Wakefulness-Promoting Agents
Receptors, Adrenergic, alpha-2
Arthritis, Rheumatoid
Receptors, Tumor Necrosis Factor
Tumor Necrosis Factor-alpha
Immunoglobulin G
alpha 1-Antitrypsin
Receptors, Adrenergic, alpha
Hormone Antagonists
Dopamine Antagonists
Dose-Response Relationship, Drug
Excitatory Amino Acid Antagonists
Neurokinin-1 Receptor Antagonists
Adrenergic alpha-1 Receptor Antagonists
Histamine H2 Antagonists
Interleukin 1 Receptor Antagonist Protein
Rats, Sprague-Dawley
Hypoxia-Inducible Factor 1, alpha Subunit
Nicotinic Antagonists
Muscarinic Antagonists
Adrenergic alpha-Antagonists
GABA Antagonists
alpha7 Nicotinic Acetylcholine Receptor
Histamine H1 Antagonists
Purinergic P1 Receptor Antagonists
Receptors, Adrenergic, alpha-1
alpha1-adrenergic receptor subtypes in human peripheral blood lymphocytes. (1/2042)
We investigated the expression of alpha1-adrenergic receptor subtypes in intact human peripheral blood lymphocytes using reverse transcription-polymerase chain reaction (RT-PCR) and radioligand binding assay techniques combined with antibodies against the three subtypes of alpha1-adrenergic receptors (alpha1A, alpha1B, and alpha1D). RT-PCR amplified in peripheral blood lymphocytes a 348-bp alpha1A-adrenergic receptor fragment, a 689-bp alpha1B-adrenergic receptor fragment, and a 540-bp alpha1D-adrenergic receptor fragment. Radioligand binding assay with [3H]prazosin as radioligand revealed a high-affinity binding with a dissociation constant value of 0. 65+/-0.05 nmol/L and a maximum density of binding sites of 175. 3+/-20.5 fmol/10(6) cells. The pharmacological profile of [3H]prazosin binding to human peripheral blood lymphocytes was consistent with the labeling of alpha1-adrenergic receptors. Antibodies against alpha1A-, alpha1B-, and alpha1D-receptor subtypes decreased [3H]prazosin binding to a different extent. This indicates that human peripheral blood lymphocytes express the three alpha1-adrenergic receptor subtypes. Of the three different alpha1-adrenergic receptor subtypes, the alpha1B is the most represented and the alpha1D, the least. Future studies should clarify the functional relevance of alpha1-adrenergic receptors expressed by peripheral blood lymphocytes. The identification of these sites may represent a step for evaluating whether they represent a marker of alpha1-adrenergic receptors in cardiovascular disorders or for assessing responses to drug treatment on these receptors. (+info)Modulation of basal intracellular calcium by inverse agonists and phorbol myristate acetate in rat-1 fibroblasts stably expressing alpha1d-adrenoceptors. (2/2042)
In rat-1 fibroblasts stably expressing alpha1d-adrenoceptors BMY 7378, phentolamine, chloroethylclonidine and 5-methyl urapidil decreased basal [Ca2+]i. WB 4101 induced a very small effect on this parameter but when added before the other antagonists it blocked their effect. All these agents inhibited the action of norepinephrine. Phorbol myristate acetate also blocked the effect of norepinephrine and decreased basal [Ca2+]i. Staurosporine inhibited these effects of the phorbol ester. Our results suggest that: (1) alpha1d-adrenoceptors exhibit spontaneous ligand-independent activity, (2) BMY 7378, phentolamine, chloroethylclonidine and 5-methyl urapidil act as inverse agonists and (3) protein kinase C activation blocks spontaneous and agonist-stimulated alpha1d-adrenoceptor activity. (+info)Transcriptional regulation of alpha1-adrenoceptor gene in the rat liver during different phases of sepsis. (3/2042)
Changes in alpha1-adrenoceptor (alpha1AR) gene expression in the rat liver during different phases of sepsis were studied. Sepsis was induced by cecal ligation and puncture (CLP). Septic rats exhibit two metabolically distinct phases: an initial hyperglycemic phase (9 h after CLP, early sepsis) followed by a hypoglycemic phase (18 h after CLP; late sepsis). The [3H]prazosin binding studies show that the density of alpha1AR was increased by 30% during the early phase while it was decreased by 24% during the late phase of sepsis. Western blot analyses reveal that alpha1AR protein level was elevated by 48% during early sepsis but was decreased by 55% during late sepsis. Northern blot analyses depict that the steady-state level of alpha1bAR mRNA was enhanced by 21% during the early phase but was declined by 29% during the late phase of sepsis. Nuclear run-off assays show that the transcription rate of alpha1bAR gene transcript was increased by 76% during early sepsis while it was decreased by 29% during late sepsis. The actinomycin D pulse-chase studies indicate that the half-life of alpha1bAR mRNA remained unaffected during the early and the late phases of sepsis. These findings demonstrate that during the early phase of sepsis, the increase in the rate of transcription of alpha1bAR gene paralleled with the elevations in the alpha1bAR mRNA abundance and alpha1AR protein level, while during the late phase of sepsis, the decrease in the rate of transcription of alpha1bAR gene coincided with the declines in the alpha1bAR mRNA abundance and the alpha1AR protein level in the rat liver. These observations indicate that the altered expression of alpha1AR genes in the rat liver during the progression of sepsis was regulated transcriptionally. (+info)Facilitation and depression of ATP and noradrenaline release from sympathetic nerves of rat tail artery. (4/2042)
1. Excitatory junction currents (EJCs) were used to measure ATP release; noradrenaline (NA) oxidation currents and fractional overflow of labelled NA, [3H]NA, were used to monitor the release of endogenous and exogenous NA, respectively, from post-ganglionic sympathetic nerves of rat tail artery. 2. During nerve stimulation with 100 pulses at 5-20 Hz the EJCs initially grew in size (maximally by 23 %, at 2-10 Hz), and then depressed, maximally by 68 % at 20 Hz. 3. The peak amplitude of NA oxidation currents in response to nerve stimulation with 100 pulses at 2-20 Hz grew in size with frequency, while the area was independent of frequency and roughly constant. 4. The size of the NA oxidation currents evoked by nerve stimulation with 4-100 pulses at 20 Hz grew linearly with train length between pulses 4-16. Between pulses 20-100 there was a train length-dependent depression of the signal. 5. Fractional overflow of [3H]NA in response to nerve stimulation with 5-100 pulses at 20 Hz behaved similarly to the EJCs. It initially grew roughly linearly between pulses 5-25, and then showed a dramatic depression similar to that of the EJCs. 6. The alpha2-adrenoceptor antagonists rauwolscine and yohimbine increased the overflow of [3H]NA and the amplitude of NA oxidation currents, but not that of the EJCs. 7. It is concluded that during high-frequency stimulation (i) the release of ATP and NA is first briefly facilitated then markedly depressed, (ii) facilitation and depression of the two transmitters are similar in magnitude and time course, and (iii) alpha2-adrenoceptor antagonists differentially modify EJCs and the NA signals. The results obtained in the absence of drugs are compatible with the hypothesis that ATP and NA are released in parallel, while the effects of alpha2-adrenoceptor antagonists seem to suggest dissociated release. (+info)Homologous regulation of the alpha2C-adrenoceptor subtype in human hepatocarcinoma, HepG2. (5/2042)
1. Previous studies of the regulation of the alpha2C-adrenoceptor in OK and in transfected cells have led to discrepant conclusions. In the present work, we examined the homologous regulation of the human alpha2C-adrenoceptor in the hepatocarcinoma cell-line, HepG2; a model which expresses this subtype spontaneously. 2. Short-period treatment of the cells with UK14304 provoked neither a diminution of the potency of the alpha2-agonist to inhibit forskolin-induced cyclic AMP-accumulation nor a change in the degree of receptor coupling to G-proteins. 3. Long-period exposure to UK14304 resulted in a large reduction of [3H]MK912 binding sites (55% decrease). The action of UK14304 was dose-dependent (EC50 = 190 +/- 45 nM), rapid (t1/2 = 4.2 h) and reversible. Receptor down-regulation was also observed with clonidine or (-)adrenaline (38 and 36% decrease, respectively) and was blocked by the addition of alpha2-antagonists. 4. Conversely to that observed with alpha2-agonists, treatment of the cells with RX821002 or yohimbine alone, but not with phentolamine, promoted a significant increase of the receptor expression. 5. The observed alterations of receptor density are not the reflection of changes at the alpha2C4 mRNA level. Estimation of the receptor protein turnover and measurement of its half-life demonstrated that down-regulation by alpha2-agonists and up-regulation by alpha2-antagonists, with inverse-agonist efficacy, are respectively the consequence of increased and decreased rate of receptor degradation. 6. In conclusion, our data show that alpha2C-adrenoceptor does not undergo desensitization but is down-regulated in HepG2. The lack of desensitization agrees with previous results obtained in cells transfected with the alpha2C4 gene, but not with observations made in OK cells. Inversely, down-regulation fits with results obtained in OK but not in transfected cells. The reasons for these discrepancies are discussed. Our results also demonstrated that certain alpha2-antagonists behave as inverse agonist on the HepG2 model and thus provide for the first time evidence of inverse efficacy of antagonists on a cellular model expressing physiological level of a wild-type alpha2-adrenoceptor. (+info)Effects of heptanol on the neurogenic and myogenic contractions of the guinea-pig vas deferens. (6/2042)
1. The effects of the putative gap junction uncoupler, 1-heptanol, on the neurogenic and myogenic contractile responses of guinea-pig vas deferens were studied in vitro. 2. Superfusion of 2.0 mM heptanol for 20-30 min produced the following reversible changes in the biphasic neurogenic contractile response (8 trials): (i) suppression of both phases; (ii) delayed development of both the first as well as the second phase, accompanied by complete temporal separation of the two phases; (iii) prominent oscillations of force during the second (noradrenergic) phase only. 3. To eliminate prejunctional effects of heptanol, myogenic contractions were evoked by field stimulation of the vas in the presence of suramin (200 microM) and prazosin (1 microM). Heptanol (2.0 mM) abolished these contractions reversibly. 4. These results show that (i) heptanol inhibits both excitatory junction potential (EJP)-dependent and non EJP-dependent contractions of the vas; (ii) a postjunctional site of action of heptanol, probably intercellular uncoupling of smooth muscle cells, contributes to the inhibition of contraction. (+info)Nitric oxide mediates sympathetic vasoconstriction at supraspinal, spinal, and synaptic levels. (7/2042)
The purposes of this study were to investigate the level of the sympathetic nervous system in which nitric oxide (NO) mediates regional sympathetic vasoconstriction and to determine whether neural mechanisms are involved in vasoconstriction after NO inhibition. Ganglionic blockade (hexamethonium), alpha1-receptor blockade (prazosin), and spinal section at T1 were used to study sympathetic involvement. NO was blocked with Nomega-nitro-L-arginine methyl ester (L-NAME). Regional blood flow in the mesenteric and renal arteries and terminal aorta was monitored by electromagnetic flowmetry in conscious rats. L-NAME (3-5 mg/kg iv) increased arterial pressure and peripheral resistance. Ganglionic blockade (25 mg/kg iv) significantly reduced the increase in resistance in the mesentery and kidney in intact and spinal-sectioned rats. Ganglionic blockade significantly decreased hindquarter resistance in intact rats but not in spinal-sectioned rats. Prazosin (200 micrograms/kg iv) significantly reduced the increased hindquarter resistance. We concluded that NO suppresses sympathetic vasoconstriction in the mesentery and kidney at the spinal level, whereas hindquarter tone is mediated at supraspinal and synaptic levels. (+info)Facilitatory beta2-adrenoceptors on cholinergic and adrenergic nerve endings of the guinea pig trachea. (8/2042)
Using electrical field stimulation of epithelium-denuded intact guinea pig tracheal tube preparations, we studied the presence and role of prejunctional beta2-adrenoceptors by measuring evoked endogenous acetylcholine (ACh) and norepinephrine (NE) release directly. Analysis of ACh and NE was through two HPLC systems with electrochemical detection. Electrical field stimulation (150 mA, 0.8 ms, 16 Hz, 5 min, biphasic pulses) released 29.1 +/- 2.5 pmol ACh/g tissue and 70.2 +/- 6.2 pmol NE/g tissue. Preincubation for 15 min with the selective beta2-adrenoceptor agonist fenoterol (1 microM) increased both ACh and NE overflow to 178 +/- 28 (P < 0.01) and 165 +/- 12% (P < 0.01), respectively, of control values, increases that were abolished completely by the selective beta2-adrenoceptor antagonist ICI-118551 (1 microM). Further experiments with increasing fenoterol concentrations (0.1-100 microM) and different preincubation periods (1, 5, and 15 min) showed a strong and concentration-dependent facilitation of NE release, with maximum response levels decreasing (from nearly 5-fold to only 2.5-fold of control value) with increasing agonist contact time. In contrast, sensitivity of facilitatory beta2-adrenoceptors on cholinergic nerves to fenoterol gradually increased when the incubation period was prolonged; in addition, a bell-shaped concentration-response relationship was found at 15 min of preincubation. Fenoterol concentration-response relationships (15-min agonist preincubation) in the presence of atropine and yohimbine (1 microM each) were similar in the case of NE release, but in the case of ACh release, the bell shape was lost. The results indicate a differential capacity and response time profile of facilitatory prejunctional beta2-adrenoceptors on adrenergic and cholinergic nerve terminals in the guinea pig trachea and suggest that the receptors on adrenergic nerves are more susceptible to desensitization. (+info)Wakefulness-promoting agents are a class of medications that are used to promote and maintain alertness and wakefulness. They work by stimulating the brain's arousal centers, increasing the release of neurotransmitters such as dopamine, norepinephrine, and histamine, which help to counteract the effects of sleep-promoting substances in the brain.
Wakefulness-promoting agents are typically used to treat excessive daytime sleepiness associated with conditions such as narcolepsy, obstructive sleep apnea, shift work sleep disorder, and other disorders that cause disrupted sleep patterns. Some examples of wakefulness-promoting agents include modafinil, armodafinil, pitolisant, and solriamfetol.
It is important to note that while these medications can help to promote alertness and reduce excessive daytime sleepiness, they are not a substitute for getting adequate amounts of quality sleep. It is also important to use them under the guidance of a healthcare provider, as they may have potential side effects and interactions with other medications.
Alpha-2 adrenergic receptors are a type of G protein-coupled receptor that binds catecholamines, such as norepinephrine and epinephrine. These receptors are widely distributed in the central and peripheral nervous system, as well as in various organs and tissues throughout the body.
Activation of alpha-2 adrenergic receptors leads to a variety of physiological responses, including inhibition of neurotransmitter release, vasoconstriction, and reduced heart rate. These receptors play important roles in regulating blood pressure, pain perception, and various cognitive and emotional processes.
There are several subtypes of alpha-2 adrenergic receptors, including alpha-2A, alpha-2B, and alpha-2C, which may have distinct physiological functions and be targeted by different drugs. For example, certain medications used to treat hypertension or opioid withdrawal target alpha-2 adrenergic receptors to produce their therapeutic effects.
Antirheumatic agents are a class of drugs used to treat rheumatoid arthritis, other inflammatory types of arthritis, and related conditions. These medications work by reducing inflammation in the body, relieving symptoms such as pain, swelling, and stiffness in the joints. They can also help slow down or prevent joint damage and disability caused by the disease.
There are several types of antirheumatic agents, including:
1. Nonsteroidal anti-inflammatory drugs (NSAIDs): These medications, such as ibuprofen and naproxen, reduce inflammation and relieve pain. They are often used to treat mild to moderate symptoms of arthritis.
2. Corticosteroids: These powerful anti-inflammatory drugs, such as prednisone and cortisone, can quickly reduce inflammation and suppress the immune system. They are usually used for short-term relief of severe symptoms or in combination with other antirheumatic agents.
3. Disease-modifying antirheumatic drugs (DMARDs): These medications, such as methotrexate and hydroxychloroquine, work by slowing down the progression of rheumatoid arthritis and preventing joint damage. They can take several weeks or months to become fully effective.
4. Biologic response modifiers (biologics): These are a newer class of DMARDs that target specific molecules involved in the immune response. They include drugs such as adalimumab, etanercept, and infliximab. Biologics are usually used in combination with other antirheumatic agents for patients who have not responded to traditional DMARD therapy.
5. Janus kinase (JAK) inhibitors: These medications, such as tofacitinib and baricitinib, work by blocking the action of enzymes called JAKs that are involved in the immune response. They are used to treat moderate to severe rheumatoid arthritis and can be used in combination with other antirheumatic agents.
It is important to note that antirheumatic agents can have significant side effects and should only be prescribed by a healthcare provider who is experienced in the management of rheumatoid arthritis. Regular monitoring and follow-up are essential to ensure safe and effective treatment.
Rheumatoid arthritis (RA) is a systemic autoimmune disease that primarily affects the joints. It is characterized by persistent inflammation, synovial hyperplasia, and subsequent damage to the articular cartilage and bone. The immune system mistakenly attacks the body's own tissues, specifically targeting the synovial membrane lining the joint capsule. This results in swelling, pain, warmth, and stiffness in affected joints, often most severely in the hands and feet.
RA can also have extra-articular manifestations, affecting other organs such as the lungs, heart, skin, eyes, and blood vessels. The exact cause of RA remains unknown, but it is believed to involve a complex interplay between genetic susceptibility and environmental triggers. Early diagnosis and treatment are crucial in managing rheumatoid arthritis to prevent joint damage, disability, and systemic complications.
Tumor Necrosis Factor (TNF) Receptors are cell surface receptors that bind to tumor necrosis factor cytokines. They play crucial roles in the regulation of a variety of immune cell functions, including inflammation, immunity, and cell survival or death (apoptosis).
There are two major types of TNF receptors: TNFR1 (also known as p55 or CD120a) and TNFR2 (also known as p75 or CD120b). TNFR1 is widely expressed in most tissues, while TNFR2 has a more restricted expression pattern and is mainly found on immune cells.
TNF receptors have an intracellular domain called the death domain, which can trigger signaling pathways leading to apoptosis when activated by TNF ligands. However, they can also activate other signaling pathways that promote cell survival, differentiation, and inflammation. Dysregulation of TNF receptor signaling has been implicated in various diseases, including cancer, autoimmune disorders, and neurodegenerative conditions.
Tumor Necrosis Factor-alpha (TNF-α) is a cytokine, a type of small signaling protein involved in immune response and inflammation. It is primarily produced by activated macrophages, although other cell types such as T-cells, natural killer cells, and mast cells can also produce it.
TNF-α plays a crucial role in the body's defense against infection and tissue injury by mediating inflammatory responses, activating immune cells, and inducing apoptosis (programmed cell death) in certain types of cells. It does this by binding to its receptors, TNFR1 and TNFR2, which are found on the surface of many cell types.
In addition to its role in the immune response, TNF-α has been implicated in the pathogenesis of several diseases, including autoimmune disorders such as rheumatoid arthritis, inflammatory bowel disease, and psoriasis, as well as cancer, where it can promote tumor growth and metastasis.
Therapeutic agents that target TNF-α, such as infliximab, adalimumab, and etanercept, have been developed to treat these conditions. However, these drugs can also increase the risk of infections and other side effects, so their use must be carefully monitored.
Monoclonal antibodies are a type of antibody that are identical because they are produced by a single clone of cells. They are laboratory-produced molecules that act like human antibodies in the immune system. They can be designed to attach to specific proteins found on the surface of cancer cells, making them useful for targeting and treating cancer. Monoclonal antibodies can also be used as a therapy for other diseases, such as autoimmune disorders and inflammatory conditions.
Monoclonal antibodies are produced by fusing a single type of immune cell, called a B cell, with a tumor cell to create a hybrid cell, or hybridoma. This hybrid cell is then able to replicate indefinitely, producing a large number of identical copies of the original antibody. These antibodies can be further modified and engineered to enhance their ability to bind to specific targets, increase their stability, and improve their effectiveness as therapeutic agents.
Monoclonal antibodies have several mechanisms of action in cancer therapy. They can directly kill cancer cells by binding to them and triggering an immune response. They can also block the signals that promote cancer growth and survival. Additionally, monoclonal antibodies can be used to deliver drugs or radiation directly to cancer cells, increasing the effectiveness of these treatments while minimizing their side effects on healthy tissues.
Monoclonal antibodies have become an important tool in modern medicine, with several approved for use in cancer therapy and other diseases. They are continuing to be studied and developed as a promising approach to treating a wide range of medical conditions.
Immunoglobulin G (IgG) is a type of antibody, which is a protective protein produced by the immune system in response to foreign substances like bacteria or viruses. IgG is the most abundant type of antibody in human blood, making up about 75-80% of all antibodies. It is found in all body fluids and plays a crucial role in fighting infections caused by bacteria, viruses, and toxins.
IgG has several important functions:
1. Neutralization: IgG can bind to the surface of bacteria or viruses, preventing them from attaching to and infecting human cells.
2. Opsonization: IgG coats the surface of pathogens, making them more recognizable and easier for immune cells like neutrophils and macrophages to phagocytose (engulf and destroy) them.
3. Complement activation: IgG can activate the complement system, a group of proteins that work together to help eliminate pathogens from the body. Activation of the complement system leads to the formation of the membrane attack complex, which creates holes in the cell membranes of bacteria, leading to their lysis (destruction).
4. Antibody-dependent cellular cytotoxicity (ADCC): IgG can bind to immune cells like natural killer (NK) cells and trigger them to release substances that cause target cells (such as virus-infected or cancerous cells) to undergo apoptosis (programmed cell death).
5. Immune complex formation: IgG can form immune complexes with antigens, which can then be removed from the body through various mechanisms, such as phagocytosis by immune cells or excretion in urine.
IgG is a critical component of adaptive immunity and provides long-lasting protection against reinfection with many pathogens. It has four subclasses (IgG1, IgG2, IgG3, and IgG4) that differ in their structure, function, and distribution in the body.
Alpha 1-antitrypsin (AAT, or α1-antiproteinase, A1AP) is a protein that is primarily produced by the liver and released into the bloodstream. It belongs to a group of proteins called serine protease inhibitors, which help regulate inflammation and protect tissues from damage caused by enzymes involved in the immune response.
Alpha 1-antitrypsin is particularly important for protecting the lungs from damage caused by neutrophil elastase, an enzyme released by white blood cells called neutrophils during inflammation. In the lungs, AAT binds to and inhibits neutrophil elastase, preventing it from degrading the extracellular matrix and damaging lung tissue.
Deficiency in alpha 1-antitrypsin can lead to chronic obstructive pulmonary disease (COPD) and liver disease. The most common cause of AAT deficiency is a genetic mutation that results in abnormal folding and accumulation of the protein within liver cells, leading to reduced levels of functional AAT in the bloodstream. This condition is called alpha 1-antitrypsin deficiency (AATD) and can be inherited in an autosomal codominant manner. Individuals with severe AATD may require augmentation therapy with intravenous infusions of purified human AAT to help prevent lung damage.
Adrenergic receptors are a type of G protein-coupled receptor that bind and respond to catecholamines, such as epinephrine (adrenaline) and norepinephrine (noradrenaline). Alpha adrenergic receptors (α-ARs) are a subtype of adrenergic receptors that are classified into two main categories: α1-ARs and α2-ARs.
The activation of α1-ARs leads to the activation of phospholipase C, which results in an increase in intracellular calcium levels and the activation of various signaling pathways that mediate diverse physiological responses such as vasoconstriction, smooth muscle contraction, and cell proliferation.
On the other hand, α2-ARs are primarily located on presynaptic nerve terminals where they function to inhibit the release of neurotransmitters, including norepinephrine. The activation of α2-ARs also leads to the inhibition of adenylyl cyclase and a decrease in intracellular cAMP levels, which can mediate various physiological responses such as sedation, analgesia, and hypotension.
Overall, α-ARs play important roles in regulating various physiological functions, including cardiovascular function, mood, and cognition, and are also involved in the pathophysiology of several diseases, such as hypertension, heart failure, and neurodegenerative disorders.
Hormone antagonists are substances or drugs that block the action of hormones by binding to their receptors without activating them, thereby preventing the hormones from exerting their effects. They can be classified into two types: receptor antagonists and enzyme inhibitors. Receptor antagonists bind directly to hormone receptors and prevent the hormone from binding, while enzyme inhibitors block the production or breakdown of hormones by inhibiting specific enzymes involved in their metabolism. Hormone antagonists are used in the treatment of various medical conditions, such as cancer, hormonal disorders, and cardiovascular diseases.
Dopamine antagonists are a class of drugs that block the action of dopamine, a neurotransmitter in the brain associated with various functions including movement, motivation, and emotion. These drugs work by binding to dopamine receptors and preventing dopamine from attaching to them, which can help to reduce the symptoms of certain medical conditions such as schizophrenia, bipolar disorder, and gastroesophageal reflux disease (GERD).
There are several types of dopamine antagonists, including:
1. Typical antipsychotics: These drugs are primarily used to treat psychosis, including schizophrenia and delusional disorders. Examples include haloperidol, chlorpromazine, and fluphenazine.
2. Atypical antipsychotics: These drugs are also used to treat psychosis but have fewer side effects than typical antipsychotics. They may also be used to treat bipolar disorder and depression. Examples include risperidone, olanzapine, and quetiapine.
3. Antiemetics: These drugs are used to treat nausea and vomiting. Examples include metoclopramide and prochlorperazine.
4. Dopamine agonists: While not technically dopamine antagonists, these drugs work by stimulating dopamine receptors and can be used to treat conditions such as Parkinson's disease. However, they can also have the opposite effect and block dopamine receptors in high doses, making them functionally similar to dopamine antagonists.
Common side effects of dopamine antagonists include sedation, weight gain, and movement disorders such as tardive dyskinesia. It's important to use these drugs under the close supervision of a healthcare provider to monitor for side effects and adjust the dosage as needed.
A dose-response relationship in the context of drugs refers to the changes in the effects or symptoms that occur as the dose of a drug is increased or decreased. Generally, as the dose of a drug is increased, the severity or intensity of its effects also increases. Conversely, as the dose is decreased, the effects of the drug become less severe or may disappear altogether.
The dose-response relationship is an important concept in pharmacology and toxicology because it helps to establish the safe and effective dosage range for a drug. By understanding how changes in the dose of a drug affect its therapeutic and adverse effects, healthcare providers can optimize treatment plans for their patients while minimizing the risk of harm.
The dose-response relationship is typically depicted as a curve that shows the relationship between the dose of a drug and its effect. The shape of the curve may vary depending on the drug and the specific effect being measured. Some drugs may have a steep dose-response curve, meaning that small changes in the dose can result in large differences in the effect. Other drugs may have a more gradual dose-response curve, where larger changes in the dose are needed to produce significant effects.
In addition to helping establish safe and effective dosages, the dose-response relationship is also used to evaluate the potential therapeutic benefits and risks of new drugs during clinical trials. By systematically testing different doses of a drug in controlled studies, researchers can identify the optimal dosage range for the drug and assess its safety and efficacy.
Excitatory amino acid antagonists are a class of drugs that block the action of excitatory neurotransmitters, particularly glutamate and aspartate, in the brain. These drugs work by binding to and blocking the receptors for these neurotransmitters, thereby reducing their ability to stimulate neurons and produce an excitatory response.
Excitatory amino acid antagonists have been studied for their potential therapeutic benefits in a variety of neurological conditions, including stroke, epilepsy, traumatic brain injury, and neurodegenerative disorders such as Alzheimer's disease and Parkinson's disease. However, their use is limited by the fact that blocking excitatory neurotransmission can also have negative effects on cognitive function and memory.
There are several types of excitatory amino acid receptors, including N-methyl-D-aspartate (NMDA), alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainite receptors. Different excitatory amino acid antagonists may target one or more of these receptor subtypes, depending on their specific mechanism of action.
Examples of excitatory amino acid antagonists include ketamine, memantine, and dextromethorphan. These drugs have been used in clinical practice for various indications, such as anesthesia, sedation, and treatment of neurological disorders. However, their use must be carefully monitored due to potential side effects and risks associated with blocking excitatory neurotransmission.
Neurokinin-1 (NK-1) receptor antagonists are a class of drugs that block the action of substance P, a neuropeptide involved in pain transmission and inflammation. These drugs work by binding to NK-1 receptors found on nerve cells, preventing substance P from activating them and transmitting pain signals. NK-1 receptor antagonists have been studied for their potential use in treating various conditions associated with pain and inflammation, such as migraine headaches, depression, and irritable bowel syndrome. Some examples of NK-1 receptor antagonists include aprepitant, fosaprepitant, and rolapitant.
Narcotic antagonists are a class of medications that block the effects of opioids, a type of narcotic pain reliever, by binding to opioid receptors in the brain and blocking the activation of these receptors by opioids. This results in the prevention or reversal of opioid-induced effects such as respiratory depression, sedation, and euphoria. Narcotic antagonists are used for a variety of medical purposes, including the treatment of opioid overdose, the management of opioid dependence, and the prevention of opioid-induced side effects in certain clinical situations. Examples of narcotic antagonists include naloxone, naltrexone, and methylnaltrexone.
Adrenergic alpha-1 receptor antagonists, also known as alpha-blockers, are a class of medications that block the effects of the neurotransmitter norepinephrine at alpha-1 receptors. These receptors are found in various tissues throughout the body, including the smooth muscle of blood vessels, the bladder, and the eye.
When norepinephrine binds to alpha-1 receptors, it causes smooth muscle to contract, leading to vasoconstriction (constriction of blood vessels), increased blood pressure, and other effects. By blocking these receptors, alpha-blockers can cause relaxation of smooth muscle, leading to vasodilation (expansion of blood vessels), decreased blood pressure, and other effects.
Alpha-blockers are used in the treatment of various medical conditions, including hypertension (high blood pressure), benign prostatic hyperplasia (enlarged prostate), and pheochromocytoma (a rare tumor of the adrenal gland). Examples of alpha-blockers include doxazosin, prazosin, and terazosin.
It's important to note that while alpha-blockers can be effective in treating certain medical conditions, they can also have side effects, such as dizziness, lightheadedness, and orthostatic hypotension (a sudden drop in blood pressure when standing up). As with any medication, it's important to use alpha-blockers under the guidance of a healthcare provider.
Histamine H2 antagonists, also known as H2 blockers, are a class of medications that work by blocking the action of histamine on the H2 receptors in the stomach. Histamine is a chemical that is released by the body during an allergic reaction and can also be released by certain cells in the stomach in response to food or other stimuli. When histamine binds to the H2 receptors in the stomach, it triggers the release of acid. By blocking the action of histamine on these receptors, H2 antagonists reduce the amount of acid produced by the stomach, which can help to relieve symptoms such as heartburn, indigestion, and stomach ulcers. Examples of H2 antagonists include ranitidine (Zantac), famotidine (Pepcid), and cimetidine (Tagamet).
Interleukin-1 Receptor Antagonist Protein (IL-1Ra) is a naturally occurring protein that acts as a competitive inhibitor of the interleukin-1 (IL-1) receptor. IL-1 is a pro-inflammatory cytokine involved in various physiological processes, including the immune response and inflammation. The binding of IL-1 to its receptor triggers a signaling cascade that leads to the activation of inflammatory genes and cellular responses.
IL-1Ra shares structural similarities with IL-1 but does not initiate the downstream signaling pathway. Instead, it binds to the same receptor site as IL-1, preventing IL-1 from interacting with its receptor and thus inhibiting the inflammatory response.
Increased levels of IL-1Ra have been found in various inflammatory conditions, such as rheumatoid arthritis, inflammatory bowel disease, and sepsis, where it acts to counterbalance the pro-inflammatory effects of IL-1. Recombinant IL-1Ra (Anakinra) is used clinically as a therapeutic agent for the treatment of rheumatoid arthritis and other inflammatory diseases.
Sprague-Dawley rats are a strain of albino laboratory rats that are widely used in scientific research. They were first developed by researchers H.H. Sprague and R.C. Dawley in the early 20th century, and have since become one of the most commonly used rat strains in biomedical research due to their relatively large size, ease of handling, and consistent genetic background.
Sprague-Dawley rats are outbred, which means that they are genetically diverse and do not suffer from the same limitations as inbred strains, which can have reduced fertility and increased susceptibility to certain diseases. They are also characterized by their docile nature and low levels of aggression, making them easier to handle and study than some other rat strains.
These rats are used in a wide variety of research areas, including toxicology, pharmacology, nutrition, cancer, and behavioral studies. Because they are genetically diverse, Sprague-Dawley rats can be used to model a range of human diseases and conditions, making them an important tool in the development of new drugs and therapies.
Hypoxia-Inducible Factor 1 (HIF-1) is a transcription factor that plays a crucial role in the body's response to low oxygen levels, also known as hypoxia. HIF-1 is a heterodimeric protein composed of two subunits: an alpha subunit (HIF-1α) and a beta subunit (HIF-1β).
The alpha subunit, HIF-1α, is the regulatory subunit that is subject to oxygen-dependent degradation. Under normal oxygen conditions (normoxia), HIF-1α is constantly produced in the cell but is rapidly degraded by proteasomes due to hydroxylation of specific proline residues by prolyl hydroxylase domain-containing proteins (PHDs). This hydroxylation reaction requires oxygen as a substrate, and under hypoxic conditions, the activity of PHDs is inhibited, leading to the stabilization and accumulation of HIF-1α.
Once stabilized, HIF-1α translocates to the nucleus, where it heterodimerizes with HIF-1β and binds to hypoxia-responsive elements (HREs) in the promoter regions of target genes. This binding results in the activation of gene transcription programs that promote cellular adaptation to low oxygen levels. These adaptive responses include increased erythropoiesis, angiogenesis, glucose metabolism, and pH regulation, among others.
Therefore, HIF-1α is a critical regulator of the body's response to hypoxia, and its dysregulation has been implicated in various pathological conditions, including cancer, cardiovascular disease, and neurodegenerative disorders.
Nicotinic antagonists are a class of drugs that block the action of nicotine at nicotinic acetylcholine receptors (nAChRs). These receptors are found in the nervous system and are activated by the neurotransmitter acetylcholine, as well as by nicotine. When nicotine binds to these receptors, it can cause the release of various neurotransmitters, including dopamine, which can lead to rewarding effects and addiction.
Nicotinic antagonists work by binding to nAChRs and preventing nicotine from activating them. This can help to reduce the rewarding effects of nicotine and may be useful in treating nicotine addiction. Examples of nicotinic antagonists include mecamylamine, varenicline, and cytisine.
It's important to note that while nicotinic antagonists can help with nicotine addiction, they can also have side effects, such as nausea, vomiting, and abnormal dreams. Additionally, some people may experience more serious side effects, such as seizures or cardiovascular problems, so it's important to use these medications under the close supervision of a healthcare provider.
Muscarinic antagonists, also known as muscarinic receptor antagonists or parasympatholytics, are a class of drugs that block the action of acetylcholine at muscarinic receptors. Acetylcholine is a neurotransmitter that plays an important role in the parasympathetic nervous system, which helps to regulate various bodily functions such as heart rate, digestion, and respiration.
Muscarinic antagonists work by binding to muscarinic receptors, which are found in various organs throughout the body, including the eyes, lungs, heart, and gastrointestinal tract. By blocking the action of acetylcholine at these receptors, muscarinic antagonists can produce a range of effects depending on the specific receptor subtype that is affected.
For example, muscarinic antagonists may be used to treat conditions such as chronic obstructive pulmonary disease (COPD) and asthma by relaxing the smooth muscle in the airways and reducing bronchoconstriction. They may also be used to treat conditions such as urinary incontinence or overactive bladder by reducing bladder contractions.
Some common muscarinic antagonists include atropine, scopolamine, ipratropium, and tiotropium. It's important to note that these drugs can have significant side effects, including dry mouth, blurred vision, constipation, and confusion, especially when used in high doses or for prolonged periods of time.
Adrenergic alpha-antagonists, also known as alpha-blockers, are a class of medications that block the effects of adrenaline and noradrenaline at alpha-adrenergic receptors. These receptors are found in various tissues throughout the body, including the smooth muscle of blood vessels, the heart, the genitourinary system, and the eyes.
When alpha-blockers bind to these receptors, they prevent the activation of the sympathetic nervous system, which is responsible for the "fight or flight" response. This results in a relaxation of the smooth muscle, leading to vasodilation (widening of blood vessels), decreased blood pressure, and increased blood flow.
Alpha-blockers are used to treat various medical conditions, such as hypertension (high blood pressure), benign prostatic hyperplasia (enlarged prostate), pheochromocytoma (a rare tumor of the adrenal gland), and certain types of glaucoma.
Examples of alpha-blockers include doxazosin, prazosin, terazosin, and tamsulosin. Side effects of alpha-blockers may include dizziness, lightheadedness, headache, weakness, and orthostatic hypotension (a sudden drop in blood pressure upon standing).
GABA (gamma-aminobutyric acid) antagonists are substances that block the action of GABA, which is the primary inhibitory neurotransmitter in the central nervous system. GABA plays a crucial role in regulating neuronal excitability and reducing the transmission of nerve impulses.
GABA antagonists work by binding to the GABA receptors without activating them, thereby preventing the normal function of GABA and increasing neuronal activity. These agents can cause excitation of the nervous system, leading to various effects depending on the specific type of GABA receptor they target.
GABA antagonists are used in medical treatments for certain conditions, such as sleep disorders, depression, and cognitive enhancement. However, they can also have adverse effects, including anxiety, agitation, seizures, and even neurotoxicity at high doses. Examples of GABA antagonists include picrotoxin, bicuculline, and flumazenil.
The alpha7 nicotinic acetylcholine receptor (α7nAChR) is a type of cholinergic receptor found in the nervous system that is activated by the neurotransmitter acetylcholine. It is a ligand-gated ion channel that is widely distributed throughout the central and peripheral nervous systems, including in the hippocampus, cortex, thalamus, and autonomic ganglia.
The α7nAChR is composed of five subunits arranged around a central pore, and it has a high permeability to calcium ions (Ca2+). When acetylcholine binds to the receptor, it triggers a conformational change that opens the ion channel, allowing Ca2+ to flow into the cell. This influx of Ca2+ can activate various intracellular signaling pathways and have excitatory or inhibitory effects on neuronal activity, depending on the location and function of the receptor.
The α7nAChR has been implicated in a variety of physiological processes, including learning and memory, attention, sensory perception, and motor control. It has also been studied as a potential therapeutic target for various neurological and psychiatric disorders, such as Alzheimer's disease, schizophrenia, and pain.
Histamine H1 antagonists, also known as H1 blockers or antihistamines, are a class of medications that work by blocking the action of histamine at the H1 receptor. Histamine is a chemical mediator released by mast cells and basophils in response to an allergic reaction or injury. It causes various symptoms such as itching, sneezing, runny nose, and wheal and flare reactions (hives).
H1 antagonists prevent the binding of histamine to its receptor, thereby alleviating these symptoms. They are commonly used to treat allergic conditions such as hay fever, hives, and eczema, as well as motion sickness and insomnia. Examples of H1 antagonists include diphenhydramine (Benadryl), loratadine (Claritin), cetirizine (Zyrtec), and doxylamine (Unisom).
Purinergic P1 receptor antagonists are a class of pharmaceutical drugs that block the activity of purinergic P1 receptors, which are a type of G-protein coupled receptor found in many tissues throughout the body. These receptors are activated by extracellular nucleotides such as adenosine and ATP, and play important roles in regulating a variety of physiological processes, including cardiovascular function, neurotransmission, and immune response.
Purinergic P1 receptor antagonists work by binding to these receptors and preventing them from being activated by nucleotides. This can have various therapeutic effects, depending on the specific receptor subtype that is targeted. For example, A1 receptor antagonists have been shown to improve cardiac function in heart failure, while A2A receptor antagonists have potential as anti-inflammatory and neuroprotective agents.
However, it's important to note that the use of purinergic P1 receptor antagonists is still an area of active research, and more studies are needed to fully understand their mechanisms of action and therapeutic potential.
Piperidines are not a medical term per se, but they are a class of organic compounds that have important applications in the pharmaceutical industry. Medically relevant piperidines include various drugs such as some antihistamines, antidepressants, and muscle relaxants.
A piperidine is a heterocyclic amine with a six-membered ring containing five carbon atoms and one nitrogen atom. The structure can be described as a cyclic secondary amine. Piperidines are found in some natural alkaloids, such as those derived from the pepper plant (Piper nigrum), which gives piperidines their name.
In a medical context, it is more common to encounter specific drugs that belong to the class of piperidines rather than the term itself.
Alpha-1 adrenergic receptors (also known as α1-adrenoreceptors) are a type of G protein-coupled receptor that binds catecholamines, such as norepinephrine and epinephrine. These receptors are primarily found in the smooth muscle of various organs, including the vasculature, heart, liver, kidneys, gastrointestinal tract, and genitourinary system.
When an alpha-1 adrenergic receptor is activated by a catecholamine, it triggers a signaling cascade that leads to the activation of phospholipase C, which in turn activates protein kinase C and increases intracellular calcium levels. This ultimately results in smooth muscle contraction, increased heart rate and force of contraction, and vasoconstriction.
Alpha-1 adrenergic receptors are also found in the central nervous system, where they play a role in regulating wakefulness, attention, and anxiety. There are three subtypes of alpha-1 adrenergic receptors (α1A, α1B, and α1D), each with distinct physiological roles and pharmacological properties.
In summary, alpha-1 adrenergic receptors are a type of G protein-coupled receptor that binds catecholamines and mediates various physiological responses, including smooth muscle contraction, increased heart rate and force of contraction, vasoconstriction, and regulation of wakefulness and anxiety.
Adrenergic alpha-2 receptor antagonists are a class of medications that block the action of norepinephrine, a neurotransmitter and hormone, at adrenergic alpha-2 receptors. These receptors are found in the central and peripheral nervous system and play a role in regulating various physiological functions such as blood pressure, heart rate, and insulin secretion.
By blocking the action of norepinephrine at these receptors, adrenergic alpha-2 receptor antagonists can increase sympathetic nervous system activity, leading to vasodilation, increased heart rate, and increased insulin secretion. These effects make them useful in the treatment of conditions such as hypotension (low blood pressure), opioid-induced sedation and respiratory depression, and diagnostic procedures that require vasodilation.
Examples of adrenergic alpha-2 receptor antagonists include yohimbine, idazoxan, and atipamezole. It's important to note that these medications can have significant side effects, including hypertension, tachycardia, and agitation, and should be used under the close supervision of a healthcare provider.
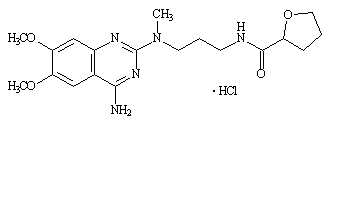
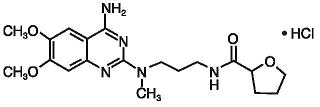
 Silodosin
Silodosin Opioid Abuse Medication: Opioid analgesics, Analgesics, Opioid Partial Agonist, Opioid Partial Agonist/Opioid Antagonist,...
Opioid Abuse Medication: Opioid analgesics, Analgesics, Opioid Partial Agonist, Opioid Partial Agonist/Opioid Antagonist,... Modulation of desynchronized sleep through microinjection of alpha 1-adrenergic agonists and antagonists in the dorsal pontine...
Modulation of desynchronized sleep through microinjection of alpha 1-adrenergic agonists and antagonists in the dorsal pontine... Erowid.org: Erowid Reference 3730 : Evidence for a Central Sympathoexcitatory Action of Alpha-2 Adrenergic Antagonists : Mccall...
Erowid.org: Erowid Reference 3730 : Evidence for a Central Sympathoexcitatory Action of Alpha-2 Adrenergic Antagonists : Mccall... Pharmacophore development for antagonists at alpha::1:: adrenergic receptor subtypes - researchr publication authors
Pharmacophore development for antagonists at alpha::1:: adrenergic receptor subtypes - researchr publication authors Noradrenergic Synaptic Function in the Bed Nucleus of the Stria Terminalis Varies in Animal Models of Anxiety and Addiction |...
Noradrenergic Synaptic Function in the Bed Nucleus of the Stria Terminalis Varies in Animal Models of Anxiety and Addiction |... DailyMed - DOXAZOSIN tablet
DailyMed - DOXAZOSIN tablet Bacteria, Beware: New Finding About E Coli Could Block Infections, Lead To Better Treatments | ScienceDaily
Bacteria, Beware: New Finding About E Coli Could Block Infections, Lead To Better Treatments | ScienceDaily Chronic Bacterial Prostatitis
Chronic Bacterial Prostatitis Multiple choice questions on drugs used in treating hypertension are presented.
Multiple choice questions on drugs used in treating hypertension are presented. High Blood Pressure
High Blood Pressure Returning Veterans With Addictions
Returning Veterans With Addictions RVL Pharmaceuticals plc Reports Fourth Quarter and Full Year 2022 Financial Results; Provides Commercial Update | BioSpace
RVL Pharmaceuticals plc Reports Fourth Quarter and Full Year 2022 Financial Results; Provides Commercial Update | BioSpace Complicated Cataract Surgery in Patients Receiving Alpha-Blockers for Benign Prostatic Hyperplasia
Complicated Cataract Surgery in Patients Receiving Alpha-Blockers for Benign Prostatic Hyperplasia Systematic review of antihypertensive therapies | CMAJ
Systematic review of antihypertensive therapies | CMAJ Pharmaceutical Tablets - DHA Extract Powder Wholesale Sellers from Surat
Pharmaceutical Tablets - DHA Extract Powder Wholesale Sellers from Surat Pheochromocytomas in Animals - Endocrine System - Merck Veterinary Manual
Pheochromocytomas in Animals - Endocrine System - Merck Veterinary Manual 'PREDICTION OF RESPONSE TO CLINICAL TREATMENT IN PATIENTS WITH BENIGN PROSTATIC HYPERPLASIA DIAGNOSED WITH PRONOUNCED MIDDLE...
'PREDICTION OF RESPONSE TO CLINICAL TREATMENT IN PATIENTS WITH BENIGN PROSTATIC HYPERPLASIA DIAGNOSED WITH PRONOUNCED MIDDLE... RAPAFLO® (silodosin) capsules - GlobalRPH
RAPAFLO® (silodosin) capsules - GlobalRPH