Autoimmune Lymphoproliferative Syndrome
Disordini Linfoproliferativi
Antigeni Cd95
Malattie Autoimmuni
Caspase 10
Anemia Emolitica Autoimmune
Tirosina Chinasi 3 Fms-Simile
Apoptosi
Mutazione
Linfociti T
L'Autoimmune Lymphoproliferative Syndrome (ALPS) è una rara malattia genetica del sistema immunitario. Si verifica quando il corpo non è in grado di regolare correttamente la produzione e l'eliminazione dei linfociti T, un tipo di globuli bianchi che aiutano a combattere le infezioni. Questa mancanza di regolazione porta all'accumulo di linfociti T nel corpo, il che può causare una varietà di sintomi, tra cui ingrossamento dei linfonodi, aumento del rischio di infezioni, anemia, trombocitopenia (ridotta conta delle piastrine) e la possibilità di sviluppare linfoma.
L'ALPS è spesso causata da mutazioni nei geni che codificano per le proteine che aiutano a regolare la morte programmata (apoptosi) dei linfociti T. Queste mutazioni portano ad un accumulo di linfociti T vitally importanti, che possono attaccere i tessuti sani del corpo, causando infiammazione e danni ai vari organi.
Il trattamento dell'ALPS dipende dai sintomi specifici e dalla gravità della malattia. Può includere farmaci per controllare l'infiammazione e la soppressione del sistema immunitario, la rimozione chirurgica dei linfonodi ingrossati e, in casi gravi, trapianto di midollo osseo.
I disordini linfoproliferativi (LPD) sono un gruppo eterogeneo di malattie che si verificano quando il sistema linfatico produce un numero eccessivo di cellule immunitarie, note come linfociti, in modo anomalo. Questi disordini possono essere classificati in base alla loro velocità di crescita e al grado di maturazione delle cellule coinvolte.
Esistono quattro principali categorie di LPD:
1. Linfomi: si tratta di tumori maligni che originano dai linfociti. I linfomi possono essere classificati in base al tipo di linfocita interessato (B o T) e alla velocità di crescita della malattia.
2. Leucemie: si tratta di tumori maligni che originano dai linfociti immaturi nel midollo osseo. Possono diffondersi rapidamente nel sangue e in altri organi ematopoietici.
3. Linfocitosi reattiva: si verifica quando il sistema immunitario produce un numero elevato di linfociti in risposta a un'infezione o ad altri stimoli, come una vaccinazione. Questa condizione è generalmente reversibile e non è considerata maligna.
4. Malattie linfoproliferative a lenta insorgenza: si tratta di condizioni croniche che comportano un'espansione clonale dei linfociti, ma che non soddisfano i criteri per la diagnosi di linfoma o leucemia. Questi disordini possono evolvere in linfomi a cellule B mature in alcuni pazienti.
I sintomi dei LPD possono variare notevolmente, a seconda del tipo e della localizzazione della malattia. Alcuni pazienti possono presentare sintomi aspecifici come stanchezza, febbre, sudorazione notturna e perdita di peso, mentre altri possono presentare segni e sintomi associati a specifiche localizzazioni della malattia, come adenopatie ingrossate, splenomegalia o lesioni cutanee.
La diagnosi dei LPD si basa sull'esame clinico, sulla storia del paziente, sui test di laboratorio e sulle indagini radiologiche. La conferma della diagnosi richiede spesso l'esecuzione di biopsie tissutali e l'analisi immunofenotipica e genetica delle cellule neoplastiche.
Il trattamento dei LPD dipende dal tipo, dallo stadio e dalla gravità della malattia. Le opzioni terapeutiche comprendono la chemioterapia, l'immunoterapia, la radioterapia e il trapianto di cellule staminali ematopoietiche. In alcuni casi, è possibile adottare un approccio osservazionale e attendista, soprattutto per le malattie a lenta insorgenza o con bassa aggressività.
La prognosi dei LPD varia notevolmente in base al tipo di malattia, allo stadio e alla risposta al trattamento. Alcuni tipi di LPD, come il linfoma follicolare a cellule B o il linfoma mantellare a cellule B, possono presentare un decorso clinico indolente e una prognosi favorevole, mentre altri, come il linfoma diffuso a grandi cellule B o il linfoma anaplastico a grandi cellule T, possono avere un decorso aggressivo e una prognosi sfavorevole.
In sintesi, i linfomi non Hodgkin sono un gruppo eterogeneo di neoplasie maligne del sistema ematolinfopoietico che comprendono diverse entità cliniche e patologiche. La diagnosi e il trattamento dei LNH richiedono una valutazione multidisciplinare e un approccio personalizzato in base al tipo, allo stadio e alla gravità della malattia. Nonostante le recenti innovazioni terapeutiche, i LNH rimangono una causa significativa di morbidità e mortalità, sottolineando l'importanza di ulteriori ricerche per migliorare la comprensione della patogenesi e lo sviluppo di nuove strategie terapeutiche.
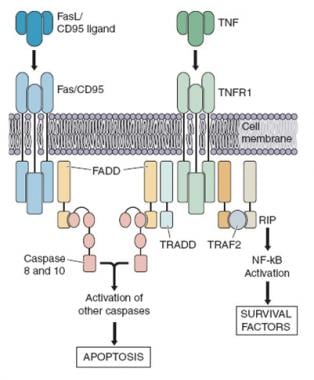
Gli antigeni CD95, anche noti come Fas (fattore di necrosi tumorale associato a una cellula T stimolata) o APO-1 (antigene correlato alla morte programmata), sono proteine transmembrana appartenenti alla superfamiglia dei recettori della morte (DR, death receptors). Sono espressi sulla superficie di molte cellule del corpo umano e svolgono un ruolo cruciale nella regolazione dell'apoptosi, o morte cellulare programmata.
L'antigene CD95 si lega al suo ligando (CD95L), che è presente sulla superficie di alcune cellule del sistema immunitario, come i linfociti T citotossici e le cellule natural killer (NK). Quando il CD95L si lega al CD95, avvia una cascata di segnalazione intracellulare che porta all'attivazione della caspasi, un gruppo di enzimi proteolitici che disgregano le proteine cellulari e innescano l'apoptosi.
Il sistema CD95/CD95L è importante per la regolazione dell'immunità e del mantenimento dell'omeostasi tissutale. Tuttavia, un'attivazione anomala o eccessiva di questo sistema può contribuire allo sviluppo di diverse patologie, tra cui malattie autoimmuni, infezioni virali e tumori.
In sintesi, gli antigeni CD95 sono proteine che mediano l'apoptosi cellulare e svolgono un ruolo cruciale nella regolazione dell'immunità e dell'omeostasi tissutale. Un'attivazione anomala o eccessiva di questo sistema può contribuire allo sviluppo di diverse patologie.
La definizione medica di "malattie autoimmuni" si riferisce a un gruppo eterogeneo di condizioni patologiche caratterizzate da una risposta immunitaria anomala dell'organismo contro i propri antigeni, ossia le proprie cellule e tessuti sani.
Normalmente, il sistema immunitario è in grado di distinguere tra agenti estranei (come batteri, virus e tossine) e componenti del corpo stesso, ed è programmato per attaccare solo i primi. Tuttavia, nelle malattie autoimmuni, questo meccanismo di difesa si altera, portando allo sviluppo di anticorpi e cellule immunitarie che attaccano i tessuti sani dell'organismo.
Le cause esatte alla base delle malattie autoimmuni non sono ancora del tutto chiare, ma sembrano coinvolgere una combinazione di fattori genetici e ambientali. Tra questi ultimi vi possono essere infezioni, traumi, stress emotivi o esposizione a sostanze chimiche tossiche.
Le malattie autoimmuni possono colpire quasi ogni organo o sistema del corpo, causando una vasta gamma di sintomi e complicazioni. Alcune delle più comuni malattie autoimmuni includono la artrite reumatoide, il lupus eritematoso sistemico, la celiachia, la tiroidite di Hashimoto, la vitiligine, la sclerosi multipla e il diabete di tipo 1.
Il trattamento delle malattie autoimmuni dipende dalla specifica condizione e dai suoi sintomi. Spesso prevede l'uso di farmaci immunosoppressori che aiutano a controllare la risposta immune anomala, riducendo così i danni ai tessuti sani. In alcuni casi, possono essere necessari anche interventi chirurgici o terapie di supporto per gestire le complicanze della malattia.
In medicina, una sindrome è generalmente definita come un insieme di segni e sintomi che insieme caratterizzano una particolare condizione o malattia. Una sindrome non è una malattia specifica, ma piuttosto un gruppo di sintomi che possono essere causati da diverse malattie o disturbi medici.
Una sindrome può essere causata da fattori genetici, ambientali o combinazioni di entrambi. Può anche derivare da una disfunzione o danno a un organo o sistema corporeo specifico. I sintomi associati a una sindrome possono variare in termini di numero, tipo e gravità, e possono influenzare diverse parti del corpo.
Esempi comuni di sindromi includono la sindrome metabolica, che è un gruppo di fattori di rischio per malattie cardiache e diabete, e la sindrome di Down, che è una condizione genetica caratterizzata da ritardo mentale e tratti fisici distintivi.
In sintesi, una sindrome è un insieme di segni e sintomi che insieme costituiscono una particolare condizione medica, ma non è una malattia specifica in sé.
La caspasi 10 è una proteina appartenente alla famiglia delle caspasi, enzimi proteolitici che svolgono un ruolo chiave nella regolazione dell'apoptosi, ossia il processo di morte cellulare programmata.
La caspasi 10 è codificata dal gene CASP10 ed è attivata in risposta a diversi stimoli apoptotici, come ad esempio la privazione di fattori di crescita o l'esposizione a radiazioni. Una volta attivata, la caspasi 10 media la rottura della membrana mitocondriale e l'attivazione di altre caspasi, portando infine alla morte cellulare.
La caspasi 10 è stata anche identificata come un possibile fattore chiave nella regolazione dell'immunità innata, in particolare nella risposta all'infezione da parte di patogeni intracellulari. Tuttavia, il suo ruolo esatto in questo contesto rimane ancora oggetto di studio e non è completamente chiaro.
In sintesi, la caspasi 10 è un enzima proteolitico che media l'apoptosi e potrebbe avere un ruolo nella regolazione dell'immunità innata.
La splenomegalia è un termine medico che si riferisce all'ingrossamento della milza oltre le sue dimensioni normali. La milza è un organo situato nella parte superiore sinistra dell'addome, vicino allo stomaco, e fa parte del sistema linfatico e immunitario. Normalmente, la milza non è palpabile al di sotto del bordo costale, ma in caso di splenomegalia, può essere avvertita come una massa durante l'esame fisico.
Le cause della splenomegalia possono essere varie e includono:
1. Infezioni: alcune infezioni batteriche, virali o parassitarie possono causare l'ingrossamento della milza, come la mononucleosi infettiva, l'epatite virale, la tubercolosi e la malaria.
2. Malattie ematologiche: alcune condizioni che colpiscono il sangue o i vasi sanguigni possono causare splenomegalia, come l'anemia falciforme, le talassemie, le leucemie e i linfomi.
3. Malattie del fegato: alcune malattie epatiche, come la cirrosi, l'epatite cronica o il tumore al fegato, possono causare l'ingrossamento della milza.
4. Condizioni cardiovascolari: alcune patologie cardiovascolari, come l'insufficienza cardiaca congestizia o l'endocardite batterica, possono determinare la splenomegalia.
5. Malattie reumatiche: alcune malattie reumatiche, come il lupus eritematoso sistemico o l'artrite reumatoide, possono causare l'ingrossamento della milza.
6. Tumori: alcuni tumori, sia benigni che maligni, possono portare all'ingrandimento della milza.
La diagnosi di splenomegalia si basa sulla storia clinica del paziente, sull'esame fisico e su esami di laboratorio e strumentali specifici. Il trattamento dipende dalla causa sottostante e può variare da un semplice monitoraggio a interventi chirurgici o chemioterapici.
L'anemia emolitica autoimmune (AEAI) è una condizione caratterizzata dalla distruzione prematura dei globuli rossi (eritrociti) nel sangue. Questo processo di distruzione è noto come emolisi. Nell'AEAI, l'emolisi è causata da anticorpi autoimmuni che attaccano e distruggono i propri globuli rossi sani.
I globuli rossi sani sono responsabili del trasporto di ossigeno dai polmoni ai tessuti corporei. Quando vengono distrutti prematuramente, l'organismo non riceve abbastanza ossigeno, il che può causare una serie di sintomi, tra cui affaticamento, debolezza, mancanza di respiro, vertigini e, in casi gravi, insufficienza d'organo.
L'AEAI può essere classificata in due tipi principali:
1. Anemia emolitica autoimmune calda: in questo tipo, gli anticorpi si legano ai globuli rossi a temperature corporee normali o elevate. L'anemia emolitica autoimmune calda è spesso associata a malattie autoimmuni come il lupus eritematoso sistemico e la sindrome di Sjogren.
2. Anemia emolitica autoimmune fredda: in questo tipo, gli anticorpi si legano ai globuli rossi a temperature più basse, ad esempio quando la temperatura corporea scende o quando le mani e i piedi sono esposti al freddo. L'anemia emolitica autoimmune fredda è spesso associata a infezioni virali o batteriche, ma può anche verificarsi senza una causa nota.
Il trattamento dell'AEAI dipende dalla gravità della condizione e dalle cause sottostanti. Può includere farmaci corticosteroidi, immunosoppressori, plasmaferesi o terapia con immunoglobuline. In alcuni casi, può essere necessario un trapianto di midollo osseo.
La tirosina chinasi 3 Fms-simile (FTK3 o FLT3) è un enzima che si trova sulla superficie delle cellule ematopoietiche e svolge un ruolo importante nella proliferazione, differenziazione e sopravvivenza di queste cellule. In particolare, FTK3 è una tirosina chinasi che si attiva quando lega il suo ligando specifico, portando all'attivazione di diversi percorsi di segnalazione cellulare che controllano la crescita e la divisione cellulare.
Le mutazioni a carico del gene FTK3 sono state identificate in una varietà di tumori ematologici, tra cui la leucemia mieloide acuta (LMA) e la leucemia linfoblastica acuta (LLA). Queste mutazioni possono comportare un'attivazione costitutiva dell'enzima, che a sua volta può portare all'aumento della proliferazione cellulare e alla ridotta apoptosi, contribuendo al sviluppo e alla progressione del tumore.
La tirosina chinasi 3 Fms-simile è quindi un bersaglio importante per la terapia mirata dei tumori ematologici, con diversi inibitori di FTK3 attualmente in fase di sviluppo clinico. Questi farmaci sono progettati per bloccare l'attività enzimatica di FTK3 e quindi ridurre la crescita e la sopravvivenza delle cellule tumorali.
L'apoptosi è un processo programmato di morte cellulare che si verifica naturalmente nelle cellule multicellulari. È un meccanismo importante per l'eliminazione delle cellule danneggiate, invecchiate o potenzialmente cancerose, e per la regolazione dello sviluppo e dell'homeostasi dei tessuti.
Il processo di apoptosi è caratterizzato da una serie di cambiamenti cellulari specifici, tra cui la contrazione del citoplasma, il ripiegamento della membrana plasmatica verso l'interno per formare vescicole (blebbing), la frammentazione del DNA e la formazione di corpi apoptotici. Questi corpi apoptotici vengono quindi fagocitati da cellule immunitarie specializzate, come i macrofagi, evitando così una risposta infiammatoria dannosa per l'organismo.
L'apoptosi può essere innescata da diversi stimoli, tra cui la privazione di fattori di crescita o di attacco del DNA, l'esposizione a tossine o radiazioni, e il rilascio di specifiche molecole segnale. Il processo è altamente regolato da una rete complessa di proteine pro- e anti-apoptotiche che interagiscono tra loro per mantenere l'equilibrio tra la sopravvivenza e la morte cellulare programmata.
Un'alterazione del processo di apoptosi è stata associata a diverse malattie, tra cui il cancro, le malattie neurodegenerative e le infezioni virali.
In campo medico e genetico, una mutazione è definita come un cambiamento permanente nel materiale genetico (DNA o RNA) di una cellula. Queste modifiche possono influenzare il modo in cui la cellula funziona e si sviluppa, compreso l'effetto sui tratti ereditari. Le mutazioni possono verificarsi naturalmente durante il processo di replicazione del DNA o come risultato di fattori ambientali dannosi come radiazioni, sostanze chimiche nocive o infezioni virali.
Le mutazioni possono essere classificate in due tipi principali:
1. Mutazioni germinali (o ereditarie): queste mutazioni si verificano nelle cellule germinali (ovuli e spermatozoi) e possono essere trasmesse dai genitori ai figli. Le mutazioni germinali possono causare malattie genetiche o predisporre a determinate condizioni mediche.
2. Mutazioni somatiche: queste mutazioni si verificano nelle cellule non riproduttive del corpo (somatiche) e di solito non vengono trasmesse alla prole. Le mutazioni somatiche possono portare a un'ampia gamma di effetti, tra cui lo sviluppo di tumori o il cambiamento delle caratteristiche cellulari.
Le mutazioni possono essere ulteriormente suddivise in base alla loro entità:
- Mutazione puntiforme: una singola base (lettera) del DNA viene modificata, eliminata o aggiunta.
- Inserzione: una o più basi vengono inserite nel DNA.
- Delezione: una o più basi vengono eliminate dal DNA.
- Duplicazione: una sezione di DNA viene duplicata.
- Inversione: una sezione di DNA viene capovolta end-to-end, mantenendo l'ordine delle basi.
- Traslocazione: due segmenti di DNA vengono scambiati tra cromosomi o all'interno dello stesso cromosoma.
Le mutazioni possono avere effetti diversi sul funzionamento delle cellule e dei geni, che vanno da quasi impercettibili a drammatici. Alcune mutazioni non hanno alcun effetto, mentre altre possono portare a malattie o disabilità.
Le malattie linfatiche si riferiscono a un gruppo diversificato di disturbi che colpiscono il sistema linfatico, un importante componente del sistema immunitario che aiuta a proteggere il corpo dalle infezioni e delle malattie. Il sistema linfatico è composto da una rete di vasi sottili (vasi linfatici), organi linfoidi (come milza, timo, tonsille e linfonodi) e tessuto linfoide diffuso nel corpo.
Le malattie linfatiche possono essere classificate in diverse categorie, tra cui:
1. Linfangite: infiammazione dei vasi linfatici che può causare gonfiore, arrossamento e dolore.
2. Linfoma: un tumore maligno che si sviluppa nei tessuti linfoidi, come i linfonodi, la milza o il midollo osseo. Esistono due principali tipi di linfomi: il linfoma di Hodgkin e il linfoma non-Hodgkin.
3. Malattie infiammatorie dei linfonodi: condizioni che causano gonfiore e infiammazione dei linfonodi, come la linfoadenopatia reattiva, la sarcoidosi e l'amiloidosi.
4. Infezioni del sistema linfatico: infezioni batteriche, virali o fungine che colpiscono il sistema linfatico, come la tubercolosi, la toxoplasmosi e la candidosi.
5. Altre malattie rare del sistema linfatico: tra cui la sindrome di Kartagener, la displasia vascolare linfatica primitiva e il lipedema.
I sintomi delle malattie linfatiche variano ampiamente a seconda della specifica condizione e del suo stadio di avanzamento. Possono includere gonfiore, dolore, arrossamento, febbre, stanchezza, perdita di peso e difficoltà respiratorie. Il trattamento dipende dalla malattia sottostante e può comprendere farmaci, terapia fisica, chirurgia o radioterapia.
I linfociti T, anche noti come cellule T, sono un sottotipo di globuli bianchi che giocano un ruolo cruciale nel sistema immunitario adattativo. Si sviluppano nel timo e sono essenziali per la risposta immunitaria cellulo-mediata. Esistono diversi sottotipi di linfociti T, tra cui i linfociti T helper (CD4+), i linfociti T citotossici (CD8+) e i linfociti T regolatori.
I linfociti T helper aiutano a coordinare la risposta immunitaria, attivando altri effettori del sistema immunitario come i linfociti B e altri linfociti T. I linfociti T citotossici, d'altra parte, sono in grado di distruggere direttamente le cellule infette o tumorali. Infine, i linfociti T regolatori svolgono un ruolo importante nel mantenere la tolleranza immunologica e prevenire l'insorgenza di malattie autoimmuni.
I linfociti T riconoscono le cellule infette o le cellule tumorali attraverso l'interazione con il complesso maggiore di istocompatibilità (MHC) presente sulla superficie delle cellule. Quando un linfocita T incontra una cellula che esprime un antigene specifico, viene attivato e inizia a secernere citochine che aiutano a coordinare la risposta immunitaria.
In sintesi, i linfociti T sono una componente fondamentale del sistema immunitario adattativo, responsabili della risposta cellulo-mediata alle infezioni e alle cellule tumorali.
 Sindrome di Evans
Sindrome di Evans
Mononucleosi infettiva
Sindrome di Evans - Wikipedia
 A novel EP300 mutation associated with Rubinstein-Taybi syndrome type 2 presenting as combined immunodeficiency
A novel EP300 mutation associated with Rubinstein-Taybi syndrome type 2 presenting as combined immunodeficiency
 Sindrome linfoproliferativa legata al cromosoma X - Immunologia; malattie allergiche - Manuali MSD Edizione Professionisti
Sindrome linfoproliferativa legata al cromosoma X - Immunologia; malattie allergiche - Manuali MSD Edizione Professionisti
Malattie autoimmuni2
- Le malattie autoimmuni preesistenti possono includere la sindrome linfoproliferativa autoimmune (ALPS), l'immunodeficienza variabile combinata (CVID), la malattia autoimmune sistemica o un altro tipo di disregolazione immunitaria. (wikipedia.org)
- Panoramica sui disturbi da immunodeficienza I disturbi da immunodeficienza vengono associati o predispongono i pazienti a varie complicanze, tra cui infezioni, malattie autoimmuni, linfomi e altri tumori. (msdmanuals.com)
Sindrome4
- La sindrome di Evans è una malattia autoimmune in cui il sistema immunitario della persona affetta attacca i suoi stessi eritrociti, leucociti e piastrine. (wikipedia.org)
- La sindrome di Evans è considerata una malattia autoimmune molto rara. (wikipedia.org)
- È stato riportato da alcuni studi che tra il 7,8% e il 23% dei pazienti affetti da anemia emolitica autoimmune presenta anche trombocitopenia e quindi sindrome di Evans. (wikipedia.org)
- Una diagnosi di sindrome di Evans secondaria viene posta in presenza di un'altra malattia autoimmune preesistente. (wikipedia.org)