Ceroid
Neuronal Ceroid-Lipofuscinoses
Serine Proteases
Thiolester Hydrolases
Dipeptidyl-Peptidases and Tripeptidyl-Peptidases
Aminopeptidases
Lipofuscin
Lipidoses
Lysosomal Storage Diseases, Nervous System
Lysosomes
Molecular Chaperones
Cathepsin F
Endopeptidases
Dolichol
Cathepsin D
Dog Diseases
Hermanski-Pudlak Syndrome
Brain
Morphological study on pigmented cells in the horse testis. (1/32)
One of the most attractive characteristics of a horse testis is the change of the weight during development. As the testicular weight changes and the number of Leydig cells decreases, pigments appear in interstitial tissues. In the present study, the characteristics of the pigments found in the interstitial tissues were examined histochemically and ultrastructurally. Specific stainings indicated that the pigmented granules showed almost all of the histological and histochemical characteristics of ceroid or ceroid-like pigment. The cells showed positive reaction for acid phosphatase while the pigmented cells contained a lot of lysosomes ultrastructurally. These results suggest that macrophages might phagocytize Leydig cells, and store their digested materials as ceroid-like pigment. (+info)Inducible nitric oxide synthase colocalizes with signs of lipid oxidation/peroxidation in human atherosclerotic plaques. (2/32)
OBJECTIVE: Advanced human atherosclerotic plaques are characterized by the abundant presence of the autofluorescent non-soluble lipid pigment ceroid, consisting of oxidized lipoproteins. The aim of the present study was to examine the topographical and cellular distribution of inducible nitric oxide synthase (iNOS or NOS II) within different stages of atherosclerosis and its colocalization with ceroid deposits and nitrotyrosine. METHODS AND RESULTS: Different stages of atherosclerosis were studied by immunohistochemistry on whole-mount longitudinal sections of carotid endarterectomy specimens. In the adaptive intimal thickening the predominant cell type were smooth muscle cells. The fatty streaks contained both smooth muscle cells and macrophages with an extremely low NOS II immunoreactivity. The advanced atherosclerotic plaques however, showed a very dense infiltration by macrophages, of which a subpopulation expressed NOS II as a vesicular immunoreactivity in their cytoplasm. These were mainly present around the necrotic core, in association with ceroid accumulation and nitrotyrosine. Fluorescence quenching microscopy showed the presence of NOS II on autofluorescent ceroid vesicles in the macrophages. Large extracellular ceroid granules were not NOS II immunoreactive. NOS II mRNA was detected by RT-PCR and the protein by Western blot in the plaque tissue but not in mammary arteries used as controls. CONCLUSION: Ceroid, nitrotyrosine and NOS II colocalized in late stages of atherosclerosis and were found around the necrotic core in the plaque. This could suggest that NOS II expression in macrophages is involved in oxidation and peroxidation of lipids, leading to ceroid formation. (+info)Proteasome inhibition by lipofuscin/ceroid during postmitotic aging of fibroblasts. (3/32)
We have studied the effects of hyperoxia and of cell loading with artificial lipofuscin or ceroid pigment on the postmitotic aging of human lung fibroblast cell cultures. Normobaric hyperoxia (40% oxygen) caused an irreversible senescence-like growth arrest after about 4 wk and shortened postmitotic life span from 1-1/2 years down to 3 months. During the first 8 wk of hyperoxia-induced 'aging', overall protein degradation (breakdown of [(35)S]methionine metabolically radiolabeled cell proteins) increased somewhat, but by 12 wk and thereafter overall proteolysis was significantly depressed. In contrast, protein synthesis rates were unaffected by 12 wk of hyperoxia. Lysosomal cathepsin-specific activity (using the fluorogenic substrate z-FR-MCA) and cytoplasmic proteasome-specific activity (measured with suc-LLVY-MCA) both declined by 80% or more over 12 wk. Hyperoxia also caused a remarkable increase in lipofuscin/ceroid formation and accumulation over 12 wk, as judged by both fluorescence measurements and FACscan methods. To test whether the association between lipofuscin/ceroid accumulation and decreased proteolysis might be causal, we next exposed cells to lipofuscin/ceroid loading under normoxic conditions. Lipofuscin/ceroid-loaded cells indeed exhibited a gradual decrease in overall protein degradation over 4 wk of treatment, whereas protein synthesis was unaffected. Proteasome specific activity decreased by 25% over this period, which is important since proteasome is normally responsible for degrading oxidized cell proteins. In contrast, an apparent increase in lysosomal cathepsin activity was actually caused by a large increase in the number of lysosomes per cell. To test whether lipofuscin/ceroid could in fact directly inhibit proteasome activity, thus causing oxidized proteins to accumulate, we incubated purified proteasome with lipofuscin/ceroid preparations in vitro. We found that proteasome is directly inhibited by lipofuscin/ceroid. Our results indicate that an accumulation of oxidized proteins (and lipids) such as lipofuscin/ceroid may actually cause further increases in damage accumulation during aging by inhibiting the proteasome. (+info)Phagocytosis and macrophage activation associated with hemorrhagic microvessels in human atherosclerosis. (4/32)
OBJECTIVE: Previously, we demonstrated that activated inducible NO synthase (iNOS)-expressing foam cells in human carotid plaques often produce autofluorescent (per)oxidized lipids (ceroid). Here, we investigate whether intraplaque microvessels can provide foam cells with lipids and trigger macrophage activation. METHODS AND RESULTS: Microvessels (von Willebrand factor [vWf] immunoreactivity), activated macrophages (iNOS immunoreactivity), and ceroid were systematically mapped in longitudinal sections of 15 human carotid endarterectomy specimens. An unbiased hierarchical cluster analysis classified vascular regions into 2 categories. One type with normal vWf expression and without inflammatory cells was seen, and another type with cuboidal endothelial cells, perivascular vWf deposits, and iNOS and ceroid-containing foam cells was seen in 4 (27%) of 15 plaques. The perivascular foam cells frequently contained platelets (glycoprotein Ibalpha) and erythrocytes (hemoglobin, iron), pointing to microhemorrhage/thrombosis and subsequent phagocytosis. Similar lipid-containing cells, expressing both ceroid and iNOS, were generated in atherosclerosis-free settings by incubating murine J774 macrophages with platelets or oxidized erythrocytes and also in vivo in organizing thrombi in normocholesterolemic rabbits. CONCLUSIONS: Focal intraplaque microhemorrhages initiate platelet and erythrocyte phagocytosis, leading to iron deposition, macrophage activation, ceroid production, and foam cell formation. Neovascularization, besides supplying plaques with leukocytes and lipoproteins, can thus promote focal plaque expansion when microvessels become thrombotic or rupture prone. (+info)A clinical variant of familial Hermansky-Pudlak syndrome. (5/32)
Hermansky-Pudlak syndrome (HPS) is an autosomal recessive inherited disease consisting of (1) partial oculocutaneous albinism (with nystagmus, strabism, and visual acuity loss), (2) platelet storage pool deficiency (with bleeding diathesis), and (3) disorder of "ceroid" metabolism with a multisystem tissue lysosomal ceroid deposition. HPS is less uncommon in Puerto Rico, where the most important studies have been performed, but is a very rare disease in Europe. HPS basic defect remains unknown, even if an HPS-causing gene was identified in chromosome segment 10q23-q23.3, and several mutations have been reported. The aim of this article is to discuss, on the basis of a review of relevant literature, a new familial HPS clinical variant observed in 2 young sisters (aged 16 and 23 years old, respectively), characterized by the typical symptoms of this syndrome. Our patients also suffered from diffuse interstitial pulmonary disease and an unexpectedly increased platelet aggregation and were prone to bacterial infections. Interestingly, we observed urinary tract abnormality in the younger HPS sister and a porencephalic cyst in the older HPS sister; both of these developmental defects have been reported in the Cross syndrome (or oculocerebral hypopigmentation syndrome). It seems that in our patients, an overlapping of the phenotypic manifestations of different rare syndromes may be present. The presence of ceroid-like autofluorescent material in urinary sediment together with the histologic aspects and the autofluorescence of oral mucosa biopsy are consistent with a ceroid-like lipofuscin storage. HPS should be carefully tested for in suspected cases to prevent the severe visual impairment, rapidly progressive pulmonary fibrosis, and other complications associated with this disorder. (+info)Ascorbic acid oxidation: a potential cause of the elevated severity of atherosclerosis in diabetes mellitus? (6/32)
The exposure of mouse peritoneal macrophages to cholesterol linoleate-containing artificial lipoproteins can lead to intracellular ceroid accumulation. This can be used as a model to study the role of oxidation in macrophage uptake of lipoproteins containing unsaturated fatty acids, considered by many as a primary event in atherosclerotic plaque formation. Our studies show that ascorbic acid can both inhibit and promote the formation of ceroid in such a model system. The transition metal copper (Cu(II)) further elevates ceroid accumulation and EDTA, a metal chelator, inhibits it. When trace levels of transition metals are present, low concentrations of ascorbic acid can elevate ceroid formation. This pro- and antioxidant characteristic of ascorbic acid was confirmed by monitoring the generation of oxidants by various concentrations of ascorbic acid, assessed by benzoic acid hydroxylation or the fragmentation of BSA. We discuss these observations in the context of an apparent increase in ascorbic acid oxidation and elevated severity of atherosclerosis in diabetes mellitus. (+info)Diagnostic usefulness of bronchoalveolar lavage in Hermansky-Pudlak syndrome: a case with double lung cancers. (7/32)
A 65-year-old man was admitted to our hospital because of dyspnea on exertion. He had oculocutaneous albinism innately and his parents were consanguineous. His chest roentgenogram on admission showed reticulo-nodular infiltrates and cystic changes throughout both lung fields, and 7 cm mass in the left middle field. Cytology of bronchoalveolar lavage fluid (BALF) revealed macrophages containing ceroid. The diagnosis of HPS was made clinically and the tumor was diagnosed as poorly differentiated adenocarcinoma of the lung. He died of respiratory failure. By autopsy, additional well-differentiated adenocarcinoma was detected. Cytology of BALF was useful to confirm ceroid accumulation in the lung. (+info)Retinal pathology in a canine model of late infantile neuronal ceroid lipofuscinosis. (8/32)
(+info)Ceroid is a term used in pathology to describe a type of inclusion body that can be found in various tissues and cells in the body. These inclusions are composed of a protein called alpha-synuclein that has become aggregated and tangled, as well as lipids and other substances. Ceroids are often seen in neurons, but they can also be found in other types of cells such as glial cells.
Ceroid deposits are associated with several neurodegenerative disorders, including Parkinson's disease, dementia with Lewy bodies, and multiple system atrophy. These conditions are characterized by the accumulation of abnormal protein aggregates in the brain, which can lead to neuronal dysfunction and death. The exact role that ceroids play in these diseases is not fully understood, but they are thought to contribute to the toxicity and degeneration of nerve cells.
It's worth noting that ceroid is sometimes used interchangeably with other terms such as "lipofuscin" or "age pigment," although there are some differences between these substances at a molecular level. Nonetheless, they all refer to the accumulation of lipid-rich inclusion bodies in cells and tissues over time.
Neuronal Ceroid-Lipofuscinoses (NCLs) are a group of inherited neurodegenerative disorders characterized by the intracellular accumulation of autofluorescent lipopigment granules, known as ceroid-lipofuscin, in various tissues including the brain and retina. This accumulation is caused by mutations in different genes involved in lysosomal function or protein degradation pathways. The condition primarily affects neurons, leading to progressive neurological deterioration, including motor and cognitive decline, seizures, visual loss, and premature death. NCLs are also known as Batten disease, and they have several subtypes classified based on the age of onset, clinical presentation, and genetic defects.
Serine proteases are a type of enzyme that cleaves peptide bonds in proteins. They have a serine residue in their active site that plays a crucial role in the catalytic mechanism. These enzymes are involved in various biological processes, including blood coagulation, fibrinolysis, inflammation, cell death, and hormone activation. Some examples of serine proteases include trypsin, chymotrypsin, thrombin, and elastase. They play a significant role in disease processes such as cancer, Alzheimer's disease, and emphysema.
Thiol esters are chemical compounds that contain a sulfur atom (from a mercapto group, -SH) linked to a carbonyl group (a carbon double-bonded to an oxygen atom, -CO-) through an ester bond. Thiolester hydrolases are enzymes that catalyze the hydrolysis of thiol esters, breaking down these compounds into a carboxylic acid and a thiol (a compound containing a mercapto group).
In biological systems, thiolester bonds play important roles in various metabolic pathways. For example, acetyl-CoA, a crucial molecule in energy metabolism, is a thiol ester that forms between coenzyme A and an acetyl group. Thiolester hydrolases help regulate the formation and breakdown of these thiol esters, allowing cells to control various biochemical reactions.
Examples of thiolester hydrolases include:
1. CoA thioesterases (CoATEs): These enzymes hydrolyze thiol esters between coenzyme A and fatty acids, releasing free coenzyme A and a fatty acid. This process is essential for fatty acid metabolism.
2. Acetyl-CoA hydrolase: This enzyme specifically breaks down the thiol ester bond in acetyl-CoA, releasing acetic acid and coenzyme A.
3. Thioesterases involved in non-ribosomal peptide synthesis (NRPS): These enzymes hydrolyze thiol esters during the biosynthesis of complex peptides, allowing for the formation of unique amino acid sequences and structures.
Understanding the function and regulation of thiolester hydrolases can provide valuable insights into various metabolic processes and potential therapeutic targets in disease treatment.
Dipeptidyl-peptidases (DPPs) and tripeptidyl-peptidases (TPPs) are two types of enzymes that belong to the class of peptidases, which are proteins that help break down other proteins into smaller peptides or individual amino acids.
Dipeptidyl-peptidases cleave dipeptides (two-amino acid units) from the N-terminus (the end with a free amino group) of polypeptides and proteins, while tripeptidyl-peptidases cleave tripeptides (three-amino acid units) from the same location.
There are several different isoforms of DPPs and TPPs that have been identified in various organisms, including humans. These enzymes play important roles in regulating various physiological processes, such as digestion, immune function, and blood glucose homeostasis.
Inhibitors of DPP-4, one specific isoform of DPPs, have been developed for the treatment of type 2 diabetes, as they help increase the levels of incretin hormones that stimulate insulin secretion and suppress glucagon production.
Aminopeptidases are a group of enzymes that catalyze the removal of amino acids from the N-terminus of polypeptides and proteins. They play important roles in various biological processes, including protein degradation, processing, and activation. Aminopeptidases are classified based on their specificity for different types of amino acids and the mechanism of their action. Some of the well-known aminopeptidases include leucine aminopeptidase, alanyl aminopeptidase, and arginine aminopeptidase. They are widely distributed in nature and found in various tissues and organisms, including bacteria, plants, and animals. In humans, aminopeptidases are involved in several physiological functions, such as digestion, immune response, and blood pressure regulation.
Lipofuscin is a type of pigment that accumulates in the lysosomes (membrane-bound organelles found inside cells) of various tissues, particularly in nerve cells and heart muscle cells. It consists of cross-linked proteins and lipids that are resistant to degradation by enzymes. The accumulation of lipofuscin is a normal part of aging but can also be associated with certain diseases such as neurodegenerative disorders.
It's often referred to as "age pigment" because it tends to increase in amount with age, and its presence in tissues has been linked to oxidative stress and cellular damage caused by free radicals. Lipofuscin is autofluorescent, meaning that it emits light when excited by certain wavelengths of light, which can be useful for its detection and quantification in research and diagnostic settings.
Lipidoses are a group of genetic disorders characterized by abnormal accumulation of lipids (fats or fat-like substances) in various tissues and cells of the body due to defects in lipid metabolism. These disorders include conditions such as Gaucher's disease, Tay-Sachs disease, Niemann-Pick disease, Fabry disease, and Wolman disease, among others. The accumulation of lipids can lead to progressive damage in multiple organs, resulting in a range of symptoms and health complications. Early diagnosis and management are essential for improving the quality of life and prognosis of affected individuals.
Lysosomal storage diseases (LSDs) are a group of rare inherited metabolic disorders caused by defects in lysosomal function. These diseases affect many different organ systems, including the nervous system. Lysosomes are membrane-bound organelles found inside cells that break down and recycle various types of cellular waste materials through the action of enzymes. In LSDs, a genetic mutation leads to a deficiency or complete lack of a specific lysosomal enzyme, resulting in the accumulation of undigested substrates within the lysosomes. This accumulation can cause progressive damage to cells and tissues throughout the body, including those in the nervous system.
There are more than 50 different types of LSDs, some of which primarily affect the nervous system:
1. Tay-Sachs disease: A severe neurological disorder caused by a deficiency of the enzyme hexosaminidase A (HEXA). The accumulation of ganglioside GM2 in neurons leads to progressive neurodegeneration, resulting in motor and cognitive decline, blindness, and early death.
2. Sandhoff disease: Similar to Tay-Sachs disease but caused by a deficiency in both HEXA and hexosaminidase B (HEXB) enzymes. This disorder affects multiple organ systems, including the nervous system, with symptoms similar to Tay-Sachs disease but often more severe and rapid progression.
3. GM1 gangliosidosis: A condition caused by a deficiency of the enzyme β-galactosidase (GLB1), leading to the accumulation of GM1 ganglioside in neurons. Symptoms include developmental delay, motor and cognitive decline, seizures, and progressive neurological deterioration.
4. Gaucher disease: A disorder caused by a deficiency of the enzyme glucocerebrosidase (GBA), resulting in the accumulation of glucocerebroside in various tissues, including the nervous system. There are three main types of Gaucher disease, with type 2 and 3 having neurological involvement.
5. Niemann-Pick disease types A and B: These disorders are caused by a deficiency of the enzyme acid sphingomyelinase (SMPD1), leading to the accumulation of sphingomyelin in various tissues, including the nervous system. Type A primarily affects the nervous system, while type B mainly involves visceral organs.
6. Fabry disease: An X-linked disorder caused by a deficiency of the enzyme α-galactosidase A (GLA), resulting in the accumulation of globotriaosylceramide (Gb3) in various tissues, including the nervous system. Symptoms include pain, gastrointestinal issues, skin lesions, and progressive renal, cardiac, and cerebrovascular complications.
7. Metachromatic leukodystrophy: A disorder caused by a deficiency of the enzyme arylsulfatase A (ARSA), leading to the accumulation of sulfatides in the white matter of the brain. Symptoms include motor and cognitive decline, seizures, and progressive neurological deterioration.
8. Krabbe disease: An autosomal recessive disorder caused by a deficiency of the enzyme galactocerebrosidase (GALC), resulting in the accumulation of psychosine in the nervous system. Symptoms include motor and cognitive decline, seizures, and progressive neurological deterioration.
9. Mucopolysaccharidoses: A group of disorders caused by deficiencies of various enzymes involved in the breakdown of glycosaminoglycans (GAGs), leading to their accumulation in tissues throughout the body, including the nervous system. Symptoms vary depending on the specific disorder and include skeletal abnormalities, cardiac complications, vision and hearing loss, and progressive neurological decline.
10. Neuronal ceroid lipofuscinoses: A group of neurodegenerative disorders caused by mutations in various genes involved in lysosomal function, leading to the accumulation of lipopigments in neurons and other cells. Symptoms include seizures, motor and cognitive decline, vision loss, and progressive neurological deterioration.
11. Peroxisomal biogenesis disorders: A group of disorders caused by mutations in genes involved in peroxisome biogenesis, leading to the accumulation of very long-chain fatty acids, phytanic acid, and pipecolic acid in tissues throughout the body, including the nervous system. Symptoms vary depending on the specific disorder and include developmental delay, hypotonia, seizures, vision loss, hearing impairment, and progressive neurological decline.
12. Congenital disorders of glycosylation: A group of disorders caused by mutations in genes involved in N-glycosylation, leading to abnormal protein folding, trafficking, and function. Symptoms vary depending on the specific disorder and include developmental delay, hypotonia, seizures, vision loss, hearing impairment, and progressive neurological decline.
13. Leukodystrophies: A group of disorders characterized by abnormalities in the white matter of the brain due to defects in myelin formation or maintenance. Symptoms vary depending on the specific disorder and include developmental delay, hypotonia, seizures, vision loss, hearing impairment, and progressive neurological decline.
14. Mitochondrial disorders: A group of disorders caused by mutations in genes involved in mitochondrial function, leading to energy production deficits and oxidative stress. Symptoms vary depending on the specific disorder and include developmental delay, hypotonia, seizures, vision loss, hearing impairment, and progressive neurological decline.
15. Neurodegenerative disorders: A group of disorders characterized by progressive degeneration of the nervous system, leading to cognitive decline, motor dysfunction, and ultimately death. Examples include Alzheimer's disease, Parkinson's disease, Huntington's disease, and amyotrophic lateral sclerosis (ALS).
16. Neurodevelopmental disorders: A group of disorders characterized by impairments in cognitive, social, and motor development, including autism spectrum disorder, attention deficit hyperactivity disorder (ADHD), intellectual disability, and specific learning disorders.
17. Epilepsy: A group of disorders characterized by recurrent seizures due to abnormal electrical activity in the brain. Epilepsy can be caused by various genetic and environmental factors, including structural brain abnormalities, infections, trauma, and metabolic imbalances.
18. Neuroinflammatory disorders: A group of disorders characterized by inflammation of the nervous system, leading to damage and dysfunction. Examples include multiple sclerosis, neuromyelitis optica, and autoimmune encephalitis.
19. Infectious diseases of the nervous system: A group of disorders caused by infectious agents such as viruses, bacteria, fungi, or parasites that affect the nervous system. Examples include meningitis, encephalitis, and HIV-associated neurological disorders.
20. Neurotoxic disorders: A group of disorders caused by exposure to neurotoxic substances such as heavy metals, pesticides, solvents, or drugs that damage the nervous system. Examples include lead poisoning, organophosphate poisoning, and methanol toxicity.
21. Neurooncological disorders: A group of disorders characterized by tumors of the nervous system, including primary brain tumors, metastatic brain tumors, and spinal cord tumors.
22. Vascular disorders of the nervous system: A group of disorders caused by disruption of blood flow to the nervous system, leading to ischemia or hemorrhage. Examples include stroke, transient ischemic attack, and subarachnoid hemorrhage.
23. Degenerative disorders of the nervous system: A group of disorders characterized by progressive degeneration of nerve cells and their supporting structures, leading to functional impairment. Examples include Alzheimer's disease, Parkinson's disease, and Huntington's disease.
24. Neurodevelopmental disorders: A group of disorders that affect the development of the nervous system, leading to cognitive, behavioral, or motor impairments. Examples include autism spectrum disorder, attention deficit hyperactivity disorder, and intellectual disability.
25. Epilepsy and seizure disorders: A group of disorders characterized by recurrent seizures, which are abnormal electrical discharges in the brain that can cause a variety of symptoms such as convulsions, altered consciousness, or sensory disturbances.
26. Neurogenetic disorders: A group of disorders caused by genetic mutations that affect the structure or function of the nervous system. Examples include fragile X syndrome, tuberous sclerosis complex, and neurofibromatosis type 1.
27. Neuromuscular
Lysosomes are membrane-bound organelles found in the cytoplasm of eukaryotic cells. They are responsible for breaking down and recycling various materials, such as waste products, foreign substances, and damaged cellular components, through a process called autophagy or phagocytosis. Lysosomes contain hydrolytic enzymes that can break down biomolecules like proteins, nucleic acids, lipids, and carbohydrates into their basic building blocks, which can then be reused by the cell. They play a crucial role in maintaining cellular homeostasis and are often referred to as the "garbage disposal system" of the cell.
Biological pigments are substances produced by living organisms that absorb certain wavelengths of light and reflect others, resulting in the perception of color. These pigments play crucial roles in various biological processes such as photosynthesis, vision, and protection against harmful radiation. Some examples of biological pigments include melanin, hemoglobin, chlorophyll, carotenoids, and flavonoids.
Melanin is a pigment responsible for the color of skin, hair, and eyes in animals, including humans. Hemoglobin is a protein found in red blood cells that contains a porphyrin ring with an iron atom at its center, which gives blood its red color and facilitates oxygen transport. Chlorophyll is a green pigment found in plants, algae, and some bacteria that absorbs light during photosynthesis to convert carbon dioxide and water into glucose and oxygen. Carotenoids are orange, yellow, or red pigments found in fruits, vegetables, and some animals that protect against oxidative stress and help maintain membrane fluidity. Flavonoids are a class of plant pigments with antioxidant properties that have been linked to various health benefits.
Molecular chaperones are a group of proteins that assist in the proper folding and assembly of other protein molecules, helping them achieve their native conformation. They play a crucial role in preventing protein misfolding and aggregation, which can lead to the formation of toxic species associated with various neurodegenerative diseases. Molecular chaperones are also involved in protein transport across membranes, degradation of misfolded proteins, and protection of cells under stress conditions. Their function is generally non-catalytic and ATP-dependent, and they often interact with their client proteins in a transient manner.
Cathepsin F is a lysosomal cysteine protease that belongs to the papain family. It is primarily expressed in hematopoietic cells, including monocytes, macrophages, and dendritic cells. Cathepsin F plays a role in various physiological processes, such as antigen presentation, bone remodeling, and extracellular matrix degradation. It is also implicated in several pathological conditions, such as cancer, neurodegenerative disorders, and infectious diseases.
Cathepsin F has a broad substrate specificity and can cleave various proteins, including collagen, elastin, and casein. Its activity is tightly regulated by endogenous inhibitors, such as cystatins and stefins, to prevent excessive protein degradation and tissue damage.
In summary, Cathepsin F is a lysosomal enzyme involved in various physiological and pathological processes, with a broad substrate specificity and regulatory mechanisms.
Endopeptidases are a type of enzyme that breaks down proteins by cleaving peptide bonds inside the polypeptide chain. They are also known as proteinases or endoproteinases. These enzymes work within the interior of the protein molecule, cutting it at specific points along its length, as opposed to exopeptidases, which remove individual amino acids from the ends of the protein chain.
Endopeptidases play a crucial role in various biological processes, such as digestion, blood coagulation, and programmed cell death (apoptosis). They are classified based on their catalytic mechanism and the structure of their active site. Some examples of endopeptidase families include serine proteases, cysteine proteases, aspartic proteases, and metalloproteases.
It is important to note that while endopeptidases are essential for normal physiological functions, they can also contribute to disease processes when their activity is unregulated or misdirected. For instance, excessive endopeptidase activity has been implicated in the pathogenesis of neurodegenerative disorders, cancer, and inflammatory conditions.
Dolichol is a type of lipid molecule that is involved in the process of protein glycosylation within the endoplasmic reticulum of eukaryotic cells. Glycosylation is the attachment of sugar molecules to proteins, and it plays a crucial role in various biological processes such as protein folding, trafficking, and cell-cell recognition.
Dolichols are long-chain polyisoprenoid alcohols that serve as carriers for the sugars during glycosylation. They consist of a hydrophobic tail made up of many isoprene units and a hydrophilic head group. The dolichol molecule is first activated by the addition of a diphosphate group to its terminal end, forming dolichyl pyrophosphate.
The sugars that will be attached to the protein are then transferred from their nucleotide sugar donors onto the dolichyl pyrophosphate carrier, creating a dolichol-linked oligosaccharide. This oligosaccharide is then transferred en bloc to the target protein in a process called "oligosaccharyltransferase" (OST) reaction.
Defects in dolichol biosynthesis or function can lead to various genetic disorders, such as congenital disorders of glycosylation (CDG), which are characterized by abnormal protein glycosylation and a wide range of clinical manifestations, including developmental delay, neurological impairment, and multi-systemic involvement.
I am not aware of a medical term called "Cystaphos." It is possible that there may be a typographical error or misspelling in the term. If you have more context about where this term was used, I may be able to provide more information. However, without further information, I cannot provide a medical definition for "Cystaphos."
Cathepsin D is a lysosomal aspartic protease that plays a role in intracellular protein degradation and turnover. It is produced as an inactive precursor and is activated by cleavage into two subunits within the acidic environment of the lysosome. Cathepsin D is also known to be secreted by certain cells, where it can contribute to extracellular matrix remodeling and tissue degradation. In addition, abnormal levels or activity of cathepsin D have been implicated in various diseases, including cancer, neurodegenerative disorders, and infectious diseases.
There is no medical definition for "dog diseases" as it is too broad a term. However, dogs can suffer from various health conditions and illnesses that are specific to their species or similar to those found in humans. Some common categories of dog diseases include:
1. Infectious Diseases: These are caused by viruses, bacteria, fungi, or parasites. Examples include distemper, parvovirus, kennel cough, Lyme disease, and heartworms.
2. Hereditary/Genetic Disorders: Some dogs may inherit certain genetic disorders from their parents. Examples include hip dysplasia, elbow dysplasia, progressive retinal atrophy (PRA), and degenerative myelopathy.
3. Age-Related Diseases: As dogs age, they become more susceptible to various health issues. Common age-related diseases in dogs include arthritis, dental disease, cancer, and cognitive dysfunction syndrome (CDS).
4. Nutritional Disorders: Malnutrition or improper feeding can lead to various health problems in dogs. Examples include obesity, malnutrition, and vitamin deficiencies.
5. Environmental Diseases: These are caused by exposure to environmental factors such as toxins, allergens, or extreme temperatures. Examples include heatstroke, frostbite, and toxicities from ingesting harmful substances.
6. Neurological Disorders: Dogs can suffer from various neurological conditions that affect their nervous system. Examples include epilepsy, intervertebral disc disease (IVDD), and vestibular disease.
7. Behavioral Disorders: Some dogs may develop behavioral issues due to various factors such as anxiety, fear, or aggression. Examples include separation anxiety, noise phobias, and resource guarding.
It's important to note that regular veterinary care, proper nutrition, exercise, and preventative measures can help reduce the risk of many dog diseases.
Hermanski-Pudlak Syndrome (HPS) is a rare genetic disorder characterized by the triad of albinism, bleeding disorders, and lysosomal storage disease. It is caused by mutations in any one of several genes involved in biogenesis of lysosome-related organelles (LROs), such as melanosomes in melanocytes, platelet dense granules, and lung lamellar bodies.
The albinism in HPS results from abnormal melanosome biogenesis, leading to decreased pigmentation in the skin, hair, and eyes. The bleeding disorder is due to defective platelet dense granules, which are necessary for normal clotting function. This can result in prolonged bleeding times and easy bruising.
The lysosomal storage disease component of HPS is characterized by the accumulation of ceroid lipofuscin within LROs, leading to progressive damage to affected tissues. The most common form of HPS (HPS-1) also involves pulmonary fibrosis, which can lead to respiratory failure and death in the third or fourth decade of life.
There are currently seven known subtypes of HPS, each caused by mutations in different genes involved in LRO biogenesis. The clinical features and severity of HPS can vary widely between subtypes and even within families with the same genetic mutation.
I'm happy to help, but I want to clarify that the term "sheep diseases" refers to a wide range of health issues that can affect sheep populations. It's not a single medical condition with a specific definition. Here are some examples of common diseases that can affect sheep:
1. Ovine Johne's Disease (OJD): A chronic, infectious disease caused by the bacterium Mycobacterium avium subspecies paratuberculosis. It affects the intestines and can cause weight loss, diarrhea, and death.
2. Footrot: A highly contagious bacterial infection that affects the feet of sheep, causing lameness, swelling, and pain. It's caused by the bacteria Dichelobacter nodosus.
3. Caseous Lymphadenitis (CL): A chronic infectious disease caused by the bacterium Corynebacterium pseudotuberculosis. It affects the lymph nodes and can cause abscesses, weight loss, and death.
4. Contagious Ecthyma (Orf): A highly contagious viral infection that affects the skin and mucous membranes of sheep, causing sores and lesions.
5. Mastitis: An inflammation of the mammary gland in sheep, usually caused by a bacterial infection. It can cause decreased milk production, fever, and loss of appetite.
6. Pneumonia: A respiratory infection that can affect sheep, causing coughing, difficulty breathing, and fever. It can be caused by various bacteria or viruses.
7. Enterotoxemia: A potentially fatal disease caused by the overproduction of toxins in the intestines of sheep, usually due to a bacterial infection with Clostridium perfringens.
8. Polioencephalomalacia (PEM): A neurological disorder that affects the brain of sheep, causing symptoms such as blindness, circling, and seizures. It's often caused by a thiamine deficiency or excessive sulfur intake.
9. Toxoplasmosis: A parasitic infection that can affect sheep, causing abortion, stillbirth, and neurological symptoms.
10. Blue tongue: A viral disease that affects sheep, causing fever, respiratory distress, and mouth ulcers. It's transmitted by insect vectors and is often associated with climate change.
The subdural space is a potential space between the dura mater, which is the outermost of the three meninges covering the brain and spinal cord, and the arachnoid mater, which is the middle meningeal layer. This space normally contains a thin film of fluid, but when it becomes filled with blood (subdural hematoma) or pus (subdural empyema), it can cause significant neurological problems due to increased pressure on the brain. The subdural space can also become widened in certain conditions such as dementia or hydrocephalus, leading to a condition called subdural hygroma.
The brain is the central organ of the nervous system, responsible for receiving and processing sensory information, regulating vital functions, and controlling behavior, movement, and cognition. It is divided into several distinct regions, each with specific functions:
1. Cerebrum: The largest part of the brain, responsible for higher cognitive functions such as thinking, learning, memory, language, and perception. It is divided into two hemispheres, each controlling the opposite side of the body.
2. Cerebellum: Located at the back of the brain, it is responsible for coordinating muscle movements, maintaining balance, and fine-tuning motor skills.
3. Brainstem: Connects the cerebrum and cerebellum to the spinal cord, controlling vital functions such as breathing, heart rate, and blood pressure. It also serves as a relay center for sensory information and motor commands between the brain and the rest of the body.
4. Diencephalon: A region that includes the thalamus (a major sensory relay station) and hypothalamus (regulates hormones, temperature, hunger, thirst, and sleep).
5. Limbic system: A group of structures involved in emotional processing, memory formation, and motivation, including the hippocampus, amygdala, and cingulate gyrus.
The brain is composed of billions of interconnected neurons that communicate through electrical and chemical signals. It is protected by the skull and surrounded by three layers of membranes called meninges, as well as cerebrospinal fluid that provides cushioning and nutrients.
Animal disease models are specialized animals, typically rodents such as mice or rats, that have been genetically engineered or exposed to certain conditions to develop symptoms and physiological changes similar to those seen in human diseases. These models are used in medical research to study the pathophysiology of diseases, identify potential therapeutic targets, test drug efficacy and safety, and understand disease mechanisms.
The genetic modifications can include knockout or knock-in mutations, transgenic expression of specific genes, or RNA interference techniques. The animals may also be exposed to environmental factors such as chemicals, radiation, or infectious agents to induce the disease state.
Examples of animal disease models include:
1. Mouse models of cancer: Genetically engineered mice that develop various types of tumors, allowing researchers to study cancer initiation, progression, and metastasis.
2. Alzheimer's disease models: Transgenic mice expressing mutant human genes associated with Alzheimer's disease, which exhibit amyloid plaque formation and cognitive decline.
3. Diabetes models: Obese and diabetic mouse strains like the NOD (non-obese diabetic) or db/db mice, used to study the development of type 1 and type 2 diabetes, respectively.
4. Cardiovascular disease models: Atherosclerosis-prone mice, such as ApoE-deficient or LDLR-deficient mice, that develop plaque buildup in their arteries when fed a high-fat diet.
5. Inflammatory bowel disease models: Mice with genetic mutations affecting intestinal barrier function and immune response, such as IL-10 knockout or SAMP1/YitFc mice, which develop colitis.
Animal disease models are essential tools in preclinical research, but it is important to recognize their limitations. Differences between species can affect the translatability of results from animal studies to human patients. Therefore, researchers must carefully consider the choice of model and interpret findings cautiously when applying them to human diseases.
 Ceroid cactus
Ceroid cactus
Neuronal ceroid lipofuscinosis
Infantile neuronal ceroid lipofuscinosis
Jansky-Bielschowsky disease
Batten disease
Degenerative disease
Myoclonic epilepsy
Sara Mole
Kufs disease
Sweat gland
List of OMIM disorder codes
Jan Janský
Frederick Batten
CLN5
Tripeptidyl peptidase I
Cerliponase alfa
Northern epilepsy syndrome
CLN6
DNAJC5
PPT1
Battenin
Major facilitator superfamily
Erika F. Augustine
Night-blooming cereus
VPS35
MFSD8
Model organism database
Juan Pedro Bolaños
CLN8
Tibetan Terrier
Ceroid cactus - Wikipedia
 Neuronal Ceroid Lipofuscinoses: Background, Etiology, Epidemiology
Neuronal Ceroid Lipofuscinoses: Background, Etiology, Epidemiology
 Retinal degeneration in motor neuron degeneration: a mouse model of ceroid lipofuscinosis
Retinal degeneration in motor neuron degeneration: a mouse model of ceroid lipofuscinosis
 Neuronal Ceroid Lipofuscinosis - Australian Shepherd Health & Genetics Institute
Neuronal Ceroid Lipofuscinosis - Australian Shepherd Health & Genetics Institute
 Neuronal Ceroid Lipofuscinosis (CLN5-Related) - Norton & Elaine Sarnoff Center for Jewish Genetics
Neuronal Ceroid Lipofuscinosis (CLN5-Related) - Norton & Elaine Sarnoff Center for Jewish Genetics
 CLN8-related neuronal ceroid lipofuscinosis | Myriad Foresight® Carrier Screen
CLN8-related neuronal ceroid lipofuscinosis | Myriad Foresight® Carrier Screen
Late Infantile Neuronal Ceroid Lipofuscinosis - CheckOrphan
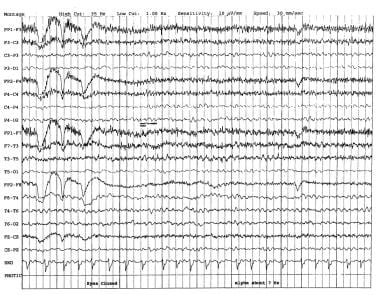
EEG in Dementia and Encephalopathy: Overview, Dementia, Vascular Dementia
 Neuronal Ceroid Lipofuscinosis 7 (NCL7) - DogWellNet
Neuronal Ceroid Lipofuscinosis 7 (NCL7) - DogWellNet
Neuronal Ceroid Lipofuscionsis (10) - Animal Genetics
 NCL1 - neuronal ceroid lipofuscinosis - Slovgen.sk
NCL1 - neuronal ceroid lipofuscinosis - Slovgen.sk
 Neuronal ceroid lipofuscinosis, 8-1 - CombiBreed
Neuronal ceroid lipofuscinosis, 8-1 - CombiBreed infantile neuronal ceroid lipofuscinosis (INCL) • BioPharma Media
infantile neuronal ceroid lipofuscinosis (INCL) • BioPharma Media
 CEROID LIPOFUSCINOSIS, NEURONAL, 2; CLN2 | MENDELIAN.CO
CEROID LIPOFUSCINOSIS, NEURONAL, 2; CLN2 | MENDELIAN.CO
The Neuronal Ceroid Lipofuscinoses (Batten Disease) | Basi6Direct
 AtlasRLeye - Ocular Manifestations of Neuronal Ceroid Lipofuscinosis
AtlasRLeye - Ocular Manifestations of Neuronal Ceroid Lipofuscinosis
 ATP13A2-related juvenile neuronal ceroid lipofuscinosis - Global Genes
ATP13A2-related juvenile neuronal ceroid lipofuscinosis - Global Genes
 Juvenile neuronal ceroid lipofuscinosis: Facts, Symptoms, Treatments | Carenity
Juvenile neuronal ceroid lipofuscinosis: Facts, Symptoms, Treatments | Carenity
 Neuronale Ceroid-Lipofiszinose (NCL)
Neuronale Ceroid-Lipofiszinose (NCL)
 Diagnosis of neuronal ceroid lipofuscinosis (Batten disease) by electron microscopy in peripheral blood specimens. | Read by...
Diagnosis of neuronal ceroid lipofuscinosis (Batten disease) by electron microscopy in peripheral blood specimens. | Read by...
 Neuronal Ceroid Lipofuscinosis 5 (Border Collie Type)
- DNA Test - Orivet
Neuronal Ceroid Lipofuscinosis 5 (Border Collie Type)
- DNA Test - Orivet
 Neuronal ceroid lipofuscinosis 8 northern epilepsy variant (Concept Id: C1864923)
- MedGen - NCBI
Neuronal ceroid lipofuscinosis 8 northern epilepsy variant (Concept Id: C1864923)
- MedGen - NCBI
 Saluki Dog Breed Health and Care | PetMD
Saluki Dog Breed Health and Care | PetMD
 Paw Print Genetics - Neuronal Ceroid Lipofuscinosis 10 in the American Pit Bull Terrier
Paw Print Genetics - Neuronal Ceroid Lipofuscinosis 10 in the American Pit Bull Terrier
 "Atypical" phenotypes of neuronal ceroid lipofuscinosis: the Argentine experience in the genomic...
"Atypical" phenotypes of neuronal ceroid lipofuscinosis: the Argentine experience in the genomic...
Paw Print Genetics - Neuronal Ceroid Lipofuscinosis 5 (Herding Dog Type) in the Koolie
 Gesellschaft Schweizer Tierärztinnen und Tierärzte GST: Die neuronale Ceroid-Lipofuszinose beim Hund: Eine Übersicht
Gesellschaft Schweizer Tierärztinnen und Tierärzte GST: Die neuronale Ceroid-Lipofuszinose beim Hund: Eine Übersicht
 Immune modulation attenuates infantile neuronal ceroid lipofuscinosis in mice before and after disease onset - Striatech
Immune modulation attenuates infantile neuronal ceroid lipofuscinosis in mice before and after disease onset - Striatech
A case of neuronal ceroid lipofuscinosis type 8 associated with central precocious puberty<...
Called ceroid lipofuscin1
- A defect in metabolism leads to a build up of a pigmented toxin called ceroid lipofuscin within cells, including those of the brain and retina. (orivet.com)
NCLs8
- The neuronal ceroid lipofuscinoses (NCLs), also known as Batten disease, are a group of neurodegenerative disorders. (medscape.com)
- The neuronal ceroid lipofuscinoses (NCLs) originally were defined by their age of onset and clinical symptoms. (medscape.com)
- The Neuronal ceroid lipofuscinosis (NCLs) are a group of inherited neurodegenerative diseases characterized by accumulation of autofluorescent cytoplasmic antibodies within cells of the nervous system. (dogwellnet.com)
- CLN8 disease is one of a group of disorders known as neuronal ceroid lipofuscinoses (NCLs), which may also be collectively referred to as Batten disease. (nih.gov)
- CLN11 disease is one of a group of disorders known as neuronal ceroid lipofuscinoses (NCLs). (medlineplus.gov)
- This article addresses the educational issues associated with rare diseases (RD) and in particular the Neuronal Ceroid Lipofuscinoses (NCLs, or CLN diseases) in the curricula of Health Sciences and Professional's Training Programs. (ox.ac.uk)
- The neuronal ceroid lipofuscinoses (NCLs) are a group of inherited neurodegenerative disorders characterized by accumulation of autofluorescent material in many tissues, especially in neurons. (biomedcentral.com)
- The neuronal ceroid lipofuscinoses (NCLs) comprise a group of human neurodegenerative disorders (CLN1-CLN8) characterized by epilepsy, visual failure, psychomotor deterioration and accumulation of autofluorescent lipopigment in many tissues, especially in neurons [ 1 ]. (biomedcentral.com)
Infantile6
- Other names for this condition are Finnish variant late infantile neuronal ceroid lipofuscinosis, Finnish vLINCL, Jansky-Bielschowsky disease, late-infantile neuronal ceroid lipofuscinosis, and neuronal ceroid lipofuscinosis 5. (jewishgenetics.org)
- Mutations in the CLN8 gene typically result in variant late-infantile neuronal ceroid lipofuscinosis (vLINCL) or Northern epilepsy (also know as Progressive Epilepsy with Mental Retardation, or EPMR). (myriad.com)
- Variant late-infantile neuronal ceroid lipofuscinosis The symptoms of vLINCL typically begin between 3 and 8 years of age. (myriad.com)
- Late Infantile Neuronal Ceroid. (checkorphan.org)
- Late infantile neuronal ceroid lipofuscinosis (also known as LINCL, Jansky-Bielschowsky and late infantile CLN2/TPP1 disorder) is part of a group of progressive degenerative neurometabolic disorders known as the ceroid lipofuscinosis neuronal (CLNs). (checkorphan.org)
- In two sibs with late-infantile neuronal ceroid lipofuscinosis (LINCL), Sleat et al. (coriell.org)
Batten5
- The mnd mouse model is similar to the juvenile onset Spielmeyer-Vogt form of ceroid lipofuscinosis (Batten disease), and provides a good model for the retinal degeneration found in these patients. (nih.gov)
- Diagnosis of neuronal ceroid lipofuscinosis (Batten disease) by electron microscopy in peripheral blood specimens. (qxmd.com)
- Neuronal ceroid lipopofuscinosis (Batten disease, NCL) represents a group of common childhood neurodegenerative diseases with a shared feature of deposition of abnormal metabolic products in neurons and other tissues, including peripheral blood lymphocytes. (qxmd.com)
- Quantifying physical decline in juvenile neuronal ceroid lipofuscinosis (Batten disease). (ac.ir)
- Enzyme replacement therapy (ERT) appears safe and effective for peripheral manifestations in patients with Gaucher disease types I and III, Fabry disease, mucopolysaccharidosis I (Hurler, Hurler-Scheie, and Scheie syndromes), mucopolysaccharidosis II (Hunter syndrome), mucopolysaccharidosis VI (Maroteaux-Lamy syndrome), Pompe disease, and recently Batten disease (neuronal ceroid lipofuscinoses, CLN2). (medscape.com)
Stored ceroid-lipofuscin in neurons1
- The stored ceroid-lipofuscin in neurons leads to an impaired cell function and subsequently to cell death. (gstsvs.ch)
Lipofuscinosis neuronal3
- Persaud-Sawin et al found that transfecting CLN1 (ceroid lipofuscinosis, neuronal 1)- or CLN2-deficient cells with CLN deoxyribonucleic acid (DNA) constructs for either CLN1 or CLN2 was somewhat protective against etoposide-induced apoptosis in both cell types. (medscape.com)
- Each disease type is given the designation "CLN," meaning ceroid lipofuscinosis, neuronal, and then a number to indicate its subtype. (nih.gov)
- Juvenile neuronal ceroid lipofuscinosis (JNCL), one of the most frequent forms of the NCL storage diseases, is known to be caused by loss-of-function mutations in ceroid-lipofuscinosis neuronal protein 3 (CLN3), but its cell function has not been fully elucidated. (ac.ir)
CLN21
- Background: Neuronal ceroid lipofuscinosis type 2 or CLN2 disease is a rare, autosomal recessive, neurodegenerative lysosomal storage disorder caused by tripeptidyl peptidase 1 deficiency. (blogspot.com)
Neuronal ceroid lipofuscinos1
- Intracerebroventricular Cerliponase Alfa for Neuronal Ceroid Lipofuscinosis Type 2 Disease: Clinical Practice Considerations From US Clinics [published online ahead of print, 2020 May 4]. (blogspot.com)
Causes neuronal ceroid lipofus2
- A mutation in canine CLN5 causes neuronal ceroid lipofuscinosis in Border collie dogs. (orivet.com)
- Mutation of the Parkinsonism Gene ATP13A2 Causes Neuronal Ceroid-Lipofuscinosis , Hum. (ucl.ac.uk)
Lipofuscin4
- NCL was later so named because of the accumulation of autofluorescent lipopigments resembling ceroid and lipofuscin seen in patients with the condition. (medscape.com)
- Neuronal Ceroid Lipofuscinosis (NCL) refers to a group of inherited lysosomal storage disorders characterized by the intracellular accumulation of ceroid-lipofuscin compounds and neurodegeneration. (unc.edu.ar)
- In consequence of a gene mutation, there is an accumulation of ceroid-lipofuscin in neurons, cells of the retina and the skin and other cells. (gstsvs.ch)
- Pulmonary fibrosis, granulomatous colitis, cardiomyopathy, and renal failure are due to the lysosomal accumulation of ceroid lipofuscin. (lu.se)
Neurodegenerative3
- Neuronal ceroid lipofuscinosis (NCL) is a neurodegenerative disorder caused by dysregulated sphingolipid metabolism, leading to progressive and severe accumulation of lipofuscins in cells of the brain and other tissues. (atlasrleye.com)
- Die Neuronale Ceroid Lipofuszinose (NCL) ist eine neurodegenerative Erkrankung aufgrund von. (labogen.com)
- BACKGROUND AND PURPOSE: Juvenile neuronal ceroid lipofuscinosis is a progressive neurodegenerative lysosomal storage disease of childhood. (uantwerpen.be)
Accumulation1
- Neuronal ceroid lipofuscinosis causes accumulation of storage material in the brain and eye, which can lead to blindness, disorientation, and seizures. (petmd.com)
Inherited disorder1
- CLN5-related neuronal ceroid lipofuscinosis (NCL) is an inherited disorder stemming from mutations in the CLN5 gene, leading to progressive brain degeneration and the gradual loss of both cognitive and motor abilities. (jewishgenetics.org)
Mutation3
- A mutation in the cathepsin D gene (CTSD) in American Bulldogs with neuronal ceroid lipofuscinosis. (animalgenetics.com)
- Almeida MR, Macario MC, Ramos L, Baldeiras I, Ribeiro MH, Santana I. Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. (medlineplus.gov)
- Homozygous TBC1 domain-containing kinase (TBCK) mutation causes a novel lysosomal storage disease - a new type of neuronal ceroid lipofuscinosis (CLN15)? (uni-tuebingen.de)
Mutations2
- Mutations in CLN5 cause a subtype of neuronal ceroid lipofuscinosis (NCL) called CLN5 disease. (frontiersin.org)
- A team of researchers led by Drs. Rita Guerreiro and Jose Bras at UCL has identified mutations in ATP13A2 as a cause of a separate disease entity called Neuronal Ceroid-Lipofuscinosis (NCL) in a large family from Belgium. (ucl.ac.uk)
Symptoms1
- There are several forms of neuronal ceroid lipofuscinosis (NCL), largely differentiated by the gene responsible and the age at which symptoms begin. (myriad.com)
Variant1
- Evans J, Katz ML, Levesque D, Shelton GD, de Lahunta A, O'Brien D. A variant form of neuronal ceroid lipofuscinosis in American bulldogs. (animalgenetics.com)
Canine1
- The present article gives a survey over the current scientific knowledge of the canine neuronal ceroid-lipofuscinosis (NCL). (gstsvs.ch)
CLN82
- CLN8-related neuronal ceroid lipofuscinosis (NCL8) is an inherited condition that causes degeneration of the brain, leading to a progressive loss of mental and motor skills, seizures, and vision impairment in some cases. (myriad.com)
- What Is the Prognosis for an Individual with CLN8-Related Neuronal Ceroid Lipofuscinosis? (myriad.com)
CLN51
- Genetic testing of the CLN5 gene will reliably determine whether a dog is a genetic Carrier of neuronal ceroid lipofuscinosis 5. (pawprintgenetics.com)
Epilepsy1
- Epilepsy Advanced Sequencing and CNV Evaluation - Neuronal Ceroid Lipofuscinosis. (mendelian.co)
Diseases2
- Guidelines for incorporating scientific knowledge and practice on rare diseases into higher education: neuronal ceroid lipofuscinoses as a model disorder. (ox.ac.uk)
- Lysosomal storage diseases are generally classified by the accumulated substrate and include the sphingolipidoses, oligosaccharidoses, mucolipidoses, mucopolysaccharidoses (MPSs), lipoprotein storage disorders, lysosomal transport defects, neuronal ceroid lipofuscinoses and others. (medscape.com)
Hund1
- Die vorliegende Arbeit gibt eine Übersicht und Zusammenfassung über den aktuellen Wissensstand der neuronalen Ceroid-Lipofuszinose (NCL) beim Hund. (gstsvs.ch)
Juvenile onset1
- A rare neuronal ceroid lipofiscinosis disorder characterized by juvenile-onset of progressive spinocerebellar ataxia bulbar syndrome (manifesting with dysarthria dysphagia and dysphonia) pyramidal and extrapyramidal involvement (including myoclonus amyotrophy unsteady gait akinesia rigidity dysarthric speech) and intellectual deterioration. (globalgenes.org)
Type1
- a new type of neuronal ceroid lipofuscinosis (CLN15)? (uni-tuebingen.de)
Disease4
- It is believed that a dog who was imported into Australia was a carrier of ceroid lipofuscinosis before anyone was aware of the disease, and that many Border collies now in Australia can trace their descent to this dog. (orivet.com)
- Neuronal ceroid lipofuscinosis 10 (NCL10) is a lysosomal storage disease affecting dogs. (pawprintgenetics.com)
- Neuronal ceroid lipofuscinosis 10 is inherited in an Autosomal Recessive manner in dogs meaning that they must receive two copies of the mutated gene (one from each parent) to develop the disease. (pawprintgenetics.com)
- Neuronal ceroid lipofuscinosis 5 (NCL5) is a lysosomal storage disease affecting Border Collies. (pawprintgenetics.com)
Classification1
- New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. (ac.ir)
Aufgrund1
- Aufgrund einer Genmutation kommt es zur Akkumulation von Ceroid-Lipofuszin in Nervenzellen, Zellen der Netzhaut, Haut und anderen Körperzellen. (gstsvs.ch)
Seizures1
- All childhood forms of ceroid lipofuscinosis are fatal and their manifestations include epileptic seizures, loss of vision, ataxia, loss of motor coordination and mental deterioration. (atlasrleye.com)
Dogs1
- Mizukami K, Chang HS, Yabuki A, Kawamichi T, Kawahara N, Hayashi D, Hossain MA, Rahman MM, Uddin MM, Yamato O. Novel rapid genotyping assays for neuronal ceroid lipofuscinosis in Border Collie dogs and high frequency of the mutant allele in Japan. (pawprintgenetics.com)