Aspartylglucosaminuria
N4-(Bêta-N-Acétylglucosaminyl)-L-Asparaginase
Maladies Lysosomiales
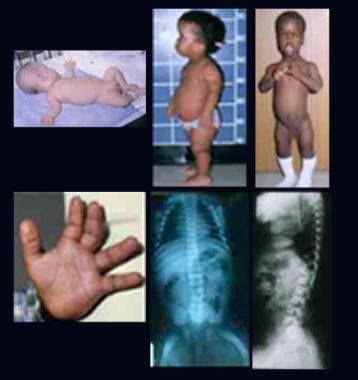
L'aspartylglucosaminuria est une maladie métabolique rare et héréditaire causée par un déficit en activité de l'enzyme aspartylglucosaminidase. Cette enzyme est responsable du catabolisme des glycoprotéines, qui sont des protéines complexes contenant des sucres. Lorsque l'activité de cette enzyme est réduite ou absente, les glycoprotéines ne peuvent pas être correctement dégradées et s'accumulent dans les cellules, entraînant une toxicité cellulaire et une variété de symptômes.
Les symptômes de l'aspartylglucosaminuria comprennent un retard de développement, des faciès caractéristiques, des problèmes osseux, une hypertrophie du foie et de la rate, des problèmes respiratoires, des convulsions, des problèmes de vision et de l'audition, et un déclin mental progressif. Ces symptômes apparaissent généralement pendant l'enfance ou l'adolescence et s'aggravent progressivement avec le temps.
Le diagnostic de l'aspartylglucosaminuria est établi par des tests d'activité enzymatique et des tests génétiques, qui peuvent confirmer la présence d'une mutation dans le gène AGA responsable de la production de l'enzyme aspartylglucosaminidase.
Actuellement, il n'existe pas de traitement curatif pour l'aspartylglucosaminuria. Le traitement est principalement axé sur les symptômes et peut inclure des médicaments pour contrôler les convulsions, une thérapie physique et occupante pour aider au développement moteur et cognitif, et une gestion des complications respiratoires et gastro-intestinales. Les patients atteints d'aspartylglucosaminuria ont généralement une espérance de vie réduite, avec un décès survenant généralement pendant l'adolescence ou au début de l'âge adulte.
Les maladies lysosomales sont un groupe de plus de 50 affections héréditaires différentes causées par des mutations dans les gènes qui codent pour des protéines spécifiques, appelées enzymes lysosomiales. Ces enzymes sont responsables du processus de dégradation et de recyclage des matières cellulaires dans les lysosomes, qui sont des organites membranaires trouvés dans la plupart des cellules du corps humain.
Lorsque ces enzymes ne fonctionnent pas correctement ou manquent complètement, les molécules et les déchets cellulaires s'accumulent dans les lysosomes, entraînant une toxicité cellulaire et des dommages aux tissus et organes. Cette accumulation de matériaux peut affecter divers systèmes corporels, y compris le cerveau, la moelle osseuse, le foie, les reins, les poumons, les yeux et la peau.
Les symptômes des maladies lysosomales varient considérablement en fonction du type de maladie et de sa gravité. Ils peuvent inclure une croissance et un développement anormaux, des anomalies squelettiques, des problèmes neurologiques, des dommages aux organes internes, des problèmes sensoriels tels que la surdité ou la cécité, et une diminution de l'espérance de vie.
Les maladies lysosomales sont généralement héritées d'une manière autosomique récessive, ce qui signifie qu'un individu doit hériter de deux copies du gène muté (une de chaque parent) pour développer la maladie. Cependant, certaines formes de ces maladies peuvent être héritées d'une manière liée à l'X, ce qui signifie qu'un seul gène muté sur le chromosome X peut entraîner la maladie.
Actuellement, il n'existe pas de traitement curatif pour les maladies lysosomales, mais des thérapies enzymatiques substitutives et d'autres approches thérapeutiques peuvent aider à gérer les symptômes et à améliorer la qualité de vie des personnes atteintes. La recherche se poursuit pour développer de nouveaux traitements et thérapies pour ces maladies rares mais graves.
L'acétylglucosamine est un monosaccharide amine dérivée de la glucose qui joue un rôle important dans la biosynthèse des glycosaminoglycanes, une composante clé des tissus conjonctifs tels que le cartilage et la peau. Il est également un constituant important des glycoprotéines et des protéoglycanes, qui sont des molécules complexes trouvées dans les membranes cellulaires et l'espace extracellulaire.
L'acétylglucosamine peut être obtenue à partir de la chitine, une substance présente dans la paroi cellulaire des champignons et des crustacés, ou elle peut être synthétisée en laboratoire. Dans le corps, il est produit à partir du glucose par un processus appelé la voie de l'hexosamine.
En médecine, l'acétylglucosamine est souvent utilisé comme un supplément nutritionnel pour aider à soutenir la santé des articulations et de la peau. Il peut également être utilisé dans le traitement de certaines conditions médicales telles que l'arthrose, la maladie inflammatoire de l'intestin et les infections respiratoires.
Cependant, il est important de noter que l'utilisation de suppléments d'acétylglucosamine doit être discutée avec un professionnel de la santé avant de commencer tout nouveau régime de supplémentation, en particulier pour les personnes atteintes de certaines conditions médicales ou qui prennent des médicaments sur ordonnance.
 Guérir - Maladie rare AGU: aspartylglucosaminurie
Guérir - Maladie rare AGU: aspartylglucosaminurie Aspartylglucosaminurie (AGU) : le traitement par molécules chaperons est (...) - Vaincre les Maladies Lysosomales
Aspartylglucosaminurie (AGU) : le traitement par molécules chaperons est (...) - Vaincre les Maladies Lysosomales