUrd was 34-47%. CONCLUSIONS: dFUrd, which selectively releases the antimetabolite FUra in tumour cells, can be given orally at doses of 600-1000 mg m-2 three times daily for 5 days. The systemic levels achieved are equivalent to those seen following continuous infusions of dFUrd or FUra. Toxicity is tolerable, and further clinical investigation of oral dFUrd is warranted. (+info)Comparison of paclitaxel-, 5-fluoro-2'-deoxyuridine-, and epidermal growth factor (EGF)-induced apoptosis. Evidence for EGF-induced anoikis. (5/548)
Epidermal growth factor (EGF), a hormone that stimulates proliferation of many cell types, induces apoptosis in some cell lines that overexpress the EGF receptor. To evaluate the mechanism of EGF-induced apoptosis, MDA-MB-468 breast cancer cells were examined by microscopy, flow cytometry, immunoblotting, enzyme assays, and affinity labeling after treatment with EGF, paclitaxel, or 5-fluoro-2'-deoxyuridine (5FUdR). Apoptosis induced by all three agents was accompanied by activation of caspases-3, -6, and -7, as indicated by disappearance of the corresponding zymogens from immunoblots, cleavage of substrate polypeptides in situ, and detection of active forms of these caspases in cytosol and nuclei using fluorogenic assays and affinity labeling. Further analysis indicated involvement of the cytochrome c/Apaf-1/caspase-9 pathway of caspase activation, but not the Fas/Fas ligand pathway. Interestingly, caspase activation was consistently lower after EGF treatment than after paclitaxel or 5FUdR treatment. Additional experiments revealed that the majority of cells detaching from the substratum after EGF (but not paclitaxel or 5FUdR) were morphologically normal and retained the capacity to readhere, suggesting that EGF-induced apoptosis involves cell detachment followed by anoikis. These observations not only indicate that EGF- and chemotherapy-induced apoptosis in this cell line involve the same downstream pathways but also suggest that detachment-induced apoptosis is responsible for the paradoxical antiproliferative effects of EGF. (+info)Inhibition of cyclin D1 expression in human pancreatic cancer cells is associated with increased chemosensitivity and decreased expression of multiple chemoresistance genes. (6/548)
Cyclin D1 belongs to a family of protein kinases that have been implicated in cell cycle regulation. Inhibition of cyclin D1 expression has been recently shown (M. Kornmann, et al., J. Clin. Invest, 101: 344-352, 1998) to suppress pancreatic cancer cell growth and increase cytotoxic actions of cisplatinum. The aim of the present study was to determine whether inhibition of cyclin D1 expression also modulates the effects of other antineoplastic drugs and whether it is associated with alterations in the level of expression of drug resistance genes. The suppression of cyclin D1 expression after the stable transfection of a cyclin D1 antisense construct in PANC-1 and COLO-357 human pancreatic cancer cells resulted in a significant increase in sensitivity to the fluoropyrimidines 5-fluorouracil and 5-fluoro-2'-deoxyuridine and to mitoxantrone. All of the antisense-expressing dones exhibited a decrease in thymidylate synthase and an increase in thymidine phosphorylase mRNA expression as determined by reverse transcription-PCR analysis and decreased levels of MDR-1 and MRP mRNA as determined by Northern blotting. These findings demonstrate that the inhibition of cyclin D1, in addition to suppressing the growth of pancreatic cancer cells, enhances their responsiveness to multiple chemotherapeutic agents and suggest that this effect may be due to the altered expression of several chemoresistance genes. (+info)Antitumor activity of ZD1694 (tomudex) against human head and neck cancer in nude mouse models: role of dosing schedule and plasma thymidine. (7/548)
We studied the antitumor activity and toxicity of ZD1694 (tomudex), a specific inhibitor of thymidylate synthase (TS), in nude mice bearing human head and neck squamous cell carcinoma A253 and FaDu xenografts. Mice were treated by single i.v. push (i.v. x 1), i.v. push once a week for 3 weeks (weekly x 3), and i.v. push once a day for 5 days (daily x 5), and the maximum tolerated doses (MTDs) of ZD1694 were 300 mg/kg, 60 mg/kg/week, and 30 mg/kg/day, respectively. ZD1694 was moderately active against both A253 and FaDu xenografts. Antitumor activity was schedule-dependent in both tumors: weekly x 3 > or = i.v. x 1 >> daily x 5. In contrast, the rank order of toxicity was daily x 5 >> weekly x 3 > or = i.v. x 1. ZD1694 at the MTD produced 20% complete tumor regression and 20% partial tumor regression (PR) with i.v. x 1 and weekly x 3 schedules and 12-day tumor growth delay with daily x 5 schedule against FaDu xenografts. No complete tumor regression was achieved with ZD1694 with any schedule against A253; a 20% PR, 40% PR, and 10-day tumor growth delay were observed with i.v. x 1, weekly x 3, and daily x 5 schedules, respectively. The data indicate that ZD1694 was slightly more effective against FaDu than against A253. Of interest and potential clinical importance was the observation that ZD1694 was still active at doses lower than the MTD (> or =1/3 MTD), which showed a high therapeutic index and wide safety margin. Study of ZD1694 compared with 5-fluorouracil and 5-fluoro-2'-deoxyuridine at the MTD revealed that the antitumor activity of ZD1694 was comparable with or superior to 5-fluorouracil and 5-fluoro-2'-deoxyuridine against both A253 and FaDu xenografts, with less toxicity. High plasma thymidine in mouse relative to human (approximately 1.3 microM and <0.1 microM, respectively) may complicate the study of antitumor activity and toxicity of TS inhibitors with human tumor xenografts grown in the mouse. To test this hypothesis, we preadministered methoxypolyethyleneglycol-conjugated thymidine phosphorylase (MPEG-TPase; 2500 units/kg/dose) to reduce mouse plasma thymidine, then treated with various doses of ZD1694 using the daily x 5 or i.v. x 1 schedules in the A253 tumor model. MPEG-TPase significantly increased the toxicity of ZD1694; the MTD of ZD1694 plus MPEG-TPase was reduced 3- and 10-fold compared with ZD1694 alone for i.v x 1 and daily x 5 schedules, respectively. However, preadministration of MPEG-TPase did not potentiate the antitumor activity of ZD1694 with either schedule. The data indicate that the study of TS inhibitors in rodent models may not be suitable for predicting a safe dose for clinical study. However, rodent models, particularly human tumor xenografts, are still useful models for evaluation of antitumor activity and schedule selection for TS inhibitors. (+info)Selective transfer of a lipophilic prodrug of 5-fluorodeoxyuridine from immunoliposomes to colon cancer cells. (8/548)
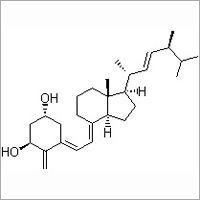
A monoclonal antibody against the rat colon carcinoma CC531 was covalently coupled to liposomes containing a dipalmitoylated derivative of the anticancer drug FUdR as a prodrug in their bilayers. We investigated the in vitro interaction of these liposomes with CC531 target cells and the mechanism by which they deliver the active drug FUdR intracellularly to the cells by monitoring the fate of the liposomal bilayer markers cholesterol-[(14)C]oleate and [(3)H]cholesteryloleylether as well as the (3)H-labeled prodrug and colloidal gold as an encapsulated liposome marker. After binding of the immunoliposomes to the cell surface, only limited amounts were internalized as demonstrated by a low level of hydrolysis of liposomal cholesterol ester and by morphological studies employing colloidal gold-labeled immunoliposomes. By contrast, already within 24 h immunoliposome-incorporated FUdR-dP was hydrolyzed virtually completely to the parent drug FUdR intracellularly. This process was inhibited by a variety of endocytosis inhibitors, indicating that the prodrug enters and is processed by the cells by a mechanism involving an endocytic process, resulting in intracellular FUdR concentrations up to 3000-fold higher than those in the medium. Immunoliposomes containing poly(ethyleneglycol) (PEG) chains on their surface, with the antibody coupled either directly to the bilayer or at the distal end of the PEG chains were able to deliver the prodrug into the tumor cells at the same rate as immunoliposomes without PEG. Based on these observations, we tentatively conclude that during the interaction of the immunoliposomes with the tumor cells the lipophilic prodrug FUdR-dP is selectively transferred to the cell surface and subsequently internalized by constitutive endocytic or pinocytic invaginations of the plasma membrane, thus ultimately delivering the prodrug to a lysosomal compartment where hydrolysis and release of parent drug takes place. This concept allows for an efficient delivery of a liposome-associated drug without the need for the liposome as such to be internalized by the cells. (+info)Floxuridine is a chemotherapeutic antimetabolite medication that is primarily used in the treatment of colon cancer. It is a fluorinated pyrimidine nucleoside analogue, which means it is similar in structure to the building blocks of DNA and RNA, and can be incorporated into these molecules during cell division, disrupting their normal function and preventing cell replication.
Floxuridine works by inhibiting the enzyme thymidylate synthase, which is necessary for the synthesis of thymidine, a nucleoside that is essential for DNA replication. By blocking this enzyme, floxuridine can prevent the growth and proliferation of cancer cells.
Floxuridine is often used in combination with other chemotherapy drugs as part of a treatment regimen for colon cancer. It may be administered intravenously or via continuous infusion, depending on the specific treatment plan. As with all chemotherapy drugs, floxuridine can have significant side effects, including nausea, vomiting, diarrhea, and myelosuppression (suppression of bone marrow function), which can lead to anemia, neutropenia, and thrombocytopenia.
Intra-arterial infusion is a medical procedure in which a liquid medication or fluid is delivered directly into an artery. This technique is used to deliver drugs directly to a specific organ or region of the body, bypassing the usual systemic circulation and allowing for higher concentrations of the drug to reach the target area. It is often used in cancer treatment to deliver chemotherapeutic agents directly to tumors, as well as in other conditions such as severe infections or inflammation.
Intra-arterial infusions are typically administered through a catheter that is inserted into an artery, usually under the guidance of imaging techniques such as fluoroscopy, CT, or MRI. The procedure requires careful monitoring and precise control to ensure proper placement of the catheter and accurate delivery of the medication.
It's important to note that intra-arterial infusions are different from intra venous (IV) infusions, where medications are delivered into a vein instead of an artery. The choice between intra-arterial and intra-venous infusion depends on various factors such as the type of medication being used, the location of the target area, and the patient's overall medical condition.
The hepatic artery is a branch of the celiac trunk or abdominal aorta that supplies oxygenated blood to the liver. It typically divides into two main branches, the right and left hepatic arteries, which further divide into smaller vessels to supply different regions of the liver. The hepatic artery also gives off branches to supply other organs such as the gallbladder, pancreas, and duodenum.
It's worth noting that there is significant variability in the anatomy of the hepatic artery, with some individuals having additional branches or variations in the origin of the vessel. This variability can have implications for surgical procedures involving the liver and surrounding organs.
A prodrug is a pharmacologically inactive substance that, once administered, is metabolized into a drug that is active. Prodrugs are designed to improve the bioavailability or delivery of a drug, to minimize adverse effects, or to target the drug to specific sites in the body. The conversion of a prodrug to its active form typically occurs through enzymatic reactions in the liver or other tissues.
Prodrugs can offer several advantages over traditional drugs, including:
* Improved absorption: Some drugs have poor bioavailability due to their chemical properties, which make them difficult to absorb from the gastrointestinal tract. Prodrugs can be designed with improved absorption characteristics, allowing for more efficient delivery of the active drug to the body.
* Reduced toxicity: By masking the active drug's chemical structure, prodrugs can reduce its interactions with sensitive tissues and organs, thereby minimizing adverse effects.
* Targeted delivery: Prodrugs can be designed to selectively release the active drug in specific areas of the body, such as tumors or sites of infection, allowing for more precise and effective therapy.
Examples of prodrugs include:
* Aspirin (acetylsalicylic acid), which is metabolized to salicylic acid in the liver.
* Enalapril, an angiotensin-converting enzyme (ACE) inhibitor used to treat hypertension and heart failure, which is metabolized to enalaprilat in the liver.
* Codeine, an opioid analgesic, which is metabolized to morphine in the liver by the enzyme CYP2D6.
It's important to note that not all prodrugs are successful, and some may even have unintended consequences. For example, if a patient has a genetic variation that affects the activity of the enzyme responsible for converting the prodrug to its active form, the drug may not be effective or may produce adverse effects. Therefore, it's essential to consider individual genetic factors when prescribing prodrugs.
Antimetabolites are a class of antineoplastic (chemotherapy) drugs that interfere with the metabolism of cancer cells and inhibit their growth and proliferation. These agents are structurally similar to naturally occurring metabolites, such as amino acids, nucleotides, and folic acid, which are essential for cellular replication and growth. Antimetabolites act as false analogs and get incorporated into the growing cells' DNA or RNA, causing disruption of the normal synthesis process, leading to cell cycle arrest and apoptosis (programmed cell death).
Examples of antimetabolite drugs include:
1. Folate antagonists: Methotrexate, Pemetrexed
2. Purine analogs: Mercaptopurine, Thioguanine, Fludarabine, Cladribine
3. Pyrimidine analogs: 5-Fluorouracil (5-FU), Capecitabine, Cytarabine, Gemcitabine
These drugs are used to treat various types of cancers, such as leukemias, lymphomas, breast, ovarian, and gastrointestinal cancers. Due to their mechanism of action, antimetabolites can also affect normal, rapidly dividing cells in the body, leading to side effects like myelosuppression (decreased production of blood cells), mucositis (inflammation and ulceration of the gastrointestinal tract), and alopecia (hair loss).
Liver neoplasms refer to abnormal growths in the liver that can be benign or malignant. Benign liver neoplasms are non-cancerous tumors that do not spread to other parts of the body, while malignant liver neoplasms are cancerous tumors that can invade and destroy surrounding tissue and spread to other organs.
Liver neoplasms can be primary, meaning they originate in the liver, or secondary, meaning they have metastasized (spread) to the liver from another part of the body. Primary liver neoplasms can be further classified into different types based on their cell of origin and behavior, including hepatocellular carcinoma, cholangiocarcinoma, and hepatic hemangioma.
The diagnosis of liver neoplasms typically involves a combination of imaging studies, such as ultrasound, CT scan, or MRI, and biopsy to confirm the type and stage of the tumor. Treatment options depend on the type and extent of the neoplasm and may include surgery, radiation therapy, chemotherapy, or liver transplantation.
Esters are organic compounds that are formed by the reaction between an alcohol and a carboxylic acid. They are widely found in nature and are used in various industries, including the production of perfumes, flavors, and pharmaceuticals. In the context of medical definitions, esters may be mentioned in relation to their use as excipients in medications or in discussions of organic chemistry and biochemistry. Esters can also be found in various natural substances such as fats and oils, which are triesters of glycerol and fatty acids.
Leucovorin is the pharmaceutical name for a form of folic acid, also known as folinic acid. It is used in medicine as a medication to reduce the toxic effects of certain chemotherapy drugs, such as methotrexate, that work by blocking the action of folic acid in the body. Leucovorin is able to bypass this blockage and restore some of the necessary functions of folic acid, helping to prevent or reduce the severity of side effects like nausea, vomiting, and damage to the mucous membranes.
Leucovorin may also be used in combination with fluorouracil chemotherapy to enhance its effectiveness in treating certain types of cancer. It is important to note that leucovorin should only be used under the supervision of a healthcare professional, as it can interact with other medications and have potentially serious side effects if not used properly.
Colorectal neoplasms refer to abnormal growths in the colon or rectum, which can be benign or malignant. These growths can arise from the inner lining (mucosa) of the colon or rectum and can take various forms such as polyps, adenomas, or carcinomas.
Benign neoplasms, such as hyperplastic polyps and inflammatory polyps, are not cancerous but may need to be removed to prevent the development of malignant tumors. Adenomas, on the other hand, are precancerous lesions that can develop into colorectal cancer if left untreated.
Colorectal cancer is a malignant neoplasm that arises from the uncontrolled growth and division of cells in the colon or rectum. It is one of the most common types of cancer worldwide and can spread to other parts of the body through the bloodstream or lymphatic system.
Regular screening for colorectal neoplasms is recommended for individuals over the age of 50, as early detection and removal of precancerous lesions can significantly reduce the risk of developing colorectal cancer.
Camptothecin is a topoisomerase I inhibitor, which is a type of chemotherapeutic agent used in cancer treatment. It works by interfering with the function of an enzyme called topoisomerase I, which helps to uncoil DNA during cell division. By inhibiting this enzyme, camptothecin prevents the cancer cells from dividing and growing, ultimately leading to their death.
Camptothecin is found naturally in the bark and stem of the Camptotheca acuminata tree, also known as the "happy tree," which is native to China. It was first isolated in 1966 and has since been developed into several synthetic derivatives, including irinotecan and topotecan, which are used clinically to treat various types of cancer, such as colon, lung, and ovarian cancers.
Like other chemotherapeutic agents, camptothecin can have significant side effects, including nausea, vomiting, diarrhea, and myelosuppression (suppression of bone marrow function). It is important for patients receiving camptothecin-based therapies to be closely monitored by their healthcare team to manage these side effects effectively.
Fluorouracil is a antineoplastic medication, which means it is used to treat cancer. It is a type of chemotherapy drug known as an antimetabolite. Fluorouracil works by interfering with the growth of cancer cells and ultimately killing them. It is often used to treat colon, esophageal, stomach, and breast cancers, as well as skin conditions such as actinic keratosis and superficial basal cell carcinoma. Fluorouracil may be given by injection or applied directly to the skin in the form of a cream.
It is important to note that fluorouracil can have serious side effects, including suppression of bone marrow function, mouth sores, stomach and intestinal ulcers, and nerve damage. It should only be used under the close supervision of a healthcare professional.
Antineoplastic combined chemotherapy protocols refer to a treatment plan for cancer that involves the use of more than one antineoplastic (chemotherapy) drug given in a specific sequence and schedule. The combination of drugs is used because they may work better together to destroy cancer cells compared to using a single agent alone. This approach can also help to reduce the likelihood of cancer cells becoming resistant to the treatment.
The choice of drugs, dose, duration, and frequency are determined by various factors such as the type and stage of cancer, patient's overall health, and potential side effects. Combination chemotherapy protocols can be used in various settings, including as a primary treatment, adjuvant therapy (given after surgery or radiation to kill any remaining cancer cells), neoadjuvant therapy (given before surgery or radiation to shrink the tumor), or palliative care (to alleviate symptoms and prolong survival).
It is important to note that while combined chemotherapy protocols can be effective in treating certain types of cancer, they can also cause significant side effects, including nausea, vomiting, hair loss, fatigue, and an increased risk of infection. Therefore, patients undergoing such treatment should be closely monitored and managed by a healthcare team experienced in administering chemotherapy.
 Floxuridine
Floxuridine Floxuridine: MedlinePlus Drug Information
Floxuridine: MedlinePlus Drug Information 00143-9270 Floxuridine - CanMED: NDC
00143-9270 Floxuridine - CanMED: NDC Floxuridine
- FUdR
Summary Report | CureHunter
Floxuridine
- FUdR
Summary Report | CureHunter FUDR (floxuridine) dosing, indications, interactions, adverse effects, and more
FUDR (floxuridine) dosing, indications, interactions, adverse effects, and more Floxuridine in: Extended Stability for Parenteral Drugs
Floxuridine in: Extended Stability for Parenteral Drugs What are some of the most common chemo drugs?
What are some of the most common chemo drugs? Chemotherapy Types: About, Side Effects, and Cancers They're Used For
Chemotherapy Types: About, Side Effects, and Cancers They're Used For UpToDate
UpToDate Hemorrhage associated with hepatic artery pseudoaneurysms after regional chemotherapy with floxuridine: case report |...
Hemorrhage associated with hepatic artery pseudoaneurysms after regional chemotherapy with floxuridine: case report |... Carriers for Prodrug Synthesis: A Review
Carriers for Prodrug Synthesis: A Review WHOCC - ATC/DDD Index
WHOCC - ATC/DDD Index Liver and Other Neoplasms - Treatment Approaches - Medical Clinical Policy Bulletins | Aetna
Liver and Other Neoplasms - Treatment Approaches - Medical Clinical Policy Bulletins | Aetna![Gramm HF[au] - Search Results - PubMed](data:image/png;base64,iVBORw0KGgoAAAANSUhEUgAAABAAAAAQCAMAAAAoLQ9TAAAARVBMVEVHcEwoU45gYmYAUpQAUpRPYGVgYmZLXnJgYmYAUZUAUpRJXnIAUpQAUpRgYmYAUpRgYmZgYmZhYmYAUpQAUpQAUpRgYmaDiPJuAAAAFXRSTlMADOJ+6QewGO8/uTRqtH7GdFJ11p1bCL3TAAAAZUlEQVQYlV2PVw7AIAxDTeney7n/UcsoldX3E+VJOAboEi7MBpHWMs1ADlG8u7UYWauwyZFeRQVPOhG2o+aiwhByJxUx91Jxhje3iJSqGfHuLKI0+0TpXvY1twCOPlFh5pa/++MB0vIOBm+1zaoAAAAASUVORK5CYII=) Gramm HF[au] - Search Results - PubMed
Gramm HF[au] - Search Results - PubMed Kidney Cancer | RxWiki
Kidney Cancer | RxWiki Hepatic Arterial Infusion Pump Chemotherapy vs Resection for Multifocal Intrahepatic Cholangiocarcinoma - The ASCO Post
Hepatic Arterial Infusion Pump Chemotherapy vs Resection for Multifocal Intrahepatic Cholangiocarcinoma - The ASCO Post Medtronic SynchroMed Implantable Infusion System Devices: Class 1 Recalls - Feed Through Failure, Failure of Priming Bolus, and...
Medtronic SynchroMed Implantable Infusion System Devices: Class 1 Recalls - Feed Through Failure, Failure of Priming Bolus, and... Nanomaterials | Free Full-Text | Lipid-Based Nanoparticles: Application and Recent Advances in Cancer Treatment
Nanomaterials | Free Full-Text | Lipid-Based Nanoparticles: Application and Recent Advances in Cancer Treatment Rectal Cancer Treatment (PDQ®) - NCI
Rectal Cancer Treatment (PDQ®) - NCI Class I Recall Issued for Medtronic Drug Pump - Parker Waichman LLP
Class I Recall Issued for Medtronic Drug Pump - Parker Waichman LLP Pancreatic disorders | PPT
Pancreatic disorders | PPT G190104-NCT04251715 | CMS
G190104-NCT04251715 | CMS