Wolman Disease
Cholesterol Ester Storage Disease
Sterol Esterase
Lipase
Metabolism, Inborn Errors
Lysosomes
Compound heterozygosity for a Wolman mutation is frequent among patients with cholesteryl ester storage disease. (1/30)
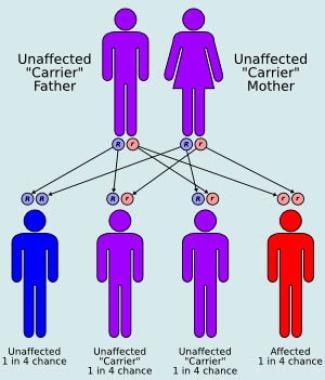
Cholesteryl ester storage disease and Wolman disease are rare autosomal recessive lipoprotein-processing disorders caused by mutations in the gene encoding human lysosomal acid lipase. Thus far we have elucidated the genetic defects in 15 unrelated CESD patients. Seven were homozygotes for the prevalent hLAL exon 8 splice junction mutation which results in incomplete exon skipping, while eight probands were compound heterozygotes for E8SJM and a rare mutation on the second chromosome. In this report, we describe the molecular basis of CESD in three compound heterozygous subjects of Czech and Irish origin. RFLP and DNA sequence analysis revealed that they were heteroallelic for the common G(934)-->A substitution in exon 8 of the hLAL gene and a mutation which, if inherited on both alleles, would be expected to result in complete loss of enzyme activity and to cause Wolman disease. In patients A. M. and J. J., two nucleotide deletions in exons 7 and 10 were detected, involving a T at position 722, 723, or 724 and a G in a stretch of five guanosines at positions 1064;-1068 of the hLAL cDNA. Both mutations result in premature termination of protein translation at residues 219 and 336, respectively, and in the production of truncated, inactive enzymes. Subject D. H., in contrast, is a compound heterozygote for the Arg(44)-->Stop mutation previously described in a French CESD proband. Combined with data in the literature, our results demonstrate that compound heterozygosity for a mutation causing Wolman disease is common among cholesteryl ester storage disease patients. (+info)Wolman disease successfully treated by bone marrow transplantation. (2/30)
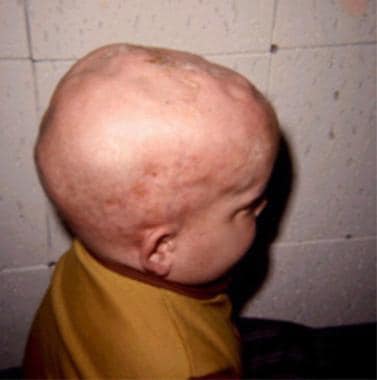
Wolman disease is characterized by severe diarrhea and malnutrition leading to death during infancy. Lysosomal acid lipase deficiency is the cause of the symptoms and signs. It is inherited in an autosomal recessive manner. All Wolman disease patients have adrenal gland calcification. Previous therapeutic attempts have failed to provide remission. We report successful long-term bone marrow engraftment in a patient with Wolman disease resulting in continued normalization of peripheral leukocyte lysosomal acid lipase enzyme activity. Diarrhea is no longer present. Now, at 4 years of age, this patient is gaining developmental milestones. Cholesterol and triglyceride levels are normal. Liver function is normal. This is the first long-term continued remission reported for Wolman disease. (+info)Lysosomal acid lipase-deficient mice: depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. (3/30)
Lysosomal acid lipase (LAL) is essential for the hydrolysis of triglycerides (TG) and cholesteryl esters (CE) in lysosomes. A mouse model created by gene targeting produces no LAL mRNA, protein, or enzyme activity. The lal-/- mice appear normal at birth, survive into adulthood, and are fertile. Massive storage of TG and CE is observed in adult liver, adrenal glands, and small intestine. The age-dependent tissue and gross progression in this mouse model are detailed here. Although lal-/- mice can be bred to give homozygous litters, they die at ages of 7 to 8 months. The lal-/- mice develop enlargement of a single mesenteric lymph node that is full of stored lipids. At 6;-8 months of age, the lal-/- mice have completely absent inguinal, interscapular, and retroperitoneal white adipose tissue. In addition, brown adipose tissue is progressively lost. The plasma free fatty acid levels are significantly higher in lal-/- mice than age-matched lal+/+ mice, and plasma insulin levels were more elevated upon glucose challenge. Energy intake was also higher in lal-/- male mice, although age-matched body weights were not significantly altered from age-matched lal+/+ mice. Early in the disease course, hepatocytes are the main storage cell in the liver; by 3;-8 months, the lipid-stored Kupffer cells progressively fill the liver. The involvement of macrophages throughout the body of lal-/- mice provide evidence for a critical nonappreciated role of LAL in cellular cholesterol and fatty acid metabolism, adipocyte differentiation, and fat mobilization. (+info)Characterization of lysosomal acid lipase mutations in the signal peptide and mature polypeptide region causing Wolman disease. (4/30)
Wolman disease results from an inherited deficiency of lysosomal acid lipase (LAL; EC 3.1.1.13). This enzyme is essential for the hydrolysis of cholesteryl esters and triacylglycerols derived from endocytosed lipoproteins. Because of a complete absence of LAL activity, Wolman patients accumulate progressive amounts of cholesteryl esters and triacylglycerols in affected tissues. To investigate the nature of the genetic defects causing this disease, mutations in the LAL gene from three subjects of Moslem-Arab and Russian descent living in Israel were determined. Two homozygotes for a novel 1-bp deletion introducing a premature in-frame termination codon at amino acid position 106 (S106X) were identified. A third subject was a homozygote for a G-5R signal peptide substitution and a G60V missense mutation. The functional significance of these mutations was tested by in vitro expression of single and double mutants in Spodoptera frugiperda cells. Single mutants G60V and S106X and double mutant G-5R/G60V displayed a virtual absence of lipase activity in cell extracts and culture medium. Signal peptide mutant G-5R retained lipase activity in cell extracts and showed a drastically reduced enzyme activity in culture supernatant, indicating that the mutation may affect secretion of active enzyme from cells. These results support the notion that Wolman disease is a genetically heterogeneous disorder of lipid metabolism. (+info)Enzyme therapy for lysosomal acid lipase deficiency in the mouse. (5/30)
Lysosomal acid lipase (LAL) is the critical enzyme for the hydrolysis of the triglycerides (TG) and cholesteryl esters (CE) delivered to lysosomes. Its deficiency produces two human phenotypes, Wolman disease (WD) and cholesteryl ester storage disease (CESD). A targeted disruption of the LAL locus produced a null (lal( -/-)) mouse model that mimics human WD/CESD. The potential for enzyme therapy was tested using mannose terminated human LAL expressed in Pichia pastoris (phLAL), purified, and administered by tail vein injections to lal( -/-) mice. Mannose receptor (MR)-dependent uptake and lysosomal targeting of phLAL were evidenced ex vivo using competitive assays with MR-positive J774E cells, a murine monocyte/macrophage line, immunofluorescence and western blots. Following (bolus) IV injection, phLAL was detected in Kupffer cells, lung macrophages and intestinal macrophages in lal( -/-) mice. Two-month-old lal( -/-) mice received phLAL (1.5 U/dose) or saline injections once every 3 days for 30 days (10 doses). The treated lal( -/-) mice showed nearly complete resolution of hepatic yellow coloration; hepatic weight decreased by approximately 36% compared to PBS-treated lal( -/-) mice. Histologic analyses of numerous tissues from phLAL-treated mice showed reductions in macrophage lipid storage. TG and cholesterol levels decreased by approximately 50% in liver, 69% in spleen and 50% in small intestine. These studies provide feasibility for LAL enzyme therapy in human WD and CESD. (+info)Wolman's disease--a case report. (6/30)
Wolman's disease is a rare autosomal recessive lysosomal storage disorder. We report a case, which we identified with foamy histiocytes in bone marrow and adrenal calcification in radiological imaging. The diagnosis can be made on minimal investigation when clinically suspected. But cytogenetic study is required to substantiate the diagnosis further. (+info)Isolated fetal ascites caused by Wolman disease. (7/30)
Wolman disease is a rare autosomal-recessive disorder caused by reduced levels of lysosomal acid lipase. It occurs in infancy and is fatal in most cases before the age of 1 year. Affected infants show signs of lipid storage in most tissues, including hepatosplenomegaly, abdominal distension, vomiting, steatorrhea, failure to thrive, and adrenal calcifications. We present a case of isolated fetal ascites diagnosed at 32 weeks of gestation, with negative work-up for immune and non-immune hydrops fetalis and congenital infections and malformations. After delivery, the diagnosis of Wolman disease was established. Although rare, storage diseases such as Wolman disease should be considered in cases of isolated fetal ascites. (+info)Familial spinal xanthomatosis with sitosterolemia. (8/30)
A family with multiple spinal xanthomas and sitosterolemia is described. A 48-year-old woman presented with paraplegia due to multiple intradural extramedullary tumors. The patient also showed marked tendon xanthomas and analysis of sterol composition in both plasma and the xanthoma established the diagnosis of the rare inherited metabolic disease, sitosterolemia and xanthomatosis. Two other siblings in the family presented with marked tendon xanthomas and coronary atherosclerosis, but did not show any neurological signs or symptoms. Magnetic resonance imaging (MRI) study revealed multiple intradural extramedullary tumors in spinal canals of the proband and her sister, but not in the other affected sibling (brother). This is the first report of familial occurrence of multiple extramedullary spinal tumors due to the inherited metabolic abnormality. (+info)Wolman disease is a rare inherited disorder of lipid metabolism, specifically affecting the enzyme acid lipase that is responsible for breaking down cholesteryl esters and triglycerides in lysosomes. This autosomal recessive condition leads to an accumulation of these fatty substances in various tissues and organs, including the liver, spleen, intestines, adrenal glands, and lymph nodes.
The symptoms of Wolman disease typically appear within the first few months of life and can include vomiting, diarrhea, failure to thrive, abdominal distention, and severe malnutrition. Other features may consist of hepatosplenomegaly (enlarged liver and spleen), calcification of adrenal glands, and progressive deterioration of the nervous system. The disease often results in death within the first two years of life if left untreated.
A related condition called acid lipase deficiency or Cholesteryl Ester Storage Disease (CESD) has a later onset and milder symptoms compared to Wolman disease, as it affects only one form of acid lipase enzyme.
Cholesteryl Ester Storage Disease (CESD) is a rare genetic disorder characterized by the accumulation of cholesteryl esters in various tissues and organs, particularly in the liver and spleen. It is caused by mutations in the gene responsible for producing lipoprotein lipase (LPL), an enzyme that helps break down fats called triglycerides in the body.
In CESD, the lack of functional LPL leads to an accumulation of cholesteryl esters in the lysosomes of cells, which can cause damage and inflammation in affected organs. Symptoms of CESD can vary widely, but often include enlargement of the liver and spleen, abdominal pain, jaundice, and fatty deposits under the skin (xanthomas).
CESD is typically diagnosed through a combination of clinical evaluation, imaging studies, and genetic testing. Treatment may involve dietary modifications to reduce the intake of fats, medications to help control lipid levels in the blood, and in some cases, liver transplantation.
A sterol esterase is an enzyme that catalyzes the hydrolysis of sterol esters, which are fatty acid esters of sterols (such as cholesterol) that are commonly found in lipoproteins and cell membranes. Sterol esterases play a crucial role in the metabolism of lipids by breaking down sterol esters into free sterols and free fatty acids, which can then be used in various biochemical processes.
There are several types of sterol esterases that have been identified, including:
1. Cholesteryl esterase (CE): This enzyme is responsible for hydrolyzing cholesteryl esters in the intestine and liver. It plays a critical role in the absorption and metabolism of dietary cholesterol.
2. Hormone-sensitive lipase (HSL): This enzyme is involved in the hydrolysis of sterol esters in adipose tissue, as well as other lipids such as triacylglycerols. It is regulated by hormones such as insulin and catecholamines.
3. Carboxylesterase (CES): This enzyme is a broad-specificity esterase that can hydrolyze various types of esters, including sterol esters. It is found in many tissues throughout the body.
Sterol esterases are important targets for drug development, as inhibiting these enzymes can have therapeutic effects in a variety of diseases, such as obesity, diabetes, and cardiovascular disease.
Lipase is an enzyme that is produced by the pancreas and found in the digestive system of most organisms. Its primary function is to catalyze the hydrolysis of fats (triglycerides) into smaller molecules, such as fatty acids and glycerol, which can then be absorbed by the intestines and utilized for energy or stored for later use.
In medical terms, lipase levels in the blood are often measured to diagnose or monitor conditions that affect the pancreas, such as pancreatitis (inflammation of the pancreas), pancreatic cancer, or cystic fibrosis. Elevated lipase levels may indicate damage to the pancreas and its ability to produce digestive enzymes.
Inborn errors of metabolism (IEM) refer to a group of genetic disorders caused by defects in enzymes or transporters that play a role in the body's metabolic processes. These disorders result in the accumulation or deficiency of specific chemicals within the body, which can lead to various clinical manifestations, such as developmental delay, intellectual disability, seizures, organ damage, and in some cases, death.
Examples of IEM include phenylketonuria (PKU), maple syrup urine disease (MSUD), galactosemia, and glycogen storage diseases, among many others. These disorders are typically inherited in an autosomal recessive manner, meaning that an affected individual has two copies of the mutated gene, one from each parent.
Early diagnosis and management of IEM are crucial to prevent or minimize complications and improve outcomes. Treatment options may include dietary modifications, supplementation with missing enzymes or cofactors, medication, and in some cases, stem cell transplantation or gene therapy.
Lysosomes are membrane-bound organelles found in the cytoplasm of eukaryotic cells. They are responsible for breaking down and recycling various materials, such as waste products, foreign substances, and damaged cellular components, through a process called autophagy or phagocytosis. Lysosomes contain hydrolytic enzymes that can break down biomolecules like proteins, nucleic acids, lipids, and carbohydrates into their basic building blocks, which can then be reused by the cell. They play a crucial role in maintaining cellular homeostasis and are often referred to as the "garbage disposal system" of the cell.
 Wolman's disease in an infant - PubMed
Wolman's disease in an infant - PubMed
 Wolman disease - About the Disease - Genetic and Rare Diseases Information Center
Wolman disease - About the Disease - Genetic and Rare Diseases Information Center
 Cholesteryl Ester Storage Disease and Wolman Disease - Pediatrics - Merck Manuals Professional Edition
Cholesteryl Ester Storage Disease and Wolman Disease - Pediatrics - Merck Manuals Professional Edition
 Wolman disease successfully treated by bone marrow transplantation - PubMed
Wolman disease successfully treated by bone marrow transplantation - PubMed
Cholesteryl Ester Storage Disease and Wolman Disease - Pediatrics - MSD Manual Professional Edition
 Wolman disease
Wolman disease
 Moshe Wolman - Wikipedia
Moshe Wolman - Wikipedia
 Lysosomal Storage Disease: Overview, Classification of Lysosomal Storage Diseases, Glycogen Storage Disease Type II
Lysosomal Storage Disease: Overview, Classification of Lysosomal Storage Diseases, Glycogen Storage Disease Type II
Treating Deadly Disease in Utero Called 'Revolutionary' Advance
 Lysosomal acid lipase deficiency: MedlinePlus Genetics
Lysosomal acid lipase deficiency: MedlinePlus Genetics
 Tests & Charges
Tests & Charges
wolman f&p application
 Copy Link
Copy Link
Lysosomal Storage Disease: Overview, Classification of Lysosomal Storage Diseases, Glycogen Storage Disease Type II
 Less than lifetime limits for N-nitrosamine mutagenic impurities
Less than lifetime limits for N-nitrosamine mutagenic impurities
 ICD-10 Chapter IV: Endocrine, nutritional and metabolic diseases
ICD-10 Chapter IV: Endocrine, nutritional and metabolic diseases
DeCS 2008 - versión 17 de Marzo de 2008
Biomarkers Search
 Lipid Storage Diseases | National Institute of Neurological Disorders and Stroke
Lipid Storage Diseases | National Institute of Neurological Disorders and Stroke
 Lysosomal acid lipase (LIPA) (NM 000235) Human Recombinant Protein - TP301637 | OriGene
Lysosomal acid lipase (LIPA) (NM 000235) Human Recombinant Protein - TP301637 | OriGene
 Rcan1 Mouse Gene Details | regulator of calcineurin 1 | International Mouse Phenotyping Consortium
Rcan1 Mouse Gene Details | regulator of calcineurin 1 | International Mouse Phenotyping Consortium
JournalTOCs
LIPA gene: MedlinePlus Genetics
 The Ehlers-Danlos syndromes | Nature Reviews Disease Primers
The Ehlers-Danlos syndromes | Nature Reviews Disease Primers
 Reactome | LIPA hydrolyses sterol esters to sterols and fatty acids
Reactome | LIPA hydrolyses sterol esters to sterols and fatty acids
HuGE Navigator|Genopedia|PHGKB
 Metabolic Testing | CENTOGENE CentoMetabolic MOx: centogene.com
Metabolic Testing | CENTOGENE CentoMetabolic MOx: centogene.com
Lipid Storage Diseases | National Institute of Neurological Disorders and Stroke
Cholesteryl Ester S6
- The early-onset form was known as Wolman disease, and the later-onset form was known as cholesteryl ester storage disease. (medlineplus.gov)
- Mutations in this gene can result in Wolman disease and cholesteryl ester storage disease. (cancerindex.org)
- The later onset form, cholesteryl ester storage disease (CESD), presents between childhood and adulthood with a more variable clinical course that ranges from insidious to symptomatic. (orpha.net)
- Deficiencies in LAL are primarily associated with two diseases: cholesteryl ester storage disease (CESD) and Wolman's disease (WD). (goldbio.com)
- Deficiency of lysosomal acid lipase causes two distinct phenotypes in humans: Wolman disease and cholesteryl ester storage disease (CESD). (bredagenetics.com)
- Cholesteryl Ester Storage Disease (CESD) is at the milder end of disease variability, it is later-onset and presents with primary hepatic involvement by foam cells (macrophages engorged with cholesteryl esters). (bredagenetics.com)
Described Wolman's disease1
- citation needed] In 1954, he described Wolman's disease. (wikipedia.org)
Infants2
- Also known as Wolman's Disease, it affects only a handful of infants, but without this vital enzyme they accumulate fats in their digestive organs, leading to organ swelling, failure and death within the first year of life. (theconversation.com)
- These findings suggest that FGF19 or perhaps another compound that acts similarly might hold promise for infants with Wolman disease, who often die at a very early age. (nih.gov)
Deficiency13
- Understanding of the chemistry involved in histological techniques enabled Moshe Wolman in the 1950s to determine that the storage disease, that was later named after him is known as lysosomal acid lipase deficiency or Wolman disease, is caused by accumulation in cells of a mixture of cholesterol and triglycerides and differed substantially from Niemann-Pick disease. (wikipedia.org)
- Background and importance Lysosomal acid lipase (LAL) deficiency is a rare metabolic disease (0.2:10 000) characterised by lysosomal accumulation of cholesterol esters and triglycerides, with a severe and rapidly progressive form known as Wolman disease (WD), usually fatal in the first 6-12 months of life. (bmj.com)
- Dietary restriction has shown promise for disorders such as lysosomal acid lipase deficiency (Wolman disease), as has incorporation of lipid-lowering drugs in the regimen along with sebelipase alpha, a recombinant enzyme replacement therapy. (medscape.com)
- A rare, progressive metabolic liver disease due to marked to complete lysosomal acid lipase deficiency and characterized by dyslipidemia and massive lipid accumulation leading to hepatomegaly and liver dysfunction, splenomegaly, accelerated atherosclerosis. (orpha.net)
- Gaucher disease is caused by a deficiency of the enzyme glucocerebrosidase. (nih.gov)
- Niemann-Pick disease type C is not caused by a deficiency of sphlingomyelinase but by a lack of the NPC1 or NPC2 proteins. (nih.gov)
- Dr. Michael Kaback and Dr. David Rimoin, who developed the West Coast study, included HIBIM as well as three diseases common to the Persian community, which are relatively easy to treat: pseudocholinesterase deficiency (anesthesia sensitivity) congenital hypoaldosteronism (salt losing disorder), and polyglandular deficiency (multiple hormone deficiency). (jta.org)
- Let's look at just one rare disease, Liposomal Acid Deficiency. (theconversation.com)
- Lysosomal acid lipase deficiency (LAL-D), also known as Wolman disease or cholesterol ester storage disease (CESD), is an inherited genetic condition in which the body does not produce enough lysosomal acid lipase enzyme to process fats and cholesterol. (panfoundation.org)
- Lysosomal acid lipase deficiency is predominantly a pediatric disease , although milder forms of the disease with possibility of a normal life-span are also possible. (bredagenetics.com)
- In 2015 the results of a phase 3 trial with sebelipase , a recombinant human lysosomal acid lipase, resulted in a reduction in multiple disease-related hepatic and lipid abnormalities in children and adults with lysosomal acid lipase deficiency. (bredagenetics.com)
- Previous studies had shown that LAL deficiency in kids with Wolman disease overactivates another signaling pathway, which could be suppressed by targeting a receptor known as FXR. (nih.gov)
- Phosphoenolpyruvate carboxykinase deficiency impairs gluconeogenesis and results in symptoms and signs similar to the hepatic forms of glycogen storage disease but without hepatic glycogen accumulation. (merckmanuals.com)
CESD3
- The variable phenotype is due to the amount of residual LAL activity, less than 1% for Wolman disease and between 1-12% for CESD. (orpha.net)
- Although CESD is relatively benign in contrast to Wolman disease, death may occur as consequence of acute hepatic failure. (bredagenetics.com)
- The difference between Wolman-related and CESD-related mutations is seemingly in that the first ones completely abolish the protein synthesis leading to undetectable LIPA activity , whereas the latter allow some sort of residual enzimatic activity . (bredagenetics.com)
Disorders14
- Lipid storage disorders are a family of diverse diseases related by their molecular pathology. (medscape.com)
- In addition, for some disorders (eg, Gaucher disease), making genotype-phenotype correlations that predict disease severity and allow more accurate genetic risk counseling is possible. (medscape.com)
- One of the most common lysosomal storage disorders is Gaucher disease, discussed below. (medscape.com)
- All lipid storage disorders are inherited in an autosomal-recessive fashion, except for Fabry disease and mucopolysaccharidosis type II (Hunter disease), which are X-linked. (medscape.com)
- Lysosomal storage diseases describe a heterogeneous group of dozens of rare inherited disorders characterized by the accumulation of undigested or partially digested macromolecules, which ultimately results in cellular dysfunction and clinical abnormalities. (medscape.com)
- Lysosomal storage diseases are generally classified by the accumulated substrate and include the sphingolipidoses, oligosaccharidoses, mucolipidoses, mucopolysaccharidoses (MPSs), lipoprotein storage disorders, lysosomal transport defects, neuronal ceroid lipofuscinoses and others. (medscape.com)
- Accumulated data indicate that hematopoietic stem cell transplantation may be effective under optimal conditions in preventing the progression of central nervous system symptoms in neuronopathic forms of lysosomal storage diseases (such as Krabbe disease), including some of the mucopolysaccharidoses, oligosaccharidoses, sphingolipidoses, and lipidoses as well as peroxisome disorders such as X-linked adrenoleukodystrophy. (medscape.com)
- In general, transplantation yields the best results when performed early in the course of the disease (ie, in an asymptomatic affected sibling of a child with a lysosomal storage disorder), in centers with experience in performing transplantations to treat inherited metabolic disorders, and in patients healthy enough to tolerate the conditioning and transplantation regimen. (medscape.com)
- Differential diagnosis includes familial hypercholesterolemia, non-alcoholic fatty liver disease, cryptogenic cirrhosis, and combined hyperlipidemia, as well as other lysosomal storage disorders. (orpha.net)
- Lipid storage diseases (also known as lipidoses) are a group of inherited metabolic disorders in which harmful amounts of fatty materials (lipids) accumulate in various cells and tissues in the body. (nih.gov)
- Disorders in which intracellular material that cannot be metabolized is stored in lysosomes are called lysosomal storage diseases. (nih.gov)
- Niemann-Pick disease is a group of autosomal recessive disorders caused by an accumulation of fat and cholesterol in cells of the liver, spleen, bone marrow, lungs, and, in some instances, brain. (nih.gov)
- IMMUNE SYSTEM DISORDERS: Genetic or acquired diseases which result in white blood cells that are not able to fight off infections. (upstatecordbloodbank.com)
- INHERITED METABOLIC DISORDERS: Genetic diseases that prevent the body from correctly processing normal substances in the body or diet. (upstatecordbloodbank.com)
Genetic7
- Background: Breast cancer is a heterogeneous disease that can be subdivided on the basis of histopathological features, genetic alterations, and gene-expression profiles. (cancerindex.org)
- Akler is also helping to develop a screening test for Persian Jewish genetic diseases for the Mount Sinai research project. (jta.org)
- Melamed, who spit into a tube to offer his saliva sample while CNN cameras rolled, says he wasn't surprised to learn that he was not a carrier of any of the four genetic mutations tested as he hadn't heard about such diseases in his immediate family, but he gladly participated "to send a message that this is not something to be scared of. (jta.org)
- And the hope is that the same philosophy that was applied in combating Tay Sachs, which has largely been eradicated, can be used to reduce the incidence of these genetic diseases, says Rimoin. (jta.org)
- Specialty drugs, usually complex biologic products created by genetic alterations of living tissues or organisms, have revolutionized patient care by creating human proteins, enzymes and antibodies that can treat diseases much more specifically than previously. (theconversation.com)
- INHERITED RED CELL ABNORMALITIES: Genetic diseases resulting in red blood cells that do not work correctly. (upstatecordbloodbank.com)
- INHERITED PLATELET ABNORMALITIES: Genetic diseases resulting in platelets that are not able to correctly form clots. (upstatecordbloodbank.com)
Mutations3
- These studies have resulted in identifying specific disease-causing mutations and have led to improved clinical and laboratory diagnosis, prenatal diagnosis, and carrier identification. (medscape.com)
- Age of onset and clinical manifestations may vary widely among patients with a given lysosomal storage disease, and significant phenotypic heterogeneity between family members carrying identical mutations has been reported. (medscape.com)
- The disease is due to mutations in the gene LIPA (10q23.2-q23.3) encoding the enzyme lysosomal acid lipase (LAL). (orpha.net)
Early-onset2
- The early-onset, rapidly progressive form, Wolman disease, presents in the neonatal or infantile period with non-specific symptoms of massive hepatosplenomegaly, liver failure, diarrhea/steatorrhea and vomiting, resulting in malabsorption, and cachexia. (orpha.net)
- The disorder is slowly progressive and show a wide clinical variability , ranging from early onset involvement with severe cirrhosis to later onset manifestations with more slowly progressive hepatic disease and survival into adulthood. (bredagenetics.com)
Acid1
- The discovery of metabolic liver diseases, bile acid synthesis defects, cryptogenic neonatal hepatitis syndromes, and the development of the Kasai portoenterostomy procedure for the treatment of biliary atresia, a condition up until then considered untreatable, were all factors that contributed to the foundation of the subspecialty of pediatric hepatology in the 1970s. (abdominalkey.com)
Wolman's1
- Prenatal diagnosis of Wolman's disease. (bmj.com)
Gene1
- A knowledge graph of biological entities such as genes, gene functions, diseases, phenotypes and chemicals. (edu.sa)
Fabry2
- Enzyme replacement therapy (ERT) appears safe and effective for peripheral manifestations in patients with Gaucher disease types I and III, Fabry disease, mucopolysaccharidosis I (Hurler, Hurler-Scheie, and Scheie syndromes), mucopolysaccharidosis II (Hunter syndrome), mucopolysaccharidosis VI (Maroteaux-Lamy syndrome), Pompe disease, and recently Batten disease (neuronal ceroid lipofuscinoses, CLN2). (medscape.com)
- Enzyme replacement therapy (ERT) appears safe and effective for peripheral manifestations in patients with Gaucher disease types I and III, Fabry disease, mucopolysaccharidosis I (Hurler, Hurler-Scheie, and Scheie syndromes), mucopolysaccharidosis II (Hunter syndrome), mucopolysaccharidosis VI (Maroteaux-Lamy syndrome), and Pompe disease. (medscape.com)
Amyloid1
- Another study dealt with the substance (or substances) called amyloid (Wolman 1971). (wikipedia.org)
Syndrome2
- There are certain diseases such as Hurler Syndrome, Gaucher's disease, Wolman disease for which the annual treatment expenses may vary from Rs 10 lakh to Rs 1 crore. (upsc247.com)
- AIDS-like syndrome: AIDS-like disease (illness) (syndrome) ARC AIDS-related complex Pre-AIDS AIDS-related conditions Prodromal-AIDS 3. (cdc.gov)
Lysosomal storage6
- Classically, lysosomal storage diseases encompassed only enzyme deficiencies of the lysosomal hydrolases. (medscape.com)
- More than 50 lysosomal storage diseases have been described, some of which are discussed in this article. (medscape.com)
- Thus far, ERT has been largely unsuccessful in improving central nervous system manifestations of the lysosomal storage diseases, putatively due to difficulty in penetrating the blood-brain barrier. (medscape.com)
- This has led to active clinical trials evaluating the safety and efficacy of intrathecal enzyme delivery in several lysosomal storage diseases (see www.ClinicalTrials.gov ). (medscape.com)
- The availability of both ERT and hematopoietic stem cell transplantation has prompted ongoing consideration of newborn screening efforts to diagnose lysosomal storage diseases. (medscape.com)
- Accumulated data indicate that hematopoietic stem cell transplantation may be effective under optimal conditions in preventing the progression of central nervous system symptoms in neuronopathic forms of lysosomal storage diseases, including some of the mucopolysaccharidoses, oligosaccharidoses, sphingolipidoses, and lipidoses. (medscape.com)
Patients8
- For example, in familial hypercholesterolemia, enzymes do not receive the signals that typically inhibit cholesterol synthesis, so that excessive production of cholesterol occurs, leading to early coronary vascular disease and strokes in patients. (newworldencyclopedia.org)
- In addition to our partnership with Galapagos for example, Gilead is collaborating with Verily Life Sciences LLC, part of Google, who will use their cutting-edge technology to analyse biological samples and clinical disease and treatment response data from patients participating in current and future Gilead clinical trials. (europeanpharmaceuticalreview.com)
- Specialty drugs can do amazing things , especially in the realm of rare diseases (those that afflict fewer than 200,000 patients) in which there had been no serious research in the past. (theconversation.com)
- Addressing infection prevention and control in the first U.S. community hospital to care for patients with Ebola virus disease: context for national recommendations and future strategies. (cdc.gov)
- Ultimately, Takebe suggests it might prove useful to grow liver organoids from individual patients with fatty liver diseases, in order to identify the underlying biological causes and test the response of those patient-specific organoids to available treatments. (nih.gov)
- For diseases listed under Group 2, State Governments can consider supporting patients of such rare diseases that can be managed with special diets or hormonal supplements or other relatively low cost interventions. (upsc247.com)
- Switzerland uses temporary authorization to accelerate access to treatments for life-threatening diseases, enabling patients to receive therapies while the developer works to meet the full approval requirements. (ris.world)
- Given the impact of pregnancy on CNS diseases, it is crucial for healthcare providers and patients alike to be aware of these potential effects. (bvsalud.org)
Lipid storage diseases1
- Some severe diseases, such as many of the lipid storage diseases, currently have no effective therapy. (newworldencyclopedia.org)
Accumulation2
- While these "organoids" closely mimic the structures of the liver and other vital organs, it's been tough to get them to represent inflammation, fibrosis, fat accumulation, and many other complex features of disease. (nih.gov)
- So, in the new study, the team applied an FXR-targeted compound called FGF19, and it prevented fat accumulation in the liver organoids derived from people with Wolman disease. (nih.gov)
Triglycerides1
- The disease is suspected on clinical presentation of hepatomegaly, elevated transaminases, total cholesterol, low-density lipoprotein, and triglycerides, and low high-density lipoprotein. (orpha.net)
Diagnosis2
- To overcome this, a hospital based National Registry for Rare Diseases has been initiated by ICMR by involving centers across the Country that are involved in diagnosis and management of Rare Diseases. (upsc247.com)
- The first two centers recognized for the diagnosis and management of pediatric liver disease were in Paris, led by Professor Daniel Alagille, and in London, led by Professor Alex Mowat. (abdominalkey.com)
Infantile1
- Type 2 (acute infantile neuropathic Gaucher disease) typically begins within three months of birth. (nih.gov)
Gaucher Disease1
- It is characterized by slowly progressive yet milder neurologic symptoms compared to type 2 Gaucher disease. (nih.gov)
Cirrhosis1
- Scar tissue quickly builds up in the liver, leading to liver disease (cirrhosis). (medlineplus.gov)
Infectious3
- Infectious disease risks associated with occupational exposure: a systematic review of the literature. (cdc.gov)
- CLASSIFICATION OF DISEASES AND INJURIES I. INFECTIOUS AND PARASITIC DISEASES (001-139) Includes: diseases generally recognized as communicable or transmissible as well as a few diseases of unknown but possibly infectious origin Excludes: acute respiratory infections (460-466) influenza (487. (cdc.gov)
- certain localized infections Note: Categories for "late effects" of infectious and parasitic diseases are to be found at 137. (cdc.gov)
Rare18
- How were you diagnosed with a rare disease? (carenity.co.uk)
- If your baby has a rare disease, should we put a limit on how much to spend on the drug that may save her? (theconversation.com)
- The situation for so-called orphan drugs, which are specialty biologic drugs used for rare diseases, is even worse. (theconversation.com)
- The term orphan drug was codified in 1983 to describe medications intended to treat diseases so rare that pharmaceutical companies are reluctant to develop them. (theconversation.com)
- We help underinsured people with life-threatening, chronic, and rare diseases get the medications and treatments they need by assisting with their out-of-pocket costs and advocating for improved access and affordability. (panfoundation.org)
- Wolman disease is very rare, with a stimated incidence of less than one in 100,000 live births . (bredagenetics.com)
- Recently, the Ministry of Health and Family Welfare has approved the National Rare Disease Policy 2021. (upsc247.com)
- Earlier, the Delhi High Court had directed the Centre to set up a Rare Diseases Committee, a Rare Diseases Fund and to finalise and notify the National Health Policy for Rare Diseases on or before 31st March, 2021. (upsc247.com)
- What is a Rare Disease? (upsc247.com)
- WHO defines rare disease as often debilitating lifelong disease or disorder with a prevalence of 1 or less, per 1000 population. (upsc247.com)
- There are 6,000-8,000 classified rare diseases , but less than 5% have therapies available to treat them. (upsc247.com)
- India, like many other developing countries, currently has no standard definition of rare diseases and data on prevalence. (upsc247.com)
- Since there is no epidemiological data, there are no figures on burden of rare diseases and morbidity and mortality associated with them. (upsc247.com)
- This will yield much needed epidemiological data for rare diseases. (upsc247.com)
- So far only about 450 rare diseases have been recorded in India from tertiary care hospitals. (upsc247.com)
- A patient registry of rare diseases is to be constituted under ICMR (Indian Council of Medical Research). (upsc247.com)
- Under the policy, certain medical institutes will be certified as Centre of Excellence for rare diseases. (upsc247.com)
- It also aims to create Administrative Committee that will develop guidelines to determine which rare diseases to fund. (upsc247.com)
Acute1
- Late onset of a metabolic disease is often triggered by acute metabolic stresses, such as infection, fasting, or consumption of a nutrient for which a metabolic intolerance exists. (newworldencyclopedia.org)
DISORDER1
- A metabolic disorder is any disease or disorder that negatively affects the biochemical reactions through which individual animal cells process nutrient molecules (such as the components of carbohydrates , proteins , and fats ) to yield energy or perform the functions necessary to sustain life (such as building complex molecules and creating cellular structure). (newworldencyclopedia.org)
Adrenal Insufficiency1
- Addison's disease, or adrenal insufficiency, is usually an autoimmune disease, resulting from a faulty immune response. (medicalnewstoday.com)
Centers2
Difficulty1
- He was one of the first to recognize that polarized light microscopy is a powerful tool, which allows the detection of structures that otherwise be obtained only with great difficulty (Wolman, 1970). (wikipedia.org)
Pediatric1
- Pediatric liver transplantation, therefore, is positioned at the very birth of the hepatic transplantation surgical specialty, which now has become the standard treatment worldwide for a large range of serious or end-stage liver diseases in adults and children. (abdominalkey.com)
Hereditary1
- If you're an Ashkenazi Jewish woman, a standard prenatal visit to the obstetrician includes testing for as many as 15 hereditary diseases that could affect your offspring. (jta.org)
Affects1
- The disease affects males and females equally. (nih.gov)
Substances1
- Substances and Disease Registry or the U.S. Department of Health and Human Services. (cdc.gov)
Tissues1
- In affected individuals, harmful amounts of fats (lipids) accumulate in cells and tissues throughout the body, which typically causes liver disease. (medlineplus.gov)
Liver diseases4
- Fatty liver diseases are an increasingly serious health problem. (nih.gov)
- Missing were other key cell types involved in the inflammatory response to fatty liver diseases. (nih.gov)
- When exposed to free fatty acids, the organoids gradually accumulated fat in a dose-dependent manner and grew inflamed, which is similar to what happens to people with fatty liver diseases. (nih.gov)
- The findings also demonstrate a promising approach to accelerating the search for new treatments for a variety of liver diseases. (nih.gov)