Utrophin
Dystrophin
Muscular Dystrophy, Duchenne
Mice, Inbred mdx
Dystrophin-Associated Proteins
Muscular Dystrophy, Animal
Dystroglycans
Cytoskeletal Proteins
GA-Binding Protein Transcription Factor
Membrane Proteins
Shadowing (Histology)
Muscle, Skeletal
Muscular Dystrophies
Sarcolemma
Characterization of dystrophin and utrophin diversity in the mouse. (1/258)
Utrophin is a 400 kDa autosomal homolog of dystrophin and a component of the submembranous cytoskeleton. While multiple dystrophin isoforms have been identified along with alternatively spliced products, to date only two different mRNA species of utrophin have been identified. To determine the degree of evolutionary conservation between dystrophin and utrophin isoforms, we have compared their expression patterns in adult mice. Northern blot analysis of multiple adult tissues confirmed that only two major sizes of transcripts are produced from each gene: 13 and 5.5 kb from utrophin and 14 and 4.8 kb from dystrophin. However, western blot analysis detected several putative short utrophin isoforms that may be homologs of the dystrophin isoforms Dp140, Dp116 and Dp71. We also identified an alternatively spliced utrophin transcript that lacks the equivalent of the alternatively spliced dystrophin exon 71. Finally, we demonstrated that the C-terminal domain of utrophin targeted to neuromuscular junctions in normal mice, but localized to the sarcolemma efficiently only in the absence of dystrophin. Our results provide further evidence for a common evolutionary origin of the utrophin and dystrophin genes. (+info)Induction of utrophin gene expression by heregulin in skeletal muscle cells: role of the N-box motif and GA binding protein. (2/258)
The modulation of utrophin gene expression in muscle by the nerve-derived factor agrin plausibly involves the trophic factor ARIA/heregulin. Here we show that heregulin treatment of mouse and human cultured myotubes caused a approximately 2.5-fold increase in utrophin mRNA levels. Transient transfection experiments with utrophin promoter-reporter gene constructs showed that this increase resulted from an enhanced transcription of the utrophin gene. In the case of the nicotinic acetylcholine receptor delta and epsilon subunit genes, heregulin was previously reported to stimulate transcription via a conserved promoter element, the N-box, which binds the multimeric Ets-related transcription factor GA binding protein (GABP). Accordingly, site-directed mutagenesis of a single N-box motif in the utrophin gene promoter abolished the transcriptional response to heregulin. In addition, overexpression of heregulin, or of the two GABP subunits in cultured myotubes, caused an N-box-dependent increase of the utrophin promoter activity. In vivo, direct gene transfer into muscle confirmed that heregulin regulates utrophin gene expression. Finally, electrophoretic mobility shift assays and supershift experiments performed with muscle extracts revealed that the N-box of the utrophin promoter binds GABP. These findings suggest that the subsynaptic activation of transcription by heregulin via the N-box motif and GABP are conserved among genes expressed at the neuromuscular junction. Because utrophin can functionally compensate for the lack of dystrophin, the elucidation of the molecular mechanisms regulating utrophin gene transcription may ultimately lead to therapies based on utrophin expression throughout the muscle fibers of Duchenne muscular dystrophy patients. (+info)Extensive but coordinated reorganization of the membrane skeleton in myofibers of dystrophic (mdx) mice. (3/258)
We used immunofluorescence techniques and confocal imaging to study the organization of the membrane skeleton of skeletal muscle fibers of mdx mice, which lack dystrophin. beta-Spectrin is normally found at the sarcolemma in costameres, a rectilinear array of longitudinal strands and elements overlying Z and M lines. However, in the skeletal muscle of mdx mice, beta-spectrin tends to be absent from the sarcolemma over M lines and the longitudinal strands may be disrupted or missing. Other proteins of the membrane and associated cytoskeleton, including syntrophin, beta-dystroglycan, vinculin, and Na,K-ATPase are also concentrated in costameres, in control myofibers, and mdx muscle. They also distribute into the same altered sarcolemmal arrays that contain beta-spectrin. Utrophin, which is expressed in mdx muscle, also codistributes with beta-spectrin at the mutant sarcolemma. By contrast, the distribution of structural and intracellular membrane proteins, including alpha-actinin, the Ca-ATPase and dihydropyridine receptors, is not affected, even at sites close to the sarcolemma. Our results suggest that in myofibers of the mdx mouse, the membrane- associated cytoskeleton, but not the nearby myoplasm, undergoes widespread coordinated changes in organization. These changes may contribute to the fragility of the sarcolemma of dystrophic muscle. (+info)Membrane targeting and stabilization of sarcospan is mediated by the sarcoglycan subcomplex. (4/258)
The dystrophin-glycoprotein complex (DGC) is a multisubunit complex that spans the muscle plasma membrane and forms a link between the F-actin cytoskeleton and the extracellular matrix. The proteins of the DGC are structurally organized into distinct subcomplexes, and genetic mutations in many individual components are manifested as muscular dystrophy. We recently identified a unique tetraspan-like dystrophin-associated protein, which we have named sarcospan (SPN) for its multiple sarcolemma spanning domains (Crosbie, R.H., J. Heighway, D.P. Venzke, J.C. Lee, and K.P. Campbell. 1997. J. Biol. Chem. 272:31221-31224). To probe molecular associations of SPN within the DGC, we investigated SPN expression in normal muscle as a baseline for comparison to SPN's expression in animal models of muscular dystrophy. We show that, in addition to its sarcolemma localization, SPN is enriched at the myotendinous junction (MTJ) and neuromuscular junction (NMJ), where it is a component of both the dystrophin- and utrophin-glycoprotein complexes. We demonstrate that SPN is preferentially associated with the sarcoglycan (SG) subcomplex, and this interaction is critical for stable localization of SPN to the sarcolemma, NMJ, and MTJ. Our experiments indicate that assembly of the SG subcomplex is a prerequisite for targeting SPN to the sarcolemma. In addition, the SG- SPN subcomplex functions to stabilize alpha-dystroglycan to the muscle plasma membrane. Taken together, our data provide important information about assembly and function of the SG-SPN subcomplex. (+info)A PDZ-containing scaffold related to the dystrophin complex at the basolateral membrane of epithelial cells. (5/258)
Membrane scaffolding complexes are key features of many cell types, serving as specialized links between the extracellular matrix and the actin cytoskeleton. An important scaffold in skeletal muscle is the dystrophin-associated protein complex. One of the proteins bound directly to dystrophin is syntrophin, a modular protein comprised entirely of interaction motifs, including PDZ (protein domain named for PSD-95, discs large, ZO-1) and pleckstrin homology (PH) domains. In skeletal muscle, the syntrophin PDZ domain recruits sodium channels and signaling molecules, such as neuronal nitric oxide synthase, to the dystrophin complex. In epithelia, we identified a variation of the dystrophin complex, in which syntrophin, and the dystrophin homologues, utrophin and dystrobrevin, are restricted to the basolateral membrane. We used exogenously expressed green fluorescent protein (GFP)-tagged fusion proteins to determine which domains of syntrophin are responsible for its polarized localization. GFP-tagged full-length syntrophin targeted to the basolateral membrane, but individual domains remained in the cytoplasm. In contrast, the second PH domain tandemly linked to a highly conserved, COOH-terminal region was sufficient for basolateral membrane targeting and association with utrophin. The results suggest an interaction between syntrophin and utrophin that leaves the PDZ domain of syntrophin available to recruit additional proteins to the epithelial basolateral membrane. The assembly of multiprotein signaling complexes at sites of membrane specialization may be a widespread function of dystrophin-related protein complexes. (+info)Activation of utrophin promoter by heregulin via the ets-related transcription factor complex GA-binding protein alpha/beta. (6/258)
Utrophin/dystrophin-related protein is the autosomal homologue of the chromosome X-encoded dystrophin protein. In adult skeletal muscle, utrophin is highly enriched at the neuromuscular junction. However, the molecular mechanisms underlying regulation of utrophin gene expression are yet to be defined. Here we demonstrate that the growth factor heregulin increases de novo utrophin transcription in muscle cell cultures. Using mutant reporter constructs of the utrophin promoter, we define the N-box region of the promoter as critical for heregulin-mediated activation. Using this region of the utrophin promoter for DNA affinity purification, immunoblots, in vitro kinase assays, electrophoretic mobility shift assays, and in vitro expression in cultured muscle cells, we demonstrate that ets-related GA-binding protein alpha/beta transcription factors are activators of the utrophin promoter. Taken together, these results suggest that the GA-binding protein alpha/beta complex of transcription factors binds and activates the utrophin promoter in response to heregulin-activated extracellular signal-regulated kinase in muscle cell cultures. These findings suggest methods for achieving utrophin up-regulation in Duchenne's muscular dystrophy as well as mechanisms by which neurite-derived growth factors such as heregulin may influence the regulation of utrophin gene expression and subsequent enrichment at the neuromuscular junction of skeletal muscle. (+info)Up71 and up140, two novel transcripts of utrophin that are homologues of short forms of dystrophin. (7/258)
Utrophin is a large protein which accumulates at the neuromuscular synapse and myotendinous junctions in adult skeletal muscle, and is widely expressed in several non-skeletal muscle tissues. Evidence from a variety of sources suggests that a successful strategy for treatment of Duchenne muscular dystrophy patients will be to increase expression of utrophin in muscle. There is still much to be learnt about utrophin gene regulation, in particular regarding alternative isoforms, their promoters and role in muscle and non-muscle tissues. Using 5"-RACE we have identified two novel transcripts of utrophin, Up71 and Up140, with unique first exons and promoters located in intron 62 and intron 44, respectively. These transcripts appear to be structural homologues of the short dystrophin transcripts, Dp140 and Dp71, emphasizing the high degree of structural conservation between the utrophin and dystrophin genes. RT-PCR shows that Up71 and Up140 are widely expressed in both human and mouse tissues, including skeletal muscle. We present evidence for transcript-specific differential mRNA splicing of exon 71, in both Up71 and Up140, similar to that described for dystrophin. No evidence for splicing of exon 78 of utrophin was found. This is in contrast to dystrophin and may reflect a subtle functional difference in patterns of phosphorylation between the two proteins. (+info)Expression of the utrophin gene during myogenic differentiation. (8/258)
The process of myogenic differentiation is known to be accompanied by large increases ( approximately 10-fold) in the expression of genes encoding cytoskeletal and membrane proteins including dystrophin and the acetylcholine receptor (AChR) subunits, via the effects of transcription factors belonging to the MyoD family. Since in skeletal muscle (i) utrophin is a synaptic homolog to dystrophin, and (ii) the utrophin promoter contains an E-box, we examined, in the present study, expression of the utrophin gene during myogenic differentiation using the mouse C2 muscle cell line. We observed that in comparison to myoblasts, the levels of utrophin and its transcript were approximately 2-fold higher in differentiated myotubes. In order to address whether a greater rate of transcription contributed to the elevated levels of utrophin transcripts, we performed nuclear run-on assays. In these studies we determined that the rate of transcription of the utrophin gene was approximately 2-fold greater in myotubes as compared to myoblasts. Finally, we examined the stability of utrophin mRNAs in muscle cultures by two separate methods: following transcription blockade with actinomycin D and by pulse-chase experiments. Under these conditions, we determined that the half-life of utrophin mRNAs in myoblasts was approximately 20 h and that it remained largely unaffected during myogenic differentiation. Altogether, these results show that in comparison to other synaptic proteins and to dystrophin, expression of the utrophin gene is only moderately increased during myogenic differentiation. (+info)Utrophin is a protein that is found in muscle cells. It is similar in structure and function to dystrophin, which is a protein that is deficient or abnormal in people with Duchenne and Becker muscular dystrophy. Utrophin is present in both fetal and adult muscle, but its expression is usually limited to the nerve endings of the muscle fibers. However, in certain conditions such as muscle injury or disease, utrophin can be upregulated and expressed more widely throughout the muscle fiber. Research has shown that increasing the levels of utrophin in muscle cells could potentially compensate for the lack of dystrophin and provide a therapeutic approach to treating muscular dystrophy.
Dystrophin is a protein that provides structural stability to muscle fibers. It is an essential component of the dystrophin-glycoprotein complex, which helps maintain the integrity of the sarcolemma (the membrane surrounding muscle cells) during muscle contraction and relaxation. Dystrophin plays a crucial role in connecting the cytoskeleton of the muscle fiber to the extracellular matrix, allowing for force transmission and protecting the muscle cell from damage.
Mutations in the DMD gene, which encodes dystrophin, can lead to various forms of muscular dystrophy, including Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD). In DMD, a severe form of the disease, genetic alterations typically result in little or no production of functional dystrophin, causing progressive muscle weakness, wasting, and degeneration. In BMD, a milder form of the disorder, partially functional dystrophin is produced, leading to less severe symptoms and later onset of the disease.
Duchenne Muscular Dystrophy (DMD) is a genetic disorder characterized by progressive muscle weakness and degeneration. It is caused by the absence of dystrophin, a protein that helps keep muscle cells intact. Without dystrophin, the muscle cells break down and are replaced with scar tissue, leading to loss of muscle function over time.
DMD primarily affects boys, as it is inherited in an X-linked recessive pattern, meaning that females who carry one affected X chromosome typically do not show symptoms but can pass the gene on to their offspring. Symptoms usually begin in early childhood and include difficulty with motor skills such as walking, running, and climbing stairs. Over time, the muscle weakness progresses and can lead to loss of ambulation, respiratory and cardiac complications, and ultimately, premature death.
Currently, there is no cure for DMD, but various treatments such as corticosteroids, physical therapy, and assisted ventilation can help manage symptoms and improve quality of life. Gene therapy approaches are also being investigated as potential treatments for this disorder.
'Mice, Inbred mdx' is a genetic strain of laboratory mice that are widely used as a model to study Duchenne muscular dystrophy (DMD), a severe and progressive muscle-wasting disorder in humans. The 'mdx' designation refers to the specific genetic mutation present in these mice, which is a point mutation in the gene encoding for dystrophin, a crucial protein involved in maintaining the structural integrity of muscle fibers.
Inbred mdx mice carry a spontaneous mutation in exon 23 of the dystrophin gene, resulting in the production of a truncated and nonfunctional form of the protein. This leads to a phenotype that closely resembles DMD in humans, including muscle weakness, degeneration, and fibrosis. The inbred nature of these mice ensures consistent genetic backgrounds and disease manifestations, making them valuable tools for studying the pathophysiology of DMD and testing potential therapies.
It is important to note that while the inbred mdx mouse model has been instrumental in advancing our understanding of DMD, it does not fully recapitulate all aspects of the human disease. Therefore, findings from these mice should be carefully interpreted and validated in more complex models or human studies before translating them into clinical applications.
Dystrophin-associated proteins (DAPs) are a group of structural and functional proteins that interact with dystrophin, a cytoskeletal protein found in muscle cells. Dystrophin helps to maintain the integrity of the muscle fiber membrane, or sarcolemma, during contractions. The dystrophin-associated protein complex (DAPC) includes dystroglycans, sarcoglycans, syntrophins, and dystrobrevins, among others.
Mutations in genes encoding DAPs can lead to various forms of muscular dystrophy, a group of genetic disorders characterized by progressive muscle weakness and degeneration. For example, mutations in the sarcoglycan gene can cause limb-girdle muscular dystrophy type 2C (LGMD2C), while defects in dystroglycan can result in congenital muscular dystrophy with mental retardation and structural brain abnormalities.
In summary, DAPs are a group of proteins that interact with dystrophin to maintain the stability and function of muscle fibers. Defects in these proteins can lead to various forms of muscular dystrophy.
I'm sorry for any confusion, but "Muscular Dystrophy, Animal" is not a standard medical term. Muscular Dystrophy is a group of genetic disorders that cause progressive weakness and loss of muscle mass. They are primarily human diseases and there are no known animal models of muscular dystrophy that directly correspond to any type of muscular dystrophy in humans.
However, scientists often use animals (like mice, dogs, and cats) as models for human diseases, including various types of muscular dystrophies. These animal models are used to study the disease process and to test potential treatments. For example, the mdx mouse is a well-known model of Duchenne Muscular Dystrophy (DMD), which is caused by a mutation in the dystrophin gene. This mouse lacks the muscle protein dystrophin, similar to humans with DMD, and shows many of the same symptoms, making it a valuable tool for research.
Dystroglycans are a type of protein that play a crucial role in the structure and function of the muscle membrane (sarcolemma). They are an essential component of the dystrophin-glycoprotein complex, which helps maintain the stability and integrity of the sarcolemma during muscle contraction and relaxation.
Dystroglycans consist of two subunits: alpha-dystroglycan and beta-dystroglycan. Alpha-dystroglycan is a large, heavily glycosylated protein that extends from the intracellular space to the extracellular matrix, where it interacts with various extracellular matrix proteins such as laminin and agrin. Beta-dystroglycan, on the other hand, spans the muscle membrane and binds to dystrophin, a cytoskeletal protein that helps maintain the structural integrity of the sarcolemma.
Mutations in genes encoding for proteins involved in the glycosylation of alpha-dystroglycan can lead to a group of genetic disorders known as congenital muscular dystrophies, which are characterized by muscle weakness, hypotonia, and developmental delays. These disorders include Walker-Warburg syndrome, Fukuyama congenital muscular dystrophy, and Muscle-Eye-Brain disease, among others.
Cytoskeletal proteins are a type of structural proteins that form the cytoskeleton, which is the internal framework of cells. The cytoskeleton provides shape, support, and structure to the cell, and plays important roles in cell division, intracellular transport, and maintenance of cell shape and integrity.
There are three main types of cytoskeletal proteins: actin filaments, intermediate filaments, and microtubules. Actin filaments are thin, rod-like structures that are involved in muscle contraction, cell motility, and cell division. Intermediate filaments are thicker than actin filaments and provide structural support to the cell. Microtubules are hollow tubes that are involved in intracellular transport, cell division, and maintenance of cell shape.
Cytoskeletal proteins are composed of different subunits that polymerize to form filamentous structures. These proteins can be dynamically assembled and disassembled, allowing cells to change their shape and move. Mutations in cytoskeletal proteins have been linked to various human diseases, including cancer, neurological disorders, and muscular dystrophies.
A GA-binding protein (GABP) transcription factor is a type of protein complex that regulates gene expression by binding to specific DNA sequences known as GATA motifs. These motifs contain the consensus sequence (T/A)GAT(A/G)(A/T). GABP is composed of two subunits, GABPα and GABPβ, which form a heterodimer that recognizes and binds to the GATA motif.
GABP plays a crucial role in various biological processes, including cell proliferation, differentiation, and survival. It is involved in the regulation of genes that are important for the function of the cardiovascular, respiratory, and immune systems. Mutations in the genes encoding GABP subunits have been associated with several human diseases, such as congenital heart defects, pulmonary hypertension, and immunodeficiency disorders.
Overall, GABP transcription factors are essential regulators of gene expression that play a critical role in maintaining normal physiological functions and homeostasis in the body.
Membrane proteins are a type of protein that are embedded in the lipid bilayer of biological membranes, such as the plasma membrane of cells or the inner membrane of mitochondria. These proteins play crucial roles in various cellular processes, including:
1. Cell-cell recognition and signaling
2. Transport of molecules across the membrane (selective permeability)
3. Enzymatic reactions at the membrane surface
4. Energy transduction and conversion
5. Mechanosensation and signal transduction
Membrane proteins can be classified into two main categories: integral membrane proteins, which are permanently associated with the lipid bilayer, and peripheral membrane proteins, which are temporarily or loosely attached to the membrane surface. Integral membrane proteins can further be divided into three subcategories based on their topology:
1. Transmembrane proteins, which span the entire width of the lipid bilayer with one or more alpha-helices or beta-barrels.
2. Lipid-anchored proteins, which are covalently attached to lipids in the membrane via a glycosylphosphatidylinositol (GPI) anchor or other lipid modifications.
3. Monotopic proteins, which are partially embedded in the membrane and have one or more domains exposed to either side of the bilayer.
Membrane proteins are essential for maintaining cellular homeostasis and are targets for various therapeutic interventions, including drug development and gene therapy. However, their structural complexity and hydrophobicity make them challenging to study using traditional biochemical methods, requiring specialized techniques such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and single-particle cryo-electron microscopy (cryo-EM).
Skeletal muscle, also known as striated or voluntary muscle, is a type of muscle that is attached to bones by tendons or aponeuroses and functions to produce movements and support the posture of the body. It is composed of long, multinucleated fibers that are arranged in parallel bundles and are characterized by alternating light and dark bands, giving them a striped appearance under a microscope. Skeletal muscle is under voluntary control, meaning that it is consciously activated through signals from the nervous system. It is responsible for activities such as walking, running, jumping, and lifting objects.
Muscular dystrophies are a group of genetic disorders that primarily affect skeletal muscles, causing progressive weakness and degeneration. They are characterized by the lack or deficiency of a protein called dystrophin, which is essential for maintaining the integrity of muscle fibers. The most common form is Duchenne muscular dystrophy (DMD), but there are many other types with varying symptoms and severity. Over time, muscle wasting and weakness can lead to disability and shortened lifespan, depending on the type and progression of the disease. Treatment typically focuses on managing symptoms, maintaining mobility, and supporting quality of life.
Sarcolemma is the medical term for the cell membrane that surrounds a muscle fiber or a skeletal muscle cell. It is responsible for providing protection and structure to the muscle fiber, as well as regulating the movement of ions and other molecules in and out of the cell. The sarcolemma plays a crucial role in the excitation-contraction coupling process that allows muscles to contract and relax.
The sarcolemma is composed of two main layers: the outer plasma membrane, which is similar to the cell membranes of other cells, and the inner basal lamina, which provides structural support and helps to anchor the muscle fiber to surrounding tissues. The sarcolemma also contains various ion channels, receptors, and transporters that are involved in regulating muscle function and communication with other cells.
Damage to the sarcolemma can lead to a variety of muscle disorders, including muscular dystrophy and myasthenia gravis.
Methylprednisolone Hemisuccinate is a synthetic glucocorticoid drug, which is a salt of Methylprednisolone with hemisuccinic acid. It is often used in the treatment of various inflammatory and autoimmune conditions due to its potent anti-inflammatory and immunosuppressive effects.
Methylprednisolone Hemisuccinate is rapidly absorbed after intravenous or intramuscular administration, with a bioavailability of nearly 100%. It has a high penetration rate into body tissues, including the central nervous system, making it useful in the treatment of conditions such as multiple sclerosis and other inflammatory diseases of the brain and spinal cord.
Like other glucocorticoids, Methylprednisolone Hemisuccinate works by binding to specific receptors in cells, which leads to a decrease in the production of pro-inflammatory cytokines and an increase in the production of anti-inflammatory mediators. This results in a reduction in inflammation, swelling, and pain, as well as a suppression of the immune system's response to various stimuli.
Methylprednisolone Hemisuccinate is available under several brand names, including Solu-Medrol and Depo-Medrol. It is typically administered in hospital settings for the treatment of severe inflammatory conditions or as part of a treatment regimen for certain autoimmune diseases. As with all medications, it should be used under the close supervision of a healthcare provider, and its benefits and risks should be carefully weighed before use.
 Utrophin
Utrophin
WW domain
PGM5
Dystrobrevin
Syntrophin, alpha 1
Dystrophin
Dystrophin-associated protein complex
Dystroglycan
SGCB
SNTB2
Delta-sarcoglycan
ZZ zinc finger
ABCA1
Sarcospan
Duchenne muscular dystrophy
Transcription factor Sp1
Ezutromid
CHRNE
Aldehyde oxidase 1
Sp3 transcription factor
GABPA
Dystrobrevin alpha
Actin-binding protein
Trauma Center: Under the Knife
Becker muscular dystrophy
Alpha-actinin-2
LifeAct Dye
Trauma Center: Second Opinion
List of acronyms: G
Guilt
Utrophin - Wikipedia
 Summit publishes preclinical data on utrophin modulation in DMD | Drug Discovery News
Summit publishes preclinical data on utrophin modulation in DMD | Drug Discovery News
Dystrophin and utrophin influence fiber type composition and post-synaptic membrane structure. - Department of Physiology,...
Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. - SCNi
Utrophin modulation for the treatment of cardiomyopathy in mdx mice - Oxford Cardiovascular Science
 Subtle neuromuscular defects in utrophin-deficient mice<...
Subtle neuromuscular defects in utrophin-deficient mice<...
 A laminin-2, dystroglycan, utrophin axis is required for compartmentalization and elongation of myelin segments. - Courtlab
A laminin-2, dystroglycan, utrophin axis is required for compartmentalization and elongation of myelin segments. - Courtlab
Discovery of small molecule utrophin modulators for the therapy of Duchenne muscular dystrophy (DMD) - Oxford Cardiovascular...
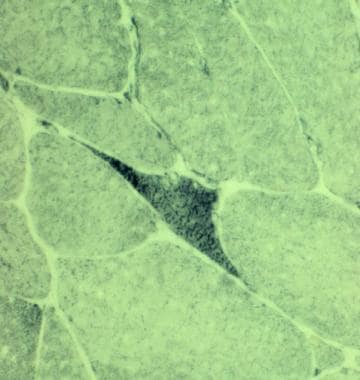
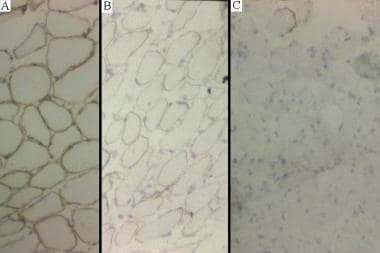
 Skeletal Muscle Pathology: Overview, Neurogenic Changes in Muscle Biopsy, Muscle Biopsy in Myopathy
Skeletal Muscle Pathology: Overview, Neurogenic Changes in Muscle Biopsy, Muscle Biopsy in Myopathy
The role of basal and myogenic factors in the transcriptional activation of utrophin promoter A: implications for therapeutic...
Non-uniform dystrophin re-expression after CRISPR-mediated exon excision in the dystrophin/utrophin double-knockout mouse model...
 Non-uniform dystrophin re-expression after CRISPR-mediated exon excision in the dystrophin/utrophin double-knockout mouse model...
Non-uniform dystrophin re-expression after CRISPR-mediated exon excision in the dystrophin/utrophin double-knockout mouse model...
 Metabolites | April 2021 - Browse Articles
Metabolites | April 2021 - Browse Articles
 12 Natural Remedies for Fibroids
12 Natural Remedies for Fibroids
Dystrophinopathies: Practice Essentials, Background, Pathophysiology
 Combined effect of cell geometry and polarity domains determines the orientation of unequal division | eLife
Combined effect of cell geometry and polarity domains determines the orientation of unequal division | eLife
 SCR 6q22.31-6q24.3 Genes
SCR 6q22.31-6q24.3 Genes
 Frontiers | Mitochondrial Dysfunction Is an Early Consequence of Partial or Complete Dystrophin Loss in mdx Mice
Frontiers | Mitochondrial Dysfunction Is an Early Consequence of Partial or Complete Dystrophin Loss in mdx Mice
Plus it
 SMART: CH domain annotation
SMART: CH domain annotation
SMART: SPEC domain annotation
Frontiers | Dystrophin Distribution and Expression in Human and Experimental Temporal Lobe Epilepsy
 CureDuchenne | Funding research to find a cure for Duchenne
CureDuchenne | Funding research to find a cure for Duchenne
 HRP Goat Anti-Mouse IgG2a, Human Adsorbed | SouthernBiotech
HRP Goat Anti-Mouse IgG2a, Human Adsorbed | SouthernBiotech
Trauma Center: Second Opinion website now open - QuickJump
 Addgene: Comparative analysis of tools for live cell imaging of actin network architecture.
Addgene: Comparative analysis of tools for live cell imaging of actin network architecture.
 Ocular Pathology of Fukuyama Congenital Muscular Dystrophy | IntechOpen
Ocular Pathology of Fukuyama Congenital Muscular Dystrophy | IntechOpen
 Macrophage Plasticity in Skeletal Muscle Repair
Macrophage Plasticity in Skeletal Muscle Repair
Located at the neuromuscular synapse a1
- In normal muscle cells, utrophin is located at the neuromuscular synapse and myotendinous junctions. (wikipedia.org)
Homologue of dystrophin1
- Utrophin is the autosomal homologue of dystrophin. (ox.ac.uk)
Dystrophin gene2
- The invention concerns in particular the treatment of dystrophies such as Duchenne's dystrophy or Becker's dystrophy in which the defective adult gene is the dystrophin gene and the homologous fetal gene is the utrophin gene. (justia.com)
- This month in Med, the description of an unusually severely affected DMD patient suffering from a large deletion in the dystrophin gene confirms that absence of utrophin worsens the dystrophy and supports the concept that utrophin upregulation ameliorates the pathology. (bvsalud.org)
Sarcolemma5
- In the human fetus during muscle differentiation, utrophin is found at the sarcolemma. (wikipedia.org)
- Here, we test the hypothesis that utrophin also brings nNOS to the sarcolemma. (ox.ac.uk)
- Despite supra-physiological utrophin expression, we did not detect nNOS at the sarcolemma. (ox.ac.uk)
- Our results suggest that full-length utrophin cannot anchor nNOS to the sarcolemma. (ox.ac.uk)
- In skeletal muscle, dystrophin is broadly distributed along the sarcolemma whereas utrophin is concentrated at the neuromuscular junction. (wustl.edu)
UTRN2
- Utrophin is a protein that in humans is encoded by the UTRN gene. (wikipedia.org)
- Materials & methods: We have isolated wild-type mesoangioblasts from aorta and tested their effectiveness in alleviating severe muscle disease in the dystrophin/utrophin knockout (mdx/utrn -l- ) mouse model for Duchenne muscular dystrophy. (illinois.edu)
Mice6
- No reports have yet associated mutation in the utrophin gene with disease, but it does not seem to play a critical role in development, since mice without utrophin develop normally. (wikipedia.org)
- In marked contrast, a dystrophin protein lacking the cysteine-rich domain, which is unable to prevent dystrophy in the mdx mouse, is able to ameliorate these abnormalities in utrophin/dystrophin-deficient mice. (ox.ac.uk)
- Full-length utrophin cDNA was expressed in dystrophin-deficient mdx mice by gutted adenovirus or via transgenic overexpression. (ox.ac.uk)
- To test these hypotheses we generated and characterized utrophin-deficient mutant mice. (wustl.edu)
- We previously demonstrated that overexpression of utrophin in the muscles of dystrophin-null transgenic mice completely prevented the phenotype arising from dystrophin deficiency. (ox.ac.uk)
- Mice homozygous for utrophin and dystrophin knockouts die prematurely with severe, progressive muscular dystrophy. (utsouthwestern.edu)
Demonstrated in dystrophin-deficient1
- By pharmacologically modulating the expression of the dystrophin-related protein utrophin, we have previously demonstrated in dystrophin-deficient mdx studies, daily SMT C1100 treatment significantly reduced muscle degeneration, leading to improved muscle function. (drugdiscoverynews.com)
Cytoskeletal protein2
- In the published report, titled "Second-generation compound for the modulation of utrophin in the therapy of DMD," the authors write that "DMD is a lethal, X-linked muscle-wasting disease caused by lack of the cytoskeletal protein dystrophin. (drugdiscoverynews.com)
- Utrophin is a large cytoskeletal protein that is homologous to dystrophin, the protein mutated in Duchenne and Becker muscular dystrophy. (wustl.edu)
Muscle19
- Utrophin expression is dramatically increased in patients with Duchenne's muscular dystrophy (and female carriers), both in those muscle fibers lacking dystrophin and in rare, revertant fibers that express dystrophin. (wikipedia.org)
- Role of dystrophin and utrophin for assembly and function of the dystrophin glycoprotein complex in non-muscle tissue" (PDF). (wikipedia.org)
- Upon modulation of utrophin protein with the second-generation utrophin modulator SMT022357, in-vivo models of DMD showed significantly improved muscle stability and a marked reduction of the muscle regeneration, necrosis and fibrosis that are at the core of DMD pathology. (drugdiscoverynews.com)
- Among the findings of the research published in the Aug. 1, 2015, issue of Human Molecular Genetics was that utrophin was expressed across the entire length of the muscle fiber, likely contributing to its ability to significantly reduce disease progression in animal models. (drugdiscoverynews.com)
- Significantly, utrophin expression is localized along the length of the muscle fiber, not just at the synapse, and is fiber-type independent, suggesting that drug treatment is modulating utrophin transcription in extra-synaptic myonuclei. (drugdiscoverynews.com)
- Utrophin is structurally and functionally similar to dystrophin, the protein which is lacking in boys with DMD, and is normally present during muscle development and repair. (drugdiscoverynews.com)
- In the reported study, SMT022357 treatment for five weeks resulted in increased utrophin expression, localized along the entire length of the muscle fiber membrane in both slow- and fast-twitch muscles. (drugdiscoverynews.com)
- To identify potential non-mechanical roles of dystrophin, we tested the ability of various truncated dystrophin transgenes to prevent any of the skeletal muscle abnormalities associated with the double knockout mouse deficient for both dystrophin and the dystrophin-related protein utrophin. (ox.ac.uk)
- We show that restoration of the DAPC with Dp71 does not prevent the structural abnormalities of the post-synaptic membrane or the abnormal oxidative properties of utrophin/dystrophin-deficient muscle. (ox.ac.uk)
- These experiments provide the first direct evidence that in addition to a mechanical role and relocalization of the DAPC, dystrophin and utrophin are able to alter both structural and biochemical properties of skeletal muscle. (ox.ac.uk)
- Furthermore, transgenic utrophin overexpression failed to protect mdx muscle from exercise-associated injury. (ox.ac.uk)
- In addition, utrophin is present in numerous non-muscle cells, suggesting that it may have a more generalized role in the maintenance of cellular integrity. (wustl.edu)
- Detailed analysis, however, revealed that the density of acetylcholine receptors and the number of junctional folds were reduced at the neuromuscular junctions in utrophin- deficient skeletal muscle. (wustl.edu)
- Two independently regulated promoters control utrophin expression and the upstream promoter (promoter A) is synaptically regulated in muscle. (ox.ac.uk)
- This study provides a basis for further understanding the regulatory mechanisms that control utrophin expression in muscle and may facilitate the development of reagents to effect therapeutic up-regulation of utrophin in DMD. (ox.ac.uk)
- Mesoangioblasts proliferate in vivo, incorporate into muscle fibers, form new fibers, and promote synthesis of clystrophin and utrophin. (illinois.edu)
- In addition, our results open the door to new approaches: 1) to improve the understanding of the mechanism underlying sarcomeric distortion in TTN-related myopathies, 2) to identify new roles for utrophin, study the mechanism of expression and assess its potential application in therapy as a protective element to delay the muscle fibre damage in certain NMDs. (ugr.es)
- Muscle utrophin and desmin expression were also evaluated using immunoblotting. (bvsalud.org)
- BACKGROUND: Utrophin, a dystrophin homolog, is consistently upregulated in muscles of patients with Duchenne muscular dystrophy (DMD) and is believed to partially compensate for the lack of dystrophin in dystrophic muscle. (bvsalud.org)
Gene3
- The 900 kb gene for utrophin is found on the long arm of human chromosome 6. (wikipedia.org)
- Among the major strategies are gene replacement, gene modification, stem cell use, inhibiting a protein called myostatin, expanding the distribution and increasing the level of a protein called utrophin, and increasing blood flow to muscles. (mda.org)
- Specifically, MK elicited a gene expression profile indicative of a more disease-resistant slow, oxidative phenotype including increased peroxisome proliferator-activated receptor ɣ coactivator-1⠺ activity and utrophin levels. (bvsalud.org)
Duchenne2
- OXFORD, U.K.- Summit Therapeutics plc , a drug discovery and development company advancing therapies for Duchenne muscular dystrophy (DMD) and C. difficile infection, recently announced the publication of preclinical data on the disease-modifying potential of utrophin modulation in the treatment of DMD. (drugdiscoverynews.com)
- The role of basal and myogenic factors in the transcriptional activation of utrophin promoter A: implications for therapeutic up-regulation in Duchenne muscular dystrophy. (ox.ac.uk)
Differentiation2
Sequence1
- Utrophin shares considerable sequence, structural and functional similarity with dystrophin. (ox.ac.uk)
Protein1
- The tertiary structure of utrophin contains a C-terminus that consists of protein-protein interaction motifs that interact with dystroglycan, a central rod region consisting of a triple coiled-coil repeat, and an actin-binding N-terminus. (wikipedia.org)
Modulation3
- These data strongly support utrophin modulation as a potentially valuable mechanism to treat DMD and highlight the importance of the continued development of second-generation, orally available utrophin modulator candidates," according to Prof. Dame Kay E. Davies of the University of Oxford. (drugdiscoverynews.com)
- There is tremendous therapeutic potential for utrophin modulation in this devastating disease because there is currently no approved disease-modifying therapy applicable to all patients with DMD, and many other candidates in clinical development are restricted to a single mutation. (drugdiscoverynews.com)
- This detailed evaluation of the SMT C1100 drug series strongly endorses the therapeutic potential of utrophin modulation as a disease-modifying therapeutic strategy for all DMD patients irrespective of their dystrophin mutation. (drugdiscoverynews.com)
Fiber-type1
- Dystrophin and utrophin influence fiber type composition and post-synaptic membrane structure. (ox.ac.uk)
Muscles1
- These studies in the mdx mouse demonstrate that oral administration of SMT022357 leads to increased utrophin expression in skeletal, respiratory and cardiac muscles. (drugdiscoverynews.com)
Expression7
- Utrophin expression in the heart and diaphragm is highly desirable in DMD as loss of function in these organs is life-limiting. (drugdiscoverynews.com)
- Treatment with SMT022357 resulted in significant increases in utrophin expression in both the heart and diaphragm. (drugdiscoverynews.com)
- A particularly attractive approach is to increase utrophin expression. (ox.ac.uk)
- Non-uniform dystrophin re-expression after CRISPR-mediated exon excision in the dystrophin/utrophin double-knockout mouse model of DMD. (ox.ac.uk)
- In conclusion, we show the important contribution of EM in the study of titinopathies and the examination of utrophin expression by IHC in the study of NMDs. (ugr.es)
- The expression of utrophin and the identification of a DUE pattern allow to guide the diagnosis, as well as to assess the persistent regenerative capacity in the tissue. (ugr.es)
- Interestingly, VPA reduced the isometric force drop following eccentric contractions in both murine models, without change in the relative eccentric maximal force and in the expression of utrophin and desmin. (bvsalud.org)
Implications1
- This finding might have important implications for the development of utrophin-based DMD therapies. (ox.ac.uk)
Clinical data1
- Even though several animal studies support the idea that utrophin can modulate DMD disease severity, human clinical data are scarce. (bvsalud.org)
Restoration1
- Here we have investigated the use of the Staphylococcus aureus CRISPR-Cas9 system and a double-cut strategy, delivered using a pair of adeno-associated virus serotype 9 (AAV9) vectors, for dystrophin restoration in the severely affected dystrophin/utrophin double-knockout (dKO) mouse. (ox.ac.uk)
Disease1
- By modifying utrophin to be continuously produced in boys with DMD, this potentially disease-modifying approach could circumvent the need for dystrophin in all patients with this devastating disease. (drugdiscoverynews.com)
Present1
- The same repeats are also present in alpha-actinin, dystrophin and utrophin. (embl.de)
Damage1
- Utrophin was found during research into Duchenne's muscular dystrophy, where boosting its production was found to prevent cellular damage from occurring. (wikipedia.org)
Skeletal muscle6
- Compensation for dystrophin-deficiency: ADAM12 overexpression in skeletal muscle results in increased alpha 7 integrin, utrophin and associated glycoproteins. (ox.ac.uk)
- 2. Haploinsufficiency of utrophin gene worsens skeletal muscle inflammation and fibrosis in mdx mice. (nih.gov)
- 5. Comparison of skeletal muscle pathology and motor function of dystrophin and utrophin deficient mouse strains. (nih.gov)
- 6. Treatment with the anti-IL-6 receptor antibody attenuates muscular dystrophy via promoting skeletal muscle regeneration in dystrophin-/utrophin-deficient mice. (nih.gov)
- Together these data show that there are four potential syntrophin-binding sites per dystrophin complex in skeletal muscle: two on dystrobrevin and two on dystrophin or utrophin. (ox.ac.uk)
- We also evaluated the therapeutic effect of utrophin upregulator drug candidates on the functionality of the skeletal muscle tissues, thus providing deeper insight into the real impact of these treatments. (biomed.news)
Homologue1
- Utrophin is a dystrophin homologue currently under investigation as a protein replacement therapy for Duchenne muscular dystrophy. (nih.gov)
Alpha-actinin2
- Alpha-actinin is a rod-like cytoskeletal protein belonging to the same family as spectrin, dystrophin and utrophin. (ihcworld.com)
- The spectrin superfamily (spectrin, alpha-actinin, utrophin and dystrophin) has in common a triple helical repeating unit of ~106 amino acid residues. (kent.ac.uk)
Dystrophin-deficient2
- Full-length utrophin cDNA was expressed in dystrophin-deficient mdx mice by gutted adenovirus or via transgenic overexpression. (ox.ac.uk)
- 13. Analysis of gene expression differences between utrophin/dystrophin-deficient vs mdx skeletal muscles reveals a specific upregulation of slow muscle genes in limb muscles. (nih.gov)
Transgenic1
- Furthermore, transgenic utrophin overexpression failed to protect mdx muscle from exercise-associated injury. (ox.ac.uk)
Spectrin1
- A profile hidden Markov model of beta-spectrin repeat 1 detects alpha-actinins, but not utrophin or dystrophin. (kent.ac.uk)
Therapeutic2
- Upregulating utrophin using small molecules could be a new therapeutic approach for Duchenne muscular dystrophy (DMD). (drugtargetreview.com)
- Utrophin modulation is a promising therapeutic strategy for Duchenne muscular dystrophy (DMD), which should be applicable to all patient populations. (ox.ac.uk)
Localization1
- Indeed, by immunostaining and immunoblotting we found an approximately 2-fold increase in expression, and distinct extrasynaptic localization, of alpha 7B integrin and utrophin, the functional homolog of dystrophin. (ox.ac.uk)
Actin2
- The tertiary structure of utrophin contains a C-terminus that consists of protein-protein interaction motifs that interact with dystroglycan, a central rod region consisting of a triple coiled-coil repeat, and an actin-binding N-terminus. (wikipedia.org)
- Here, using live imaging of an F-actin-specific probe, superfolder GFP-tagged utrophin (UtrCH-sfGFP), we demonstrated the dynamics of nuclear actin in zebrafish (Danio rerio) embryos. (genomyx.ch)
Functional3
- We're using an approach that attempts to increase utrophin levels in the body because it has functional characteristics and a genetic structure similar to dystrophin. (drugtargetreview.com)
- Utrophin shares considerable sequence, structural and functional similarity with dystrophin. (ox.ac.uk)
- 4. Utrophin haploinsufficiency does not worsen the functional performance, resistance to eccentric contractions and force production of dystrophic mice. (nih.gov)
Neuromuscular1
- In normal muscle cells, utrophin is located at the neuromuscular synapse and myotendinous junctions. (wikipedia.org)
Full-length1
- While utrophin is homologous with dystrophin from a molecular and biochemical perspective, we have recently shown that full-length utrophin expressed in eukaryotic cells is stiffer than what has been reported for dystrophin fragments expressed in bacteria. (nih.gov)
Development2
- Following on from ezutromid, the first-generation utrophin modulator, we describe the development of a second generation of utrophin modulators, based on the bioisosteric replacement of the sulfone group with a phosphinate ester and substitution of the metabolically labile naphthalene with a haloaryl substituent. (ox.ac.uk)
- This finding might have important implications for the development of utrophin-based DMD therapies. (ox.ac.uk)
Increase1
- This is a completely new approach to increase utrophin for this condition and we're very keen to test it further and eventually bring it to clinical trials. (drugtargetreview.com)
Therapy1
- While 30 was found to have dose-limiting hepatotoxicity, 27 and its enantiomers exhibited limited off-target effects, resulting in a safe profile and highlighting their potential utility as next-generation utrophin modulators suitable for progression toward a future DMD therapy. (ox.ac.uk)
Mouse2
Reports1
- A paper, published in Scientific Reports , states that utrophin, a protein naturally produced by the body but usually downregulated, could be substituted for dystrophin and reverse the symptoms of DMD. (drugtargetreview.com)
Function1
- Role of dystrophin and utrophin for assembly and function of the dystrophin glycoprotein complex in non-muscle tissue" (PDF). (wikipedia.org)