Oxidative Phosphorylation
Mitochondria
Uncoupling Agents
Adenosine Triphosphate
Oxygen Consumption
Phosphorylation
Serine
Glycolysis
Oligomycins
Mitochondria, Liver
Tyrosine
Cell Respiration
Oxidative Phosphorylation Coupling Factors
Protein-Serine-Threonine Kinases
Mitochondrial Diseases
Adenosine Diphosphate
Enzyme Activation
Electron Transport
Energy Metabolism
Phosphoproteins
Protein Kinases
Molecular Sequence Data
Mitochondrial Proteins
Amino Acid Sequence
Mitochondria, Muscle
DNA, Mitochondrial
Electron Transport Complex I
2,4-Dinitrophenol
Cells, Cultured
Mutation
Threonine
Mitochondrial ADP, ATP Translocases
Mitochondrial Proton-Translocating ATPases
Antimycin A
Protein-Tyrosine Kinases
Electron Transport Complex IV
Enzyme Inhibitors
Atractyloside
Succinates
Models, Biological
Protein Kinase C
Carbonyl Cyanide p-Trifluoromethoxyphenylhydrazone
Protein Binding
Blotting, Western
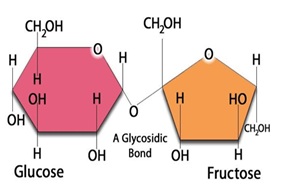
Glucose
Proton-Translocating ATPases
Cyclic AMP-Dependent Protein Kinases
Calcium
Proto-Oncogene Proteins c-akt
Phosphopeptides
Phosphotyrosine
Signal Transduction
Oxygen
NAD
Reactive Oxygen Species
Carbonyl Cyanide m-Chlorophenyl Hydrazone
Muscle, Skeletal
Adenine Nucleotide Translocator 1
Transfection
Citric Acid Cycle
Phosphocreatine
Cyanides
Polarography
Mitogen-Activated Protein Kinases
Binding Sites
Membrane Potential, Mitochondrial
Oxidation-Reduction
Phosphothreonine
Protein Processing, Post-Translational
Adenosine Triphosphatases
Electron Transport Chain Complex Proteins
Submitochondrial Particles
Malates
Carrier Proteins
ATP Synthetase Complexes
Peptide Mapping
NADH Dehydrogenase
Casein Kinase II
Immunoblotting
Proteins
Gene Expression Regulation
src-Family Kinases
HeLa Cells
Apoptosis
Cattle
Protein Structure, Tertiary
Membrane Proteins
Myocardium
Cell Nucleus
Phosphorus Radioisotopes
Transcription Factors
Phosphoprotein Phosphatases
Proto-Oncogene Proteins
Rats, Wistar
Electron Transport Complex III
Mitogen-Activated Protein Kinase 1
Intracellular Signaling Peptides and Proteins
Electrophoresis, Polyacrylamide Gel
DNA-Binding Proteins
Mutagenesis, Site-Directed
Tetramethylphenylenediamine
Protons
Calcium-Calmodulin-Dependent Protein Kinases
Phosphatidylinositol 3-Kinases
Succinic Acid
Glycogen Synthase Kinase 3
Mitogen-Activated Protein Kinase 3
Cell Membrane
Mitochondrial Swelling
Rats, Sprague-Dawley
Adenine Nucleotides
Fibroblasts
Hydrogen-Ion Concentration
Pyruvic Acid
Lactic Acid
Extracellular Signal-Regulated MAP Kinases
Precipitin Tests
Base Sequence
Hexokinase
Amobarbital
Insulin
MAP Kinase Signaling System
Immunoprecipitation
Substrate Specificity
Sodium Cyanide
Dose-Response Relationship, Drug
Sodium Azide
Protein Transport
Isoenzymes
Mitochondrial Membranes
Transcription, Genetic
Tumor Cells, Cultured
Cyclic AMP
Adaptor Proteins, Signal Transducing
Intracellular Membranes
COS Cells
Succinate Dehydrogenase
Liver
p38 Mitogen-Activated Protein Kinases
RNA, Small Interfering
Cell Cycle Proteins
Electron Transport Complex II
Electrophoresis, Gel, Two-Dimensional
Recombinant Fusion Proteins
AMP-Activated Protein Kinases
Myosin Light Chains
Nuclear Proteins
Tetradecanoylphorbol Acetate
Adenylate Kinase
Multienzyme Complexes
Okadaic Acid
Oxidative Stress
Pyruvates
Saccharomyces cerevisiae
RNA, Messenger
Membrane Potentials
Cell Survival
Antimetabolites
CDC2 Protein Kinase
Cytosol
Mice, Knockout
Trans-Activators
Rabbits
Biological Transport
Dicyclohexylcarbodiimide
Valinomycin
Amino Acid Substitution
Trityl Compounds
Mice, Inbred C57BL
Potassium Cyanide
Antibodies, Phospho-Specific
Protein Tyrosine Phosphatases
Focal Adhesion Protein-Tyrosine Kinases
Ribosomal Protein S6 Kinases, 90-kDa
3T3 Cells
14-3-3 Proteins
Down-Regulation
Mass Spectrometry
Protein Biosynthesis
Cell Cycle
Sequence Homology, Amino Acid
Proton Pumps
Paxillin
Vanadates
Protein Phosphatase 2
Cricetinae
Escherichia coli
Cyanide poisoning: pathophysiology and treatment recommendations. (1/2221)
This paper aims to assess and compare currently available antidotes for cyanide poisoning. Such evaluation, however, is difficult. Thus, extrapolation from the results of animal studies has potential pitfalls, as significant inter-species differences in response may exist, and these experiments often involve administration of toxin and antidote almost simultaneously, rather than incorporating a more realistic time delay before initiation of treatment. Direct inference from human case reports is also problematic; either because of uncertainties over the exposure levels involved (and hence the likely outcome without treatment), or because of difficulties in identifying the specific contribution of a particular antidote within the overall treatment regimen. Certainly an effort to compare the relative efficacy of cyanide antidotes produces equivocal findings, with no single regimen clearly standing out. Indeed, factors such as the risks of antidote toxicity to various individuals and other practical issues, may be more important considerations. There is therefore no single treatment regimen which is best for all situations. Besides individual risk factors for antidote toxicity, the nature of the exposure and hence its likely severity, the evolving clinical features and the number of persons involved and their proximity to hospital facilities, all need to be considered. Clinically mild poisoning may be treated by rest, oxygen and amyl nitrite. Intravenous antidotes are indicated for moderate poisoning. Where the diagnosis is uncertain, sodium thiosulphate may be the first choice. With severe poisoning, an additional agent is required. Given the various risks with methaemoglobin formers or with unselective use of kelocyanor, hydroxocobalamin may be preferred from a purely risk-benefit perspective. However the former alternatives will likely remain important. (+info)Nitric oxide inhibits cardiac energy production via inhibition of mitochondrial creatine kinase. (2/2221)
Nitric oxide biosynthesis in cardiac muscle leads to a decreased oxygen consumption and lower ATP synthesis. It is suggested that this effect of nitric oxide is mainly due to the inhibition of the mitochondrial respiratory chain enzyme, cytochrome c oxidase. However, this work demonstrates that nitric oxide is able to inhibit soluble mitochondrial creatine kinase (CK), mitochondrial CK bound in purified mitochondria, CK in situ in skinned fibres as well as the functional activity of mitochondrial CK in situ in skinned fibres. Since mitochondrial isoenzyme is functionally coupled to oxidative phosphorylation, its inhibition also leads to decreased sensitivity of mitochondrial respiration to ADP and thus decreases ATP synthesis and oxygen consumption under physiological ADP concentrations. (+info)Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. (3/2221)
Growth factor withdrawal is associated with a metabolic arrest that can result in apoptosis. Cell death is preceded by loss of outer mitochondrial membrane integrity and cytochrome c release. These mitochondrial events appear to follow a relative increase in mitochondrial membrane potential. This change in membrane potential results from the failure of the adenine nucleotide translocator (ANT)/voltage-dependent anion channel (VDAC) complex to maintain ATP/ADP exchange. Bcl-xL expression allows growth factor-deprived cells to maintain sufficient mitochondrial ATP/ADP exchange to sustain coupled respiration. These data demonstrate that mitochondrial adenylate transport is under active regulation. Efficient exchange of ADP for ATP is promoted by Bcl-xL expression permitting oxidative phosphorylation to be regulated by cellular ATP/ADP levels and allowing mitochondria to adapt to changes in metabolic demand. (+info)Nitric-oxide-induced apoptosis in human leukemic lines requires mitochondrial lipid degradation and cytochrome C release. (4/2221)
We have previously shown that nitric oxide (NO) stimulates apoptosis in different human neoplastic lymphoid cell lines through activation of caspases not only via CD95/CD95L interaction, but also independently of such death receptors. Here we investigated mitochondria-dependent mechanisms of NO-induced apoptosis in Jurkat leukemic cells. NO donor glycerol trinitrate (at the concentration, which induces apoptotic cell death) caused (1) a significant decrease in the concentration of cardiolipin, a major mitochondrial lipid; (2) a downregulation in respiratory chain complex activities; (3) a release of the mitochondrial protein cytochrome c into the cytosol; and (4) an activation of caspase-9 and caspase-3. These changes were accompanied by an increase in the number of cells with low mitochondrial transmembrane potential and with a high level of reactive oxygen species production. Higher resistance of the CD95-resistant Jurkat subclone (APO-R) cells to NO-mediated apoptosis correlated with the absence of cytochrome c release and with less alterations in other mitochondrial parameters. An inhibitor of lipid peroxidation, trolox, significantly suppressed NO-mediated apoptosis in APO-S Jurkat cells, whereas bongkrekic acid (BA), which blocks mitochondrial permeability transition, provided only a moderate antiapoptotic effect. Transfection of Jurkat cells with bcl-2 led to a complete block of apoptosis due to the prevention of changes in mitochondrial functions. We suggest that the mitochondrial damage (in particular, cardiolipin degradation and cytochrome c release) induced by NO in human leukemia cells plays a crucial role in the subsequent activation of caspase and apoptosis. (+info)Changes in mitochondrial phosphorylative activity and adenylate energy charge of regenerating rabbit liver. (5/2221)
The changes in the cellular concentrations of ATP, ADP, and AMP and in oxidative phosphorylation of mitochondria were investigated in the remaining liver of partially hepatectomized rabbits. The energy charge (defined as half of the average number of anhydride-bonded phosphate groups per adenosine moiety) of the liver remnant decreased from 0.866 to 0.767 (p less than 0.01) within 24 hr after hepatectomy, and then increased to a substantially higher level than normal within 7 days. On the other hand, the mitochondrial phosphyorylative activity increased rapidly to 170 per cent of the control within 12 hr and then retruned to normal within 7 days. The mitochondrial phosphorylative activity was inversely correlated with energy charge of the liver remnant (r = -0.75, p less less than 0.01). The maximal enhancement of mitochondrial phosphorylative activity was found in mitochondria obtained from the liver remnant with the lowest level of energy charge, suggesting a response of mitochondria in vivo involving enhanced biosynthetic ATP-utilizing reactions at an early stage of the regenerating process. The enhancement of phosphorylative activity was accompanied by a rise in the respiratory control ratio, P/O ratio and state 3 respiration. The adenylate kinase [EC 2.7.4.3] activity in the liver remnant increased to more than 160% of the control within 2 days after partial hepatectomy, while the pyruvate kinase [EC 2.7.1.40] activity decreased remarkably. However, the changes in the two enzyme activities did not correlate with those of mitochondrial phosphorylative activity or the energy charge of the liver remnant. (+info)Efficiency of oxidative phosphorylation and energy dissipation by H+ ion recycling in rat-liver mitochondrial metabolizing pyruvate. (6/2221)
A method was developed for the calculation of metabolic fluxes through individual enzymatic reactions of pyruvate metabolism including the citric acid cycle in rat liver mitochondrial incubated at metabolic states between state 4 and state 3. This method is based on the measurement of the specific radioactivities of the products formed from [2-14C]pyruvate. With this procedure the energy balance of mitochondria incubated in the presence of [2-14C]pyruvate, ATP, bicarbonate and phosphate at different ATP/ADP ratios in the medium was calculated. The ATP/ADP ratios were maintained at a steady state with creatine kinase plus creatine as a phosphoryl acceptor. The calculations revealed that by adding increasing concentrations of creatine up to 20 mM the energy dissipated by the mitochondria decreased but showed a local maximum at 13mM creatine. Omission of bicarbonate from the medium led to a shift of this maximum. When energy dissipation was minimal the overall P/O ratio was maximal. The amount of energy dissipated was paralleled by the magnitude of the pH gradient across the inner membrane. From these results it was concluded that the recycling of H+ ions which consists of a passive leakage of H+ ions into the matrix and an active extrusion of these ions out of this compartment, is an important energy dissipating process. The H+ ion recycling is thus one of the processes which give rise to the state 4 respiration in mitochondria. (+info)Influence of bioenergetic stress on heat shock protein gene expression in nucleated red blood cells of fish. (7/2221)
The physiological and biochemical signals that induce stress protein (HSP) synthesis remain conjectural. In this study, we used the nucleated red blood cells from rainbow trout, Oncorhynchus mykiss, to address the interaction between energy status and HSP gene expression. Heat shock (25 degrees C) did not significantly affect ATP levels but resulted in an increase in HSP70 mRNA. Hypoxia alone did not induce HSP transcription in these cells despite a significant depression in ATP. Inhibition of oxidative phosphorylation with azide, in the absence of thermal stress, decreased ATP by 56% and increased lactate production by 62% but did not induce HSP gene transcription. Inhibition of oxidative phosphorylation and glycolysis with azide and iodoacetic acid respectively, decreased ATP by 79% and prevented lactate production, but did not induce either HSP70 or HSP30 gene transcription in these cells. This study demonstrates that a reduction in the cellular energy status will not induce stress protein gene transcription in rainbow trout red blood cells and may, in fact, limit induction during extreme metabolic inhibition. (+info)Uncouplers of oxidative phosphorylation can enhance a Fas death signal. (8/2221)
Recent work suggests a participation of mitochondria in apoptotic cell death. This role includes the release of apoptogenic molecules into the cytosol preceding or after a loss of mitochondrial membrane potential DeltaPsim. The two uncouplers of oxidative phosphorylation carbonyl cyanide m-chlorophenylhydrazone (CCCP) and 2, 4-dinitrophenol (DNP) reduce DeltaPsim by direct attack of the proton gradient across the inner mitochondrial membrane. Here we show that both compounds enhance the apoptosis-inducing capacity of Fas/APO-1/CD95 signaling in Jurkat and CEM cells without causing apoptotic changes on their own account. This amplification occurred upstream or at the level of caspases and was not inhibited by Bcl-2. The effect could be blocked by the cowpox protein CrmA and is thus likely to require caspase 8 activity. Apoptosis induction by staurosporine in Jurkat cells as well as by Fas in SKW6 cells was unaffected by CCCP and DNP. The role of cytochrome c during Fas-DNP signaling was investigated. No early cytochrome c release from mitochondria was detected by Western blotting. Functional assays with cytoplasmic preparations from Fas-DNP-treated cells also indicated that there was no major contribution by cytochrome c or caspase 9 to the activation of effector caspases. Furthermore, an increase of rhodamine-123 uptake into intact cells, which has been explained by mitochondrial swelling, occurred considerably later than the caspase activation and was blocked by Z-VAD-fmk. These data show that uncouplers of oxidative phosphorylation can presensitize some but not all cells for a Fas death signal and provide information about the existence of separate pathways in the induction of apoptosis. (+info)Oxidative phosphorylation is the metabolic process by which cells use enzymes to generate energy in the form of adenosine triphosphate (ATP) from the oxidation of nutrients, such as glucose or fatty acids. This process occurs in the inner mitochondrial membrane of eukaryotic cells and is facilitated by the electron transport chain, which consists of a series of protein complexes that transfer electrons from donor molecules to acceptor molecules. As the electrons are passed along the chain, they release energy that is used to pump protons across the membrane, creating a gradient. The ATP synthase enzyme then uses the flow of protons back across the membrane to generate ATP, which serves as the main energy currency for cellular processes.
Mitochondria are specialized structures located inside cells that convert the energy from food into ATP (adenosine triphosphate), which is the primary form of energy used by cells. They are often referred to as the "powerhouses" of the cell because they generate most of the cell's supply of chemical energy. Mitochondria are also involved in various other cellular processes, such as signaling, differentiation, and apoptosis (programmed cell death).
Mitochondria have their own DNA, known as mitochondrial DNA (mtDNA), which is inherited maternally. This means that mtDNA is passed down from the mother to her offspring through the egg cells. Mitochondrial dysfunction has been linked to a variety of diseases and conditions, including neurodegenerative disorders, diabetes, and aging.
Uncoupling agents are chemicals that interfere with the normal process of oxidative phosphorylation in cells. In this process, the energy from food is converted into ATP (adenosine triphosphate), which is the main source of energy for cellular functions. Uncouplers disrupt this process by preventing the transfer of high-energy electrons to oxygen, which normally drives the production of ATP.
Instead, the energy from these electrons is released as heat, leading to an increase in body temperature. This effect is similar to what happens during shivering or exercise, when the body generates heat to maintain its core temperature. Uncoupling agents are therefore also known as "mitochondrial protonophores" because they allow protons to leak across the inner mitochondrial membrane, bypassing the ATP synthase enzyme that would normally use the energy from this proton gradient to produce ATP.
Uncoupling agents have been studied for their potential therapeutic uses, such as in weight loss and the treatment of metabolic disorders. However, they can also be toxic at high doses, and their long-term effects on health are not well understood.
Adenosine Triphosphate (ATP) is a high-energy molecule that stores and transports energy within cells. It is the main source of energy for most cellular processes, including muscle contraction, nerve impulse transmission, and protein synthesis. ATP is composed of a base (adenine), a sugar (ribose), and three phosphate groups. The bonds between these phosphate groups contain a significant amount of energy, which can be released when the bond between the second and third phosphate group is broken, resulting in the formation of adenosine diphosphate (ADP) and inorganic phosphate. This process is known as hydrolysis and can be catalyzed by various enzymes to drive a wide range of cellular functions. ATP can also be regenerated from ADP through various metabolic pathways, such as oxidative phosphorylation or substrate-level phosphorylation, allowing for the continuous supply of energy to cells.
Oxygen consumption, also known as oxygen uptake, is the amount of oxygen that is consumed or utilized by the body during a specific period of time, usually measured in liters per minute (L/min). It is a common measurement used in exercise physiology and critical care medicine to assess an individual's aerobic metabolism and overall health status.
In clinical settings, oxygen consumption is often measured during cardiopulmonary exercise testing (CPET) to evaluate cardiovascular function, pulmonary function, and exercise capacity in patients with various medical conditions such as heart failure, chronic obstructive pulmonary disease (COPD), and other respiratory or cardiac disorders.
During exercise, oxygen is consumed by the muscles to generate energy through a process called oxidative phosphorylation. The amount of oxygen consumed during exercise can provide important information about an individual's fitness level, exercise capacity, and overall health status. Additionally, measuring oxygen consumption can help healthcare providers assess the effectiveness of treatments and rehabilitation programs in patients with various medical conditions.
Phosphorylation is the process of adding a phosphate group (a molecule consisting of one phosphorus atom and four oxygen atoms) to a protein or other organic molecule, which is usually done by enzymes called kinases. This post-translational modification can change the function, localization, or activity of the target molecule, playing a crucial role in various cellular processes such as signal transduction, metabolism, and regulation of gene expression. Phosphorylation is reversible, and the removal of the phosphate group is facilitated by enzymes called phosphatases.
Serine is an amino acid, which is a building block of proteins. More specifically, it is a non-essential amino acid, meaning that the body can produce it from other compounds, and it does not need to be obtained through diet. Serine plays important roles in the body, such as contributing to the formation of the protective covering of nerve fibers (myelin sheath), helping to synthesize another amino acid called tryptophan, and taking part in the metabolism of fatty acids. It is also involved in the production of muscle tissues, the immune system, and the forming of cell structures. Serine can be found in various foods such as soy, eggs, cheese, meat, peanuts, lentils, and many others.
Glycolysis is a fundamental metabolic pathway that occurs in the cytoplasm of cells, consisting of a series of biochemical reactions. It's the process by which a six-carbon glucose molecule is broken down into two three-carbon pyruvate molecules. This process generates a net gain of two ATP molecules (the main energy currency in cells), two NADH molecules, and two water molecules.
Glycolysis can be divided into two stages: the preparatory phase (or 'energy investment' phase) and the payoff phase (or 'energy generation' phase). During the preparatory phase, glucose is phosphorylated twice to form glucose-6-phosphate and then converted to fructose-1,6-bisphosphate. These reactions consume two ATP molecules but set up the subsequent breakdown of fructose-1,6-bisphosphate into triose phosphates in the payoff phase. In this second stage, each triose phosphate is further oxidized and degraded to produce one pyruvate molecule, one NADH molecule, and one ATP molecule through substrate-level phosphorylation.
Glycolysis does not require oxygen to proceed; thus, it can occur under both aerobic (with oxygen) and anaerobic (without oxygen) conditions. In the absence of oxygen, the pyruvate produced during glycolysis is further metabolized through fermentation pathways such as lactic acid fermentation or alcohol fermentation to regenerate NAD+, which is necessary for glycolysis to continue.
In summary, glycolysis is a crucial process in cellular energy metabolism, allowing cells to convert glucose into ATP and other essential molecules while also serving as a starting point for various other biochemical pathways.
Oligomycins are a group of antibiotics produced by various species of Streptomyces bacteria. They are characterized by their ability to inhibit the function of ATP synthase, an enzyme that plays a crucial role in energy production within cells. By binding to the F1 component of ATP synthase, oligomycins prevent the synthesis of ATP, which is a key source of energy for cellular processes.
These antibiotics have been used in research to study the mechanisms of ATP synthase and mitochondrial function. However, their therapeutic use as antibiotics is limited due to their toxicity to mammalian cells. Oligomycin A is one of the most well-known and studied members of this group of antibiotics.
I'd be happy to help! However, I believe there might be a slight misunderstanding in your question. Mitochondria are organelles found in the cells of all complex living organisms, including humans, while the liver is a large, solid organ located in the upper right portion of the abdomen. They don't have a medical definition together. I can certainly provide you with separate definitions for each:
1. Mitochondria: These are double-membrane-bound cellular organelles that generate most of the chemical energy needed to power the cell's biochemical reactions. Commonly known as the "powerhouse of the cell," mitochondria convert organic substrates, such as glucose, fatty acids, and amino acids, into adenosine triphosphate (ATP) through a process called oxidative phosphorylation. Mitochondria are dynamic structures that can change their shape, size, and number through fission (division) and fusion (merging) processes. They play essential roles in various cellular functions, including calcium signaling, apoptosis (programmed cell death), and the regulation of cellular metabolism.
2. Liver: The liver is a large, lobulated organ that lies mainly in the upper right portion of the abdominal cavity, just below the diaphragm. It plays a crucial role in various physiological functions, such as detoxification, protein synthesis, metabolism, and nutrient storage. The liver is responsible for removing toxins from the bloodstream, producing bile to aid in digestion, regulating glucose levels, synthesizing plasma proteins, and storing glycogen, vitamins, and minerals. It also contributes to the metabolism of carbohydrates, lipids, and amino acids, helping maintain energy homeostasis in the body.
I hope this clarifies any confusion! If you have any further questions or need more information, please don't hesitate to ask.
Tyrosine is an non-essential amino acid, which means that it can be synthesized by the human body from another amino acid called phenylalanine. Its name is derived from the Greek word "tyros," which means cheese, as it was first isolated from casein, a protein found in cheese.
Tyrosine plays a crucial role in the production of several important substances in the body, including neurotransmitters such as dopamine, norepinephrine, and epinephrine, which are involved in various physiological processes, including mood regulation, stress response, and cognitive functions. It also serves as a precursor to melanin, the pigment responsible for skin, hair, and eye color.
In addition, tyrosine is involved in the structure of proteins and is essential for normal growth and development. Some individuals may require tyrosine supplementation if they have a genetic disorder that affects tyrosine metabolism or if they are phenylketonurics (PKU), who cannot metabolize phenylalanine, which can lead to elevated tyrosine levels in the blood. However, it is important to consult with a healthcare professional before starting any supplementation regimen.
Cell respiration is the process by which cells convert biochemical energy from nutrients into adenosine triphosphate (ATP), and then release waste products. The three main stages of cell respiration are glycolysis, the citric acid cycle (also known as the Krebs cycle), and the electron transport chain.
During glycolysis, which takes place in the cytoplasm, glucose is broken down into two molecules of pyruvate, producing a small amount of ATP and reducing power in the form of NADH.
The citric acid cycle occurs in the mitochondria and involves the breakdown of acetyl-CoA (formed from pyruvate) to produce more ATP, NADH, and FADH2.
Finally, the electron transport chain, also located in the mitochondria, uses the energy from NADH and FADH2 to pump protons across the inner mitochondrial membrane, creating a proton gradient. The flow of protons back across the membrane drives the synthesis of ATP, which is used as a source of energy by the cell.
Cell respiration is a crucial process that allows cells to generate the energy they need to perform various functions and maintain homeostasis.
Oxidative phosphorylation coupling factors are a group of proteins that play a crucial role in the process of oxidative phosphorylation, which is a metabolic pathway that generates energy in the form of ATP (adenosine triphosphate) through the transfer of electrons from NADH or FADH2 to oxygen, resulting in water.
The coupling factors are involved in the regulation and coordination of two key processes: electron transport and phosphorylation of ADP to ATP. These factors include:
1. Complex I (NADH-Q reductase): This complex transfers electrons from NADH to coenzyme Q10, and also contributes to the generation of a proton gradient across the inner mitochondrial membrane.
2. Complex II (Succinate-Q reductase): This complex transfers electrons from FADH2 to coenzyme Q10 and also participates in the citric acid cycle.
3. Complex III (Q-cytochrome c reductase): This complex transfers electrons from coenzyme Q10 to cytochrome c, and also contributes to the generation of a proton gradient across the inner mitochondrial membrane.
4. Complex IV (Cytochrome c oxidase): This complex transfers electrons from cytochrome c to oxygen, generating water and contributing to the generation of a proton gradient across the inner mitochondrial membrane.
5. ATP synthase: Also known as Complex V, this enzyme uses the energy generated by the proton gradient to synthesize ATP from ADP and inorganic phosphate.
Together, these coupling factors work to efficiently convert the energy stored in nutrients into a form that can be used by cells for various functions, such as muscle contraction, nerve impulse transmission, and biosynthesis.
Protein-Serine-Threonine Kinases (PSTKs) are a type of protein kinase that catalyzes the transfer of a phosphate group from ATP to the hydroxyl side chains of serine or threonine residues on target proteins. This phosphorylation process plays a crucial role in various cellular signaling pathways, including regulation of metabolism, gene expression, cell cycle progression, and apoptosis. PSTKs are involved in many physiological and pathological processes, and their dysregulation has been implicated in several diseases, such as cancer, diabetes, and neurodegenerative disorders.
Mitochondrial diseases are a group of disorders caused by dysfunctions in the mitochondria, which are the energy-producing structures in cells. These diseases can affect people of any age and can manifest in various ways, depending on which organs or systems are affected. Common symptoms include muscle weakness, neurological problems, cardiac disease, diabetes, and vision/hearing loss. Mitochondrial diseases can be inherited from either the mother's or father's side, or they can occur spontaneously due to genetic mutations. They can range from mild to severe and can even be life-threatening in some cases.
Adenosine diphosphate (ADP) is a chemical compound that plays a crucial role in energy transfer within cells. It is a nucleotide, which consists of a adenosine molecule (a sugar molecule called ribose attached to a nitrogenous base called adenine) and two phosphate groups.
In the cell, ADP functions as an intermediate in the conversion of energy from one form to another. When a high-energy phosphate bond in ADP is broken, energy is released and ADP is converted to adenosine triphosphate (ATP), which serves as the main energy currency of the cell. Conversely, when ATP donates a phosphate group to another molecule, it is converted back to ADP, releasing energy for the cell to use.
ADP also plays a role in blood clotting and other physiological processes. In the coagulation cascade, ADP released from damaged red blood cells can help activate platelets and initiate the formation of a blood clot.
Enzyme activation refers to the process by which an enzyme becomes biologically active and capable of carrying out its specific chemical or biological reaction. This is often achieved through various post-translational modifications, such as proteolytic cleavage, phosphorylation, or addition of cofactors or prosthetic groups to the enzyme molecule. These modifications can change the conformation or structure of the enzyme, exposing or creating a binding site for the substrate and allowing the enzymatic reaction to occur.
For example, in the case of proteolytic cleavage, an inactive precursor enzyme, known as a zymogen, is cleaved into its active form by a specific protease. This is seen in enzymes such as trypsin and chymotrypsin, which are initially produced in the pancreas as inactive precursors called trypsinogen and chymotrypsinogen, respectively. Once they reach the small intestine, they are activated by enteropeptidase, a protease that cleaves a specific peptide bond, releasing the active enzyme.
Phosphorylation is another common mechanism of enzyme activation, where a phosphate group is added to a specific serine, threonine, or tyrosine residue on the enzyme by a protein kinase. This modification can alter the conformation of the enzyme and create a binding site for the substrate, allowing the enzymatic reaction to occur.
Enzyme activation is a crucial process in many biological pathways, as it allows for precise control over when and where specific reactions take place. It also provides a mechanism for regulating enzyme activity in response to various signals and stimuli, such as hormones, neurotransmitters, or changes in the intracellular environment.
A cell line is a culture of cells that are grown in a laboratory for use in research. These cells are usually taken from a single cell or group of cells, and they are able to divide and grow continuously in the lab. Cell lines can come from many different sources, including animals, plants, and humans. They are often used in scientific research to study cellular processes, disease mechanisms, and to test new drugs or treatments. Some common types of human cell lines include HeLa cells (which come from a cancer patient named Henrietta Lacks), HEK293 cells (which come from embryonic kidney cells), and HUVEC cells (which come from umbilical vein endothelial cells). It is important to note that cell lines are not the same as primary cells, which are cells that are taken directly from a living organism and have not been grown in the lab.
The Electron Transport Chain (ETC) is a series of complexes in the inner mitochondrial membrane that are involved in the process of cellular respiration. It is the final pathway for electrons derived from the oxidation of nutrients such as glucose, fatty acids, and amino acids to be transferred to molecular oxygen. This transfer of electrons drives the generation of a proton gradient across the inner mitochondrial membrane, which is then used by ATP synthase to produce ATP, the main energy currency of the cell.
The electron transport chain consists of four complexes (I-IV) and two mobile electron carriers (ubiquinone and cytochrome c). Electrons from NADH and FADH2 are transferred to Complex I and Complex II respectively, which then pass them along to ubiquinone. Ubiquinone then transfers the electrons to Complex III, which passes them on to cytochrome c. Finally, cytochrome c transfers the electrons to Complex IV, where they combine with oxygen and protons to form water.
The transfer of electrons through the ETC is accompanied by the pumping of protons from the mitochondrial matrix to the intermembrane space, creating a proton gradient. The flow of protons back across the inner membrane through ATP synthase drives the synthesis of ATP from ADP and inorganic phosphate.
Overall, the electron transport chain is a crucial process for generating energy in the form of ATP in the cell, and it plays a key role in many metabolic pathways.
Dinitrophenols (DNP) are a class of chemical compounds that contain two nitro groups (-NO2) attached to a phenol group. Dinitrophenols have been used in the past as industrial dyes, wood preservatives, and pesticides. However, they have also been misused as weight loss supplements due to their ability to increase metabolic rate and cause weight loss.
The use of DNP for weight loss is dangerous and has been linked to several fatalities. DNP works by disrupting the normal functioning of the mitochondria in cells, which are responsible for producing energy. This disruption causes an increase in metabolic rate, leading to a rapid breakdown of fat and carbohydrates, and ultimately weight loss. However, this increased metabolism can also produce excessive heat, leading to hyperthermia, dehydration, and damage to organs such as the heart, liver, and kidneys.
Due to their potential for serious harm, DNP-containing products are banned in many countries, including the United States. Medical professionals should be aware of the dangers associated with DNP use and advise patients accordingly.
Energy metabolism is the process by which living organisms produce and consume energy to maintain life. It involves a series of chemical reactions that convert nutrients from food, such as carbohydrates, fats, and proteins, into energy in the form of adenosine triphosphate (ATP).
The process of energy metabolism can be divided into two main categories: catabolism and anabolism. Catabolism is the breakdown of nutrients to release energy, while anabolism is the synthesis of complex molecules from simpler ones using energy.
There are three main stages of energy metabolism: glycolysis, the citric acid cycle (also known as the Krebs cycle), and oxidative phosphorylation. Glycolysis occurs in the cytoplasm of the cell and involves the breakdown of glucose into pyruvate, producing a small amount of ATP and nicotinamide adenine dinucleotide (NADH). The citric acid cycle takes place in the mitochondria and involves the further breakdown of pyruvate to produce more ATP, NADH, and carbon dioxide. Oxidative phosphorylation is the final stage of energy metabolism and occurs in the inner mitochondrial membrane. It involves the transfer of electrons from NADH and other electron carriers to oxygen, which generates a proton gradient across the membrane. This gradient drives the synthesis of ATP, producing the majority of the cell's energy.
Overall, energy metabolism is a complex and essential process that allows organisms to grow, reproduce, and maintain their bodily functions. Disruptions in energy metabolism can lead to various diseases, including diabetes, obesity, and neurodegenerative disorders.
In the context of medicine and pharmacology, "kinetics" refers to the study of how a drug moves throughout the body, including its absorption, distribution, metabolism, and excretion (often abbreviated as ADME). This field is called "pharmacokinetics."
1. Absorption: This is the process of a drug moving from its site of administration into the bloodstream. Factors such as the route of administration (e.g., oral, intravenous, etc.), formulation, and individual physiological differences can affect absorption.
2. Distribution: Once a drug is in the bloodstream, it gets distributed throughout the body to various tissues and organs. This process is influenced by factors like blood flow, protein binding, and lipid solubility of the drug.
3. Metabolism: Drugs are often chemically modified in the body, typically in the liver, through processes known as metabolism. These changes can lead to the formation of active or inactive metabolites, which may then be further distributed, excreted, or undergo additional metabolic transformations.
4. Excretion: This is the process by which drugs and their metabolites are eliminated from the body, primarily through the kidneys (urine) and the liver (bile).
Understanding the kinetics of a drug is crucial for determining its optimal dosing regimen, potential interactions with other medications or foods, and any necessary adjustments for special populations like pediatric or geriatric patients, or those with impaired renal or hepatic function.
Phosphoproteins are proteins that have been post-translationally modified by the addition of a phosphate group (-PO3H2) onto specific amino acid residues, most commonly serine, threonine, or tyrosine. This process is known as phosphorylation and is mediated by enzymes called kinases. Phosphoproteins play crucial roles in various cellular processes such as signal transduction, cell cycle regulation, metabolism, and gene expression. The addition or removal of a phosphate group can activate or inhibit the function of a protein, thereby serving as a switch to control its activity. Phosphoproteins can be detected and quantified using techniques such as Western blotting, mass spectrometry, and immunofluorescence.
Protein kinases are a group of enzymes that play a crucial role in many cellular processes by adding phosphate groups to other proteins, a process known as phosphorylation. This modification can activate or deactivate the target protein's function, thereby regulating various signaling pathways within the cell. Protein kinases are essential for numerous biological functions, including metabolism, signal transduction, cell cycle progression, and apoptosis (programmed cell death). Abnormal regulation of protein kinases has been implicated in several diseases, such as cancer, diabetes, and neurological disorders.
Molecular sequence data refers to the specific arrangement of molecules, most commonly nucleotides in DNA or RNA, or amino acids in proteins, that make up a biological macromolecule. This data is generated through laboratory techniques such as sequencing, and provides information about the exact order of the constituent molecules. This data is crucial in various fields of biology, including genetics, evolution, and molecular biology, allowing for comparisons between different organisms, identification of genetic variations, and studies of gene function and regulation.
Mitochondrial proteins are any proteins that are encoded by the nuclear genome or mitochondrial genome and are located within the mitochondria, an organelle found in eukaryotic cells. These proteins play crucial roles in various cellular processes including energy production, metabolism of lipids, amino acids, and steroids, regulation of calcium homeostasis, and programmed cell death or apoptosis.
Mitochondrial proteins can be classified into two main categories based on their origin:
1. Nuclear-encoded mitochondrial proteins (NEMPs): These are proteins that are encoded by genes located in the nucleus, synthesized in the cytoplasm, and then imported into the mitochondria through specific import pathways. NEMPs make up about 99% of all mitochondrial proteins and are involved in various functions such as oxidative phosphorylation, tricarboxylic acid (TCA) cycle, fatty acid oxidation, and mitochondrial dynamics.
2. Mitochondrial DNA-encoded proteins (MEPs): These are proteins that are encoded by the mitochondrial genome, synthesized within the mitochondria, and play essential roles in the electron transport chain (ETC), a key component of oxidative phosphorylation. The human mitochondrial genome encodes only 13 proteins, all of which are subunits of complexes I, III, IV, and V of the ETC.
Defects in mitochondrial proteins can lead to various mitochondrial disorders, which often manifest as neurological, muscular, or metabolic symptoms due to impaired energy production. These disorders are usually caused by mutations in either nuclear or mitochondrial genes that encode mitochondrial proteins.
An amino acid sequence is the specific order of amino acids in a protein or peptide molecule, formed by the linking of the amino group (-NH2) of one amino acid to the carboxyl group (-COOH) of another amino acid through a peptide bond. The sequence is determined by the genetic code and is unique to each type of protein or peptide. It plays a crucial role in determining the three-dimensional structure and function of proteins.
Mitochondria in muscle, also known as the "powerhouses" of the cell, are organelles that play a crucial role in generating energy for muscle cells through a process called cellular respiration. They convert the chemical energy found in glucose and oxygen into ATP (adenosine triphosphate), which is the main source of energy used by cells.
Muscle cells contain a high number of mitochondria due to their high energy demands for muscle contraction and relaxation. The number and size of mitochondria in muscle fibers can vary depending on the type of muscle fiber, with slow-twitch, aerobic fibers having more numerous and larger mitochondria than fast-twitch, anaerobic fibers.
Mitochondrial dysfunction has been linked to various muscle disorders, including mitochondrial myopathies, which are characterized by muscle weakness, exercise intolerance, and other symptoms related to impaired energy production in the muscle cells.
I'm happy to help! However, it seems like there are two terms combined into one request: "Mitochondria" and "Heart." I will provide separate definitions for each.
Mitochondrion (singular) or Mitochondria (plural): These are specialized structures found in the cytoplasm of most eukaryotic cells (cells with a true nucleus), including human cells. They are often referred to as the "powerhouse" of the cell because they generate energy in the form of ATP (adenosine triphosphate) through a process called oxidative phosphorylation. Mitochondria contain their own DNA, which is distinct from the nuclear DNA, and are believed to have originated from ancient bacteria that established a symbiotic relationship with primitive eukaryotic cells.
Heart: In human anatomy, the heart is a muscular organ responsible for pumping blood throughout the body. It is located in the thoracic cavity, slightly left of the center, and is enclosed by the pericardium, a double-walled sac that provides protection and lubrication for the heart's movement. The human heart is divided into four chambers: two atria on the top and two ventricles on the bottom. The right side of the heart receives deoxygenated blood from the body and pumps it to the lungs, while the left side receives oxygenated blood from the lungs and pumps it to the rest of the body. The heart's pumping action is regulated by electrical signals that originate in a group of specialized cardiac muscle cells called the sinoatrial node (SA node).
Mitochondrial DNA (mtDNA) is the genetic material present in the mitochondria, which are specialized structures within cells that generate energy. Unlike nuclear DNA, which is present in the cell nucleus and inherited from both parents, mtDNA is inherited solely from the mother.
MtDNA is a circular molecule that contains 37 genes, including 13 genes that encode for proteins involved in oxidative phosphorylation, a process that generates energy in the form of ATP. The remaining genes encode for rRNAs and tRNAs, which are necessary for protein synthesis within the mitochondria.
Mutations in mtDNA can lead to a variety of genetic disorders, including mitochondrial diseases, which can affect any organ system in the body. These mutations can also be used in forensic science to identify individuals and establish biological relationships.
Electron Transport Complex I, also known as NADH:ubiquinone oxidoreductase, is a large protein complex located in the inner mitochondrial membrane of eukaryotic cells and the cytoplasmic membrane of prokaryotic cells. It is the first complex in the electron transport chain, a series of protein complexes that transfer electrons from NADH to oxygen, driving the synthesis of ATP through chemiosmosis.
Complex I consists of multiple subunits, including a flavin mononucleotide (FMN) cofactor and several iron-sulfur clusters, which facilitate the oxidation of NADH and the reduction of ubiquinone (coenzyme Q). The energy released during this electron transfer process is used to pump protons across the membrane, creating a proton gradient that drives ATP synthesis.
Defects in Complex I can lead to various mitochondrial diseases, including neurological disorders and muscle weakness.
2,4-Dinitrophenol (DNP) is a chemical compound with the formula C6H4N2O5. It is an organic compound that contains two nitro groups (-NO2) attached to a phenol molecule. DNP is a yellow, crystalline solid that is slightly soluble in water and more soluble in organic solvents.
In the medical field, DNP has been used in the past as a weight loss agent due to its ability to disrupt mitochondrial function and increase metabolic rate. However, its use as a weight loss drug was banned in the United States in the 1930s due to serious side effects, including cataracts, skin lesions, and hyperthermia, which can lead to death.
Exposure to DNP can occur through ingestion, inhalation, or skin contact. Acute exposure to high levels of DNP can cause symptoms such as nausea, vomiting, sweating, dizziness, headache, and rapid heartbeat. Chronic exposure to lower levels of DNP can lead to cataracts, skin lesions, and damage to the nervous system, liver, and kidneys.
It is important to note that DNP is not approved for use as a weight loss agent or any other medical purpose in the United States. Its use as a dietary supplement or weight loss aid is illegal and can be dangerous.
Phosphoserine is not a medical term per se, but rather a biochemical term. It refers to a post-translationally modified amino acid called serine that has a phosphate group attached to its side chain. This modification plays a crucial role in various cellular processes, including signal transduction and regulation of protein function. In medical contexts, abnormalities in the regulation of phosphorylation (the addition of a phosphate group) and dephosphorylation (the removal of a phosphate group) have been implicated in several diseases, such as cancer and neurological disorders.
"Cells, cultured" is a medical term that refers to cells that have been removed from an organism and grown in controlled laboratory conditions outside of the body. This process is called cell culture and it allows scientists to study cells in a more controlled and accessible environment than they would have inside the body. Cultured cells can be derived from a variety of sources, including tissues, organs, or fluids from humans, animals, or cell lines that have been previously established in the laboratory.
Cell culture involves several steps, including isolation of the cells from the tissue, purification and characterization of the cells, and maintenance of the cells in appropriate growth conditions. The cells are typically grown in specialized media that contain nutrients, growth factors, and other components necessary for their survival and proliferation. Cultured cells can be used for a variety of purposes, including basic research, drug development and testing, and production of biological products such as vaccines and gene therapies.
It is important to note that cultured cells may behave differently than they do in the body, and results obtained from cell culture studies may not always translate directly to human physiology or disease. Therefore, it is essential to validate findings from cell culture experiments using additional models and ultimately in clinical trials involving human subjects.
A mutation is a permanent change in the DNA sequence of an organism's genome. Mutations can occur spontaneously or be caused by environmental factors such as exposure to radiation, chemicals, or viruses. They may have various effects on the organism, ranging from benign to harmful, depending on where they occur and whether they alter the function of essential proteins. In some cases, mutations can increase an individual's susceptibility to certain diseases or disorders, while in others, they may confer a survival advantage. Mutations are the driving force behind evolution, as they introduce new genetic variability into populations, which can then be acted upon by natural selection.
Threonine is an essential amino acid, meaning it cannot be synthesized by the human body and must be obtained through the diet. Its chemical formula is HO2CCH(NH2)CH(OH)CH3. Threonine plays a crucial role in various biological processes, including protein synthesis, immune function, and fat metabolism. It is particularly important for maintaining the structural integrity of proteins, as it is often found in their hydroxyl-containing regions. Foods rich in threonine include animal proteins such as meat, dairy products, and eggs, as well as plant-based sources like lentils and soybeans.
Mitochondrial ADP/ATP translocases, also known as adenine nucleotide translocators (ANT), are a group of proteins located in the inner mitochondrial membrane that play a crucial role in cellular energy production. These translocases facilitate the exchange of adenosine diphosphate (ADP) and adenosine triphosphate (ATP) across the mitochondrial membrane, which is essential for oxidative phosphorylation and thus, energy homeostasis in the cell.
In more detail, during oxidative phosphorylation, ATP is produced within the mitochondria as a result of the electron transport chain's activity. This ATP must be exported to the cytosol for use by the cell's various processes. Simultaneously, the mitochondria need a continuous supply of ADP to sustain the production of ATP. The mitochondrial ADP/ATP translocases facilitate this exchange, allowing for the import of ADP into the mitochondria and the export of ATP to the cytosol.
There are multiple isoforms of the ADP/ATP translocase in humans (ANT1, ANT2, ANT3, and ANT4), encoded by different genes, with varying tissue distributions and functions. Dysfunction of these translocases has been implicated in several pathological conditions, including neurodegenerative diseases, ischemia-reperfusion injury, and cancer.
Mitochondrial proton-translocating ATPases, also known as F1F0-ATP synthase or complex V, are enzyme complexes found in the inner mitochondrial membrane of eukaryotic cells. They play a crucial role in the process of oxidative phosphorylation, which generates ATP (adenosine triphosphate), the primary energy currency of the cell.
These enzyme complexes consist of two main parts: F1 and F0. The F1 portion is located on the matrix side of the inner mitochondrial membrane and contains the catalytic sites for ATP synthesis. It is composed of three α, three β, and one γ subunits, along with additional subunits that regulate its activity.
The F0 portion spans the inner mitochondrial membrane and functions as a proton channel. It is composed of multiple subunits, including a, b, and c subunits, which form a rotor-stator structure. As protons flow through this channel due to the electrochemical gradient established by the electron transport chain, the rotation of the F0 rotor drives the synthesis of ATP in the F1 portion.
Mitochondrial proton-translocating ATPases are highly conserved across different species and play a vital role in maintaining energy homeostasis within the cell. Dysfunction in these enzyme complexes can lead to various mitochondrial disorders and diseases, such as neurodegenerative disorders, muscle weakness, and metabolic abnormalities.
Antimycin A is an antibiotic substance produced by various species of Streptomyces bacteria. It is known to inhibit the electron transport chain in mitochondria, which can lead to cellular dysfunction and death. Antimycin A has been used in research to study the mechanisms of cellular respiration and oxidative phosphorylation.
In a medical context, antimycin A is not used as a therapeutic agent due to its toxicity to mammalian cells. However, it may be used in laboratory settings to investigate various biological processes or to develop new therapies for diseases related to mitochondrial dysfunction.
Protein-Tyrosine Kinases (PTKs) are a type of enzyme that plays a crucial role in various cellular functions, including signal transduction, cell growth, differentiation, and metabolism. They catalyze the transfer of a phosphate group from ATP to the tyrosine residues of proteins, thereby modifying their activity, localization, or interaction with other molecules.
PTKs can be divided into two main categories: receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinases (NRTKs). RTKs are transmembrane proteins that become activated upon binding to specific ligands, such as growth factors or hormones. NRTKs, on the other hand, are intracellular enzymes that can be activated by various signals, including receptor-mediated signaling and intracellular messengers.
Dysregulation of PTK activity has been implicated in several diseases, such as cancer, diabetes, and inflammatory disorders. Therefore, PTKs are important targets for drug development and therapy.
Electron Transport Complex IV is also known as Cytochrome c oxidase. It is the last complex in the electron transport chain, located in the inner mitochondrial membrane of eukaryotic cells and the plasma membrane of prokaryotic cells. This complex contains 13 subunits, two heme groups (a and a3), and three copper centers (A, B, and C).
In the electron transport chain, Complex IV receives electrons from cytochrome c and transfers them to molecular oxygen, reducing it to water. This process is accompanied by the pumping of protons across the membrane, contributing to the generation of a proton gradient that drives ATP synthesis via ATP synthase (Complex V). The overall reaction catalyzed by Complex IV can be summarized as follows:
4e- + 4H+ + O2 → 2H2O
Defects in Cytochrome c oxidase can lead to various diseases, including mitochondrial encephalomyopathies and neurodegenerative disorders.
Enzyme inhibitors are substances that bind to an enzyme and decrease its activity, preventing it from catalyzing a chemical reaction in the body. They can work by several mechanisms, including blocking the active site where the substrate binds, or binding to another site on the enzyme to change its shape and prevent substrate binding. Enzyme inhibitors are often used as drugs to treat various medical conditions, such as high blood pressure, abnormal heart rhythms, and bacterial infections. They can also be found naturally in some foods and plants, and can be used in research to understand enzyme function and regulation.
Atractyloside is a toxic diterpene compound that can be found in various plants, including Atractylis gummifera (commonly known as gum cistus or rabbit-ear cistus) and other members of the Asteraceae family. This toxin is known to inhibit the mitochondrial ADP/ATP translocase, which plays a crucial role in cellular energy production.
Inhibition of this translocase leads to a disruption in the balance of adenine nucleotides inside the mitochondria, resulting in a decrease in ATP synthesis and an increase in the formation of reactive oxygen species (ROS). This can ultimately cause cell damage and even cell death.
Atractyloside poisoning can lead to various symptoms, such as gastrointestinal distress, liver and kidney damage, neurological issues, and, in severe cases, multi-organ failure. It is essential to seek immediate medical attention if atractyloside poisoning is suspected.
Succinates, in a medical context, most commonly refer to the salts or esters of succinic acid. Succinic acid is a dicarboxylic acid that is involved in the Krebs cycle, which is a key metabolic pathway in cells that generates energy through the oxidation of acetyl-CoA derived from carbohydrates, fats, and proteins.
Succinates can also be used as a buffer in medical solutions and as a pharmaceutical intermediate in the synthesis of various drugs. In some cases, succinate may be used as a nutritional supplement or as a component of parenteral nutrition formulations to provide energy and help maintain acid-base balance in patients who are unable to eat normally.
It's worth noting that there is also a condition called "succinic semialdehyde dehydrogenase deficiency" which is a genetic disorder that affects the metabolism of the amino acid gamma-aminobutyric acid (GABA). This condition can lead to an accumulation of succinic semialdehyde and other metabolic byproducts, which can cause neurological symptoms such as developmental delay, hypotonia, and seizures.
Biological models, also known as physiological models or organismal models, are simplified representations of biological systems, processes, or mechanisms that are used to understand and explain the underlying principles and relationships. These models can be theoretical (conceptual or mathematical) or physical (such as anatomical models, cell cultures, or animal models). They are widely used in biomedical research to study various phenomena, including disease pathophysiology, drug action, and therapeutic interventions.
Examples of biological models include:
1. Mathematical models: These use mathematical equations and formulas to describe complex biological systems or processes, such as population dynamics, metabolic pathways, or gene regulation networks. They can help predict the behavior of these systems under different conditions and test hypotheses about their underlying mechanisms.
2. Cell cultures: These are collections of cells grown in a controlled environment, typically in a laboratory dish or flask. They can be used to study cellular processes, such as signal transduction, gene expression, or metabolism, and to test the effects of drugs or other treatments on these processes.
3. Animal models: These are living organisms, usually vertebrates like mice, rats, or non-human primates, that are used to study various aspects of human biology and disease. They can provide valuable insights into the pathophysiology of diseases, the mechanisms of drug action, and the safety and efficacy of new therapies.
4. Anatomical models: These are physical representations of biological structures or systems, such as plastic models of organs or tissues, that can be used for educational purposes or to plan surgical procedures. They can also serve as a basis for developing more sophisticated models, such as computer simulations or 3D-printed replicas.
Overall, biological models play a crucial role in advancing our understanding of biology and medicine, helping to identify new targets for therapeutic intervention, develop novel drugs and treatments, and improve human health.
Protein Kinase C (PKC) is a family of serine-threonine kinases that play crucial roles in various cellular signaling pathways. These enzymes are activated by second messengers such as diacylglycerol (DAG) and calcium ions (Ca2+), which result from the activation of cell surface receptors like G protein-coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs).
Once activated, PKC proteins phosphorylate downstream target proteins, thereby modulating their activities. This regulation is involved in numerous cellular processes, including cell growth, differentiation, apoptosis, and membrane trafficking. There are at least 10 isoforms of PKC, classified into three subfamilies based on their second messenger requirements and structural features: conventional (cPKC; α, βI, βII, and γ), novel (nPKC; δ, ε, η, and θ), and atypical (aPKC; ζ and ι/λ). Dysregulation of PKC signaling has been implicated in several diseases, such as cancer, diabetes, and neurological disorders.
Phosphates, in a medical context, refer to the salts or esters of phosphoric acid. Phosphates play crucial roles in various biological processes within the human body. They are essential components of bones and teeth, where they combine with calcium to form hydroxyapatite crystals. Phosphates also participate in energy transfer reactions as phosphate groups attached to adenosine diphosphate (ADP) and adenosine triphosphate (ATP). Additionally, they contribute to buffer systems that help maintain normal pH levels in the body.
Abnormal levels of phosphates in the blood can indicate certain medical conditions. High phosphate levels (hyperphosphatemia) may be associated with kidney dysfunction, hyperparathyroidism, or excessive intake of phosphate-containing products. Low phosphate levels (hypophosphatemia) might result from malnutrition, vitamin D deficiency, or certain diseases affecting the small intestine or kidneys. Both hypophosphatemia and hyperphosphatemia can have significant impacts on various organ systems and may require medical intervention.
Carbonyl cyanide p-trifluoromethoxyphenylhydrazone (CCP) is a chemical compound that functions as an ionophore, which is a type of molecule that can transport ions across biological membranes. CCP is specifically known to transport protons (H+) and has been used in research as a tool to study the role of proton transport in various cellular processes.
CCP is also a potent mitochondrial uncoupler, which means that it disrupts the normal functioning of the mitochondria, the energy-producing structures in cells. By doing so, CCP can cause a rapid and irreversible decline in ATP (adenosine triphosphate) production, leading to cell death.
Due to its potent toxicity, CCP is not used as a therapeutic agent but rather as a research tool to study mitochondrial function and cellular metabolism. It is important to handle this compound with care and follow appropriate safety protocols when working with it in the laboratory.
Protein binding, in the context of medical and biological sciences, refers to the interaction between a protein and another molecule (known as the ligand) that results in a stable complex. This process is often reversible and can be influenced by various factors such as pH, temperature, and concentration of the involved molecules.
In clinical chemistry, protein binding is particularly important when it comes to drugs, as many of them bind to proteins (especially albumin) in the bloodstream. The degree of protein binding can affect a drug's distribution, metabolism, and excretion, which in turn influence its therapeutic effectiveness and potential side effects.
Protein-bound drugs may be less available for interaction with their target tissues, as only the unbound or "free" fraction of the drug is active. Therefore, understanding protein binding can help optimize dosing regimens and minimize adverse reactions.
Western blotting is a laboratory technique used in molecular biology to detect and quantify specific proteins in a mixture of many different proteins. This technique is commonly used to confirm the expression of a protein of interest, determine its size, and investigate its post-translational modifications. The name "Western" blotting distinguishes this technique from Southern blotting (for DNA) and Northern blotting (for RNA).
The Western blotting procedure involves several steps:
1. Protein extraction: The sample containing the proteins of interest is first extracted, often by breaking open cells or tissues and using a buffer to extract the proteins.
2. Separation of proteins by electrophoresis: The extracted proteins are then separated based on their size by loading them onto a polyacrylamide gel and running an electric current through the gel (a process called sodium dodecyl sulfate-polyacrylamide gel electrophoresis or SDS-PAGE). This separates the proteins according to their molecular weight, with smaller proteins migrating faster than larger ones.
3. Transfer of proteins to a membrane: After separation, the proteins are transferred from the gel onto a nitrocellulose or polyvinylidene fluoride (PVDF) membrane using an electric current in a process called blotting. This creates a replica of the protein pattern on the gel but now immobilized on the membrane for further analysis.
4. Blocking: The membrane is then blocked with a blocking agent, such as non-fat dry milk or bovine serum albumin (BSA), to prevent non-specific binding of antibodies in subsequent steps.
5. Primary antibody incubation: A primary antibody that specifically recognizes the protein of interest is added and allowed to bind to its target protein on the membrane. This step may be performed at room temperature or 4°C overnight, depending on the antibody's properties.
6. Washing: The membrane is washed with a buffer to remove unbound primary antibodies.
7. Secondary antibody incubation: A secondary antibody that recognizes the primary antibody (often coupled to an enzyme or fluorophore) is added and allowed to bind to the primary antibody. This step may involve using a horseradish peroxidase (HRP)-conjugated or alkaline phosphatase (AP)-conjugated secondary antibody, depending on the detection method used later.
8. Washing: The membrane is washed again to remove unbound secondary antibodies.
9. Detection: A detection reagent is added to visualize the protein of interest by detecting the signal generated from the enzyme-conjugated or fluorophore-conjugated secondary antibody. This can be done using chemiluminescent, colorimetric, or fluorescent methods.
10. Analysis: The resulting image is analyzed to determine the presence and quantity of the protein of interest in the sample.
Western blotting is a powerful technique for identifying and quantifying specific proteins within complex mixtures. It can be used to study protein expression, post-translational modifications, protein-protein interactions, and more. However, it requires careful optimization and validation to ensure accurate and reproducible results.
Glucose is a simple monosaccharide (or single sugar) that serves as the primary source of energy for living organisms. It's a fundamental molecule in biology, often referred to as "dextrose" or "grape sugar." Glucose has the molecular formula C6H12O6 and is vital to the functioning of cells, especially those in the brain and nervous system.
In the body, glucose is derived from the digestion of carbohydrates in food, and it's transported around the body via the bloodstream to cells where it can be used for energy. Cells convert glucose into a usable form through a process called cellular respiration, which involves a series of metabolic reactions that generate adenosine triphosphate (ATP)—the main currency of energy in cells.
Glucose is also stored in the liver and muscles as glycogen, a polysaccharide (multiple sugar) that can be broken down back into glucose when needed for energy between meals or during physical activity. Maintaining appropriate blood glucose levels is crucial for overall health, and imbalances can lead to conditions such as diabetes mellitus.
Rotenone is not strictly a medical term, but it is a pesticide that is used in some medical situations. According to the National Pesticide Information Center, rotenone is a pesticide derived from the roots and stems of several plants, including Derris Eliptica, Lonchocarpus utilis, and Tephrosia vogelii. It is used as a pesticide to control insects, mites, and fish in both agricultural and residential settings.
In medical contexts, rotenone has been studied for its potential effects on human health, particularly in relation to Parkinson's disease. Some research suggests that exposure to rotenone may increase the risk of developing Parkinson's disease, although more studies are needed to confirm this link. Rotenone works by inhibiting the mitochondria in cells, which can lead to cell death and neurodegeneration.
It is important to note that rotenone is highly toxic and should be handled with care. It can cause skin and eye irritation, respiratory problems, and gastrointestinal symptoms if ingested or inhaled. Therefore, it is recommended to use personal protective equipment when handling rotenone and to follow all label instructions carefully.
Proton-translocating ATPases are complex, multi-subunit enzymes found in the membranes of many organisms, from bacteria to humans. They play a crucial role in energy transduction processes within cells.
In simpler terms, these enzymes help convert chemical energy into a form that can be used to perform mechanical work, such as moving molecules across membranes against their concentration gradients. This is achieved through a process called chemiosmosis, where the movement of ions (in this case, protons or hydrogen ions) down their electrochemical gradient drives the synthesis of ATP, an essential energy currency for cellular functions.
Proton-translocating ATPases consist of two main domains: a catalytic domain responsible for ATP binding and hydrolysis, and a membrane domain that contains the ion transport channel. The enzyme operates in either direction depending on the energy status of the cell: it can use ATP to pump protons out of the cell when there's an excess of chemical energy or utilize the proton gradient to generate ATP during times of energy deficit.
These enzymes are essential for various biological processes, including nutrient uptake, pH regulation, and maintaining ion homeostasis across membranes. In humans, they are primarily located in the inner mitochondrial membrane (forming the F0F1-ATP synthase) and plasma membranes of certain cells (as V-type ATPases). Dysfunction of these enzymes has been linked to several diseases, including neurological disorders and cancer.
Cyclic AMP (cAMP)-dependent protein kinases, also known as protein kinase A (PKA), are a family of enzymes that play a crucial role in intracellular signaling pathways. These enzymes are responsible for the regulation of various cellular processes, including metabolism, gene expression, and cell growth and differentiation.
PKA is composed of two regulatory subunits and two catalytic subunits. When cAMP binds to the regulatory subunits, it causes a conformational change that leads to the dissociation of the catalytic subunits. The freed catalytic subunits then phosphorylate specific serine and threonine residues on target proteins, thereby modulating their activity.
The cAMP-dependent protein kinases are activated in response to a variety of extracellular signals, such as hormones and neurotransmitters, that bind to G protein-coupled receptors (GPCRs) or receptor tyrosine kinases (RTKs). These signals lead to the activation of adenylyl cyclase, which catalyzes the conversion of ATP to cAMP. The resulting increase in intracellular cAMP levels triggers the activation of PKA and the downstream phosphorylation of target proteins.
Overall, cAMP-dependent protein kinases are essential regulators of many fundamental cellular processes and play a critical role in maintaining normal physiology and homeostasis. Dysregulation of these enzymes has been implicated in various diseases, including cancer, diabetes, and neurological disorders.
Calcium is an essential mineral that is vital for various physiological processes in the human body. The medical definition of calcium is as follows:
Calcium (Ca2+) is a crucial cation and the most abundant mineral in the human body, with approximately 99% of it found in bones and teeth. It plays a vital role in maintaining structural integrity, nerve impulse transmission, muscle contraction, hormonal secretion, blood coagulation, and enzyme activation.
Calcium homeostasis is tightly regulated through the interplay of several hormones, including parathyroid hormone (PTH), calcitonin, and vitamin D. Dietary calcium intake, absorption, and excretion are also critical factors in maintaining optimal calcium levels in the body.
Hypocalcemia refers to low serum calcium levels, while hypercalcemia indicates high serum calcium levels. Both conditions can have detrimental effects on various organ systems and require medical intervention to correct.
Mitochondrial myopathies are a group of genetic disorders caused by mutations in the mitochondrial DNA or nuclear DNA that affect the function of the mitochondria, which are the energy-producing structures in cells. These mutations can result in impaired muscle function and other symptoms, depending on the specific type and severity of the disorder.
Mitochondrial myopathies can present at any age and can cause a range of symptoms, including muscle weakness, exercise intolerance, fatigue, muscle pain, and difficulty with coordination and balance. Some people with mitochondrial myopathies may also experience neurological symptoms such as seizures, developmental delays, and hearing or vision loss.
The diagnosis of mitochondrial myopathies typically involves a combination of clinical evaluation, muscle biopsy, genetic testing, and other diagnostic tests to assess mitochondrial function. Treatment is generally supportive and may include physical therapy, medications to manage symptoms, and nutritional support. In some cases, specific therapies such as vitamin or coenzyme Q10 supplementation may be recommended based on the underlying genetic defect.
Protein-kinase B, also known as AKT, is a group of intracellular proteins that play a crucial role in various cellular processes such as glucose metabolism, apoptosis, cell proliferation, transcription, and cell migration. The AKT family includes three isoforms: AKT1, AKT2, and AKT3, which are encoded by the genes PKBalpha, PKBbeta, and PKBgamma, respectively.
Proto-oncogene proteins c-AKT refer to the normal, non-mutated forms of these proteins that are involved in the regulation of cell growth and survival under physiological conditions. However, when these genes are mutated or overexpressed, they can become oncogenes, leading to uncontrolled cell growth and cancer development.
Activation of c-AKT occurs through a signaling cascade that begins with the binding of extracellular ligands such as insulin-like growth factor 1 (IGF-1) or epidermal growth factor (EGF) to their respective receptors on the cell surface. This triggers a series of phosphorylation events that ultimately lead to the activation of c-AKT, which then phosphorylates downstream targets involved in various cellular processes.
In summary, proto-oncogene proteins c-AKT are normal intracellular proteins that play essential roles in regulating cell growth and survival under physiological conditions. However, their dysregulation can contribute to cancer development and progression.
Phosphopeptides are short peptide sequences that contain one or more phosphorylated amino acid residues, most commonly serine, threonine, or tyrosine. Phosphorylation is a post-translational modification that plays a crucial role in regulating various cellular processes such as signal transduction, protein-protein interactions, enzyme activity, and protein degradation. The addition of a phosphate group to a peptide can alter its charge, conformation, stability, and interaction with other molecules, thereby modulating its function in the cell. Phosphopeptides are often generated by proteolytic digestion of phosphorylated proteins and are used as biomarkers or probes to study protein phosphorylation and signaling pathways in various biological systems.
Phosphotyrosine is not a medical term per se, but rather a biochemical term used in the field of medicine and life sciences.
Phosphotyrosine is a post-translational modification of tyrosine residues in proteins, where a phosphate group is added to the hydroxyl side chain of tyrosine by protein kinases. This modification plays a crucial role in intracellular signaling pathways and regulates various cellular processes such as cell growth, differentiation, and apoptosis. Abnormalities in phosphotyrosine-mediated signaling have been implicated in several diseases, including cancer and diabetes.
Signal transduction is the process by which a cell converts an extracellular signal, such as a hormone or neurotransmitter, into an intracellular response. This involves a series of molecular events that transmit the signal from the cell surface to the interior of the cell, ultimately resulting in changes in gene expression, protein activity, or metabolism.
The process typically begins with the binding of the extracellular signal to a receptor located on the cell membrane. This binding event activates the receptor, which then triggers a cascade of intracellular signaling molecules, such as second messengers, protein kinases, and ion channels. These molecules amplify and propagate the signal, ultimately leading to the activation or inhibition of specific cellular responses.
Signal transduction pathways are highly regulated and can be modulated by various factors, including other signaling molecules, post-translational modifications, and feedback mechanisms. Dysregulation of these pathways has been implicated in a variety of diseases, including cancer, diabetes, and neurological disorders.
Oxygen is a colorless, odorless, tasteless gas that constitutes about 21% of the earth's atmosphere. It is a crucial element for human and most living organisms as it is vital for respiration. Inhaled oxygen enters the lungs and binds to hemoglobin in red blood cells, which carries it to tissues throughout the body where it is used to convert nutrients into energy and carbon dioxide, a waste product that is exhaled.
Medically, supplemental oxygen therapy may be provided to patients with conditions such as chronic obstructive pulmonary disease (COPD), pneumonia, heart failure, or other medical conditions that impair the body's ability to extract sufficient oxygen from the air. Oxygen can be administered through various devices, including nasal cannulas, face masks, and ventilators.
NAD (Nicotinamide Adenine Dinucleotide) is a coenzyme found in all living cells. It plays an essential role in cellular metabolism, particularly in redox reactions, where it acts as an electron carrier. NAD exists in two forms: NAD+, which accepts electrons and becomes reduced to NADH. This pairing of NAD+/NADH is involved in many fundamental biological processes such as generating energy in the form of ATP during cellular respiration, and serving as a critical cofactor for various enzymes that regulate cellular functions like DNA repair, gene expression, and cell death.
Maintaining optimal levels of NAD+/NADH is crucial for overall health and longevity, as it declines with age and in certain disease states. Therefore, strategies to boost NAD+ levels are being actively researched for their potential therapeutic benefits in various conditions such as aging, neurodegenerative disorders, and metabolic diseases.
Reactive Oxygen Species (ROS) are highly reactive molecules containing oxygen, including peroxides, superoxide, hydroxyl radical, and singlet oxygen. They are naturally produced as byproducts of normal cellular metabolism in the mitochondria, and can also be generated by external sources such as ionizing radiation, tobacco smoke, and air pollutants. At low or moderate concentrations, ROS play important roles in cell signaling and homeostasis, but at high concentrations, they can cause significant damage to cell structures, including lipids, proteins, and DNA, leading to oxidative stress and potential cell death.
Carbonyl cyanide m-chlorophenyl hydrazone (CCCP) is a chemical compound that is often used in research and scientific studies. It is an ionophore, which is a type of molecule that can transport ions across biological membranes. CCCP specifically transports protons (H+ ions) across membranes.
In biochemistry and cell biology, CCCP is commonly used as an uncoupler of oxidative phosphorylation. This is a process by which cells generate energy in the form of ATP (adenosine triphosphate) using the energy from the electron transport chain. By disrupting the proton gradient across the inner mitochondrial membrane, CCCP prevents the synthesis of ATP and causes a rapid depletion of cellular energy stores.
The medical relevance of CCCP is primarily limited to its use as a research tool in laboratory studies. It is not used as a therapeutic agent in clinical medicine.
Skeletal muscle, also known as striated or voluntary muscle, is a type of muscle that is attached to bones by tendons or aponeuroses and functions to produce movements and support the posture of the body. It is composed of long, multinucleated fibers that are arranged in parallel bundles and are characterized by alternating light and dark bands, giving them a striped appearance under a microscope. Skeletal muscle is under voluntary control, meaning that it is consciously activated through signals from the nervous system. It is responsible for activities such as walking, running, jumping, and lifting objects.
Adenine Nucleotide Translocator 1 (ANT1) is a protein found in the inner mitochondrial membrane of cells. It plays a crucial role in cellular energy metabolism by facilitating the exchange of adenosine diphosphate (ADP) and adenosine triphosphate (ATP) across the mitochondrial membrane.
In simpler terms, ANT1 helps to transport ATP, which is a major source of energy for cells, out of the mitochondria and exchange it for ADP, which can be converted back into ATP through cellular respiration. This process is essential for maintaining the energy balance within the cell and supporting various physiological functions.
Mutations in the gene that encodes ANT1 have been associated with certain mitochondrial disorders, such as autosomal recessive progressive external ophthalmoplegia (arPEO) and maternally inherited diabetes and deafness (MIDD). These genetic conditions can result in a range of symptoms, including muscle weakness, exercise intolerance, and neurological problems.
Transfection is a term used in molecular biology that refers to the process of deliberately introducing foreign genetic material (DNA, RNA or artificial gene constructs) into cells. This is typically done using chemical or physical methods, such as lipofection or electroporation. Transfection is widely used in research and medical settings for various purposes, including studying gene function, producing proteins, developing gene therapies, and creating genetically modified organisms. It's important to note that transfection is different from transduction, which is the process of introducing genetic material into cells using viruses as vectors.
The Citric Acid Cycle, also known as the Krebs cycle or tricarboxylic acid (TCA) cycle, is a crucial metabolic pathway in the cell's powerhouse, the mitochondria. It plays a central role in the oxidation of acetyl-CoA derived from carbohydrates, fats, and proteins, into carbon dioxide and high-energy electrons. This process generates energy in the form of ATP (adenosine triphosphate), reducing equivalents (NADH and FADH2), and water.
The cycle begins with the condensation of acetyl-CoA with oxaloacetate, forming citrate. Through a series of enzyme-catalyzed reactions, citrate is converted back to oxaloacetate, releasing two molecules of carbon dioxide, one GTP (guanosine triphosphate), three NADH, one FADH2, and regenerating oxaloacetate to continue the cycle. The reduced coenzymes (NADH and FADH2) then donate their electrons to the electron transport chain, driving ATP synthesis through chemiosmosis. Overall, the Citric Acid Cycle is a vital part of cellular respiration, connecting various catabolic pathways and generating energy for the cell's metabolic needs.
Phosphocreatine (PCr) is a high-energy phosphate compound found in the skeletal muscles, cardiac muscle, and brain. It plays a crucial role in energy metabolism and storage within cells. Phosphocreatine serves as an immediate energy reserve that helps regenerate ATP (adenosine triphosphate), the primary source of cellular energy, during short bursts of intense activity or stress. This process is facilitated by the enzyme creatine kinase, which catalyzes the transfer of a phosphate group from phosphocreatine to ADP (adenosine diphosphate) to form ATP.
In a medical context, phosphocreatine levels may be assessed in muscle biopsies or magnetic resonance spectroscopy (MRS) imaging to evaluate muscle energy metabolism and potential mitochondrial dysfunction in conditions such as muscular dystrophies, mitochondrial disorders, and neuromuscular diseases. Additionally, phosphocreatine depletion has been implicated in various pathological processes, including ischemia-reperfusion injury, neurodegenerative disorders, and heart failure.
Cyanides are a group of chemical compounds that contain the cyano group, -CN, which consists of a carbon atom triple-bonded to a nitrogen atom. They are highly toxic and can cause rapid death due to the inhibition of cellular respiration. Cyanide ions (CN-) bind to the ferric iron in cytochrome c oxidase, a crucial enzyme in the electron transport chain, preventing the flow of electrons and the production of ATP, leading to cellular asphyxiation.
Common sources of cyanides include industrial chemicals such as hydrogen cyanide (HCN) and potassium cyanide (KCN), as well as natural sources like certain fruits, nuts, and plants. Exposure to high levels of cyanides can occur through inhalation, ingestion, or skin absorption, leading to symptoms such as headache, dizziness, nausea, vomiting, rapid heartbeat, seizures, coma, and ultimately death. Treatment for cyanide poisoning typically involves the use of antidotes that bind to cyanide ions and convert them into less toxic forms, such as thiosulfate and rhodanese.
Polarography is a type of electrochemical analysis technique used to determine the concentration of an ion or electron-transferring species in a solution. It involves measuring the current that flows through an electrode as the voltage is varied, which can provide information about the redox potential and the number of electrons transferred during a reaction. The technique is particularly useful for analyzing complex mixtures and for detecting trace amounts of substances.
In polarography, a dropping mercury electrode (DME) is typically used as the working electrode. As the mercury droplets fall from the electrode, they create fresh surfaces for analysis, which helps to minimize interference from surface-adsorbed species. The DME is immersed in a solution containing the analyte along with a supporting electrolyte, and a potential is applied between the DME and a reference electrode.
As the potential is scanned, reduction or oxidation of the analyte occurs at the DME surface, leading to a current that can be measured. The resulting polarogram (a plot of current vs. voltage) shows peaks or waves corresponding to the redox potentials of the analyte, which can be used to identify and quantify the species present in the solution.
Polarography is a sensitive and selective technique that has been widely used in fields such as environmental analysis, pharmaceuticals, and biochemistry. However, it has largely been replaced by more modern electrochemical techniques, such as cyclic voltammetry and differential pulse voltammetry, which offer higher sensitivity and better resolution of complex mixtures.
Mitogen-Activated Protein Kinases (MAPKs) are a family of serine/threonine protein kinases that play crucial roles in various cellular processes, including proliferation, differentiation, transformation, and apoptosis, in response to diverse stimuli such as mitogens, growth factors, hormones, cytokines, and environmental stresses. They are highly conserved across eukaryotes and consist of a three-tiered kinase module composed of MAPK kinase kinases (MAP3Ks), MAPK kinases (MKKs or MAP2Ks), and MAPKs.
Activation of MAPKs occurs through a sequential phosphorylation and activation cascade, where MAP3Ks phosphorylate and activate MKKs, which in turn phosphorylate and activate MAPKs at specific residues (Thr-X-Tyr or Ser-Pro motifs). Once activated, MAPKs can further phosphorylate and regulate various downstream targets, including transcription factors and other protein kinases.
There are four major groups of MAPKs in mammals: extracellular signal-regulated kinases (ERK1/2), c-Jun N-terminal kinases (JNK1/2/3), p38 MAPKs (p38α/β/γ/δ), and ERK5/BMK1. Each group of MAPKs has distinct upstream activators, downstream targets, and cellular functions, allowing for a high degree of specificity in signal transduction and cellular responses. Dysregulation of MAPK signaling pathways has been implicated in various human diseases, including cancer, diabetes, neurodegenerative disorders, and inflammatory diseases.
A cell line that is derived from tumor cells and has been adapted to grow in culture. These cell lines are often used in research to study the characteristics of cancer cells, including their growth patterns, genetic changes, and responses to various treatments. They can be established from many different types of tumors, such as carcinomas, sarcomas, and leukemias. Once established, these cell lines can be grown and maintained indefinitely in the laboratory, allowing researchers to conduct experiments and studies that would not be feasible using primary tumor cells. It is important to note that tumor cell lines may not always accurately represent the behavior of the original tumor, as they can undergo genetic changes during their time in culture.
In the context of medical and biological sciences, a "binding site" refers to a specific location on a protein, molecule, or cell where another molecule can attach or bind. This binding interaction can lead to various functional changes in the original protein or molecule. The other molecule that binds to the binding site is often referred to as a ligand, which can be a small molecule, ion, or even another protein.
The binding between a ligand and its target binding site can be specific and selective, meaning that only certain ligands can bind to particular binding sites with high affinity. This specificity plays a crucial role in various biological processes, such as signal transduction, enzyme catalysis, or drug action.
In the case of drug development, understanding the location and properties of binding sites on target proteins is essential for designing drugs that can selectively bind to these sites and modulate protein function. This knowledge can help create more effective and safer therapeutic options for various diseases.
Mitochondrial membrane potential is the electric potential difference (voltage) across the inner mitochondrial membrane. It is negative inside the mitochondria and positive outside. This electrical gradient is established by the active transport of hydrogen ions (protons) out of the mitochondrial matrix and into the intermembrane space by complexes in the electron transport chain during oxidative phosphorylation. The energy stored in this electrochemical gradient is used to generate ATP, which is the main source of energy for cellular metabolism.
Oxidation-Reduction (redox) reactions are a type of chemical reaction involving a transfer of electrons between two species. The substance that loses electrons in the reaction is oxidized, and the substance that gains electrons is reduced. Oxidation and reduction always occur together in a redox reaction, hence the term "oxidation-reduction."
In biological systems, redox reactions play a crucial role in many cellular processes, including energy production, metabolism, and signaling. The transfer of electrons in these reactions is often facilitated by specialized molecules called electron carriers, such as nicotinamide adenine dinucleotide (NAD+/NADH) and flavin adenine dinucleotide (FAD/FADH2).
The oxidation state of an element in a compound is a measure of the number of electrons that have been gained or lost relative to its neutral state. In redox reactions, the oxidation state of one or more elements changes as they gain or lose electrons. The substance that is oxidized has a higher oxidation state, while the substance that is reduced has a lower oxidation state.
Overall, oxidation-reduction reactions are fundamental to the functioning of living organisms and are involved in many important biological processes.
Phosphothreonine is not a medical term per se, but rather a biochemical term that refers to a specific post-translational modification of the amino acid threonine. In this modification, a phosphate group is added to the hydroxyl side chain of threonine, which can affect the function and regulation of proteins in which it occurs.
In medical or clinical contexts, phosphothreonine may be mentioned in relation to various disease processes or signaling pathways that involve protein kinases, enzymes that add phosphate groups to specific amino acids (including threonine) in proteins. For example, abnormal regulation of protein kinases and phosphatases (enzymes that remove phosphate groups) can contribute to the development of cancer, neurological disorders, and other diseases.
Post-translational protein processing refers to the modifications and changes that proteins undergo after their synthesis on ribosomes, which are complex molecular machines responsible for protein synthesis. These modifications occur through various biochemical processes and play a crucial role in determining the final structure, function, and stability of the protein.
The process begins with the translation of messenger RNA (mRNA) into a linear polypeptide chain, which is then subjected to several post-translational modifications. These modifications can include:
1. Proteolytic cleavage: The removal of specific segments or domains from the polypeptide chain by proteases, resulting in the formation of mature, functional protein subunits.
2. Chemical modifications: Addition or modification of chemical groups to the side chains of amino acids, such as phosphorylation (addition of a phosphate group), glycosylation (addition of sugar moieties), methylation (addition of a methyl group), acetylation (addition of an acetyl group), and ubiquitination (addition of a ubiquitin protein).
3. Disulfide bond formation: The oxidation of specific cysteine residues within the polypeptide chain, leading to the formation of disulfide bonds between them. This process helps stabilize the three-dimensional structure of proteins, particularly in extracellular environments.
4. Folding and assembly: The acquisition of a specific three-dimensional conformation by the polypeptide chain, which is essential for its function. Chaperone proteins assist in this process to ensure proper folding and prevent aggregation.
5. Protein targeting: The directed transport of proteins to their appropriate cellular locations, such as the nucleus, mitochondria, endoplasmic reticulum, or plasma membrane. This is often facilitated by specific signal sequences within the protein that are recognized and bound by transport machinery.
Collectively, these post-translational modifications contribute to the functional diversity of proteins in living organisms, allowing them to perform a wide range of cellular processes, including signaling, catalysis, regulation, and structural support.
Adenosine triphosphatases (ATPases) are a group of enzymes that catalyze the conversion of adenosine triphosphate (ATP) into adenosine diphosphate (ADP) and inorganic phosphate. This reaction releases energy, which is used to drive various cellular processes such as muscle contraction, transport of ions across membranes, and synthesis of proteins and nucleic acids.
ATPases are classified into several types based on their structure, function, and mechanism of action. Some examples include:
1. P-type ATPases: These ATPases form a phosphorylated intermediate during the reaction cycle and are involved in the transport of ions across membranes, such as the sodium-potassium pump and calcium pumps.
2. F-type ATPases: These ATPases are found in mitochondria, chloroplasts, and bacteria, and are responsible for generating a proton gradient across the membrane, which is used to synthesize ATP.
3. V-type ATPases: These ATPases are found in vacuolar membranes and endomembranes, and are involved in acidification of intracellular compartments.
4. A-type ATPases: These ATPases are found in the plasma membrane and are involved in various functions such as cell signaling and ion transport.
Overall, ATPases play a crucial role in maintaining the energy balance of cells and regulating various physiological processes.
The Electron Transport Chain (ETC) is a series of complexes in the inner mitochondrial membrane that are involved in the process of cellular respiration, through which the majority of energy is generated for the cell. The ETC complex proteins are a group of transmembrane protein complexes that facilitate the transfer of electrons from electron donors to electron acceptors via redox reactions. This transfer of electrons drives the generation of a proton gradient across the inner mitochondrial membrane, which is then used by ATP synthase to generate ATP, the primary energy currency of the cell.
The ETC complex proteins consist of four main complexes: Complex I (NADH-Q oxidoreductase), Complex II (succinate-Q oxidoreductase), Complex III (cytochrome bc1 complex or CoQ:cytochrome c oxidoreductase), and Complex IV (cytochrome c oxidase). Each complex contains a number of subunits, many of which are encoded by both the nuclear and mitochondrial genomes.
In summary, Electron Transport Chain Complex Proteins are a group of transmembrane protein complexes located in the inner mitochondrial membrane that facilitate the transfer of electrons from electron donors to electron acceptors, driving the generation of a proton gradient and ultimately ATP synthesis during cellular respiration.
Submitochondrial particles, also known as "submitochondrial vesicles" or "inner membrane fragments," are small particles that consist of the inner mitochondrial membrane and the associated components. They are obtained through sonication or other methods of disrupting mitochondria, which results in breaking down the outer membrane while leaving the inner membrane intact. These particles can be used in various biochemical studies to investigate the structure, function, and composition of the inner mitochondrial membrane and its components, such as the electron transport chain and ATP synthase complexes.
I'm sorry for any confusion, but "Malates" is not a recognized term in medical terminology. It's possible there may be a spelling mistake or it could be a slang term or an abbreviation that is not widely recognized. If you have more context or information, I'd be happy to try and help further.
Carrier proteins, also known as transport proteins, are a type of protein that facilitates the movement of molecules across cell membranes. They are responsible for the selective and active transport of ions, sugars, amino acids, and other molecules from one side of the membrane to the other, against their concentration gradient. This process requires energy, usually in the form of ATP (adenosine triphosphate).
Carrier proteins have a specific binding site for the molecule they transport, and undergo conformational changes upon binding, which allows them to move the molecule across the membrane. Once the molecule has been transported, the carrier protein returns to its original conformation, ready to bind and transport another molecule.
Carrier proteins play a crucial role in maintaining the balance of ions and other molecules inside and outside of cells, and are essential for many physiological processes, including nerve impulse transmission, muscle contraction, and nutrient uptake.
ATP synthetase complexes are molecular machines found in the inner membrane of mitochondria and the bacterial cell membrane that catalyze the synthesis of Adenosine Triphosphate (ATP) from ADP and inorganic phosphate. They are also known as F1F0-ATPases or complex V of the electron transport chain.
The complex is composed of two main parts: the F1 portion, which is located on the matrix side of the inner mitochondrial membrane and contains the catalytic site for ATP synthesis; and the F0 portion, which is embedded in the membrane and acts as a proton channel.
The process of ATP synthesis is coupled to the flow of protons across the membrane, driven by the electrochemical gradient generated by the electron transport chain. As protons flow through the F0 portion, they cause the F1 portion to rotate, which in turn drives the synthesis of ATP from ADP and Pi at the catalytic site.
ATP synthetase complexes are essential for cellular energy production and are conserved across all kingdoms of life.
Peptide mapping is a technique used in proteomics and analytical chemistry to analyze and identify the sequence and structure of peptides or proteins. This method involves breaking down a protein into smaller peptide fragments using enzymatic or chemical digestion, followed by separation and identification of these fragments through various analytical techniques such as liquid chromatography (LC) and mass spectrometry (MS).
The resulting peptide map serves as a "fingerprint" of the protein, providing information about its sequence, modifications, and structure. Peptide mapping can be used for a variety of applications, including protein identification, characterization of post-translational modifications, and monitoring of protein degradation or cleavage.
In summary, peptide mapping is a powerful tool in proteomics that enables the analysis and identification of proteins and their modifications at the peptide level.
NADH dehydrogenase, also known as Complex I, is an enzyme complex in the electron transport chain located in the inner mitochondrial membrane. It catalyzes the oxidation of NADH to NAD+ and the reduction of coenzyme Q to ubiquinol, playing a crucial role in cellular respiration and energy production. The reaction involves the transfer of electrons from NADH to coenzyme Q, which contributes to the generation of a proton gradient across the membrane, ultimately leading to ATP synthesis. Defects in NADH dehydrogenase can result in various mitochondrial diseases and disorders.
Casein Kinase II (CK2) is a serine/threonine protein kinase that is widely expressed in eukaryotic cells and is involved in the regulation of various cellular processes. It is a heterotetrameric enzyme, consisting of two catalytic subunits (alpha and alpha') and two regulatory subunits (beta).
CK2 phosphorylates a wide range of substrates, including transcription factors, signaling proteins, and other kinases. It is known to play roles in cell cycle regulation, apoptosis, DNA damage response, and protein stability, among others. CK2 activity is often found to be elevated in various types of cancer, making it a potential target for cancer therapy.
Immunoblotting, also known as western blotting, is a laboratory technique used in molecular biology and immunogenetics to detect and quantify specific proteins in a complex mixture. This technique combines the electrophoretic separation of proteins by gel electrophoresis with their detection using antibodies that recognize specific epitopes (protein fragments) on the target protein.
The process involves several steps: first, the protein sample is separated based on size through sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Next, the separated proteins are transferred onto a nitrocellulose or polyvinylidene fluoride (PVDF) membrane using an electric field. The membrane is then blocked with a blocking agent to prevent non-specific binding of antibodies.
After blocking, the membrane is incubated with a primary antibody that specifically recognizes the target protein. Following this, the membrane is washed to remove unbound primary antibodies and then incubated with a secondary antibody conjugated to an enzyme such as horseradish peroxidase (HRP) or alkaline phosphatase (AP). The enzyme catalyzes a colorimetric or chemiluminescent reaction that allows for the detection of the target protein.
Immunoblotting is widely used in research and clinical settings to study protein expression, post-translational modifications, protein-protein interactions, and disease biomarkers. It provides high specificity and sensitivity, making it a valuable tool for identifying and quantifying proteins in various biological samples.
Proteins are complex, large molecules that play critical roles in the body's functions. They are made up of amino acids, which are organic compounds that are the building blocks of proteins. Proteins are required for the structure, function, and regulation of the body's tissues and organs. They are essential for the growth, repair, and maintenance of body tissues, and they play a crucial role in many biological processes, including metabolism, immune response, and cellular signaling. Proteins can be classified into different types based on their structure and function, such as enzymes, hormones, antibodies, and structural proteins. They are found in various foods, especially animal-derived products like meat, dairy, and eggs, as well as plant-based sources like beans, nuts, and grains.
'Gene expression regulation' refers to the processes that control whether, when, and where a particular gene is expressed, meaning the production of a specific protein or functional RNA encoded by that gene. This complex mechanism can be influenced by various factors such as transcription factors, chromatin remodeling, DNA methylation, non-coding RNAs, and post-transcriptional modifications, among others. Proper regulation of gene expression is crucial for normal cellular function, development, and maintaining homeostasis in living organisms. Dysregulation of gene expression can lead to various diseases, including cancer and genetic disorders.
In the field of medicine, "time factors" refer to the duration of symptoms or time elapsed since the onset of a medical condition, which can have significant implications for diagnosis and treatment. Understanding time factors is crucial in determining the progression of a disease, evaluating the effectiveness of treatments, and making critical decisions regarding patient care.
For example, in stroke management, "time is brain," meaning that rapid intervention within a specific time frame (usually within 4.5 hours) is essential to administering tissue plasminogen activator (tPA), a clot-busting drug that can minimize brain damage and improve patient outcomes. Similarly, in trauma care, the "golden hour" concept emphasizes the importance of providing definitive care within the first 60 minutes after injury to increase survival rates and reduce morbidity.
Time factors also play a role in monitoring the progression of chronic conditions like diabetes or heart disease, where regular follow-ups and assessments help determine appropriate treatment adjustments and prevent complications. In infectious diseases, time factors are crucial for initiating antibiotic therapy and identifying potential outbreaks to control their spread.
Overall, "time factors" encompass the significance of recognizing and acting promptly in various medical scenarios to optimize patient outcomes and provide effective care.
SRC-family kinases (SFKs) are a group of non-receptor tyrosine kinases that play important roles in various cellular processes, including cell proliferation, differentiation, survival, and migration. They are named after the founding member, SRC, which was first identified as an oncogene in Rous sarcoma virus.
SFKs share a common structure, consisting of an N-terminal unique domain, a SH3 domain, a SH2 domain, a catalytic kinase domain, and a C-terminal regulatory tail with a negative regulatory tyrosine residue (Y527 in human SRC). In their inactive state, SFKs are maintained in a closed conformation through intramolecular interactions between the SH3 domain, SH2 domain, and the phosphorylated C-terminal tyrosine.
Upon activation by various signals, such as growth factors, cytokines, or integrin engagement, SFKs are activated through a series of events that involve dephosphorylation of the regulatory tyrosine residue, recruitment to membrane receptors via their SH2 and SH3 domains, and trans-autophosphorylation of the activation loop in the kinase domain.
Once activated, SFKs can phosphorylate a wide range of downstream substrates, including other protein kinases, adaptor proteins, and cytoskeletal components, thereby regulating various signaling pathways that control cell behavior. Dysregulation of SFK activity has been implicated in various diseases, including cancer, inflammation, and neurological disorders.
HeLa cells are a type of immortalized cell line used in scientific research. They are derived from a cancer that developed in the cervical tissue of Henrietta Lacks, an African-American woman, in 1951. After her death, cells taken from her tumor were found to be capable of continuous division and growth in a laboratory setting, making them an invaluable resource for medical research.
HeLa cells have been used in a wide range of scientific studies, including research on cancer, viruses, genetics, and drug development. They were the first human cell line to be successfully cloned and are able to grow rapidly in culture, doubling their population every 20-24 hours. This has made them an essential tool for many areas of biomedical research.
It is important to note that while HeLa cells have been instrumental in numerous scientific breakthroughs, the story of their origin raises ethical questions about informed consent and the use of human tissue in research.
Recombinant proteins are artificially created proteins produced through the use of recombinant DNA technology. This process involves combining DNA molecules from different sources to create a new set of genes that encode for a specific protein. The resulting recombinant protein can then be expressed, purified, and used for various applications in research, medicine, and industry.
Recombinant proteins are widely used in biomedical research to study protein function, structure, and interactions. They are also used in the development of diagnostic tests, vaccines, and therapeutic drugs. For example, recombinant insulin is a common treatment for diabetes, while recombinant human growth hormone is used to treat growth disorders.
The production of recombinant proteins typically involves the use of host cells, such as bacteria, yeast, or mammalian cells, which are engineered to express the desired protein. The host cells are transformed with a plasmid vector containing the gene of interest, along with regulatory elements that control its expression. Once the host cells are cultured and the protein is expressed, it can be purified using various chromatography techniques.
Overall, recombinant proteins have revolutionized many areas of biology and medicine, enabling researchers to study and manipulate proteins in ways that were previously impossible.
Apoptosis is a programmed and controlled cell death process that occurs in multicellular organisms. It is a natural process that helps maintain tissue homeostasis by eliminating damaged, infected, or unwanted cells. During apoptosis, the cell undergoes a series of morphological changes, including cell shrinkage, chromatin condensation, and fragmentation into membrane-bound vesicles called apoptotic bodies. These bodies are then recognized and engulfed by neighboring cells or phagocytic cells, preventing an inflammatory response. Apoptosis is regulated by a complex network of intracellular signaling pathways that involve proteins such as caspases, Bcl-2 family members, and inhibitors of apoptosis (IAPs).
"Cattle" is a term used in the agricultural and veterinary fields to refer to domesticated animals of the genus *Bos*, primarily *Bos taurus* (European cattle) and *Bos indicus* (Zebu). These animals are often raised for meat, milk, leather, and labor. They are also known as bovines or cows (for females), bulls (intact males), and steers/bullocks (castrated males). However, in a strict medical definition, "cattle" does not apply to humans or other animals.
Tertiary protein structure refers to the three-dimensional arrangement of all the elements (polypeptide chains) of a single protein molecule. It is the highest level of structural organization and results from interactions between various side chains (R groups) of the amino acids that make up the protein. These interactions, which include hydrogen bonds, ionic bonds, van der Waals forces, and disulfide bridges, give the protein its unique shape and stability, which in turn determines its function. The tertiary structure of a protein can be stabilized by various factors such as temperature, pH, and the presence of certain ions. Any changes in these factors can lead to denaturation, where the protein loses its tertiary structure and thus its function.
Membrane proteins are a type of protein that are embedded in the lipid bilayer of biological membranes, such as the plasma membrane of cells or the inner membrane of mitochondria. These proteins play crucial roles in various cellular processes, including:
1. Cell-cell recognition and signaling
2. Transport of molecules across the membrane (selective permeability)
3. Enzymatic reactions at the membrane surface
4. Energy transduction and conversion
5. Mechanosensation and signal transduction
Membrane proteins can be classified into two main categories: integral membrane proteins, which are permanently associated with the lipid bilayer, and peripheral membrane proteins, which are temporarily or loosely attached to the membrane surface. Integral membrane proteins can further be divided into three subcategories based on their topology:
1. Transmembrane proteins, which span the entire width of the lipid bilayer with one or more alpha-helices or beta-barrels.
2. Lipid-anchored proteins, which are covalently attached to lipids in the membrane via a glycosylphosphatidylinositol (GPI) anchor or other lipid modifications.
3. Monotopic proteins, which are partially embedded in the membrane and have one or more domains exposed to either side of the bilayer.
Membrane proteins are essential for maintaining cellular homeostasis and are targets for various therapeutic interventions, including drug development and gene therapy. However, their structural complexity and hydrophobicity make them challenging to study using traditional biochemical methods, requiring specialized techniques such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and single-particle cryo-electron microscopy (cryo-EM).
The myocardium is the middle layer of the heart wall, composed of specialized cardiac muscle cells that are responsible for pumping blood throughout the body. It forms the thickest part of the heart wall and is divided into two sections: the left ventricle, which pumps oxygenated blood to the rest of the body, and the right ventricle, which pumps deoxygenated blood to the lungs.
The myocardium contains several types of cells, including cardiac muscle fibers, connective tissue, nerves, and blood vessels. The muscle fibers are arranged in a highly organized pattern that allows them to contract in a coordinated manner, generating the force necessary to pump blood through the heart and circulatory system.
Damage to the myocardium can occur due to various factors such as ischemia (reduced blood flow), infection, inflammation, or genetic disorders. This damage can lead to several cardiac conditions, including heart failure, arrhythmias, and cardiomyopathy.
The cell nucleus is a membrane-bound organelle found in the eukaryotic cells (cells with a true nucleus). It contains most of the cell's genetic material, organized as DNA molecules in complex with proteins, RNA molecules, and histones to form chromosomes.
The primary function of the cell nucleus is to regulate and control the activities of the cell, including growth, metabolism, protein synthesis, and reproduction. It also plays a crucial role in the process of mitosis (cell division) by separating and protecting the genetic material during this process. The nuclear membrane, or nuclear envelope, surrounding the nucleus is composed of two lipid bilayers with numerous pores that allow for the selective transport of molecules between the nucleoplasm (nucleus interior) and the cytoplasm (cell exterior).
The cell nucleus is a vital structure in eukaryotic cells, and its dysfunction can lead to various diseases, including cancer and genetic disorders.
Phosphorus radioisotopes are radioactive isotopes or variants of the element phosphorus that emit radiation. Phosphorus has several radioisotopes, with the most common ones being phosphorus-32 (^32P) and phosphorus-33 (^33P). These radioisotopes are used in various medical applications such as cancer treatment and diagnostic procedures.
Phosphorus-32 has a half-life of approximately 14.3 days and emits beta particles, making it useful for treating certain types of cancer, such as leukemia and lymphoma. It can also be used in brachytherapy, a type of radiation therapy that involves placing a radioactive source close to the tumor.
Phosphorus-33 has a shorter half-life of approximately 25.4 days and emits both beta particles and gamma rays. This makes it useful for diagnostic procedures, such as positron emission tomography (PET) scans, where the gamma rays can be detected and used to create images of the body's internal structures.
It is important to note that handling and using radioisotopes requires specialized training and equipment to ensure safety and prevent radiation exposure.
Transcription factors are proteins that play a crucial role in regulating gene expression by controlling the transcription of DNA to messenger RNA (mRNA). They function by binding to specific DNA sequences, known as response elements, located in the promoter region or enhancer regions of target genes. This binding can either activate or repress the initiation of transcription, depending on the properties and interactions of the particular transcription factor. Transcription factors often act as part of a complex network of regulatory proteins that determine the precise spatiotemporal patterns of gene expression during development, differentiation, and homeostasis in an organism.
Phosphoprotein phosphatases (PPPs) are a family of enzymes that play a crucial role in the regulation of various cellular processes by removing phosphate groups from serine, threonine, and tyrosine residues on proteins. Phosphorylation is a post-translational modification that regulates protein function, localization, and stability, and dephosphorylation by PPPs is essential for maintaining the balance of this regulation.
The PPP family includes several subfamilies, such as PP1, PP2A, PP2B (also known as calcineurin), PP4, PP5, and PP6. Each subfamily has distinct substrate specificities and regulatory mechanisms. For example, PP1 and PP2A are involved in the regulation of metabolism, signal transduction, and cell cycle progression, while PP2B is involved in immune response and calcium signaling.
Dysregulation of PPPs has been implicated in various diseases, including cancer, neurodegenerative disorders, and cardiovascular disease. Therefore, understanding the function and regulation of PPPs is important for developing therapeutic strategies to target these diseases.
Proto-oncogene proteins are normal cellular proteins that play crucial roles in various cellular processes, such as signal transduction, cell cycle regulation, and apoptosis (programmed cell death). They are involved in the regulation of cell growth, differentiation, and survival under physiological conditions.
When proto-oncogene proteins undergo mutations or aberrations in their expression levels, they can transform into oncogenic forms, leading to uncontrolled cell growth and division. These altered proteins are then referred to as oncogene products or oncoproteins. Oncogenic mutations can occur due to various factors, including genetic predisposition, environmental exposures, and aging.
Examples of proto-oncogene proteins include:
1. Ras proteins: Involved in signal transduction pathways that regulate cell growth and differentiation. Activating mutations in Ras genes are found in various human cancers.
2. Myc proteins: Regulate gene expression related to cell cycle progression, apoptosis, and metabolism. Overexpression of Myc proteins is associated with several types of cancer.
3. EGFR (Epidermal Growth Factor Receptor): A transmembrane receptor tyrosine kinase that regulates cell proliferation, survival, and differentiation. Mutations or overexpression of EGFR are linked to various malignancies, such as lung cancer and glioblastoma.
4. Src family kinases: Intracellular tyrosine kinases that regulate signal transduction pathways involved in cell proliferation, survival, and migration. Dysregulation of Src family kinases is implicated in several types of cancer.
5. Abl kinases: Cytoplasmic tyrosine kinases that regulate various cellular processes, including cell growth, differentiation, and stress responses. Aberrant activation of Abl kinases, as seen in chronic myelogenous leukemia (CML), leads to uncontrolled cell proliferation.
Understanding the roles of proto-oncogene proteins and their dysregulation in cancer development is essential for developing targeted cancer therapies that aim to inhibit or modulate these aberrant signaling pathways.
"Wistar rats" are a strain of albino rats that are widely used in laboratory research. They were developed at the Wistar Institute in Philadelphia, USA, and were first introduced in 1906. Wistar rats are outbred, which means that they are genetically diverse and do not have a fixed set of genetic characteristics like inbred strains.
Wistar rats are commonly used as animal models in biomedical research because of their size, ease of handling, and relatively low cost. They are used in a wide range of research areas, including toxicology, pharmacology, nutrition, cancer, cardiovascular disease, and behavioral studies. Wistar rats are also used in safety testing of drugs, medical devices, and other products.
Wistar rats are typically larger than many other rat strains, with males weighing between 500-700 grams and females weighing between 250-350 grams. They have a lifespan of approximately 2-3 years. Wistar rats are also known for their docile and friendly nature, making them easy to handle and work with in the laboratory setting.
Electron Transport Complex III, also known as cytochrome bc1 complex or ubiquinol-cytochrome c reductase, is a protein complex located in the inner mitochondrial membrane of eukaryotic cells and the cytoplasmic membrane of prokaryotic cells. It plays a crucial role in the electron transport chain (ETC), a series of complexes that generate energy in the form of ATP through a process called oxidative phosphorylation.
In ETC, Electron Transport Complex III accepts electrons from ubiquinol and transfers them to cytochrome c. This electron transfer is coupled with the translocation of protons (H+ ions) across the membrane, creating an electrochemical gradient. The energy stored in this gradient drives the synthesis of ATP by ATP synthase.
Electron Transport Complex III consists of several subunits, including cytochrome b, cytochrome c1, and the Rieske iron-sulfur protein. These subunits work together to facilitate the electron transfer and proton translocation processes.
Mitogen-Activated Protein Kinase 1 (MAPK1), also known as Extracellular Signal-Regulated Kinase 2 (ERK2), is a protein kinase that plays a crucial role in intracellular signal transduction pathways. It is a member of the MAPK family, which regulates various cellular processes such as proliferation, differentiation, apoptosis, and stress response.
MAPK1 is activated by a cascade of phosphorylation events initiated by upstream activators like MAPKK (Mitogen-Activated Protein Kinase Kinase) in response to various extracellular signals such as growth factors, hormones, and mitogens. Once activated, MAPK1 phosphorylates downstream targets, including transcription factors and other protein kinases, thereby modulating their activities and ultimately influencing gene expression and cellular responses.
MAPK1 is widely expressed in various tissues and cells, and its dysregulation has been implicated in several pathological conditions, including cancer, inflammation, and neurodegenerative diseases. Therefore, understanding the regulation and function of MAPK1 signaling pathways has important implications for developing therapeutic strategies to treat these disorders.
Intracellular signaling peptides and proteins are molecules that play a crucial role in transmitting signals within cells, which ultimately lead to changes in cell behavior or function. These signals can originate from outside the cell (extracellular) or within the cell itself. Intracellular signaling molecules include various types of peptides and proteins, such as:
1. G-protein coupled receptors (GPCRs): These are seven-transmembrane domain receptors that bind to extracellular signaling molecules like hormones, neurotransmitters, or chemokines. Upon activation, they initiate a cascade of intracellular signals through G proteins and secondary messengers.
2. Receptor tyrosine kinases (RTKs): These are transmembrane receptors that bind to growth factors, cytokines, or hormones. Activation of RTKs leads to autophosphorylation of specific tyrosine residues, creating binding sites for intracellular signaling proteins such as adapter proteins, phosphatases, and enzymes like Ras, PI3K, and Src family kinases.
3. Second messenger systems: Intracellular second messengers are small molecules that amplify and propagate signals within the cell. Examples include cyclic adenosine monophosphate (cAMP), cyclic guanosine monophosphate (cGMP), diacylglycerol (DAG), inositol triphosphate (IP3), calcium ions (Ca2+), and nitric oxide (NO). These second messengers activate or inhibit various downstream effectors, leading to changes in cellular responses.
4. Signal transduction cascades: Intracellular signaling proteins often form complex networks of interacting molecules that relay signals from the plasma membrane to the nucleus. These cascades involve kinases (protein kinases A, B, C, etc.), phosphatases, and adapter proteins, which ultimately regulate gene expression, cell cycle progression, metabolism, and other cellular processes.
5. Ubiquitination and proteasome degradation: Intracellular signaling pathways can also control protein stability by modulating ubiquitin-proteasome degradation. E3 ubiquitin ligases recognize specific substrates and conjugate them with ubiquitin molecules, targeting them for proteasomal degradation. This process regulates the abundance of key signaling proteins and contributes to signal termination or amplification.
In summary, intracellular signaling pathways involve a complex network of interacting proteins that relay signals from the plasma membrane to various cellular compartments, ultimately regulating gene expression, metabolism, and other cellular processes. Dysregulation of these pathways can contribute to disease development and progression, making them attractive targets for therapeutic intervention.
Electrophoresis, polyacrylamide gel (EPG) is a laboratory technique used to separate and analyze complex mixtures of proteins or nucleic acids (DNA or RNA) based on their size and electrical charge. This technique utilizes a matrix made of cross-linked polyacrylamide, a type of gel, which provides a stable and uniform environment for the separation of molecules.
In this process:
1. The polyacrylamide gel is prepared by mixing acrylamide monomers with a cross-linking agent (bis-acrylamide) and a catalyst (ammonium persulfate) in the presence of a buffer solution.
2. The gel is then poured into a mold and allowed to polymerize, forming a solid matrix with uniform pore sizes that depend on the concentration of acrylamide used. Higher concentrations result in smaller pores, providing better resolution for separating smaller molecules.
3. Once the gel has set, it is placed in an electrophoresis apparatus containing a buffer solution. Samples containing the mixture of proteins or nucleic acids are loaded into wells on the top of the gel.
4. An electric field is applied across the gel, causing the negatively charged molecules to migrate towards the positive electrode (anode) while positively charged molecules move toward the negative electrode (cathode). The rate of migration depends on the size, charge, and shape of the molecules.
5. Smaller molecules move faster through the gel matrix and will migrate farther from the origin compared to larger molecules, resulting in separation based on size. Proteins and nucleic acids can be selectively stained after electrophoresis to visualize the separated bands.
EPG is widely used in various research fields, including molecular biology, genetics, proteomics, and forensic science, for applications such as protein characterization, DNA fragment analysis, cloning, mutation detection, and quality control of nucleic acid or protein samples.
DNA-binding proteins are a type of protein that have the ability to bind to DNA (deoxyribonucleic acid), the genetic material of organisms. These proteins play crucial roles in various biological processes, such as regulation of gene expression, DNA replication, repair and recombination.
The binding of DNA-binding proteins to specific DNA sequences is mediated by non-covalent interactions, including electrostatic, hydrogen bonding, and van der Waals forces. The specificity of binding is determined by the recognition of particular nucleotide sequences or structural features of the DNA molecule.
DNA-binding proteins can be classified into several categories based on their structure and function, such as transcription factors, histones, and restriction enzymes. Transcription factors are a major class of DNA-binding proteins that regulate gene expression by binding to specific DNA sequences in the promoter region of genes and recruiting other proteins to modulate transcription. Histones are DNA-binding proteins that package DNA into nucleosomes, the basic unit of chromatin structure. Restriction enzymes are DNA-binding proteins that recognize and cleave specific DNA sequences, and are widely used in molecular biology research and biotechnology applications.
Site-directed mutagenesis is a molecular biology technique used to introduce specific and targeted changes to a specific DNA sequence. This process involves creating a new variant of a gene or a specific region of interest within a DNA molecule by introducing a planned, deliberate change, or mutation, at a predetermined site within the DNA sequence.
The methodology typically involves the use of molecular tools such as PCR (polymerase chain reaction), restriction enzymes, and/or ligases to introduce the desired mutation(s) into a plasmid or other vector containing the target DNA sequence. The resulting modified DNA molecule can then be used to transform host cells, allowing for the production of large quantities of the mutated gene or protein for further study.
Site-directed mutagenesis is a valuable tool in basic research, drug discovery, and biotechnology applications where specific changes to a DNA sequence are required to understand gene function, investigate protein structure/function relationships, or engineer novel biological properties into existing genes or proteins.
Tetramethylphenylenediamine (TMPD) is not typically considered a medical term, but it is a chemical compound that is used in some scientific and research contexts. It's a type of aromatic amine, which is a class of organic compounds characterized by the presence of one or more amino groups (-NH2) attached to an aromatic hydrocarbon ring.
In biochemistry and molecular biology, TMPD is sometimes used as an electron carrier in experiments that involve redox reactions, such as those that occur during cellular respiration. It can also be used as a catalyst or reagent in various chemical reactions. However, it's important to note that TMPD is not a substance that is typically encountered in medical practice or patient care.
In the context of medicine, particularly in relation to cancer treatment, protons refer to positively charged subatomic particles found in the nucleus of an atom. Proton therapy, a type of radiation therapy, uses a beam of protons to target and destroy cancer cells with high precision, minimizing damage to surrounding healthy tissue. The concentrated dose of radiation is delivered directly to the tumor site, reducing side effects and improving quality of life during treatment.
Calcium-calmodulin-dependent protein kinases (CAMKs) are a family of enzymes that play a crucial role in intracellular signaling pathways. They are activated by the binding of calcium ions and calmodulin, a ubiquitous calcium-binding protein, to their regulatory domain.
Once activated, CAMKs phosphorylate specific serine or threonine residues on target proteins, thereby modulating their activity, localization, or stability. This post-translational modification is essential for various cellular processes, including synaptic plasticity, gene expression, metabolism, and cell cycle regulation.
There are several subfamilies of CAMKs, including CaMKI, CaMKII, CaMKIII (also known as CaMKIV), and CaMK kinase (CaMKK). Each subfamily has distinct structural features, substrate specificity, and regulatory mechanisms. Dysregulation of CAMK signaling has been implicated in various pathological conditions, such as neurodegenerative diseases, cancer, and cardiovascular disorders.
Phosphatidylinositol 3-Kinases (PI3Ks) are a family of enzymes that play a crucial role in intracellular signal transduction. They phosphorylate the 3-hydroxyl group of the inositol ring in phosphatidylinositol and its derivatives, which results in the production of second messengers that regulate various cellular processes such as cell growth, proliferation, differentiation, motility, and survival.
PI3Ks are divided into three classes based on their structure and substrate specificity. Class I PI3Ks are further subdivided into two categories: class IA and class IB. Class IA PI3Ks are heterodimers consisting of a catalytic subunit (p110α, p110β, or p110δ) and a regulatory subunit (p85α, p85β, p55γ, or p50γ). They are primarily activated by receptor tyrosine kinases and G protein-coupled receptors. Class IB PI3Ks consist of a catalytic subunit (p110γ) and a regulatory subunit (p101 or p84/87). They are mainly activated by G protein-coupled receptors.
Dysregulation of PI3K signaling has been implicated in various human diseases, including cancer, diabetes, and autoimmune disorders. Therefore, PI3Ks have emerged as important targets for drug development in these areas.
Succinic acid, also known as butanedioic acid, is an organic compound with the chemical formula HOOC(CH2)2COOH. It is a white crystalline powder that is soluble in water and has a slightly acerbic taste. In medicine, succinic acid is not used as a treatment for any specific condition. However, it is a naturally occurring substance found in the body and plays a role in the citric acid cycle, which is a key process in energy production within cells. It can also be found in some foods and is used in the manufacturing of various products such as pharmaceuticals, resins, and perfumes.
Glycogen Synthase Kinase 3 (GSK-3) is a serine/threonine protein kinase that plays a crucial role in the regulation of several cellular processes, including glycogen metabolism, cell signaling, gene transcription, and apoptosis. It was initially discovered as a key enzyme involved in glycogen metabolism due to its ability to phosphorylate and inhibit glycogen synthase, an enzyme responsible for the synthesis of glycogen from glucose.
GSK-3 exists in two isoforms, GSK-3α and GSK-3β, which share a high degree of sequence similarity and are widely expressed in various tissues. Both isoforms are constitutively active under normal conditions and are regulated through inhibitory phosphorylation by several upstream signaling pathways, such as insulin, Wnt, and Hedgehog signaling.
Dysregulation of GSK-3 has been implicated in the pathogenesis of various diseases, including diabetes, neurodegenerative disorders, and cancer. In recent years, GSK-3 has emerged as an attractive therapeutic target for the development of novel drugs to treat these conditions.
Mitogen-Activated Protein Kinase 3 (MAPK3), also known as extracellular signal-regulated kinase 1 (ERK1), is a serine/threonine protein kinase that plays a crucial role in intracellular signal transduction pathways. It is involved in the regulation of various cellular processes, including proliferation, differentiation, and survival, in response to extracellular stimuli such as growth factors, hormones, and stress.
MAPK3 is activated through a phosphorylation cascade that involves the activation of upstream MAPK kinases (MKK or MEK). Once activated, MAPK3 can phosphorylate and activate various downstream targets, including transcription factors, to regulate gene expression. Dysregulation of MAPK3 signaling has been implicated in several diseases, including cancer and neurological disorders.
A cell membrane, also known as the plasma membrane, is a thin semi-permeable phospholipid bilayer that surrounds all cells in animals, plants, and microorganisms. It functions as a barrier to control the movement of substances in and out of the cell, allowing necessary molecules such as nutrients, oxygen, and signaling molecules to enter while keeping out harmful substances and waste products. The cell membrane is composed mainly of phospholipids, which have hydrophilic (water-loving) heads and hydrophobic (water-fearing) tails. This unique structure allows the membrane to be flexible and fluid, yet selectively permeable. Additionally, various proteins are embedded in the membrane that serve as channels, pumps, receptors, and enzymes, contributing to the cell's overall functionality and communication with its environment.
Mitochondrial swelling is a pathological change in the structure of mitochondria, which are the energy-producing organelles found in cells. This condition is characterized by an increase in the volume of the mitochondrial matrix, which is the space inside the mitochondrion that contains enzymes and other molecules involved in energy production.
Mitochondrial swelling can occur as a result of various cellular stressors, such as oxidative damage, calcium overload, or decreased levels of adenosine triphosphate (ATP), which is the primary energy currency of the cell. This swelling can lead to disruption of the mitochondrial membrane and release of cytochrome c, a protein involved in apoptosis or programmed cell death.
Mitochondrial swelling has been implicated in several diseases, including neurodegenerative disorders, ischemia-reperfusion injury, and drug toxicity. It can be observed under an electron microscope as part of an ultrastructural analysis of tissue samples or detected through biochemical assays that measure changes in mitochondrial membrane potential or matrix volume.
Sprague-Dawley rats are a strain of albino laboratory rats that are widely used in scientific research. They were first developed by researchers H.H. Sprague and R.C. Dawley in the early 20th century, and have since become one of the most commonly used rat strains in biomedical research due to their relatively large size, ease of handling, and consistent genetic background.
Sprague-Dawley rats are outbred, which means that they are genetically diverse and do not suffer from the same limitations as inbred strains, which can have reduced fertility and increased susceptibility to certain diseases. They are also characterized by their docile nature and low levels of aggression, making them easier to handle and study than some other rat strains.
These rats are used in a wide variety of research areas, including toxicology, pharmacology, nutrition, cancer, and behavioral studies. Because they are genetically diverse, Sprague-Dawley rats can be used to model a range of human diseases and conditions, making them an important tool in the development of new drugs and therapies.
Adenine nucleotides are molecules that consist of a nitrogenous base called adenine, which is linked to a sugar molecule (ribose in the case of adenosine monophosphate or AMP, and deoxyribose in the case of adenosine diphosphate or ADP and adenosine triphosphate or ATP) and one, two, or three phosphate groups. These molecules play a crucial role in energy transfer and metabolism within cells.
AMP contains one phosphate group, while ADP contains two phosphate groups, and ATP contains three phosphate groups. When a phosphate group is removed from ATP, energy is released, which can be used to power various cellular processes such as muscle contraction, nerve impulse transmission, and protein synthesis. The reverse reaction, in which a phosphate group is added back to ADP or AMP to form ATP, requires energy input and often involves the breakdown of nutrients such as glucose or fatty acids.
In addition to their role in energy metabolism, adenine nucleotides also serve as precursors for other important molecules, including DNA and RNA, coenzymes, and signaling molecules.
Fibroblasts are specialized cells that play a critical role in the body's immune response and wound healing process. They are responsible for producing and maintaining the extracellular matrix (ECM), which is the non-cellular component present within all tissues and organs, providing structural support and biochemical signals for surrounding cells.
Fibroblasts produce various ECM proteins such as collagens, elastin, fibronectin, and laminins, forming a complex network of fibers that give tissues their strength and flexibility. They also help in the regulation of tissue homeostasis by controlling the turnover of ECM components through the process of remodeling.
In response to injury or infection, fibroblasts become activated and start to proliferate rapidly, migrating towards the site of damage. Here, they participate in the inflammatory response, releasing cytokines and chemokines that attract immune cells to the area. Additionally, they deposit new ECM components to help repair the damaged tissue and restore its functionality.
Dysregulation of fibroblast activity has been implicated in several pathological conditions, including fibrosis (excessive scarring), cancer (where they can contribute to tumor growth and progression), and autoimmune diseases (such as rheumatoid arthritis).
Hydrogen-ion concentration, also known as pH, is a measure of the acidity or basicity of a solution. It is defined as the negative logarithm (to the base 10) of the hydrogen ion activity in a solution. The standard unit of measurement is the pH unit. A pH of 7 is neutral, less than 7 is acidic, and greater than 7 is basic.
In medical terms, hydrogen-ion concentration is important for maintaining homeostasis within the body. For example, in the stomach, a high hydrogen-ion concentration (low pH) is necessary for the digestion of food. However, in other parts of the body such as blood, a high hydrogen-ion concentration can be harmful and lead to acidosis. Conversely, a low hydrogen-ion concentration (high pH) in the blood can lead to alkalosis. Both acidosis and alkalosis can have serious consequences on various organ systems if not corrected.
Pyruvic acid, also known as 2-oxopropanoic acid, is a key metabolic intermediate in both anaerobic and aerobic respiration. It is a carboxylic acid with a ketone functional group, making it a β-ketoacid. In the cytosol, pyruvate is produced from glucose during glycolysis, where it serves as a crucial link between the anaerobic breakdown of glucose and the aerobic process of cellular respiration in the mitochondria.
During low oxygen availability or high energy demands, pyruvate can be converted into lactate through anaerobic glycolysis, allowing for the continued production of ATP (adenosine triphosphate) without oxygen. In the presence of adequate oxygen and functional mitochondria, pyruvate is transported into the mitochondrial matrix where it undergoes oxidative decarboxylation to form acetyl-CoA by the enzyme pyruvate dehydrogenase complex (PDC). This reaction also involves the reduction of NAD+ to NADH and the release of CO2. Acetyl-CoA then enters the citric acid cycle, where it is further oxidized to produce energy in the form of ATP, NADH, FADH2, and GTP (guanosine triphosphate) through a series of enzymatic reactions.
In summary, pyruvic acid is a vital metabolic intermediate that plays a significant role in energy production pathways, connecting glycolysis to both anaerobic and aerobic respiration.
Lactic acid, also known as 2-hydroxypropanoic acid, is a chemical compound that plays a significant role in various biological processes. In the context of medicine and biochemistry, lactic acid is primarily discussed in relation to muscle metabolism and cellular energy production. Here's a medical definition for lactic acid:
Lactic acid (LA): A carboxylic acid with the molecular formula C3H6O3 that plays a crucial role in anaerobic respiration, particularly during strenuous exercise or conditions of reduced oxygen availability. It is formed through the conversion of pyruvate, catalyzed by the enzyme lactate dehydrogenase (LDH), when there is insufficient oxygen to complete the final step of cellular respiration in the Krebs cycle. The accumulation of lactic acid can lead to acidosis and muscle fatigue. Additionally, lactic acid serves as a vital intermediary in various metabolic pathways and is involved in the production of glucose through gluconeogenesis in the liver.
Extracellular signal-regulated mitogen-activated protein kinases (ERKs or Extracellular signal-regulated kinases) are a subfamily of the MAPK (mitogen-activated protein kinase) family, which are serine/threonine protein kinases that regulate various cellular processes such as proliferation, differentiation, migration, and survival in response to extracellular signals.
ERKs are activated by a cascade of phosphorylation events initiated by the binding of growth factors, hormones, or other extracellular molecules to their respective receptors. This activation results in the formation of a complex signaling pathway that involves the sequential activation of several protein kinases, including Ras, Raf, MEK (MAPK/ERK kinase), and ERK.
Once activated, ERKs translocate to the nucleus where they phosphorylate and activate various transcription factors, leading to changes in gene expression that ultimately result in the appropriate cellular response. Dysregulation of the ERK signaling pathway has been implicated in a variety of diseases, including cancer, diabetes, and neurological disorders.
A precipitin test is a type of immunodiagnostic test used to detect and measure the presence of specific antibodies or antigens in a patient's serum. The test is based on the principle of antigen-antibody interaction, where the addition of an antigen to a solution containing its corresponding antibody results in the formation of an insoluble immune complex known as a precipitin.
In this test, a small amount of the patient's serum is added to a solution containing a known antigen or antibody. If the patient has antibodies or antigens that correspond to the added reagent, they will bind and form a visible precipitate. The size and density of the precipitate can be used to quantify the amount of antibody or antigen present in the sample.
Precipitin tests are commonly used in the diagnosis of various infectious diseases, autoimmune disorders, and allergies. They can also be used in forensic science to identify biological samples. However, they have largely been replaced by more modern immunological techniques such as enzyme-linked immunosorbent assays (ELISAs) and radioimmunoassays (RIAs).
A base sequence in the context of molecular biology refers to the specific order of nucleotides in a DNA or RNA molecule. In DNA, these nucleotides are adenine (A), guanine (G), cytosine (C), and thymine (T). In RNA, uracil (U) takes the place of thymine. The base sequence contains genetic information that is transcribed into RNA and ultimately translated into proteins. It is the exact order of these bases that determines the genetic code and thus the function of the DNA or RNA molecule.
Hexokinase is an enzyme that plays a crucial role in the initial step of glucose metabolism, which is the phosphorylation of glucose to form glucose-6-phosphate. This reaction is the first step in most glucose catabolic pathways, including glycolysis, pentose phosphate pathway, and glycogen synthesis.
Hexokinase has a high affinity for glucose, meaning it can bind and phosphorylate glucose even at low concentrations. This property makes hexokinase an important regulator of glucose metabolism in cells. There are four isoforms of hexokinase (I-IV) found in different tissues, with hexokinase IV (also known as glucokinase) being primarily expressed in the liver and pancreas.
In summary, hexokinase is a vital enzyme involved in glucose metabolism, catalyzing the conversion of glucose to glucose-6-phosphate, and playing a crucial role in regulating cellular energy homeostasis.
Amobarbital is a barbiturate drug that is primarily used as a sedative and sleep aid. It works by depressing the central nervous system, which can lead to relaxation, drowsiness, and reduced anxiety. Amobarbital is also sometimes used as an anticonvulsant to help control seizures.
Like other barbiturates, amobarbital has a high potential for abuse and addiction, and it can be dangerous or even fatal when taken in large doses or mixed with alcohol or other drugs. It is typically prescribed only for short-term use due to the risk of tolerance and dependence.
It's important to note that the use of barbiturates like amobarbital has declined in recent years due to the development of safer and more effective alternatives, such as benzodiazepines and non-benzodiazepine sleep aids.
Insulin is a hormone produced by the beta cells of the pancreatic islets, primarily in response to elevated levels of glucose in the circulating blood. It plays a crucial role in regulating blood glucose levels and facilitating the uptake and utilization of glucose by peripheral tissues, such as muscle and adipose tissue, for energy production and storage. Insulin also inhibits glucose production in the liver and promotes the storage of excess glucose as glycogen or triglycerides.
Deficiency in insulin secretion or action leads to impaired glucose regulation and can result in conditions such as diabetes mellitus, characterized by chronic hyperglycemia and associated complications. Exogenous insulin is used as a replacement therapy in individuals with diabetes to help manage their blood glucose levels and prevent long-term complications.
Mitogen-activated protein kinase (MAPK) signaling system is a crucial pathway for the transmission and regulation of various cellular responses in eukaryotic cells. It plays a significant role in several biological processes, including proliferation, differentiation, apoptosis, inflammation, and stress response. The MAPK cascade consists of three main components: MAP kinase kinase kinase (MAP3K or MEKK), MAP kinase kinase (MAP2K or MEK), and MAP kinase (MAPK).
The signaling system is activated by various extracellular stimuli, such as growth factors, cytokines, hormones, and stress signals. These stimuli initiate a phosphorylation cascade that ultimately leads to the activation of MAPKs. The activated MAPKs then translocate into the nucleus and regulate gene expression by phosphorylating various transcription factors and other regulatory proteins.
There are four major MAPK families: extracellular signal-regulated kinases (ERK1/2), c-Jun N-terminal kinases (JNK1/2/3), p38 MAPKs (p38α/β/γ/δ), and ERK5. Each family has distinct functions, substrates, and upstream activators. Dysregulation of the MAPK signaling system can lead to various diseases, including cancer, diabetes, cardiovascular diseases, and neurological disorders. Therefore, understanding the molecular mechanisms underlying this pathway is crucial for developing novel therapeutic strategies.
Immunoprecipitation (IP) is a research technique used in molecular biology and immunology to isolate specific antigens or antibodies from a mixture. It involves the use of an antibody that recognizes and binds to a specific antigen, which is then precipitated out of solution using various methods, such as centrifugation or chemical cross-linking.
In this technique, an antibody is first incubated with a sample containing the antigen of interest. The antibody specifically binds to the antigen, forming an immune complex. This complex can then be captured by adding protein A or G agarose beads, which bind to the constant region of the antibody. The beads are then washed to remove any unbound proteins, leaving behind the precipitated antigen-antibody complex.
Immunoprecipitation is a powerful tool for studying protein-protein interactions, post-translational modifications, and signal transduction pathways. It can also be used to detect and quantify specific proteins in biological samples, such as cells or tissues, and to identify potential biomarkers of disease.
Substrate specificity in the context of medical biochemistry and enzymology refers to the ability of an enzyme to selectively bind and catalyze a chemical reaction with a particular substrate (or a group of similar substrates) while discriminating against other molecules that are not substrates. This specificity arises from the three-dimensional structure of the enzyme, which has evolved to match the shape, charge distribution, and functional groups of its physiological substrate(s).
Substrate specificity is a fundamental property of enzymes that enables them to carry out highly selective chemical transformations in the complex cellular environment. The active site of an enzyme, where the catalysis takes place, has a unique conformation that complements the shape and charge distribution of its substrate(s). This ensures efficient recognition, binding, and conversion of the substrate into the desired product while minimizing unwanted side reactions with other molecules.
Substrate specificity can be categorized as:
1. Absolute specificity: An enzyme that can only act on a single substrate or a very narrow group of structurally related substrates, showing no activity towards any other molecule.
2. Group specificity: An enzyme that prefers to act on a particular functional group or class of compounds but can still accommodate minor structural variations within the substrate.
3. Broad or promiscuous specificity: An enzyme that can act on a wide range of structurally diverse substrates, albeit with varying catalytic efficiencies.
Understanding substrate specificity is crucial for elucidating enzymatic mechanisms, designing drugs that target specific enzymes or pathways, and developing biotechnological applications that rely on the controlled manipulation of enzyme activities.
Sodium cyanide is a highly toxic chemical compound with the formula NaCN. It is a white solid that is readily soluble in water, and it has a bitter, almond-like odor that some people can detect. Sodium cyanide is used in various industrial processes, including metal cleaning and electroplating, but it is perhaps best known as a poison.
Cyanide ions (CN-) are extremely toxic because they bind to the ferric iron (Fe3+) in cytochrome c oxidase, a crucial enzyme in the mitochondria that is responsible for cellular respiration and energy production. When cyanide ions bind to this enzyme, it becomes unable to function, leading to a rapid depletion of ATP (adenosine triphosphate) and an accumulation of lactic acid, which can cause metabolic acidosis, coma, and death within minutes to hours.
It is important to note that sodium cyanide should be handled with extreme care and only by trained professionals who are familiar with its hazards and proper safety protocols. Exposure to this compound can cause severe health effects, including respiratory failure, convulsions, and cardiac arrest.
A dose-response relationship in the context of drugs refers to the changes in the effects or symptoms that occur as the dose of a drug is increased or decreased. Generally, as the dose of a drug is increased, the severity or intensity of its effects also increases. Conversely, as the dose is decreased, the effects of the drug become less severe or may disappear altogether.
The dose-response relationship is an important concept in pharmacology and toxicology because it helps to establish the safe and effective dosage range for a drug. By understanding how changes in the dose of a drug affect its therapeutic and adverse effects, healthcare providers can optimize treatment plans for their patients while minimizing the risk of harm.
The dose-response relationship is typically depicted as a curve that shows the relationship between the dose of a drug and its effect. The shape of the curve may vary depending on the drug and the specific effect being measured. Some drugs may have a steep dose-response curve, meaning that small changes in the dose can result in large differences in the effect. Other drugs may have a more gradual dose-response curve, where larger changes in the dose are needed to produce significant effects.
In addition to helping establish safe and effective dosages, the dose-response relationship is also used to evaluate the potential therapeutic benefits and risks of new drugs during clinical trials. By systematically testing different doses of a drug in controlled studies, researchers can identify the optimal dosage range for the drug and assess its safety and efficacy.
Sodium azide is a chemical compound with the formula NaN3. Medically, it is not used as a treatment, but it can be found in some pharmaceutical and laboratory settings. It is a white crystalline powder that is highly soluble in water and has a relatively low melting point.
Sodium azide is well known for its ability to release nitrogen gas upon decomposition, which makes it useful as a propellant in airbags and as a preservative in laboratory settings to prevent bacterial growth. However, this property also makes it highly toxic to both animals and humans if ingested or inhaled, as it can cause rapid respiratory failure due to the release of nitrogen gas in the body. Therefore, it should be handled with great care and appropriate safety measures.
Protein transport, in the context of cellular biology, refers to the process by which proteins are actively moved from one location to another within or between cells. This is a crucial mechanism for maintaining proper cell function and regulation.
Intracellular protein transport involves the movement of proteins within a single cell. Proteins can be transported across membranes (such as the nuclear envelope, endoplasmic reticulum, Golgi apparatus, or plasma membrane) via specialized transport systems like vesicles and transport channels.
Intercellular protein transport refers to the movement of proteins from one cell to another, often facilitated by exocytosis (release of proteins in vesicles) and endocytosis (uptake of extracellular substances via membrane-bound vesicles). This is essential for communication between cells, immune response, and other physiological processes.
It's important to note that any disruption in protein transport can lead to various diseases, including neurological disorders, cancer, and metabolic conditions.
Isoenzymes, also known as isoforms, are multiple forms of an enzyme that catalyze the same chemical reaction but differ in their amino acid sequence, structure, and/or kinetic properties. They are encoded by different genes or alternative splicing of the same gene. Isoenzymes can be found in various tissues and organs, and they play a crucial role in biological processes such as metabolism, detoxification, and cell signaling. Measurement of isoenzyme levels in body fluids (such as blood) can provide valuable diagnostic information for certain medical conditions, including tissue damage, inflammation, and various diseases.
Mitochondrial membranes refer to the double-layered structure that surrounds the mitochondrion, an organelle found in the cells of most eukaryotes. The outer mitochondrial membrane is a smooth, porous membrane that allows small molecules and ions to pass through freely, while the inner mitochondrial membrane is highly folded and selectively permeable, controlling the movement of larger molecules and maintaining the electrochemical gradient necessary for ATP synthesis. The space between the two membranes is called the intermembrane space, and the space within the inner membrane is called the matrix. Together, these membranes play a crucial role in energy production, metabolism, and cellular homeostasis.
Genetic transcription is the process by which the information in a strand of DNA is used to create a complementary RNA molecule. This process is the first step in gene expression, where the genetic code in DNA is converted into a form that can be used to produce proteins or functional RNAs.
During transcription, an enzyme called RNA polymerase binds to the DNA template strand and reads the sequence of nucleotide bases. As it moves along the template, it adds complementary RNA nucleotides to the growing RNA chain, creating a single-stranded RNA molecule that is complementary to the DNA template strand. Once transcription is complete, the RNA molecule may undergo further processing before it can be translated into protein or perform its functional role in the cell.
Transcription can be either "constitutive" or "regulated." Constitutive transcription occurs at a relatively constant rate and produces essential proteins that are required for basic cellular functions. Regulated transcription, on the other hand, is subject to control by various intracellular and extracellular signals, allowing cells to respond to changing environmental conditions or developmental cues.
'Tumor cells, cultured' refers to the process of removing cancerous cells from a tumor and growing them in controlled laboratory conditions. This is typically done by isolating the tumor cells from a patient's tissue sample, then placing them in a nutrient-rich environment that promotes their growth and multiplication.
The resulting cultured tumor cells can be used for various research purposes, including the study of cancer biology, drug development, and toxicity testing. They provide a valuable tool for researchers to better understand the behavior and characteristics of cancer cells outside of the human body, which can lead to the development of more effective cancer treatments.
It is important to note that cultured tumor cells may not always behave exactly the same way as they do in the human body, so findings from cell culture studies must be validated through further research, such as animal models or clinical trials.
Cyclic adenosine monophosphate (cAMP) is a key secondary messenger in many biological processes, including the regulation of metabolism, gene expression, and cellular excitability. It is synthesized from adenosine triphosphate (ATP) by the enzyme adenylyl cyclase and is degraded by the enzyme phosphodiesterase.
In the body, cAMP plays a crucial role in mediating the effects of hormones and neurotransmitters on target cells. For example, when a hormone binds to its receptor on the surface of a cell, it can activate a G protein, which in turn activates adenylyl cyclase to produce cAMP. The increased levels of cAMP then activate various effector proteins, such as protein kinases, which go on to regulate various cellular processes.
Overall, the regulation of cAMP levels is critical for maintaining proper cellular function and homeostasis, and abnormalities in cAMP signaling have been implicated in a variety of diseases, including cancer, diabetes, and neurological disorders.
Adaptor proteins are a type of protein that play a crucial role in intracellular signaling pathways by serving as a link between different components of the signaling complex. Specifically, "signal transducing adaptor proteins" refer to those adaptor proteins that are involved in signal transduction processes, where they help to transmit signals from the cell surface receptors to various intracellular effectors. These proteins typically contain modular domains that allow them to interact with multiple partners, thereby facilitating the formation of large signaling complexes and enabling the integration of signals from different pathways.
Signal transducing adaptor proteins can be classified into several families based on their structural features, including the Src homology 2 (SH2) domain, the Src homology 3 (SH3) domain, and the phosphotyrosine-binding (PTB) domain. These domains enable the adaptor proteins to recognize and bind to specific motifs on other signaling molecules, such as receptor tyrosine kinases, G protein-coupled receptors, and cytokine receptors.
One well-known example of a signal transducing adaptor protein is the growth factor receptor-bound protein 2 (Grb2), which contains an SH2 domain that binds to phosphotyrosine residues on activated receptor tyrosine kinases. Grb2 also contains an SH3 domain that interacts with proline-rich motifs on other signaling proteins, such as the guanine nucleotide exchange factor SOS. This interaction facilitates the activation of the Ras small GTPase and downstream signaling pathways involved in cell growth, differentiation, and survival.
Overall, signal transducing adaptor proteins play a critical role in regulating various cellular processes by modulating intracellular signaling pathways in response to extracellular stimuli. Dysregulation of these proteins has been implicated in various diseases, including cancer and inflammatory disorders.
Intracellular membranes refer to the membrane structures that exist within a eukaryotic cell (excluding bacteria and archaea, which are prokaryotic and do not have intracellular membranes). These membranes compartmentalize the cell, creating distinct organelles or functional regions with specific roles in various cellular processes.
Major types of intracellular membranes include:
1. Nuclear membrane (nuclear envelope): A double-membraned structure that surrounds and protects the genetic material within the nucleus. It consists of an outer and inner membrane, perforated by nuclear pores that regulate the transport of molecules between the nucleus and cytoplasm.
2. Endoplasmic reticulum (ER): An extensive network of interconnected tubules and sacs that serve as a major site for protein folding, modification, and lipid synthesis. The ER has two types: rough ER (with ribosomes on its surface) and smooth ER (without ribosomes).
3. Golgi apparatus/Golgi complex: A series of stacked membrane-bound compartments that process, sort, and modify proteins and lipids before they are transported to their final destinations within the cell or secreted out of the cell.
4. Lysosomes: Membrane-bound organelles containing hydrolytic enzymes for breaking down various biomolecules (proteins, carbohydrates, lipids, and nucleic acids) in the process called autophagy or from outside the cell via endocytosis.
5. Peroxisomes: Single-membrane organelles involved in various metabolic processes, such as fatty acid oxidation and detoxification of harmful substances like hydrogen peroxide.
6. Vacuoles: Membrane-bound compartments that store and transport various molecules, including nutrients, waste products, and enzymes. Plant cells have a large central vacuole for maintaining turgor pressure and storing metabolites.
7. Mitochondria: Double-membraned organelles responsible for generating energy (ATP) through oxidative phosphorylation and other metabolic processes, such as the citric acid cycle and fatty acid synthesis.
8. Chloroplasts: Double-membraned organelles found in plant cells that convert light energy into chemical energy during photosynthesis, producing oxygen and organic compounds (glucose) from carbon dioxide and water.
9. Endoplasmic reticulum (ER): A network of interconnected membrane-bound tubules involved in protein folding, modification, and transport; it is divided into two types: rough ER (with ribosomes on the surface) and smooth ER (without ribosomes).
10. Nucleus: Double-membraned organelle containing genetic material (DNA) and associated proteins involved in replication, transcription, RNA processing, and DNA repair. The nuclear membrane separates the nucleoplasm from the cytoplasm and contains nuclear pores for transporting molecules between the two compartments.
COS cells are a type of cell line that are commonly used in molecular biology and genetic research. The name "COS" is an acronym for "CV-1 in Origin," as these cells were originally derived from the African green monkey kidney cell line CV-1. COS cells have been modified through genetic engineering to express high levels of a protein called SV40 large T antigen, which allows them to efficiently take up and replicate exogenous DNA.
There are several different types of COS cells that are commonly used in research, including COS-1, COS-3, and COS-7 cells. These cells are widely used for the production of recombinant proteins, as well as for studies of gene expression, protein localization, and signal transduction.
It is important to note that while COS cells have been a valuable tool in scientific research, they are not without their limitations. For example, because they are derived from monkey kidney cells, there may be differences in the way that human genes are expressed or regulated in these cells compared to human cells. Additionally, because COS cells express SV40 large T antigen, they may have altered cell cycle regulation and other phenotypic changes that could affect experimental results. Therefore, it is important to carefully consider the choice of cell line when designing experiments and interpreting results.
An azide is a chemical compound that contains the functional group -N=N+=N-, which consists of three nitrogen atoms joined by covalent bonds. In organic chemistry, azides are often used as reagents in various chemical reactions, such as the azide-alkyne cycloaddition (also known as the "click reaction").
In medical terminology, azides may refer to a class of drugs that contain an azido group and are used for their pharmacological effects. For example, sodium nitroprusside is a vasodilator drug that contains an azido group and is used to treat hypertensive emergencies.
However, it's worth noting that azides can also be toxic and potentially explosive under certain conditions, so they must be handled with care in laboratory settings.
Succinate dehydrogenase (SDH) is an enzyme complex that plays a crucial role in the process of cellular respiration, specifically in the citric acid cycle (also known as the Krebs cycle) and the electron transport chain. It is located in the inner mitochondrial membrane of eukaryotic cells.
SDH catalyzes the oxidation of succinate to fumarate, converting it into a molecule of fadaquate in the process. During this reaction, two electrons are transferred from succinate to the FAD cofactor within the SDH enzyme complex, reducing it to FADH2. These electrons are then passed on to ubiquinone (CoQ), which is a mobile electron carrier in the electron transport chain, leading to the generation of ATP, the main energy currency of the cell.
SDH is also known as mitochondrial complex II because it is the second complex in the electron transport chain. Mutations in the genes encoding SDH subunits or associated proteins have been linked to various human diseases, including hereditary paragangliomas, pheochromocytomas, gastrointestinal stromal tumors (GISTs), and some forms of neurodegenerative disorders.
The liver is a large, solid organ located in the upper right portion of the abdomen, beneath the diaphragm and above the stomach. It plays a vital role in several bodily functions, including:
1. Metabolism: The liver helps to metabolize carbohydrates, fats, and proteins from the food we eat into energy and nutrients that our bodies can use.
2. Detoxification: The liver detoxifies harmful substances in the body by breaking them down into less toxic forms or excreting them through bile.
3. Synthesis: The liver synthesizes important proteins, such as albumin and clotting factors, that are necessary for proper bodily function.
4. Storage: The liver stores glucose, vitamins, and minerals that can be released when the body needs them.
5. Bile production: The liver produces bile, a digestive juice that helps to break down fats in the small intestine.
6. Immune function: The liver plays a role in the immune system by filtering out bacteria and other harmful substances from the blood.
Overall, the liver is an essential organ that plays a critical role in maintaining overall health and well-being.
p38 Mitogen-Activated Protein Kinases (p38 MAPKs) are a family of conserved serine-threonine protein kinases that play crucial roles in various cellular processes, including inflammation, immune response, differentiation, apoptosis, and stress responses. They are activated by diverse stimuli such as cytokines, ultraviolet radiation, heat shock, osmotic stress, and lipopolysaccharides (LPS).
Once activated, p38 MAPKs phosphorylate and regulate several downstream targets, including transcription factors and other protein kinases. This regulation leads to the expression of genes involved in inflammation, cell cycle arrest, and apoptosis. Dysregulation of p38 MAPK signaling has been implicated in various diseases, such as cancer, neurodegenerative disorders, and autoimmune diseases. Therefore, p38 MAPKs are considered promising targets for developing new therapeutic strategies to treat these conditions.
Small interfering RNA (siRNA) is a type of short, double-stranded RNA molecule that plays a role in the RNA interference (RNAi) pathway. The RNAi pathway is a natural cellular process that regulates gene expression by targeting and destroying specific messenger RNA (mRNA) molecules, thereby preventing the translation of those mRNAs into proteins.
SiRNAs are typically 20-25 base pairs in length and are generated from longer double-stranded RNA precursors called hairpin RNAs or dsRNAs by an enzyme called Dicer. Once generated, siRNAs associate with a protein complex called the RNA-induced silencing complex (RISC), which uses one strand of the siRNA (the guide strand) to recognize and bind to complementary sequences in the target mRNA. The RISC then cleaves the target mRNA, leading to its degradation and the inhibition of protein synthesis.
SiRNAs have emerged as a powerful tool for studying gene function and have shown promise as therapeutic agents for a variety of diseases, including viral infections, cancer, and genetic disorders. However, their use as therapeutics is still in the early stages of development, and there are challenges associated with delivering siRNAs to specific cells and tissues in the body.
Cell cycle proteins are a group of regulatory proteins that control the progression of the cell cycle, which is the series of events that take place in a eukaryotic cell leading to its division and duplication. These proteins can be classified into several categories based on their functions during different stages of the cell cycle.
The major groups of cell cycle proteins include:
1. Cyclin-dependent kinases (CDKs): CDKs are serine/threonine protein kinases that regulate key transitions in the cell cycle. They require binding to a regulatory subunit called cyclin to become active. Different CDK-cyclin complexes are activated at different stages of the cell cycle.
2. Cyclins: Cyclins are a family of regulatory proteins that bind and activate CDKs. Their levels fluctuate throughout the cell cycle, with specific cyclins expressed during particular phases. For example, cyclin D is important for the G1 to S phase transition, while cyclin B is required for the G2 to M phase transition.
3. CDK inhibitors (CKIs): CKIs are regulatory proteins that bind to and inhibit CDKs, thereby preventing their activation. CKIs can be divided into two main families: the INK4 family and the Cip/Kip family. INK4 family members specifically inhibit CDK4 and CDK6, while Cip/Kip family members inhibit a broader range of CDKs.
4. Anaphase-promoting complex/cyclosome (APC/C): APC/C is an E3 ubiquitin ligase that targets specific proteins for degradation by the 26S proteasome. During the cell cycle, APC/C regulates the metaphase to anaphase transition and the exit from mitosis by targeting securin and cyclin B for degradation.
5. Other regulatory proteins: Several other proteins play crucial roles in regulating the cell cycle, such as p53, a transcription factor that responds to DNA damage and arrests the cell cycle, and the polo-like kinases (PLKs), which are involved in various aspects of mitosis.
Overall, cell cycle proteins work together to ensure the proper progression of the cell cycle, maintain genomic stability, and prevent uncontrolled cell growth, which can lead to cancer.
Electron Transport Complex II, also known as succinate-Q oxidoreductase, is a key component of the electron transport chain in the inner mitochondrial membrane. It plays a crucial role in the process of cellular respiration, where it facilitates the transfer of electrons from succinate to ubiquinone (Q), thereby generating a proton gradient across the membrane. This gradient drives the synthesis of ATP, which is the primary source of energy for the cell.
The complex is composed of four core subunits: flavoprotein (Fp), iron-sulfur protein (Ip), cytochrome b (Cyb), and ubiquinone-binding protein (Qp). Electrons from succinate are accepted by FAD in the Fp subunit, and then transferred to the Ip subunit containing iron-sulfur clusters. From there, the electrons are moved to heme groups in the Cyb subunit, and finally passed on to ubiquinone at the Qp subunit.
In addition to its role in the electron transport chain, Complex II has been implicated in various cellular processes such as regulation of reactive oxygen species (ROS) production and modulation of apoptosis. Mutations in genes encoding Complex II subunits have been associated with several human diseases, including neurodegenerative disorders and cancer.
Two-dimensional (2D) gel electrophoresis is a type of electrophoretic technique used in the separation and analysis of complex protein mixtures. This method combines two types of electrophoresis – isoelectric focusing (IEF) and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) – to separate proteins based on their unique physical and chemical properties in two dimensions.
In the first dimension, IEF separates proteins according to their isoelectric points (pI), which is the pH at which a protein carries no net electrical charge. The proteins are focused into narrow zones along a pH gradient established within a gel strip. In the second dimension, SDS-PAGE separates the proteins based on their molecular weights by applying an electric field perpendicular to the first dimension.
The separated proteins form distinct spots on the 2D gel, which can be visualized using various staining techniques. The resulting protein pattern provides valuable information about the composition and modifications of the protein mixture, enabling researchers to identify and compare different proteins in various samples. Two-dimensional gel electrophoresis is widely used in proteomics research, biomarker discovery, and quality control in protein production.
Recombinant fusion proteins are artificially created biomolecules that combine the functional domains or properties of two or more different proteins into a single protein entity. They are generated through recombinant DNA technology, where the genes encoding the desired protein domains are linked together and expressed as a single, chimeric gene in a host organism, such as bacteria, yeast, or mammalian cells.
The resulting fusion protein retains the functional properties of its individual constituent proteins, allowing for novel applications in research, diagnostics, and therapeutics. For instance, recombinant fusion proteins can be designed to enhance protein stability, solubility, or immunogenicity, making them valuable tools for studying protein-protein interactions, developing targeted therapies, or generating vaccines against infectious diseases or cancer.
Examples of recombinant fusion proteins include:
1. Etaglunatide (ABT-523): A soluble Fc fusion protein that combines the heavy chain fragment crystallizable region (Fc) of an immunoglobulin with the extracellular domain of the human interleukin-6 receptor (IL-6R). This fusion protein functions as a decoy receptor, neutralizing IL-6 and its downstream signaling pathways in rheumatoid arthritis.
2. Etanercept (Enbrel): A soluble TNF receptor p75 Fc fusion protein that binds to tumor necrosis factor-alpha (TNF-α) and inhibits its proinflammatory activity, making it a valuable therapeutic option for treating autoimmune diseases like rheumatoid arthritis, ankylosing spondylitis, and psoriasis.
3. Abatacept (Orencia): A fusion protein consisting of the extracellular domain of cytotoxic T-lymphocyte antigen 4 (CTLA-4) linked to the Fc region of an immunoglobulin, which downregulates T-cell activation and proliferation in autoimmune diseases like rheumatoid arthritis.
4. Belimumab (Benlysta): A monoclonal antibody that targets B-lymphocyte stimulator (BLyS) protein, preventing its interaction with the B-cell surface receptor and inhibiting B-cell activation in systemic lupus erythematosus (SLE).
5. Romiplostim (Nplate): A fusion protein consisting of a thrombopoietin receptor agonist peptide linked to an immunoglobulin Fc region, which stimulates platelet production in patients with chronic immune thrombocytopenia (ITP).
6. Darbepoetin alfa (Aranesp): A hyperglycosylated erythropoiesis-stimulating protein that functions as a longer-acting form of recombinant human erythropoietin, used to treat anemia in patients with chronic kidney disease or cancer.
7. Palivizumab (Synagis): A monoclonal antibody directed against the F protein of respiratory syncytial virus (RSV), which prevents RSV infection and is administered prophylactically to high-risk infants during the RSV season.
8. Ranibizumab (Lucentis): A recombinant humanized monoclonal antibody fragment that binds and inhibits vascular endothelial growth factor A (VEGF-A), used in the treatment of age-related macular degeneration, diabetic retinopathy, and other ocular disorders.
9. Cetuximab (Erbitux): A chimeric monoclonal antibody that binds to epidermal growth factor receptor (EGFR), used in the treatment of colorectal cancer and head and neck squamous cell carcinoma.
10. Adalimumab (Humira): A fully humanized monoclonal antibody that targets tumor necrosis factor-alpha (TNF-α), used in the treatment of various inflammatory diseases, including rheumatoid arthritis, psoriasis, and Crohn's disease.
11. Bevacizumab (Avastin): A recombinant humanized monoclonal antibody that binds to VEGF-A, used in the treatment of various cancers, including colorectal, lung, breast, and kidney cancer.
12. Trastuzumab (Herceptin): A humanized monoclonal antibody that targets HER2/neu receptor, used in the treatment of breast cancer.
13. Rituximab (Rituxan): A chimeric monoclonal antibody that binds to CD20 antigen on B cells, used in the treatment of non-Hodgkin's lymphoma and rheumatoid arthritis.
14. Palivizumab (Synagis): A humanized monoclonal antibody that binds to the F protein of respiratory syncytial virus, used in the prevention of respiratory syncytial virus infection in high-risk infants.
15. Infliximab (Remicade): A chimeric monoclonal antibody that targets TNF-α, used in the treatment of various inflammatory diseases, including Crohn's disease, ulcerative colitis, rheumatoid arthritis, and ankylosing spondylitis.
16. Natalizumab (Tysabri): A humanized monoclonal antibody that binds to α4β1 integrin, used in the treatment of multiple sclerosis and Crohn's disease.
17. Adalimumab (Humira): A fully human monoclonal antibody that targets TNF-α, used in the treatment of various inflammatory diseases, including rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn's disease, and ulcerative colitis.
18. Golimumab (Simponi): A fully human monoclonal antibody that targets TNF-α, used in the treatment of rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, and ulcerative colitis.
19. Certolizumab pegol (Cimzia): A PEGylated Fab' fragment of a humanized monoclonal antibody that targets TNF-α, used in the treatment of rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, and Crohn's disease.
20. Ustekinumab (Stelara): A fully human monoclonal antibody that targets IL-12 and IL-23, used in the treatment of psoriasis, psoriatic arthritis, and Crohn's disease.
21. Secukinumab (Cosentyx): A fully human monoclonal antibody that targets IL-17A, used in the treatment of psoriasis, psoriatic arthritis, and ankylosing spondylitis.
22. Ixekizumab (Taltz): A fully human monoclonal antibody that targets IL-17A, used in the treatment of psoriasis and psoriatic arthritis.
23. Brodalumab (Siliq): A fully human monoclonal antibody that targets IL-17 receptor A, used in the treatment of psoriasis.
24. Sarilumab (Kevzara): A fully human monoclonal antibody that targets the IL-6 receptor, used in the treatment of rheumatoid arthritis.
25. Tocilizumab (Actemra): A humanized monoclonal antibody that targets the IL-6 receptor, used in the treatment of rheumatoid arthritis, systemic juvenile idiopathic arthritis, polyarticular juvenile idiopathic arthritis, giant cell arteritis, and chimeric antigen receptor T-cell-induced cytokine release syndrome.
26. Siltuximab (Sylvant): A chimeric monoclonal antibody that targets IL-6, used in the treatment of multicentric Castleman disease.
27. Satralizumab (Enspryng): A humanized monoclonal antibody that targets IL-6 receptor alpha, used in the treatment of neuromyelitis optica spectrum disorder.
28. Sirukumab (Plivensia): A human monoclonal antibody that targets IL-6, used in the treatment
AMP-activated protein kinases (AMPK) are a group of heterotrimeric enzymes that play a crucial role in cellular energy homeostasis. They are composed of a catalytic subunit (α) and two regulatory subunits (β and γ). AMPK is activated under conditions of low energy charge, such as ATP depletion, hypoxia, or exercise, through an increase in the AMP:ATP ratio.
Once activated, AMPK phosphorylates and regulates various downstream targets involved in metabolic pathways, including glycolysis, fatty acid oxidation, and protein synthesis. This results in the inhibition of energy-consuming processes and the promotion of energy-producing processes, ultimately helping to restore cellular energy balance.
AMPK has been implicated in a variety of physiological processes, including glucose and lipid metabolism, autophagy, mitochondrial biogenesis, and inflammation. Dysregulation of AMPK activity has been linked to several diseases, such as diabetes, obesity, cancer, and neurodegenerative disorders. Therefore, AMPK is an attractive target for therapeutic interventions in these conditions.
Myosin light chains are regulatory proteins that bind to the myosin head region of myosin molecules, which are involved in muscle contraction. There are two types of myosin light chains, essential and regulatory, that have different functions. The essential light chains are necessary for the assembly and stability of the myosin filaments, while the regulatory light chains control the calcium-sensitive activation of the myosin ATPase activity during muscle contraction. Phosphorylation of the regulatory light chains plays a critical role in regulating muscle contraction and relaxation.
Cell proliferation is the process by which cells increase in number, typically through the process of cell division. In the context of biology and medicine, it refers to the reproduction of cells that makes up living tissue, allowing growth, maintenance, and repair. It involves several stages including the transition from a phase of quiescence (G0 phase) to an active phase (G1 phase), DNA replication in the S phase, and mitosis or M phase, where the cell divides into two daughter cells.
Abnormal or uncontrolled cell proliferation is a characteristic feature of many diseases, including cancer, where deregulated cell cycle control leads to excessive and unregulated growth of cells, forming tumors that can invade surrounding tissues and metastasize to distant sites in the body.
Nuclear proteins are a category of proteins that are primarily found in the nucleus of a eukaryotic cell. They play crucial roles in various nuclear functions, such as DNA replication, transcription, repair, and RNA processing. This group includes structural proteins like lamins, which form the nuclear lamina, and regulatory proteins, such as histones and transcription factors, that are involved in gene expression. Nuclear localization signals (NLS) often help target these proteins to the nucleus by interacting with importin proteins during active transport across the nuclear membrane.
Tetradecanoylphorbol acetate (TPA) is defined as a pharmacological agent that is a derivative of the phorbol ester family. It is a potent tumor promoter and activator of protein kinase C (PKC), a group of enzymes that play a role in various cellular processes such as signal transduction, proliferation, and differentiation. TPA has been widely used in research to study PKC-mediated signaling pathways and its role in cancer development and progression. It is also used in topical treatments for skin conditions such as psoriasis.
Adenylate kinase is an enzyme (EC 2.7.4.3) that catalyzes the reversible transfer of a phosphate group between adenine nucleotides, specifically between ATP and AMP to form two ADP molecules. This reaction plays a crucial role in maintaining the energy charge of the cell by interconverting these important energy currency molecules.
The general reaction catalyzed by adenylate kinase is:
AMP + ATP ↔ 2ADP
This enzyme is widely distributed in various organisms and tissues, including mammalian cells. In humans, there are several isoforms of adenylate kinase, located in different cellular compartments such as the cytosol, mitochondria, and nucleus. These isoforms have distinct roles in maintaining energy homeostasis and protecting cells under stress conditions. Dysregulation of adenylate kinase activity has been implicated in several pathological processes, including neurodegenerative diseases, ischemia-reperfusion injury, and cancer.
Multienzyme complexes are specialized protein structures that consist of multiple enzymes closely associated or bound together, often with other cofactors and regulatory subunits. These complexes facilitate the sequential transfer of substrates along a series of enzymatic reactions, also known as a metabolic pathway. By keeping the enzymes in close proximity, multienzyme complexes enhance reaction efficiency, improve substrate specificity, and maintain proper stoichiometry between different enzymes involved in the pathway. Examples of multienzyme complexes include the pyruvate dehydrogenase complex, the citrate synthase complex, and the fatty acid synthetase complex.
Okadaic acid is a type of toxin that is produced by certain species of marine algae, including Dinophysis and Prorocentrum. It is a potent inhibitor of protein phosphatases 1 and 2A, which are important enzymes that help regulate cellular processes in the body.
Okadaic acid can accumulate in shellfish that feed on these algae, and consumption of contaminated seafood can lead to a serious illness known as diarrhetic shellfish poisoning (DSP). Symptoms of DSP include nausea, vomiting, diarrhea, and abdominal cramps. In severe cases, it can also cause neurological symptoms such as dizziness, disorientation, and tingling or numbness in the lips, tongue, and fingers.
It is important to note that okadaic acid is not only a marine toxin but also used in scientific research as a tool to study the role of protein phosphatases in cellular processes. However, exposure to this compound should be avoided due to its toxic effects.
Oxidative stress is defined as an imbalance between the production of reactive oxygen species (free radicals) and the body's ability to detoxify them or repair the damage they cause. This imbalance can lead to cellular damage, oxidation of proteins, lipids, and DNA, disruption of cellular functions, and activation of inflammatory responses. Prolonged or excessive oxidative stress has been linked to various health conditions, including cancer, cardiovascular diseases, neurodegenerative disorders, and aging-related diseases.
Pyruvate is a negatively charged ion or group of atoms, called anion, with the chemical formula C3H3O3-. It is formed from the decomposition of glucose and other sugars in the process of cellular respiration. Pyruvate plays a crucial role in the metabolic pathways that generate energy for cells.
In the cytoplasm, pyruvate is produced through glycolysis, where one molecule of glucose is broken down into two molecules of pyruvate, releasing energy and producing ATP (adenosine triphosphate) and NADH (reduced nicotinamide adenine dinucleotide).
In the mitochondria, pyruvate can be further metabolized through the citric acid cycle (also known as the Krebs cycle) to produce more ATP. The process involves the conversion of pyruvate into acetyl-CoA, which then enters the citric acid cycle and undergoes a series of reactions that generate energy in the form of ATP, NADH, and FADH2 (reduced flavin adenine dinucleotide).
Overall, pyruvate is an important intermediate in cellular respiration and plays a central role in the production of energy for cells.
"Saccharomyces cerevisiae" is not typically considered a medical term, but it is a scientific name used in the field of microbiology. It refers to a species of yeast that is commonly used in various industrial processes, such as baking and brewing. It's also widely used in scientific research due to its genetic tractability and eukaryotic cellular organization.
However, it does have some relevance to medical fields like medicine and nutrition. For example, certain strains of S. cerevisiae are used as probiotics, which can provide health benefits when consumed. They may help support gut health, enhance the immune system, and even assist in the digestion of certain nutrients.
In summary, "Saccharomyces cerevisiae" is a species of yeast with various industrial and potential medical applications.
Messenger RNA (mRNA) is a type of RNA (ribonucleic acid) that carries genetic information copied from DNA in the form of a series of three-base code "words," each of which specifies a particular amino acid. This information is used by the cell's machinery to construct proteins, a process known as translation. After being transcribed from DNA, mRNA travels out of the nucleus to the ribosomes in the cytoplasm where protein synthesis occurs. Once the protein has been synthesized, the mRNA may be degraded and recycled. Post-transcriptional modifications can also occur to mRNA, such as alternative splicing and addition of a 5' cap and a poly(A) tail, which can affect its stability, localization, and translation efficiency.
Membrane potential is the electrical potential difference across a cell membrane, typically for excitable cells such as nerve and muscle cells. It is the difference in electric charge between the inside and outside of a cell, created by the selective permeability of the cell membrane to different ions. The resting membrane potential of a typical animal cell is around -70 mV, with the interior being negative relative to the exterior. This potential is generated and maintained by the active transport of ions across the membrane, primarily through the action of the sodium-potassium pump. Membrane potentials play a crucial role in many physiological processes, including the transmission of nerve impulses and the contraction of muscle cells.
Cell survival refers to the ability of a cell to continue living and functioning normally, despite being exposed to potentially harmful conditions or treatments. This can include exposure to toxins, radiation, chemotherapeutic drugs, or other stressors that can damage cells or interfere with their normal processes.
In scientific research, measures of cell survival are often used to evaluate the effectiveness of various therapies or treatments. For example, researchers may expose cells to a particular drug or treatment and then measure the percentage of cells that survive to assess its potential therapeutic value. Similarly, in toxicology studies, measures of cell survival can help to determine the safety of various chemicals or substances.
It's important to note that cell survival is not the same as cell proliferation, which refers to the ability of cells to divide and multiply. While some treatments may promote cell survival, they may also inhibit cell proliferation, making them useful for treating diseases such as cancer. Conversely, other treatments may be designed to specifically target and kill cancer cells, even if it means sacrificing some healthy cells in the process.
Antimetabolites are a class of drugs that interfere with the normal metabolic processes of cells, particularly those involved in DNA replication and cell division. They are commonly used as chemotherapeutic agents to treat various types of cancer because many cancer cells divide more rapidly than normal cells. Antimetabolites work by mimicking natural substances needed for cell growth and division, such as nucleotides or amino acids, and getting incorporated into the growing cells' DNA or protein structures, which ultimately leads to the termination of cell division and death of the cancer cells. Examples of antimetabolites include methotrexate, 5-fluorouracil, and capecitabine.
CDC2 protein kinase, also known as cell division cycle 2 or CDK1, is a type of enzyme that plays a crucial role in the regulation of the cell cycle. The cell cycle is the series of events that cells undergo as they grow, replicate their DNA, and divide into two daughter cells.
CDC2 protein kinase is a member of the cyclin-dependent kinase (CDK) family, which are serine/threonine protein kinases that are activated by binding to regulatory subunits called cyclins. CDC2 protein kinase is primarily associated with the regulation of the G2 phase and the entry into mitosis, the stage of the cell cycle where nuclear and cytoplasmic division occur.
CDC2 protein kinase functions by phosphorylating various target proteins, which alters their activity and contributes to the coordination of the different events that occur during the cell cycle. The activity of CDC2 protein kinase is tightly regulated through a variety of mechanisms, including phosphorylation and dephosphorylation, as well as the binding and destruction of cyclin subunits.
Dysregulation of CDC2 protein kinase has been implicated in various human diseases, including cancer, where uncontrolled cell division can lead to the formation of tumors. Therefore, understanding the regulation and function of CDC2 protein kinase is an important area of research in molecular biology and medicine.
Cytosol refers to the liquid portion of the cytoplasm found within a eukaryotic cell, excluding the organelles and structures suspended in it. It is the site of various metabolic activities and contains a variety of ions, small molecules, and enzymes. The cytosol is where many biochemical reactions take place, including glycolysis, protein synthesis, and the regulation of cellular pH. It is also where some organelles, such as ribosomes and vesicles, are located. In contrast to the cytosol, the term "cytoplasm" refers to the entire contents of a cell, including both the cytosol and the organelles suspended within it.
A "knockout" mouse is a genetically engineered mouse in which one or more genes have been deleted or "knocked out" using molecular biology techniques. This allows researchers to study the function of specific genes and their role in various biological processes, as well as potential associations with human diseases. The mice are generated by introducing targeted DNA modifications into embryonic stem cells, which are then used to create a live animal. Knockout mice have been widely used in biomedical research to investigate gene function, disease mechanisms, and potential therapeutic targets.
Arsenates are salts or esters of arsenic acid (AsO4). They contain the anion AsO4(3-), which consists of an arsenic atom bonded to four oxygen atoms in a tetrahedral arrangement. Arsenates can be found in various minerals, and they have been used in pesticides, wood preservatives, and other industrial applications. However, arsenic is highly toxic to humans and animals, so exposure to arsenates should be limited. Long-term exposure to arsenic can cause skin lesions, cancer, and damage to the nervous system, among other health problems.
Trans-activators are proteins that increase the transcriptional activity of a gene or a set of genes. They do this by binding to specific DNA sequences and interacting with the transcription machinery, thereby enhancing the recruitment and assembly of the complexes needed for transcription. In some cases, trans-activators can also modulate the chromatin structure to make the template more accessible to the transcription machinery.
In the context of HIV (Human Immunodeficiency Virus) infection, the term "trans-activator" is often used specifically to refer to the Tat protein. The Tat protein is a viral regulatory protein that plays a critical role in the replication of HIV by activating the transcription of the viral genome. It does this by binding to a specific RNA structure called the Trans-Activation Response Element (TAR) located at the 5' end of all nascent HIV transcripts, and recruiting cellular cofactors that enhance the processivity and efficiency of RNA polymerase II, leading to increased viral gene expression.
I believe there may be some confusion in your question. "Rabbits" is a common name used to refer to the Lagomorpha species, particularly members of the family Leporidae. They are small mammals known for their long ears, strong legs, and quick reproduction.
However, if you're referring to "rabbits" in a medical context, there is a term called "rabbit syndrome," which is a rare movement disorder characterized by repetitive, involuntary movements of the fingers, resembling those of a rabbit chewing. It is also known as "finger-chewing chorea." This condition is usually associated with certain medications, particularly antipsychotics, and typically resolves when the medication is stopped or adjusted.
Biological transport refers to the movement of molecules, ions, or solutes across biological membranes or through cells in living organisms. This process is essential for maintaining homeostasis, regulating cellular functions, and enabling communication between cells. There are two main types of biological transport: passive transport and active transport.
Passive transport does not require the input of energy and includes:
1. Diffusion: The random movement of molecules from an area of high concentration to an area of low concentration until equilibrium is reached.
2. Osmosis: The diffusion of solvent molecules (usually water) across a semi-permeable membrane from an area of lower solute concentration to an area of higher solute concentration.
3. Facilitated diffusion: The assisted passage of polar or charged substances through protein channels or carriers in the cell membrane, which increases the rate of diffusion without consuming energy.
Active transport requires the input of energy (in the form of ATP) and includes:
1. Primary active transport: The direct use of ATP to move molecules against their concentration gradient, often driven by specific transport proteins called pumps.
2. Secondary active transport: The coupling of the movement of one substance down its electrochemical gradient with the uphill transport of another substance, mediated by a shared transport protein. This process is also known as co-transport or counter-transport.
Dicyclohexylcarbodiimide (DCC) is a chemical compound with the formula (C6H11)2NCO. It is a white to off-white solid that is used as a dehydrating agent in organic synthesis, particularly in the formation of peptide bonds. DCC works by activating carboxylic acids to form an active ester intermediate, which can then react with amines to form amides.
It's important to note that Dicyclohexylcarbodiimide is a hazardous chemical and should be handled with appropriate safety precautions, including the use of personal protective equipment (PPE) such as gloves, lab coats, and eye protection. It can cause skin and eye irritation, and prolonged exposure can lead to respiratory problems. Additionally, it can react violently with water and strong oxidizing agents.
It's also important to note that Dicyclohexylcarbodiimide is not a medical term or a substance used in medical treatment, but rather a chemical reagent used in laboratory settings for research purposes.
Valinomycin is not a medical condition or treatment, but rather it is a naturally occurring antibiotic compound that is produced by certain strains of bacteria. Valinomycin is a cyclic depsipeptide, which means it is made up of a ring of amino acids and alcohols.
Valinomycin is known for its ability to selectively bind to potassium ions (K+) with high affinity and transport them across biological membranes. This property makes valinomycin useful in laboratory research as a tool for studying ion transport and membrane permeability. However, it has no direct medical application in humans or animals.
An amino acid substitution is a type of mutation in which one amino acid in a protein is replaced by another. This occurs when there is a change in the DNA sequence that codes for a particular amino acid in a protein. The genetic code is redundant, meaning that most amino acids are encoded by more than one codon (a sequence of three nucleotides). As a result, a single base pair change in the DNA sequence may not necessarily lead to an amino acid substitution. However, if a change does occur, it can have a variety of effects on the protein's structure and function, depending on the nature of the substituted amino acids. Some substitutions may be harmless, while others may alter the protein's activity or stability, leading to disease.
Protein kinase inhibitors (PKIs) are a class of drugs that work by interfering with the function of protein kinases. Protein kinases are enzymes that play a crucial role in many cellular processes by adding a phosphate group to specific proteins, thereby modifying their activity, localization, or interaction with other molecules. This process of adding a phosphate group is known as phosphorylation and is a key mechanism for regulating various cellular functions, including signal transduction, metabolism, and cell division.
In some diseases, such as cancer, protein kinases can become overactive or mutated, leading to uncontrolled cell growth and division. Protein kinase inhibitors are designed to block the activity of these dysregulated kinases, thereby preventing or slowing down the progression of the disease. These drugs can be highly specific, targeting individual protein kinases or families of kinases, making them valuable tools for targeted therapy in cancer and other diseases.
Protein kinase inhibitors can work in various ways to block the activity of protein kinases. Some bind directly to the active site of the enzyme, preventing it from interacting with its substrates. Others bind to allosteric sites, changing the conformation of the enzyme and making it inactive. Still, others target upstream regulators of protein kinases or interfere with their ability to form functional complexes.
Examples of protein kinase inhibitors include imatinib (Gleevec), which targets the BCR-ABL kinase in chronic myeloid leukemia, and gefitinib (Iressa), which inhibits the EGFR kinase in non-small cell lung cancer. These drugs have shown significant clinical benefits in treating these diseases and have become important components of modern cancer therapy.
I'm sorry for any confusion, but "Trityl compounds" are not a recognized medical term or concept in the field of medicine. Trityl groups or compounds are terms used in chemistry and biochemistry to refer to organic compounds that contain a trityl group (Ph3C=), where Ph represents a phenyl group (a benzene ring).
Trityl groups are often used as protecting groups in chemical reactions, particularly in the synthesis of complex carbohydrates, nucleotides, and other biomolecules. They can also be used in various applications such as radiopharmaceuticals for medical imaging. However, they do not have a specific medical definition or relevance on their own.
If you have any questions related to the chemistry or use of trityl compounds, I would recommend consulting a chemistry or biochemistry resource or expert.
C57BL/6 (C57 Black 6) is an inbred strain of laboratory mouse that is widely used in biomedical research. The term "inbred" refers to a strain of animals where matings have been carried out between siblings or other closely related individuals for many generations, resulting in a population that is highly homozygous at most genetic loci.
The C57BL/6 strain was established in 1920 by crossing a female mouse from the dilute brown (DBA) strain with a male mouse from the black strain. The resulting offspring were then interbred for many generations to create the inbred C57BL/6 strain.
C57BL/6 mice are known for their robust health, longevity, and ease of handling, making them a popular choice for researchers. They have been used in a wide range of biomedical research areas, including studies of cancer, immunology, neuroscience, cardiovascular disease, and metabolism.
One of the most notable features of the C57BL/6 strain is its sensitivity to certain genetic modifications, such as the introduction of mutations that lead to obesity or impaired glucose tolerance. This has made it a valuable tool for studying the genetic basis of complex diseases and traits.
Overall, the C57BL/6 inbred mouse strain is an important model organism in biomedical research, providing a valuable resource for understanding the genetic and molecular mechanisms underlying human health and disease.
Potassium Cyanide (C6H5KN) is defined as a white, water-soluble, crystalline salt that is highly toxic. It is used in fumigation, electroplating, and metal cleaning. When combined with acids, it releases the deadly gas hydrogen cyanide. It can cause immediate death by inhibiting cellular respiration. It is also known as Cyanide of Potassium or Potassium Salt of Hydrocyanic Acid.
'Phospho-specific antibodies' are a type of antibody that binds to a specific phosphorylated amino acid residue on a protein. Phosphorylation is a post-translational modification that plays a crucial role in regulating various cellular processes, such as signal transduction, cell cycle progression, and metabolism. The addition of a phosphate group to a protein can change its structure, function, and interactions with other molecules, thereby modulating its activity.
Phospho-specific antibodies are used in research and diagnostic applications to detect and quantify the levels of specific phosphorylated proteins or phosphorylation sites in biological samples. These antibodies are generated by immunizing animals with a synthetic peptide or protein that contains a phosphorylated residue, which stimulates the production of antibodies that recognize and bind to the phosphorylated epitope. Phospho-specific antibodies can be used in various techniques, such as Western blotting, immunoprecipitation, immunofluorescence, and enzyme-linked immunosorbent assays (ELISAs), to study the regulation and function of phosphorylation in cells and tissues.
It is important to note that the specificity and sensitivity of phospho-specific antibodies can vary depending on several factors, such as the quality and purity of the antigen used for immunization, the affinity and cross-reactivity of the generated antibodies, and the experimental conditions used for detection. Therefore, it is essential to validate and optimize the use of phospho-specific antibodies for each application to ensure accurate and reliable results.
Protein Tyrosine Phosphatases (PTPs) are a group of enzymes that play a crucial role in the regulation of various cellular processes, including cell growth, differentiation, and signal transduction. PTPs function by removing phosphate groups from tyrosine residues on proteins, thereby counteracting the effects of tyrosine kinases, which add phosphate groups to tyrosine residues to activate proteins.
PTPs are classified into several subfamilies based on their structure and function, including classical PTPs, dual-specificity PTPs (DSPs), and low molecular weight PTPs (LMW-PTPs). Each subfamily has distinct substrate specificities and regulatory mechanisms.
Classical PTPs are further divided into receptor-like PTPs (RPTPs) and non-receptor PTPs (NRPTPs). RPTPs contain a transmembrane domain and extracellular regions that mediate cell-cell interactions, while NRPTPs are soluble enzymes located in the cytoplasm.
DSPs can dephosphorylate both tyrosine and serine/threonine residues on proteins and play a critical role in regulating various signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway.
LMW-PTPs are a group of small molecular weight PTPs that localize to different cellular compartments, such as the endoplasmic reticulum and mitochondria, and regulate various cellular processes, including protein folding and apoptosis.
Overall, PTPs play a critical role in maintaining the balance of phosphorylation and dephosphorylation events in cells, and dysregulation of PTP activity has been implicated in various diseases, including cancer, diabetes, and neurological disorders.
Focal adhesion protein-tyrosine kinases (FAKs) are a group of non-receptor tyrosine kinases that play crucial roles in the regulation of various cellular processes, including cell adhesion, migration, proliferation, and survival. They are primarily localized at focal adhesions, which are specialized structures formed at the sites of integrin-mediated attachment of cells to the extracellular matrix (ECM).
FAKs consist of two major domains: an N-terminal FERM (4.1 protein, ezrin, radixin, moesin) domain and a C-terminal kinase domain. The FERM domain is responsible for the interaction with various proteins, including integrins, growth factor receptors, and cytoskeletal components, while the kinase domain possesses enzymatic activity that phosphorylates tyrosine residues on target proteins.
FAKs are activated in response to various extracellular signals, such as ECM stiffness, growth factors, and integrin engagement. Once activated, FAKs initiate a cascade of intracellular signaling events that ultimately regulate cell behavior. Dysregulation of FAK signaling has been implicated in several pathological conditions, including cancer, fibrosis, and cardiovascular diseases.
In summary, focal adhesion protein-tyrosine kinases are essential regulators of cellular processes that localize to focal adhesions and modulate intracellular signaling pathways in response to extracellular cues.
Ribosomal Protein S6 Kinases, 90-kDa (RSKs) are a group of serine/threonine protein kinases that play a crucial role in signal transduction pathways linked to cell growth, proliferation, and survival. They are so named because they were initially discovered as protein kinases that phosphorylate the 40S ribosomal protein S6, a component of the ribosome involved in translation regulation.
RSKs consist of four isoforms (RSK1-4) encoded by separate genes but sharing similar structures and functions. They have an N-terminal kinase domain, a C-terminal kinase domain, and a linker region containing several regulatory phosphorylation sites. RSKs are activated through the Ras/MAPK (Mitogen-Activated Protein Kinase) signaling cascade, where Ras activates Raf, which in turn activates MEK, ultimately leading to the activation of ERK. Activated ERK then phosphorylates and activates RSKs by promoting a conformational change that allows for autophosphorylation and full kinase activity.
Once activated, RSKs can phosphorylate various substrates involved in transcriptional regulation, cytoskeletal reorganization, protein synthesis, and cell cycle progression. Dysregulation of RSK signaling has been implicated in several diseases, including cancer, where they contribute to tumor growth, metastasis, and drug resistance. Therefore, RSKs are considered potential therapeutic targets for cancer treatment.
3T3 cells are a type of cell line that is commonly used in scientific research. The name "3T3" is derived from the fact that these cells were developed by treating mouse embryo cells with a chemical called trypsin and then culturing them in a flask at a temperature of 37 degrees Celsius.
Specifically, 3T3 cells are a type of fibroblast, which is a type of cell that is responsible for producing connective tissue in the body. They are often used in studies involving cell growth and proliferation, as well as in toxicity tests and drug screening assays.
One particularly well-known use of 3T3 cells is in the 3T3-L1 cell line, which is a subtype of 3T3 cells that can be differentiated into adipocytes (fat cells) under certain conditions. These cells are often used in studies of adipose tissue biology and obesity.
It's important to note that because 3T3 cells are a type of immortalized cell line, they do not always behave exactly the same way as primary cells (cells that are taken directly from a living organism). As such, researchers must be careful when interpreting results obtained using 3T3 cells and consider any potential limitations or artifacts that may arise due to their use.
14-3-3 proteins are a family of conserved regulatory molecules found in eukaryotic cells. They are involved in various cellular processes, such as signal transduction, cell cycle regulation, and apoptosis (programmed cell death). These proteins bind to specific phosphoserine-containing motifs on their target proteins, thereby modulating their activity, localization, or stability. Dysregulation of 14-3-3 proteins has been implicated in several human diseases, including cancer, neurodegenerative disorders, and diabetes.
Down-regulation is a process that occurs in response to various stimuli, where the number or sensitivity of cell surface receptors or the expression of specific genes is decreased. This process helps maintain homeostasis within cells and tissues by reducing the ability of cells to respond to certain signals or molecules.
In the context of cell surface receptors, down-regulation can occur through several mechanisms:
1. Receptor internalization: After binding to their ligands, receptors can be internalized into the cell through endocytosis. Once inside the cell, these receptors may be degraded or recycled back to the cell surface in smaller numbers.
2. Reduced receptor synthesis: Down-regulation can also occur at the transcriptional level, where the expression of genes encoding for specific receptors is decreased, leading to fewer receptors being produced.
3. Receptor desensitization: Prolonged exposure to a ligand can lead to a decrease in receptor sensitivity or affinity, making it more difficult for the cell to respond to the signal.
In the context of gene expression, down-regulation refers to the decreased transcription and/or stability of specific mRNAs, leading to reduced protein levels. This process can be induced by various factors, including microRNA (miRNA)-mediated regulation, histone modification, or DNA methylation.
Down-regulation is an essential mechanism in many physiological processes and can also contribute to the development of several diseases, such as cancer and neurodegenerative disorders.
Mass spectrometry (MS) is an analytical technique used to identify and quantify the chemical components of a mixture or compound. It works by ionizing the sample, generating charged molecules or fragments, and then measuring their mass-to-charge ratio in a vacuum. The resulting mass spectrum provides information about the molecular weight and structure of the analytes, allowing for identification and characterization.
In simpler terms, mass spectrometry is a method used to determine what chemicals are present in a sample and in what quantities, by converting the chemicals into ions, measuring their masses, and generating a spectrum that shows the relative abundances of each ion type.
Protein biosynthesis is the process by which cells generate new proteins. It involves two major steps: transcription and translation. Transcription is the process of creating a complementary RNA copy of a sequence of DNA. This RNA copy, or messenger RNA (mRNA), carries the genetic information to the site of protein synthesis, the ribosome. During translation, the mRNA is read by transfer RNA (tRNA) molecules, which bring specific amino acids to the ribosome based on the sequence of nucleotides in the mRNA. The ribosome then links these amino acids together in the correct order to form a polypeptide chain, which may then fold into a functional protein. Protein biosynthesis is essential for the growth and maintenance of all living organisms.
The cell cycle is a series of events that take place in a cell leading to its division and duplication. It consists of four main phases: G1 phase, S phase, G2 phase, and M phase.
During the G1 phase, the cell grows in size and synthesizes mRNA and proteins in preparation for DNA replication. In the S phase, the cell's DNA is copied, resulting in two complete sets of chromosomes. During the G2 phase, the cell continues to grow and produces more proteins and organelles necessary for cell division.
The M phase is the final stage of the cell cycle and consists of mitosis (nuclear division) and cytokinesis (cytoplasmic division). Mitosis results in two genetically identical daughter nuclei, while cytokinesis divides the cytoplasm and creates two separate daughter cells.
The cell cycle is regulated by various checkpoints that ensure the proper completion of each phase before progressing to the next. These checkpoints help prevent errors in DNA replication and division, which can lead to mutations and cancer.
Sequence homology, amino acid, refers to the similarity in the order of amino acids in a protein or a portion of a protein between two or more species. This similarity can be used to infer evolutionary relationships and functional similarities between proteins. The higher the degree of sequence homology, the more likely it is that the proteins are related and have similar functions. Sequence homology can be determined through various methods such as pairwise alignment or multiple sequence alignment, which compare the sequences and calculate a score based on the number and type of matching amino acids.
A proton pump is a specialized protein structure that functions as an enzyme, known as a proton pump ATPase, which actively transports hydrogen ions (protons) across a membrane. This process creates a gradient of hydrogen ions, resulting in an electrochemical potential difference, also known as a proton motive force. The main function of proton pumps is to generate and maintain this gradient, which can be used for various purposes, such as driving the synthesis of ATP (adenosine triphosphate) or transporting other molecules against their concentration gradients.
In the context of gastric physiology, the term "proton pump" often refers to the H+/K+-ATPase present in the parietal cells of the stomach. This proton pump is responsible for secreting hydrochloric acid into the stomach lumen, contributing to the digestion and sterilization of ingested food. Inhibiting this specific proton pump with medications like proton pump inhibitors (PPIs) is a common treatment strategy for gastric acid-related disorders such as gastroesophageal reflux disease (GERD), peptic ulcers, and Zollinger-Ellison syndrome.
Paxillin is a adaptor protein that plays a crucial role in the organization of signaling complexes at focal adhesions, which are specialized structures formed at sites of integrin-mediated cell attachment to the extracellular matrix. It contains multiple binding sites for various proteins involved in signal transduction, cytoskeletal organization, and cell adhesion. Paxillin has been implicated in several biological processes such as cell migration, proliferation, differentiation, and survival, and its dysregulation has been associated with the development of various diseases including cancer.
Vanadates are salts or esters of vanadic acid (HVO3), which contains the vanadium(V) ion. They contain the vanadate ion (VO3-), which consists of one vanadium atom and three oxygen atoms. Vanadates have been studied for their potential insulin-mimetic and antidiabetic effects, as well as their possible cardiovascular benefits. However, more research is needed to fully understand their mechanisms of action and potential therapeutic uses in medicine.
Protein Phosphatase 2 (PP2A) is a type of serine/threonine protein phosphatase that plays a crucial role in the regulation of various cellular processes, including signal transduction, cell cycle progression, and metabolism. PP2A is a heterotrimeric enzyme composed of a catalytic subunit (C), a regulatory subunit A (A), and a variable regulatory subunit B (B). The different combinations of the B subunits confer specificity to PP2A, allowing it to regulate a diverse array of cellular targets.
PP2A is responsible for dephosphorylating many proteins that have been previously phosphorylated by protein kinases. This function is essential for maintaining the balance of phosphorylation and dephosphorylation in cells, which is necessary for proper protein function and cell signaling. Dysregulation of PP2A has been implicated in various diseases, including cancer, neurodegenerative disorders, and cardiovascular disease.
Cricetinae is a subfamily of rodents that includes hamsters, gerbils, and relatives. These small mammals are characterized by having short limbs, compact bodies, and cheek pouches for storing food. They are native to various parts of the world, particularly in Europe, Asia, and Africa. Some species are popular pets due to their small size, easy care, and friendly nature. In a medical context, understanding the biology and behavior of Cricetinae species can be important for individuals who keep them as pets or for researchers studying their physiology.
'Escherichia coli' (E. coli) is a type of gram-negative, facultatively anaerobic, rod-shaped bacterium that commonly inhabits the intestinal tract of humans and warm-blooded animals. It is a member of the family Enterobacteriaceae and one of the most well-studied prokaryotic model organisms in molecular biology.
While most E. coli strains are harmless and even beneficial to their hosts, some serotypes can cause various forms of gastrointestinal and extraintestinal illnesses in humans and animals. These pathogenic strains possess virulence factors that enable them to colonize and damage host tissues, leading to diseases such as diarrhea, urinary tract infections, pneumonia, and sepsis.
E. coli is a versatile organism with remarkable genetic diversity, which allows it to adapt to various environmental niches. It can be found in water, soil, food, and various man-made environments, making it an essential indicator of fecal contamination and a common cause of foodborne illnesses. The study of E. coli has contributed significantly to our understanding of fundamental biological processes, including DNA replication, gene regulation, and protein synthesis.
Focal Adhesion Kinase 1 (FAK1), also known as Protein Tyrosine Kinase 2 (PTK2), is a cytoplasmic tyrosine kinase that plays a crucial role in cellular processes such as cell adhesion, migration, and survival. It is recruited to focal adhesions, which are specialized structures that form at the sites of integrin-mediated attachment of the cell to the extracellular matrix (ECM).
FAK1 becomes activated through autophosphorylation upon integrin clustering and ECM binding. Once activated, FAK1 can phosphorylate various downstream substrates, leading to the activation of several signaling pathways that regulate cell behavior. These pathways include the Ras/MAPK, PI3K/AKT, and JNK signaling cascades, which are involved in cell proliferation, survival, and motility.
FAK1 has been implicated in various physiological and pathological processes, including embryonic development, wound healing, angiogenesis, and tumorigenesis. Dysregulation of FAK1 signaling has been associated with several diseases, such as cancer, fibrosis, and neurological disorders. Therefore, FAK1 is considered a potential therapeutic target for the treatment of these conditions.
"Mycobacterium phlei" is not a recognized medical condition or disease. Mycobacterium phlei is actually a species of non-tuberculous mycobacteria (NTM) that is commonly found in the environment, such as in soil and water. It is often used in laboratory settings as a reference strain for mycobacterial identification and research. This bacterium is not known to cause disease in humans and is generally considered to be non-pathogenic.
Protein Phosphatase 1 (PP1) is a type of serine/threonine protein phosphatase that plays a crucial role in the regulation of various cellular processes, including metabolism, signal transduction, and cell cycle progression. PP1 functions by removing phosphate groups from specific serine and threonine residues on target proteins, thereby reversing the effects of protein kinases and controlling protein activity, localization, and stability.
PP1 is a highly conserved enzyme found in eukaryotic cells and is composed of a catalytic subunit associated with one or more regulatory subunits that determine its substrate specificity, subcellular localization, and regulation. The human genome encodes several isoforms of the PP1 catalytic subunit, including PP1α, PP1β/δ, and PP1γ, which share a high degree of sequence similarity and functional redundancy.
PP1 has been implicated in various physiological processes, such as muscle contraction, glycogen metabolism, DNA replication, transcription, and RNA processing. Dysregulation of PP1 activity has been associated with several pathological conditions, including neurodegenerative diseases, cancer, and diabetes. Therefore, understanding the molecular mechanisms that regulate PP1 function is essential for developing novel therapeutic strategies to treat these disorders.
Homeostasis is a fundamental concept in the field of medicine and physiology, referring to the body's ability to maintain a stable internal environment, despite changes in external conditions. It is the process by which biological systems regulate their internal environment to remain in a state of dynamic equilibrium. This is achieved through various feedback mechanisms that involve sensors, control centers, and effectors, working together to detect, interpret, and respond to disturbances in the system.
For example, the body maintains homeostasis through mechanisms such as temperature regulation (through sweating or shivering), fluid balance (through kidney function and thirst), and blood glucose levels (through insulin and glucagon secretion). When homeostasis is disrupted, it can lead to disease or dysfunction in the body.
In summary, homeostasis is the maintenance of a stable internal environment within biological systems, through various regulatory mechanisms that respond to changes in external conditions.
Trialkyltin compounds are a category of organotin (oceanic) chemicals, characterized by the presence of three alkyl groups bonded to a tin atom. The general formula for these compounds is (CnH2n+1)3Sn, where n represents the number of carbon atoms in each alkyl group.
These compounds have been used in various industrial applications such as biocides, heat stabilizers, and PVC plasticizers. However, due to their high toxicity, environmental persistence, and potential bioaccumulation, their use has been restricted or banned in many countries.
Examples of trialkyltin compounds include tributyltin (TBT) and triphenyltin (TPT). TBT was widely used as an antifouling agent in marine paints to prevent the growth of barnacles, algae, and other organisms on ship hulls. However, due to its detrimental effects on marine life, particularly on shellfish and mollusks, its use has been largely phased out.
Trialkyltin compounds can have toxic effects on both aquatic and terrestrial organisms, including humans. They can cause neurological damage, impaired immune function, reproductive issues, and developmental abnormalities in various species.
Cytoplasm is the material within a eukaryotic cell (a cell with a true nucleus) that lies between the nuclear membrane and the cell membrane. It is composed of an aqueous solution called cytosol, in which various organelles such as mitochondria, ribosomes, endoplasmic reticulum, Golgi apparatus, lysosomes, and vacuoles are suspended. Cytoplasm also contains a variety of dissolved nutrients, metabolites, ions, and enzymes that are involved in various cellular processes such as metabolism, signaling, and transport. It is where most of the cell's metabolic activities take place, and it plays a crucial role in maintaining the structure and function of the cell.
I'm sorry for any confusion, but "Venturicidins" is not a recognized term in medical terminology or pharmacology. It appears to be a made-up term, possibly related to "ventricular" (referring to the heart's lower chambers) and "-cidin" (a suffix often used in antibiotics, meaning "killing"). However, without more context, it is impossible to provide an accurate definition or explanation. If you have more information about where this term comes from or its intended meaning, I'd be happy to try to help further!
Magnetic Resonance Spectroscopy (MRS) is a non-invasive diagnostic technique that provides information about the biochemical composition of tissues, including their metabolic state. It is often used in conjunction with Magnetic Resonance Imaging (MRI) to analyze various metabolites within body tissues, such as the brain, heart, liver, and muscles.
During MRS, a strong magnetic field, radio waves, and a computer are used to produce detailed images and data about the concentration of specific metabolites in the targeted tissue or organ. This technique can help detect abnormalities related to energy metabolism, neurotransmitter levels, pH balance, and other biochemical processes, which can be useful for diagnosing and monitoring various medical conditions, including cancer, neurological disorders, and metabolic diseases.
There are different types of MRS, such as Proton (^1^H) MRS, Phosphorus-31 (^31^P) MRS, and Carbon-13 (^13^C) MRS, each focusing on specific elements or metabolites within the body. The choice of MRS technique depends on the clinical question being addressed and the type of information needed for diagnosis or monitoring purposes.
Mitochondrial genes are a type of gene that is located in the DNA (deoxyribonucleic acid) found in the mitochondria, which are small organelles present in the cytoplasm of eukaryotic cells (cells with a true nucleus). Mitochondria are responsible for generating energy for the cell through a process called oxidative phosphorylation.
The human mitochondrial genome is a circular DNA molecule that contains 37 genes, including 13 genes that encode for proteins involved in oxidative phosphorylation, 22 genes that encode for transfer RNAs (tRNAs), and 2 genes that encode for ribosomal RNAs (rRNAs). Mutations in mitochondrial genes can lead to a variety of inherited mitochondrial disorders, which can affect any organ system in the body and can present at any age.
Mitochondrial DNA is maternally inherited, meaning that it is passed down from the mother to her offspring through the egg cell. This is because during fertilization, only the sperm's nucleus enters the egg, while the mitochondria remain outside. As a result, all of an individual's mitochondrial DNA comes from their mother.
RNA interference (RNAi) is a biological process in which RNA molecules inhibit the expression of specific genes. This process is mediated by small RNA molecules, including microRNAs (miRNAs) and small interfering RNAs (siRNAs), that bind to complementary sequences on messenger RNA (mRNA) molecules, leading to their degradation or translation inhibition.
RNAi plays a crucial role in regulating gene expression and defending against foreign genetic elements, such as viruses and transposons. It has also emerged as an important tool for studying gene function and developing therapeutic strategies for various diseases, including cancer and viral infections.
Iodoacetates are salts or esters of iodoacetic acid, an organic compound containing iodine. In medicine, iodoacetates have been used as topical antiseptics and anti-inflammatory agents. However, their use is limited due to potential skin irritation and the availability of safer alternatives.
In a broader context, iodoacetates are also known for their chemical properties. They can act as alkylating agents, which means they can react with proteins and enzymes in living organisms, disrupting their function. This property has been exploited in research to study various cellular processes.
Cytochromes are a type of hemeprotein found in the mitochondria and other cellular membranes of organisms. They contain a heme group, which is a prosthetic group composed of an iron atom surrounded by a porphyrin ring. This structure allows cytochromes to participate in redox reactions, acting as electron carriers in various biological processes.
There are several types of cytochromes, classified based on the type of heme they contain and their absorption spectra. Some of the most well-known cytochromes include:
* Cytochrome c: a small, mobile protein found in the inner mitochondrial membrane that plays a crucial role in the electron transport chain during cellular respiration.
* Cytochrome P450: a large family of enzymes involved in the metabolism of drugs, toxins, and other xenobiotics. They are found in various tissues, including the liver, lungs, and skin.
* Cytochrome b: a component of several electron transport chains, including those found in mitochondria, bacteria, and chloroplasts.
Cytochromes play essential roles in energy production, detoxification, and other metabolic processes, making them vital for the survival and function of living organisms.
A protein subunit refers to a distinct and independently folding polypeptide chain that makes up a larger protein complex. Proteins are often composed of multiple subunits, which can be identical or different, that come together to form the functional unit of the protein. These subunits can interact with each other through non-covalent interactions such as hydrogen bonds, ionic bonds, and van der Waals forces, as well as covalent bonds like disulfide bridges. The arrangement and interaction of these subunits contribute to the overall structure and function of the protein.
Biological transport, active is the process by which cells use energy to move materials across their membranes from an area of lower concentration to an area of higher concentration. This type of transport is facilitated by specialized proteins called transporters or pumps that are located in the cell membrane. These proteins undergo conformational changes to physically carry the molecules through the lipid bilayer of the membrane, often against their concentration gradient.
Active transport requires energy because it works against the natural tendency of molecules to move from an area of higher concentration to an area of lower concentration, a process known as diffusion. Cells obtain this energy in the form of ATP (adenosine triphosphate), which is produced through cellular respiration.
Examples of active transport include the uptake of glucose and amino acids into cells, as well as the secretion of hormones and neurotransmitters. The sodium-potassium pump, which helps maintain resting membrane potential in nerve and muscle cells, is a classic example of an active transporter.
Cell division is the process by which a single eukaryotic cell (a cell with a true nucleus) divides into two identical daughter cells. This complex process involves several stages, including replication of DNA, separation of chromosomes, and division of the cytoplasm. There are two main types of cell division: mitosis and meiosis.
Mitosis is the type of cell division that results in two genetically identical daughter cells. It is a fundamental process for growth, development, and tissue repair in multicellular organisms. The stages of mitosis include prophase, prometaphase, metaphase, anaphase, and telophase, followed by cytokinesis, which divides the cytoplasm.
Meiosis, on the other hand, is a type of cell division that occurs in the gonads (ovaries and testes) during the production of gametes (sex cells). Meiosis results in four genetically unique daughter cells, each with half the number of chromosomes as the parent cell. This process is essential for sexual reproduction and genetic diversity. The stages of meiosis include meiosis I and meiosis II, which are further divided into prophase, prometaphase, metaphase, anaphase, and telophase.
In summary, cell division is the process by which a single cell divides into two daughter cells, either through mitosis or meiosis. This process is critical for growth, development, tissue repair, and sexual reproduction in multicellular organisms.
Fluorescence microscopy is a type of microscopy that uses fluorescent dyes or proteins to highlight and visualize specific components within a sample. In this technique, the sample is illuminated with high-energy light, typically ultraviolet (UV) or blue light, which excites the fluorescent molecules causing them to emit lower-energy, longer-wavelength light, usually visible light in the form of various colors. This emitted light is then collected by the microscope and detected to produce an image.
Fluorescence microscopy has several advantages over traditional brightfield microscopy, including the ability to visualize specific structures or molecules within a complex sample, increased sensitivity, and the potential for quantitative analysis. It is widely used in various fields of biology and medicine, such as cell biology, neuroscience, and pathology, to study the structure, function, and interactions of cells and proteins.
There are several types of fluorescence microscopy techniques, including widefield fluorescence microscopy, confocal microscopy, two-photon microscopy, and total internal reflection fluorescence (TIRF) microscopy, each with its own strengths and limitations. These techniques can provide valuable insights into the behavior of cells and proteins in health and disease.
Molecular weight, also known as molecular mass, is the mass of a molecule. It is expressed in units of atomic mass units (amu) or daltons (Da). Molecular weight is calculated by adding up the atomic weights of each atom in a molecule. It is a useful property in chemistry and biology, as it can be used to determine the concentration of a substance in a solution, or to calculate the amount of a substance that will react with another in a chemical reaction.
"Inbred strains of rats" are genetically identical rodents that have been produced through many generations of brother-sister mating. This results in a high degree of homozygosity, where the genes at any particular locus in the genome are identical in all members of the strain.
Inbred strains of rats are widely used in biomedical research because they provide a consistent and reproducible genetic background for studying various biological phenomena, including the effects of drugs, environmental factors, and genetic mutations on health and disease. Additionally, inbred strains can be used to create genetically modified models of human diseases by introducing specific mutations into their genomes.
Some commonly used inbred strains of rats include the Wistar Kyoto (WKY), Sprague-Dawley (SD), and Fischer 344 (F344) rat strains. Each strain has its own unique genetic characteristics, making them suitable for different types of research.
Gene expression is the process by which the information encoded in a gene is used to synthesize a functional gene product, such as a protein or RNA molecule. This process involves several steps: transcription, RNA processing, and translation. During transcription, the genetic information in DNA is copied into a complementary RNA molecule, known as messenger RNA (mRNA). The mRNA then undergoes RNA processing, which includes adding a cap and tail to the mRNA and splicing out non-coding regions called introns. The resulting mature mRNA is then translated into a protein on ribosomes in the cytoplasm through the process of translation.
The regulation of gene expression is a complex and highly controlled process that allows cells to respond to changes in their environment, such as growth factors, hormones, and stress signals. This regulation can occur at various stages of gene expression, including transcriptional activation or repression, RNA processing, mRNA stability, and translation. Dysregulation of gene expression has been implicated in many diseases, including cancer, genetic disorders, and neurological conditions.
Mitosis is a type of cell division in which the genetic material of a single cell, called the mother cell, is equally distributed into two identical daughter cells. It's a fundamental process that occurs in multicellular organisms for growth, maintenance, and repair, as well as in unicellular organisms for reproduction.
The process of mitosis can be broken down into several stages: prophase, prometaphase, metaphase, anaphase, and telophase. During prophase, the chromosomes condense and become visible, and the nuclear envelope breaks down. In prometaphase, the nuclear membrane is completely disassembled, and the mitotic spindle fibers attach to the chromosomes at their centromeres.
During metaphase, the chromosomes align at the metaphase plate, an imaginary line equidistant from the two spindle poles. In anaphase, sister chromatids are pulled apart by the spindle fibers and move toward opposite poles of the cell. Finally, in telophase, new nuclear envelopes form around each set of chromosomes, and the chromosomes decondense and become less visible.
Mitosis is followed by cytokinesis, a process that divides the cytoplasm of the mother cell into two separate daughter cells. The result of mitosis and cytokinesis is two genetically identical cells, each with the same number and kind of chromosomes as the original parent cell.
Creatine kinase (CK) is a muscle enzyme that is normally present in small amounts in the blood. It is primarily found in tissues that require a lot of energy, such as the heart, brain, and skeletal muscles. When these tissues are damaged or injured, CK is released into the bloodstream, causing the levels to rise.
Creatine kinase exists in several forms, known as isoenzymes, which can be measured in the blood to help identify the location of tissue damage. The three main isoenzymes are:
1. CK-MM: Found primarily in skeletal muscle
2. CK-MB: Found primarily in heart muscle
3. CK-BB: Found primarily in the brain
Elevated levels of creatine kinase, particularly CK-MB, can indicate damage to the heart muscle, such as occurs with a heart attack. Similarly, elevated levels of CK-BB may suggest brain injury or disease. Overall, measuring creatine kinase levels is a useful diagnostic tool for assessing tissue damage and determining the severity of injuries or illnesses.
Reverse Transcriptase Polymerase Chain Reaction (RT-PCR) is a laboratory technique used in molecular biology to amplify and detect specific DNA sequences. This technique is particularly useful for the detection and quantification of RNA viruses, as well as for the analysis of gene expression.
The process involves two main steps: reverse transcription and polymerase chain reaction (PCR). In the first step, reverse transcriptase enzyme is used to convert RNA into complementary DNA (cDNA) by reading the template provided by the RNA molecule. This cDNA then serves as a template for the PCR amplification step.
In the second step, the PCR reaction uses two primers that flank the target DNA sequence and a thermostable polymerase enzyme to repeatedly copy the targeted cDNA sequence. The reaction mixture is heated and cooled in cycles, allowing the primers to anneal to the template, and the polymerase to extend the new strand. This results in exponential amplification of the target DNA sequence, making it possible to detect even small amounts of RNA or cDNA.
RT-PCR is a sensitive and specific technique that has many applications in medical research and diagnostics, including the detection of viruses such as HIV, hepatitis C virus, and SARS-CoV-2 (the virus that causes COVID-19). It can also be used to study gene expression, identify genetic mutations, and diagnose genetic disorders.
Mitogen-Activated Protein Kinase Kinases (MAP2K or MEK) are a group of protein kinases that play a crucial role in intracellular signal transduction pathways. They are so named because they are activated by mitogens, which are substances that stimulate cell division, and other extracellular signals.
MAP2Ks are positioned upstream of the Mitogen-Activated Protein Kinases (MAPK) in a three-tiered kinase cascade. Once activated, MAP2Ks phosphorylate and activate MAPKs, which then go on to regulate various cellular processes such as proliferation, differentiation, survival, and apoptosis.
There are several subfamilies of MAP2Ks, including MEK1/2, MEK3/6 (also known as MKK3/6), MEK4/7 (also known as MKK4/7), and MEK5. Each MAP2K is specific to activating a particular MAPK, and they are activated by different MAP3Ks (MAP kinase kinase kinases) in response to various extracellular signals.
Dysregulation of the MAPK/MAP2K signaling pathways has been implicated in numerous diseases, including cancer, cardiovascular disease, and neurological disorders. Therefore, targeting these pathways with therapeutic agents has emerged as a promising strategy for treating various diseases.
Saccharomyces cerevisiae proteins are the proteins that are produced by the budding yeast, Saccharomyces cerevisiae. This organism is a single-celled eukaryote that has been widely used as a model organism in scientific research for many years due to its relatively simple genetic makeup and its similarity to higher eukaryotic cells.
The genome of Saccharomyces cerevisiae has been fully sequenced, and it is estimated to contain approximately 6,000 genes that encode proteins. These proteins play a wide variety of roles in the cell, including catalyzing metabolic reactions, regulating gene expression, maintaining the structure of the cell, and responding to environmental stimuli.
Many Saccharomyces cerevisiae proteins have human homologs and are involved in similar biological processes, making this organism a valuable tool for studying human disease. For example, many of the proteins involved in DNA replication, repair, and recombination in yeast have human counterparts that are associated with cancer and other diseases. By studying these proteins in yeast, researchers can gain insights into their function and regulation in humans, which may lead to new treatments for disease.
Aerobiosis is the process of living, growing, and functioning in the presence of oxygen. It refers to the metabolic processes that require oxygen to break down nutrients and produce energy in cells. This is in contrast to anaerobiosis, which is the ability to live and grow in the absence of oxygen.
In medical terms, aerobiosis is often used to describe the growth of microorganisms, such as bacteria and fungi, that require oxygen to survive and multiply. These organisms are called aerobic organisms, and they play an important role in many biological processes, including decomposition and waste breakdown.
However, some microorganisms are unable to grow in the presence of oxygen and are instead restricted to environments where oxygen is absent or limited. These organisms are called anaerobic organisms, and their growth and metabolism are referred to as anaerobiosis.
The Adenine Nucleotide Translocator 3 (ANT3) is a protein found in the inner mitochondrial membrane. It plays a crucial role in cellular energy metabolism by facilitating the exchange of adenosine diphosphate (ADP) and adenosine triphosphate (ATP) across the mitochondrial membrane.
More specifically, ANT3 transports ATP from the mitochondrial matrix to the cytoplasm, where it can be used for various cellular processes, while simultaneously transporting ADP in the opposite direction. This exchange is essential for maintaining the balance of adenine nucleotides and ensuring the proper functioning of the energy-producing machinery within the mitochondria.
ANT3 has also been implicated in the regulation of apoptosis or programmed cell death, as it can interact with pro-apoptotic proteins to facilitate the release of cytochrome c from the mitochondria, leading to caspase activation and cell death. Dysregulation of ANT3 function has been linked to various pathological conditions, including neurodegenerative diseases and cancer.
DNA primers are short single-stranded DNA molecules that serve as a starting point for DNA synthesis. They are typically used in laboratory techniques such as the polymerase chain reaction (PCR) and DNA sequencing. The primer binds to a complementary sequence on the DNA template through base pairing, providing a free 3'-hydroxyl group for the DNA polymerase enzyme to add nucleotides and synthesize a new strand of DNA. This allows for specific and targeted amplification or analysis of a particular region of interest within a larger DNA molecule.
HEK293 cells, also known as human embryonic kidney 293 cells, are a line of cells used in scientific research. They were originally derived from human embryonic kidney cells and have been adapted to grow in a lab setting. HEK293 cells are widely used in molecular biology and biochemistry because they can be easily transfected (a process by which DNA is introduced into cells) and highly express foreign genes. As a result, they are often used to produce proteins for structural and functional studies. It's important to note that while HEK293 cells are derived from human tissue, they have been grown in the lab for many generations and do not retain the characteristics of the original embryonic kidney cells.
The brain is the central organ of the nervous system, responsible for receiving and processing sensory information, regulating vital functions, and controlling behavior, movement, and cognition. It is divided into several distinct regions, each with specific functions:
1. Cerebrum: The largest part of the brain, responsible for higher cognitive functions such as thinking, learning, memory, language, and perception. It is divided into two hemispheres, each controlling the opposite side of the body.
2. Cerebellum: Located at the back of the brain, it is responsible for coordinating muscle movements, maintaining balance, and fine-tuning motor skills.
3. Brainstem: Connects the cerebrum and cerebellum to the spinal cord, controlling vital functions such as breathing, heart rate, and blood pressure. It also serves as a relay center for sensory information and motor commands between the brain and the rest of the body.
4. Diencephalon: A region that includes the thalamus (a major sensory relay station) and hypothalamus (regulates hormones, temperature, hunger, thirst, and sleep).
5. Limbic system: A group of structures involved in emotional processing, memory formation, and motivation, including the hippocampus, amygdala, and cingulate gyrus.
The brain is composed of billions of interconnected neurons that communicate through electrical and chemical signals. It is protected by the skull and surrounded by three layers of membranes called meninges, as well as cerebrospinal fluid that provides cushioning and nutrients.
Ubiquinone, also known as coenzyme Q10 (CoQ10), is a lipid-soluble benzoquinone that plays a crucial role in the mitochondrial electron transport chain as an essential component of Complexes I, II, and III. It functions as an electron carrier, assisting in the transfer of electrons from reduced nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) to molecular oxygen during oxidative phosphorylation, thereby contributing to the generation of adenosine triphosphate (ATP), the primary energy currency of the cell.
Additionally, ubiquinone acts as a potent antioxidant in both membranes and lipoproteins, protecting against lipid peroxidation and oxidative damage to proteins and DNA. Its antioxidant properties stem from its ability to donate electrons and regenerate other antioxidants like vitamin E. Ubiquinone is synthesized endogenously in all human cells, with the highest concentrations found in tissues with high energy demands, such as the heart, liver, kidneys, and skeletal muscles.
Deficiency in ubiquinone can result from genetic disorders, aging, or certain medications (such as statins), leading to impaired mitochondrial function and increased oxidative stress. Supplementation with ubiquinone has been explored as a potential therapeutic strategy for various conditions associated with mitochondrial dysfunction and oxidative stress, including cardiovascular diseases, neurodegenerative disorders, and cancer.
Lactates, also known as lactic acid, are compounds that are produced by muscles during intense exercise or other conditions of low oxygen supply. They are formed from the breakdown of glucose in the absence of adequate oxygen to complete the full process of cellular respiration. This results in the production of lactate and a hydrogen ion, which can lead to a decrease in pH and muscle fatigue.
In a medical context, lactates may be measured in the blood as an indicator of tissue oxygenation and metabolic status. Elevated levels of lactate in the blood, known as lactic acidosis, can indicate poor tissue perfusion or hypoxia, and may be seen in conditions such as sepsis, cardiac arrest, and severe shock. It is important to note that lactates are not the primary cause of acidemia (low pH) in lactic acidosis, but rather a marker of the underlying process.
Casein kinases are a family of protein kinases that play important roles in various cellular processes, including signal transduction, cell cycle regulation, and DNA damage repair. These enzymes phosphorylate serine and threonine residues on their target proteins by transferring a phosphate group from ATP to the hydroxyl side chain of these amino acids.
There are several isoforms of casein kinases, including Casein Kinase 1 (CK1) and Casein Kinase 2 (CK2), which differ in their structure, substrate specificity, and cellular functions. CK1 is involved in various signaling pathways, such as the Wnt signaling pathway, and regulates processes such as gene transcription, DNA repair, and circadian rhythm. CK2, on the other hand, is a highly conserved serine/threonine protein kinase that plays a role in many cellular processes, including cell division, apoptosis, and transcriptional regulation.
Dysregulation of casein kinases has been implicated in various diseases, including cancer, neurodegenerative disorders, and cardiovascular disease. Therefore, these enzymes are considered important targets for the development of new therapeutic strategies.
Amino acid motifs are recurring patterns or sequences of amino acids in a protein molecule. These motifs can be identified through various sequence analysis techniques and often have functional or structural significance. They can be as short as two amino acids in length, but typically contain at least three to five residues.
Some common examples of amino acid motifs include:
1. Active site motifs: These are specific sequences of amino acids that form the active site of an enzyme and participate in catalyzing chemical reactions. For example, the catalytic triad in serine proteases consists of three residues (serine, histidine, and aspartate) that work together to hydrolyze peptide bonds.
2. Signal peptide motifs: These are sequences of amino acids that target proteins for secretion or localization to specific organelles within the cell. For example, a typical signal peptide consists of a positively charged n-region, a hydrophobic h-region, and a polar c-region that directs the protein to the endoplasmic reticulum membrane for translocation.
3. Zinc finger motifs: These are structural domains that contain conserved sequences of amino acids that bind zinc ions and play important roles in DNA recognition and regulation of gene expression.
4. Transmembrane motifs: These are sequences of hydrophobic amino acids that span the lipid bilayer of cell membranes and anchor transmembrane proteins in place.
5. Phosphorylation sites: These are specific serine, threonine, or tyrosine residues that can be phosphorylated by protein kinases to regulate protein function.
Understanding amino acid motifs is important for predicting protein structure and function, as well as for identifying potential drug targets in disease-associated proteins.
Epidermal Growth Factor (EGF) is a small polypeptide that plays a significant role in various biological processes, including cell growth, proliferation, differentiation, and survival. It primarily binds to the Epidermal Growth Factor Receptor (EGFR) on the surface of target cells, leading to the activation of intracellular signaling pathways that regulate these functions.
EGF is naturally produced in various tissues, such as the skin, and is involved in wound healing, tissue regeneration, and maintaining the integrity of epithelial tissues. In addition to its physiological roles, EGF has been implicated in several pathological conditions, including cancer, where it can contribute to tumor growth and progression by promoting cell proliferation and survival.
As a result, EGF and its signaling pathways have become targets for therapeutic interventions in various diseases, particularly cancer. Inhibitors of EGFR or downstream signaling components are used in the treatment of several types of malignancies, such as non-small cell lung cancer, colorectal cancer, and head and neck cancer.
Metabolism is the complex network of chemical reactions that occur within our bodies to maintain life. It involves two main types of processes: catabolism, which is the breaking down of molecules to release energy, and anabolism, which is the building up of molecules using energy. These reactions are necessary for the body to grow, reproduce, respond to environmental changes, and repair itself. Metabolism is a continuous process that occurs at the cellular level and is regulated by enzymes, hormones, and other signaling molecules. It is influenced by various factors such as age, genetics, diet, physical activity, and overall health status.
Ribosomal Protein S6 Kinases (RSKs) are a family of serine/threonine protein kinases that play a crucial role in the regulation of cell growth, proliferation, and survival. They are so named because they phosphorylate and regulate the function of the ribosomal protein S6, which is a component of the 40S ribosomal subunit involved in protein synthesis.
RSKs are activated by various signals, including growth factors, hormones, and mitogens, through a cascade of phosphorylation events involving several upstream kinases such as MAPK/ERK kinase (MEK) and extracellular signal-regulated kinase (ERK). Once activated, RSKs phosphorylate a wide range of downstream targets, including transcription factors, regulators of translation, and cytoskeletal proteins, thereby modulating their activities and functions.
There are four isoforms of RSKs in humans, namely RSK1, RSK2, RSK3, and RSK4, which share a common structural organization and functional domains, including an N-terminal kinase domain, a C-terminal kinase domain, and a linker region that contains several regulatory motifs. Dysregulation of RSKs has been implicated in various pathological conditions, including cancer, cardiovascular diseases, neurological disorders, and diabetes, making them attractive targets for therapeutic intervention.
Insulin Receptor Substrate (IRS) proteins are a family of cytoplasmic signaling proteins that play a crucial role in the insulin signaling pathway. There are four main isoforms in humans, namely IRS-1, IRS-2, IRS-3, and IRS-4, which contain several conserved domains for interacting with various signaling molecules.
When insulin binds to its receptor, the intracellular tyrosine kinase domain of the receptor becomes activated and phosphorylates specific tyrosine residues on IRS proteins. This leads to the recruitment and activation of downstream effectors, such as PI3K and Grb2/SOS, which ultimately result in metabolic responses (e.g., glucose uptake, glycogen synthesis) and mitogenic responses (e.g., cell proliferation, differentiation).
Dysregulation of the IRS-mediated insulin signaling pathway has been implicated in several pathological conditions, including insulin resistance, type 2 diabetes, and certain types of cancer.
A peptide fragment is a short chain of amino acids that is derived from a larger peptide or protein through various biological or chemical processes. These fragments can result from the natural breakdown of proteins in the body during regular physiological processes, such as digestion, or they can be produced experimentally in a laboratory setting for research or therapeutic purposes.
Peptide fragments are often used in research to map the structure and function of larger peptides and proteins, as well as to study their interactions with other molecules. In some cases, peptide fragments may also have biological activity of their own and can be developed into drugs or diagnostic tools. For example, certain peptide fragments derived from hormones or neurotransmitters may bind to receptors in the body and mimic or block the effects of the full-length molecule.
Cyclin-dependent kinases (CDKs) are a family of serine/threonine protein kinases that play crucial roles in regulating the cell cycle, transcription, and other cellular processes. They are activated by binding to cyclin proteins, which accumulate and degrade at specific stages of the cell cycle. The activation of CDKs leads to phosphorylation of various downstream target proteins, resulting in the promotion or inhibition of different cell cycle events. Dysregulation of CDKs has been implicated in several human diseases, including cancer, and they are considered important targets for drug development.
Up-regulation is a term used in molecular biology and medicine to describe an increase in the expression or activity of a gene, protein, or receptor in response to a stimulus. This can occur through various mechanisms such as increased transcription, translation, or reduced degradation of the molecule. Up-regulation can have important functional consequences, for example, enhancing the sensitivity or response of a cell to a hormone, neurotransmitter, or drug. It is a normal physiological process that can also be induced by disease or pharmacological interventions.
Adenosine monophosphate (AMP) is a nucleotide that is the monophosphate ester of adenosine, consisting of the nitrogenous base adenine attached to the 1' carbon atom of ribose via a β-N9-glycosidic bond, which in turn is esterified to a phosphate group. It is an important molecule in biological systems as it plays a key role in cellular energy transfer and storage, serving as a precursor to other nucleotides such as ADP and ATP. AMP is also involved in various signaling pathways and can act as a neurotransmitter in the central nervous system.
Gene expression profiling is a laboratory technique used to measure the activity (expression) of thousands of genes at once. This technique allows researchers and clinicians to identify which genes are turned on or off in a particular cell, tissue, or organism under specific conditions, such as during health, disease, development, or in response to various treatments.
The process typically involves isolating RNA from the cells or tissues of interest, converting it into complementary DNA (cDNA), and then using microarray or high-throughput sequencing technologies to determine which genes are expressed and at what levels. The resulting data can be used to identify patterns of gene expression that are associated with specific biological states or processes, providing valuable insights into the underlying molecular mechanisms of diseases and potential targets for therapeutic intervention.
In recent years, gene expression profiling has become an essential tool in various fields, including cancer research, drug discovery, and personalized medicine, where it is used to identify biomarkers of disease, predict patient outcomes, and guide treatment decisions.
JNK (c-Jun N-terminal kinase) Mitogen-Activated Protein Kinases are a subgroup of the Ser/Thr protein kinases that are activated by stress stimuli and play important roles in various cellular processes, including inflammation, apoptosis, and differentiation. They are involved in the regulation of gene expression through phosphorylation of transcription factors such as c-Jun. JNKs are activated by a variety of upstream kinases, including MAP2Ks (MKK4/SEK1 and MKK7), which are in turn activated by MAP3Ks (such as ASK1, MEKK1, MLKs, and TAK1). JNK signaling pathways have been implicated in various diseases, including cancer, neurodegenerative disorders, and inflammatory diseases.
TOR (Target Of Rapamycin) Serine-Threonine Kinases are a family of conserved protein kinases that play crucial roles in the regulation of cell growth, proliferation, and metabolism in response to various environmental cues such as nutrients, growth factors, and energy status. They are named after their ability to phosphorylate serine and threonine residues on target proteins.
Mammalian cells express two distinct TOR kinases, mTORC1 and mTORC2, which have different protein compositions and functions. mTORC1 is rapamycin-sensitive and regulates cell growth, proliferation, and metabolism by phosphorylating downstream targets such as p70S6 kinase and 4E-BP1, thereby controlling protein synthesis, autophagy, and lysosome biogenesis. mTORC2 is rapamycin-insensitive and regulates cell survival, cytoskeleton organization, and metabolism by phosphorylating AGC kinases such as AKT and PKCα.
Dysregulation of TOR Serine-Threonine Kinases has been implicated in various human diseases, including cancer, diabetes, and neurological disorders. Therefore, targeting TOR kinases has emerged as a promising therapeutic strategy for the treatment of these diseases.
Cardiolipins are a type of phospholipid that are primarily found in the inner mitochondrial membrane of cells. They play a crucial role in several important cellular processes, including energy production, apoptosis (programmed cell death), and maintenance of the structural integrity of the mitochondria.
Cardiolipins are unique because they contain four fatty acid chains, whereas most other phospholipids contain only two. This gives cardiolipins a distinctive conical shape that is important for their function in maintaining the curvature and stability of the inner mitochondrial membrane.
Cardiolipins have also been implicated in various diseases, including neurodegenerative disorders, cancer, and bacterial infections. For example, changes in cardiolipin composition or distribution have been linked to mitochondrial dysfunction in Parkinson's disease and other neurological conditions. Additionally, certain bacteria, such as Neisseria gonorrhoeae and Chlamydia trachomatis, can manipulate host cell cardiolipins to facilitate their own survival and replication.
In summary, cardiolipins are essential phospholipids found in the inner mitochondrial membrane that play a critical role in several cellular processes, and have been implicated in various diseases.
Ion channels are specialized transmembrane proteins that form hydrophilic pores or gaps in the lipid bilayer of cell membranes. They regulate the movement of ions (such as sodium, potassium, calcium, and chloride) across the cell membrane by allowing these charged particles to pass through selectively in response to various stimuli, including voltage changes, ligand binding, mechanical stress, or temperature changes. This ion movement is essential for many physiological processes, including electrical signaling, neurotransmission, muscle contraction, and maintenance of resting membrane potential. Ion channels can be categorized based on their activation mechanisms, ion selectivity, and structural features. Dysfunction of ion channels can lead to various diseases, making them important targets for drug development.
Ribosomal Protein S6 (RP S6) is a protein component of the 40S subunit of eukaryotic ribosomes, which are complexes responsible for protein synthesis in cells. Specifically, RP S6 is part of the heterodimer that makes up the head of the 40S subunit.
RP S6 plays a role in regulating translation, the process by which mRNA (messenger RNA) molecules are decoded to produce proteins. It has been found to be involved in the initiation and elongation steps of translation, particularly in response to various cellular signals such as growth factors, hormones, and nutrients.
Phosphorylation of RP S6 is a key regulatory mechanism that modulates its activity during translation. This phosphorylation can be mediated by several kinases, including the p70S6 kinase (p70S6K), which is activated in response to growth factor signaling and nutrient availability.
Abnormalities in RP S6 regulation have been implicated in various diseases, such as cancer, where increased RP S6 phosphorylation has been observed in many tumor types, suggesting a role in promoting cell proliferation and survival.
Pentachlorophenol is not primarily a medical term, but rather a chemical compound with some uses and applications in the medical field. Medically, it's important to understand what pentachlorophenol is due to its potential health implications.
Pentachlorophenol (PCP) is an organochlorine compound that has been widely used as a pesticide, wood preservative, and disinfectant. Its chemical formula is C6HCl5O. It is a white crystalline solid with a distinct, somewhat unpleasant odor. In the environment, pentachlorophenol can be found in soil, water, and air as well as in various organisms, including humans.
Pentachlorophenol has been associated with several potential health risks. It is classified as a probable human carcinogen by the International Agency for Research on Cancer (IARC) and as a possible human carcinogen by the United States Environmental Protection Agency (EPA). Exposure to pentachlorophenol can occur through inhalation, skin contact, or ingestion. Potential health effects include irritation of the skin, eyes, and respiratory tract; damage to the liver and kidneys; neurological issues; and reproductive problems.
In a medical context, pentachlorophenol might be relevant in cases where individuals have been exposed to this compound through occupational or environmental sources. Medical professionals may need to assess potential health risks, diagnose related health issues, and provide appropriate treatment.
"Malonates" is not a recognized medical term. However, in chemistry, malonates refer to salts or esters of malonic acid, a dicarboxylic acid with the formula CH2(COOH)2. Malonic acid and its derivatives have been used in the synthesis of various pharmaceuticals and chemicals, but they are not typically associated with any specific medical condition or treatment. If you have encountered the term "malonates" in a medical context, it may be helpful to provide more information or seek clarification from the source.
Hydrogen peroxide (H2O2) is a colorless, odorless, clear liquid with a slightly sweet taste, although drinking it is harmful and can cause poisoning. It is a weak oxidizing agent and is used as an antiseptic and a bleaching agent. In diluted form, it is used to disinfect wounds and kill bacteria and viruses on the skin; in higher concentrations, it can be used to bleach hair or remove stains from clothing. It is also used as a propellant in rocketry and in certain industrial processes. Chemically, hydrogen peroxide is composed of two hydrogen atoms and two oxygen atoms, and it is structurally similar to water (H2O), with an extra oxygen atom. This gives it its oxidizing properties, as the additional oxygen can be released and used to react with other substances.
Dicumarol is an anticoagulant medication that belongs to a class of compounds known as coumarins. It works by inhibiting the action of vitamin K, which is necessary for the production of certain clotting factors in the liver. This results in a decrease in blood clotting ability and helps prevent the formation of harmful blood clots.
Dicumarol is primarily used to treat and prevent deep vein thrombosis (DVT), pulmonary embolism, and other conditions that may require anticoagulation therapy. It is also used in the management of atrial fibrillation, valvular heart disease, and certain types of heart attacks.
It's important to note that dicumarol has a narrow therapeutic index, meaning that the difference between an effective dose and a toxic dose is relatively small. Therefore, it requires careful monitoring of blood clotting times (INR) to ensure that the drug is working effectively without causing excessive bleeding.
Dicumarol is available in oral form and is typically taken once or twice daily. Common side effects include nausea, vomiting, diarrhea, skin rash, and abnormal liver function tests. Rare but serious side effects include severe bleeding, necrosis of the skin and other tissues, and allergic reactions.
Dicumarol is a prescription medication that should only be used under the guidance of a healthcare professional. It interacts with many other medications and foods, so it's important to inform your doctor about all the drugs you are taking and any dietary changes you may make while on this medication.
Actin is a type of protein that forms part of the contractile apparatus in muscle cells, and is also found in various other cell types. It is a globular protein that polymerizes to form long filaments, which are important for many cellular processes such as cell division, cell motility, and the maintenance of cell shape. In muscle cells, actin filaments interact with another type of protein called myosin to enable muscle contraction. Actins can be further divided into different subtypes, including alpha-actin, beta-actin, and gamma-actin, which have distinct functions and expression patterns in the body.
DNA damage refers to any alteration in the structure or composition of deoxyribonucleic acid (DNA), which is the genetic material present in cells. DNA damage can result from various internal and external factors, including environmental exposures such as ultraviolet radiation, tobacco smoke, and certain chemicals, as well as normal cellular processes such as replication and oxidative metabolism.
Examples of DNA damage include base modifications, base deletions or insertions, single-strand breaks, double-strand breaks, and crosslinks between the two strands of the DNA helix. These types of damage can lead to mutations, genomic instability, and chromosomal aberrations, which can contribute to the development of diseases such as cancer, neurodegenerative disorders, and aging-related conditions.
The body has several mechanisms for repairing DNA damage, including base excision repair, nucleotide excision repair, mismatch repair, and double-strand break repair. However, if the damage is too extensive or the repair mechanisms are impaired, the cell may undergo apoptosis (programmed cell death) to prevent the propagation of potentially harmful mutations.
Deoxyglucose is a glucose molecule that has had one oxygen atom removed, resulting in the absence of a hydroxyl group (-OH) at the 2' position of the carbon chain. It is used in research and medical settings as a metabolic tracer to study glucose uptake and metabolism in cells and organisms.
Deoxyglucose can be taken up by cells through glucose transporters, but it cannot be further metabolized by glycolysis or other glucose-utilizing pathways. This leads to the accumulation of deoxyglucose within the cell, which can interfere with normal cellular processes and cause toxicity in high concentrations.
In medical research, deoxyglucose is sometimes labeled with radioactive isotopes such as carbon-14 or fluorine-18 to create radiolabeled deoxyglucose (FDG), which can be used in positron emission tomography (PET) scans to visualize and measure glucose uptake in tissues. This technique is commonly used in cancer imaging, as tumors often have increased glucose metabolism compared to normal tissue.
Iodoacetic acid is not typically defined in the context of medical terminology, but rather it is a chemical compound with the formula CH2ICO2H. It is a colorless, oily liquid that is used in organic synthesis as an alkylating agent and also has been studied for its potential antibacterial and antifungal properties.
In medical contexts, iodoacetic acid may be mentioned in relation to its use in research or in the discussion of certain chemical reactions that may occur in the body. For example, it can inhibit the enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which plays a crucial role in energy metabolism. However, iodoacetic acid itself is not a medical treatment or therapy.
Subcellular fractions refer to the separation and collection of specific parts or components of a cell, including organelles, membranes, and other structures, through various laboratory techniques such as centrifugation and ultracentrifugation. These fractions can be used in further biochemical and molecular analyses to study the structure, function, and interactions of individual cellular components. Examples of subcellular fractions include nuclear extracts, mitochondrial fractions, microsomal fractions (membrane vesicles), and cytosolic fractions (cytoplasmic extracts).
A phenotype is the physical or biochemical expression of an organism's genes, or the observable traits and characteristics resulting from the interaction of its genetic constitution (genotype) with environmental factors. These characteristics can include appearance, development, behavior, and resistance to disease, among others. Phenotypes can vary widely, even among individuals with identical genotypes, due to differences in environmental influences, gene expression, and genetic interactions.
Cytoskeletal proteins are a type of structural proteins that form the cytoskeleton, which is the internal framework of cells. The cytoskeleton provides shape, support, and structure to the cell, and plays important roles in cell division, intracellular transport, and maintenance of cell shape and integrity.
There are three main types of cytoskeletal proteins: actin filaments, intermediate filaments, and microtubules. Actin filaments are thin, rod-like structures that are involved in muscle contraction, cell motility, and cell division. Intermediate filaments are thicker than actin filaments and provide structural support to the cell. Microtubules are hollow tubes that are involved in intracellular transport, cell division, and maintenance of cell shape.
Cytoskeletal proteins are composed of different subunits that polymerize to form filamentous structures. These proteins can be dynamically assembled and disassembled, allowing cells to change their shape and move. Mutations in cytoskeletal proteins have been linked to various human diseases, including cancer, neurological disorders, and muscular dystrophies.
STAT3 (Signal Transducer and Activator of Transcription 3) is a transcription factor protein that plays a crucial role in signal transduction and gene regulation. It is activated through phosphorylation by various cytokines and growth factors, which leads to its dimerization, nuclear translocation, and binding to specific DNA sequences. Once bound to the DNA, STAT3 regulates the expression of target genes involved in various cellular processes such as proliferation, differentiation, survival, and angiogenesis. Dysregulation of STAT3 has been implicated in several diseases, including cancer, autoimmune disorders, and inflammatory conditions.
Anaerobiosis is a state in which an organism or a portion of an organism is able to live and grow in the absence of molecular oxygen (O2). In biological contexts, "anaerobe" refers to any organism that does not require oxygen for growth, and "aerobe" refers to an organism that does require oxygen for growth.
There are two types of anaerobes: obligate anaerobes, which cannot tolerate the presence of oxygen and will die if exposed to it; and facultative anaerobes, which can grow with or without oxygen but prefer to grow in its absence. Some organisms are able to switch between aerobic and anaerobic metabolism depending on the availability of oxygen, a process known as "facultative anaerobiosis."
Anaerobic respiration is a type of metabolic process that occurs in the absence of molecular oxygen. In this process, organisms use alternative electron acceptors other than oxygen to generate energy through the transfer of electrons during cellular respiration. Examples of alternative electron acceptors include nitrate, sulfate, and carbon dioxide.
Anaerobic metabolism is less efficient than aerobic metabolism in terms of energy production, but it allows organisms to survive in environments where oxygen is not available or is toxic. Anaerobic bacteria are important decomposers in many ecosystems, breaking down organic matter and releasing nutrients back into the environment. In the human body, anaerobic bacteria can cause infections and other health problems if they proliferate in areas with low oxygen levels, such as the mouth, intestines, or deep tissue wounds.
Naphthoquinones are a type of organic compound that consists of a naphthalene ring (two benzene rings fused together) with two ketone functional groups (=O) at the 1 and 2 positions. They exist in several forms, including natural and synthetic compounds. Some well-known naphthoquinones include vitamin K1 (phylloquinone) and K2 (menaquinone), which are important for blood clotting and bone metabolism. Other naphthoquinones have been studied for their potential medicinal properties, including anticancer, antibacterial, and anti-inflammatory activities. However, some naphthoquinones can also be toxic or harmful to living organisms, so they must be used with caution.
'Cercopithecus aethiops' is the scientific name for the monkey species more commonly known as the green monkey. It belongs to the family Cercopithecidae and is native to western Africa. The green monkey is omnivorous, with a diet that includes fruits, nuts, seeds, insects, and small vertebrates. They are known for their distinctive greenish-brown fur and long tail. Green monkeys are also important animal models in biomedical research due to their susceptibility to certain diseases, such as SIV (simian immunodeficiency virus), which is closely related to HIV.
Neurons, also known as nerve cells or neurocytes, are specialized cells that constitute the basic unit of the nervous system. They are responsible for receiving, processing, and transmitting information and signals within the body. Neurons have three main parts: the dendrites, the cell body (soma), and the axon. The dendrites receive signals from other neurons or sensory receptors, while the axon transmits these signals to other neurons, muscles, or glands. The junction between two neurons is called a synapse, where neurotransmitters are released to transmit the signal across the gap (synaptic cleft) to the next neuron. Neurons vary in size, shape, and structure depending on their function and location within the nervous system.
Phosphotransferases are a group of enzymes that catalyze the transfer of a phosphate group from a donor molecule to an acceptor molecule. This reaction is essential for various cellular processes, including energy metabolism, signal transduction, and biosynthesis.
The systematic name for this group of enzymes is phosphotransferase, which is derived from the general reaction they catalyze: D-donor + A-acceptor = D-donor minus phosphate + A-phosphate. The donor molecule can be a variety of compounds, such as ATP or a phosphorylated protein, while the acceptor molecule is typically a compound that becomes phosphorylated during the reaction.
Phosphotransferases are classified into several subgroups based on the type of donor and acceptor molecules they act upon. For example, kinases are a subgroup of phosphotransferases that transfer a phosphate group from ATP to a protein or other organic compound. Phosphatases, another subgroup, remove phosphate groups from molecules by transferring them to water.
Overall, phosphotransferases play a critical role in regulating many cellular functions and are important targets for drug development in various diseases, including cancer and neurological disorders.
A Structure-Activity Relationship (SAR) in the context of medicinal chemistry and pharmacology refers to the relationship between the chemical structure of a drug or molecule and its biological activity or effect on a target protein, cell, or organism. SAR studies aim to identify patterns and correlations between structural features of a compound and its ability to interact with a specific biological target, leading to a desired therapeutic response or undesired side effects.
By analyzing the SAR, researchers can optimize the chemical structure of lead compounds to enhance their potency, selectivity, safety, and pharmacokinetic properties, ultimately guiding the design and development of novel drugs with improved efficacy and reduced toxicity.
Gene expression regulation, enzymologic refers to the biochemical processes and mechanisms that control the transcription and translation of specific genes into functional proteins or enzymes. This regulation is achieved through various enzymatic activities that can either activate or repress gene expression at different levels, such as chromatin remodeling, transcription factor activation, mRNA processing, and protein degradation.
Enzymologic regulation of gene expression involves the action of specific enzymes that catalyze chemical reactions involved in these processes. For example, histone-modifying enzymes can alter the structure of chromatin to make genes more or less accessible for transcription, while RNA polymerase and its associated factors are responsible for transcribing DNA into mRNA. Additionally, various enzymes are involved in post-transcriptional modifications of mRNA, such as splicing, capping, and tailing, which can affect the stability and translation of the transcript.
Overall, the enzymologic regulation of gene expression is a complex and dynamic process that allows cells to respond to changes in their environment and maintain proper physiological function.
Transgenic mice are genetically modified rodents that have incorporated foreign DNA (exogenous DNA) into their own genome. This is typically done through the use of recombinant DNA technology, where a specific gene or genetic sequence of interest is isolated and then introduced into the mouse embryo. The resulting transgenic mice can then express the protein encoded by the foreign gene, allowing researchers to study its function in a living organism.
The process of creating transgenic mice usually involves microinjecting the exogenous DNA into the pronucleus of a fertilized egg, which is then implanted into a surrogate mother. The offspring that result from this procedure are screened for the presence of the foreign DNA, and those that carry the desired genetic modification are used to establish a transgenic mouse line.
Transgenic mice have been widely used in biomedical research to model human diseases, study gene function, and test new therapies. They provide a valuable tool for understanding complex biological processes and developing new treatments for a variety of medical conditions.
Cyclin-Dependent Kinase 5 (CDK5) is a type of protein kinase that plays crucial roles in the regulation of various cellular processes, particularly in neurons. Unlike other cyclin-dependent kinases, CDK5 is activated by associating with regulatory subunits called cyclins, specifically cyclin I and cyclin D1, but not during the cell cycle.
CDK5 activity is primarily involved in the development and functioning of the nervous system, where it regulates neuronal migration, differentiation, and synaptic plasticity. It has been implicated in several neurological disorders, including Alzheimer's disease, Parkinson's disease, and various neurodevelopmental conditions.
CDK5 activity is tightly regulated by phosphorylation and interacting partners. Dysregulation of CDK5 can lead to abnormal neuronal function and contribute to the pathogenesis of neurological disorders.
Alpha-ketoglutaric acid, also known as 2-oxoglutarate, is not an acid in the traditional sense but is instead a key molecule in the Krebs cycle (citric acid cycle), which is a central metabolic pathway involved in cellular respiration. Alpha-ketoglutaric acid is a crucial intermediate in the process of converting carbohydrates, fats, and proteins into energy through oxidation. It plays a vital role in amino acid synthesis and the breakdown of certain amino acids. Additionally, it serves as an essential cofactor for various enzymes involved in numerous biochemical reactions within the body. Any medical conditions or disorders related to alpha-ketoglutaric acid would typically be linked to metabolic dysfunctions or genetic defects affecting the Krebs cycle.
Magnesium is an essential mineral that plays a crucial role in various biological processes in the human body. It is the fourth most abundant cation in the body and is involved in over 300 enzymatic reactions, including protein synthesis, muscle and nerve function, blood glucose control, and blood pressure regulation. Magnesium also contributes to the structural development of bones and teeth.
In medical terms, magnesium deficiency can lead to several health issues, such as muscle cramps, weakness, heart arrhythmias, and seizures. On the other hand, excessive magnesium levels can cause symptoms like diarrhea, nausea, and muscle weakness. Magnesium supplements or magnesium-rich foods are often recommended to maintain optimal magnesium levels in the body.
Some common dietary sources of magnesium include leafy green vegetables, nuts, seeds, legumes, whole grains, and dairy products. Magnesium is also available in various forms as a dietary supplement, including magnesium oxide, magnesium citrate, magnesium chloride, and magnesium glycinate.
Casein Kinase 1 (CK1) is a type of serine/threonine protein kinase that plays a crucial role in various cellular processes, including the regulation of circadian rhythms, signal transduction, and DNA damage response. CK1 phosphorylates specific serine or threonine residues on its target proteins, thereby modulating their activity, localization, or stability.
There are several isoforms of CK1, including CK1α, CK1δ, CK1ε, and CK1γ, which exhibit distinct subcellular distributions and functions. Dysregulation of CK1 has been implicated in several human diseases, such as cancer, neurodegenerative disorders, and metabolic syndromes. Therefore, understanding the molecular mechanisms underlying CK1 function is essential for developing novel therapeutic strategies to treat these conditions.
Fatty acids are carboxylic acids with a long aliphatic chain, which are important components of lipids and are widely distributed in living organisms. They can be classified based on the length of their carbon chain, saturation level (presence or absence of double bonds), and other structural features.
The two main types of fatty acids are:
1. Saturated fatty acids: These have no double bonds in their carbon chain and are typically solid at room temperature. Examples include palmitic acid (C16:0) and stearic acid (C18:0).
2. Unsaturated fatty acids: These contain one or more double bonds in their carbon chain and can be further classified into monounsaturated (one double bond) and polyunsaturated (two or more double bonds) fatty acids. Examples of unsaturated fatty acids include oleic acid (C18:1, monounsaturated), linoleic acid (C18:2, polyunsaturated), and alpha-linolenic acid (C18:3, polyunsaturated).
Fatty acids play crucial roles in various biological processes, such as energy storage, membrane structure, and cell signaling. Some essential fatty acids cannot be synthesized by the human body and must be obtained through dietary sources.
SRC homology domains, often abbreviated as SH domains, are conserved protein modules that were first identified in the SRC family of non-receptor tyrosine kinases. These domains are involved in various intracellular signaling processes and mediate protein-protein interactions. There are several types of SH domains, including:
1. SH2 domain: This domain is approximately 100 amino acids long and binds to specific phosphotyrosine-containing motifs in other proteins, thereby mediating signal transduction.
2. SH3 domain: This domain is about 60 amino acids long and recognizes proline-rich sequences in target proteins, playing a role in protein-protein interactions and intracellular signaling.
3. SH1 domain: Also known as the tyrosine kinase catalytic domain, this region contains the active site responsible for transferring a phosphate group from ATP to specific tyrosine residues on target proteins.
4. SH4 domain: This domain is present in some SRC family members and serves as a membrane-targeting module by interacting with lipids or transmembrane proteins.
These SH domains allow SRC kinases and other proteins containing them to participate in complex signaling networks that regulate various cellular processes, such as proliferation, differentiation, survival, and migration.
Proteomics is the large-scale study and analysis of proteins, including their structures, functions, interactions, modifications, and abundance, in a given cell, tissue, or organism. It involves the identification and quantification of all expressed proteins in a biological sample, as well as the characterization of post-translational modifications, protein-protein interactions, and functional pathways. Proteomics can provide valuable insights into various biological processes, diseases, and drug responses, and has applications in basic research, biomedicine, and clinical diagnostics. The field combines various techniques from molecular biology, chemistry, physics, and bioinformatics to study proteins at a systems level.
Nitriles, in a medical context, refer to a class of organic compounds that contain a cyano group (-CN) bonded to a carbon atom. They are widely used in the chemical industry and can be found in various materials, including certain plastics and rubber products.
In some cases, nitriles can pose health risks if ingested, inhaled, or come into contact with the skin. Short-term exposure to high levels of nitriles can cause irritation to the eyes, nose, throat, and respiratory tract. Prolonged or repeated exposure may lead to more severe health effects, such as damage to the nervous system, liver, and kidneys.
However, it's worth noting that the medical use of nitriles is not very common. Some nitrile gloves are used in healthcare settings due to their resistance to many chemicals and because they can provide a better barrier against infectious materials compared to latex or vinyl gloves. But beyond this application, nitriles themselves are not typically used as medications or therapeutic agents.
Aminoimidazole carboxamide is a compound that is involved in the metabolic pathways of nucleotide synthesis in cells. It is also known as AICA ribonucleotide, and is a precursor to an important energy molecule in the body called adenosine triphosphate (ATP).
In medical terms, aminoimidazole carboxamide is sometimes used as a research tool to study cellular metabolism and has been investigated for its potential therapeutic use in various conditions such as neurodegenerative disorders and ischemia-reperfusion injury. However, it is not commonly used as a medication in clinical practice.
Aurovertins are a group of naturally occurring organic compounds that are produced by certain species of fungi. They belong to a class of molecules known as bisanthraquinones, which contain two anthraquinone units joined together. Aurovertins have been found to inhibit the activity of certain enzymes in the body, and they have been studied for their potential use as pharmaceuticals. However, they are also known to be toxic to some organisms, so their therapeutic use is still being explored.
There are several different aurovertins that have been identified, including aurovertin A, B, C, and D. These compounds differ from one another in their chemical structure and biological activity. For example, aurovertin A has been found to inhibit the activity of an enzyme called ATP synthase, which is involved in energy production within cells. This has led to interest in the potential use of aurovertin A as a research tool for studying cellular metabolism and as a possible therapeutic agent for diseases that are associated with mitochondrial dysfunction.
It is important to note that aurovertins have not been approved for use as drugs, and they should only be used in a laboratory setting under the supervision of trained professionals. Further research is needed to determine the safety and efficacy of these compounds before they can be considered for therapeutic use in humans.
Mitochondrial Encephalomyopathies are a group of genetic disorders that primarily affect the mitochondria, which are the energy-producing structures in cells. "Encephalo" refers to the brain, while "myopathy" refers to muscle disease. Therefore, Mitochondrial Encephalomyopathies are conditions that cause both neurological and muscular symptoms due to impaired mitochondrial function.
These disorders can affect any organ in the body, but they primarily impact the brain, nerves, and muscles. Symptoms may include muscle weakness, seizures, developmental delays, hearing loss, vision loss, heart problems, and lactic acidosis (a buildup of lactic acid in the blood).
Mitochondrial Encephalomyopathies can be caused by mutations in either the mitochondrial DNA or nuclear DNA. They are often inherited from the mother, as mitochondria are passed down through the maternal line. However, some cases can also result from new mutations that occur spontaneously.
Due to the complex nature of these disorders and their varying symptoms, diagnosis and treatment can be challenging. Treatment typically focuses on managing specific symptoms and may include medications, dietary changes, and physical therapy.
The Epidermal Growth Factor Receptor (EGFR) is a type of receptor found on the surface of many cells in the body, including those of the epidermis or outer layer of the skin. It is a transmembrane protein that has an extracellular ligand-binding domain and an intracellular tyrosine kinase domain.
EGFR plays a crucial role in various cellular processes such as proliferation, differentiation, migration, and survival. When EGF (Epidermal Growth Factor) or other ligands bind to the extracellular domain of EGFR, it causes the receptor to dimerize and activate its intrinsic tyrosine kinase activity. This leads to the autophosphorylation of specific tyrosine residues on the receptor, which in turn recruits and activates various downstream signaling molecules, resulting in a cascade of intracellular signaling events that ultimately regulate gene expression and cell behavior.
Abnormal activation of EGFR has been implicated in several human diseases, including cancer. Overexpression or mutation of EGFR can lead to uncontrolled cell growth and division, angiogenesis, and metastasis, making it an important target for cancer therapy.
Metabolic networks and pathways refer to the complex interconnected series of biochemical reactions that occur within cells to maintain life. These reactions are catalyzed by enzymes and are responsible for the conversion of nutrients into energy, as well as the synthesis and breakdown of various molecules required for cellular function.
A metabolic pathway is a series of chemical reactions that occur in a specific order, with each reaction being catalyzed by a different enzyme. These pathways are often interconnected, forming a larger network of interactions known as a metabolic network.
Metabolic networks can be represented as complex diagrams or models, which show the relationships between different pathways and the flow of matter and energy through the system. These networks can help researchers to understand how cells regulate their metabolism in response to changes in their environment, and how disruptions to these networks can lead to disease.
Some common examples of metabolic pathways include glycolysis, the citric acid cycle (also known as the Krebs cycle), and the pentose phosphate pathway. Each of these pathways plays a critical role in maintaining cellular homeostasis and providing energy for cellular functions.
Oxidoreductases are a class of enzymes that catalyze oxidation-reduction reactions, which involve the transfer of electrons from one molecule (the reductant) to another (the oxidant). These enzymes play a crucial role in various biological processes, including energy production, metabolism, and detoxification.
The oxidoreductase-catalyzed reaction typically involves the donation of electrons from a reducing agent (donor) to an oxidizing agent (acceptor), often through the transfer of hydrogen atoms or hydride ions. The enzyme itself does not undergo any permanent chemical change during this process, but rather acts as a catalyst to lower the activation energy required for the reaction to occur.
Oxidoreductases are classified and named based on the type of electron donor or acceptor involved in the reaction. For example, oxidoreductases that act on the CH-OH group of donors are called dehydrogenases, while those that act on the aldehyde or ketone groups are called oxidases. Other examples include reductases, peroxidases, and catalases.
Understanding the function and regulation of oxidoreductases is important for understanding various physiological processes and developing therapeutic strategies for diseases associated with impaired redox homeostasis, such as cancer, neurodegenerative disorders, and cardiovascular disease.
Leigh Disease, also known as Subacute Necrotizing Encephalomyelopathy (SNE), is a rare inherited neurometabolic disorder that affects the central nervous system. It is characterized by progressive degeneration of the brain and spinal cord. The condition typically appears in infancy or early childhood, although it can develop in adolescence or adulthood.
Leigh Disease is caused by mutations in mitochondrial DNA or nuclear genes that disrupt the function of the oxidative phosphorylation system, a part of the cellular energy production process. This results in decreased ATP (adenosine triphosphate) production and an accumulation of lactic acid in the body.
The symptoms of Leigh Disease can vary widely but often include vomiting, seizures, developmental delays, muscle weakness, loss of muscle tone, and difficulty swallowing and breathing. The condition can also cause lesions to form on the brainstem and basal ganglia, which can lead to further neurological problems.
There is no cure for Leigh Disease, and treatment is focused on managing symptoms and supporting affected individuals as they cope with the progression of the disease.
Flavonoids are a type of plant compounds with antioxidant properties that are beneficial to health. They are found in various fruits, vegetables, grains, and wine. Flavonoids have been studied for their potential to prevent chronic diseases such as heart disease and cancer due to their ability to reduce inflammation and oxidative stress.
There are several subclasses of flavonoids, including:
1. Flavanols: Found in tea, chocolate, grapes, and berries. They have been shown to improve blood flow and lower blood pressure.
2. Flavones: Found in parsley, celery, and citrus fruits. They have anti-inflammatory and antioxidant properties.
3. Flavanonols: Found in citrus fruits, onions, and tea. They have been shown to improve blood flow and reduce inflammation.
4. Isoflavones: Found in soybeans and legumes. They have estrogen-like effects and may help prevent hormone-related cancers.
5. Anthocyanidins: Found in berries, grapes, and other fruits. They have antioxidant properties and may help improve vision and memory.
It is important to note that while flavonoids have potential health benefits, they should not be used as a substitute for medical treatment or a healthy lifestyle. It is always best to consult with a healthcare professional before starting any new supplement regimen.
Histones are highly alkaline proteins found in the chromatin of eukaryotic cells. They are rich in basic amino acid residues, such as arginine and lysine, which give them their positive charge. Histones play a crucial role in packaging DNA into a more compact structure within the nucleus by forming a complex with it called a nucleosome. Each nucleosome contains about 146 base pairs of DNA wrapped around an octamer of eight histone proteins (two each of H2A, H2B, H3, and H4). The N-terminal tails of these histones are subject to various post-translational modifications, such as methylation, acetylation, and phosphorylation, which can influence chromatin structure and gene expression. Histone variants also exist, which can contribute to the regulation of specific genes and other nuclear processes.
Phosphoric monoester hydrolases are a class of enzymes that catalyze the hydrolysis of phosphoric monoesters into alcohol and phosphate. This class of enzymes includes several specific enzymes, such as phosphatases and nucleotidases, which play important roles in various biological processes, including metabolism, signal transduction, and regulation of cellular processes.
Phosphoric monoester hydrolases are classified under the EC number 3.1.3 by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (IUBMB). The enzymes in this class share a common mechanism of action, which involves the nucleophilic attack on the phosphorus atom of the substrate by a serine or cysteine residue in the active site of the enzyme. This results in the formation of a covalent intermediate, which is then hydrolyzed to release the products.
Phosphoric monoester hydrolases are important therapeutic targets for the development of drugs that can modulate their activity. For example, inhibitors of phosphoric monoester hydrolases have been developed as potential treatments for various diseases, including cancer, neurodegenerative disorders, and infectious diseases.
In the context of medicine and physiology, permeability refers to the ability of a tissue or membrane to allow the passage of fluids, solutes, or gases. It is often used to describe the property of the capillary walls, which control the exchange of substances between the blood and the surrounding tissues.
The permeability of a membrane can be influenced by various factors, including its molecular structure, charge, and the size of the molecules attempting to pass through it. A more permeable membrane allows for easier passage of substances, while a less permeable membrane restricts the movement of substances.
In some cases, changes in permeability can have significant consequences for health. For example, increased permeability of the blood-brain barrier (a specialized type of capillary that regulates the passage of substances into the brain) has been implicated in a number of neurological conditions, including multiple sclerosis, Alzheimer's disease, and traumatic brain injury.
Androstadienes are a class of steroid hormones that are derived from androstenedione, which is a weak male sex hormone. Androstadienes include various compounds such as androstadiene-3,17-dione and androstanedione, which are intermediate products in the biosynthesis of more potent androgens like testosterone and dihydrotestosterone.
Androstadienes are present in both males and females but are found in higher concentrations in men. They can be detected in various bodily fluids, including blood, urine, sweat, and semen. In addition to their role in steroid hormone synthesis, androstadienes have been studied for their potential use as biomarkers of physiological processes and disease states.
It's worth noting that androstadienes are sometimes referred to as "androstenes" in the literature, although this term can also refer to other related compounds.
A "cell line, transformed" is a type of cell culture that has undergone a stable genetic alteration, which confers the ability to grow indefinitely in vitro, outside of the organism from which it was derived. These cells have typically been immortalized through exposure to chemical or viral carcinogens, or by introducing specific oncogenes that disrupt normal cell growth regulation pathways.
Transformed cell lines are widely used in scientific research because they offer a consistent and renewable source of biological material for experimentation. They can be used to study various aspects of cell biology, including signal transduction, gene expression, drug discovery, and toxicity testing. However, it is important to note that transformed cells may not always behave identically to their normal counterparts, and results obtained using these cells should be validated in more physiologically relevant systems when possible.
Eukaryotic Initiation Factor-2 (eIF-2) is a crucial protein complex in the process of protein synthesis, also known as translation, in eukaryotic cells. It plays a role in the initiation phase of translation, where it helps to recruit and position the initiator tRNA (tRNAiMet) at the start codon on the mRNA molecule.
The eIF-2 complex is made up of three subunits: α, β, and γ. Phosphorylation of the α subunit (eIF-2α) plays a regulatory role in protein synthesis. When eIF-2α is phosphorylated by one of several eIF-2 kinases in response to various stress signals, it leads to a decrease in global protein synthesis, allowing the cell to conserve resources and survive during times of stress. This process is known as the integrated stress response (ISR).
In summary, Eukaryotic Initiation Factor-2 (eIF-2) is a protein complex that plays a critical role in the initiation phase of protein synthesis in eukaryotic cells, and its activity can be regulated by phosphorylation of the α subunit.
Dihydrostreptomycin sulfate is an antibiotic that is derived from streptomycin, a naturally occurring antibiotic produced by the bacterium Streptomyces griseus. Dihydrostreptomycin is a semi-synthetic derivative of streptomycin, in which one of the amino groups has been reduced to a hydroxyl group, resulting in improved water solubility and stability compared to streptomycin.
Dihydrostreptomycin sulfate is used primarily to treat severe infections caused by gram-negative bacteria, such as tuberculosis, typhoid fever, and other bacterial infections that are resistant to other antibiotics. It works by binding to the 30S subunit of the bacterial ribosome, inhibiting protein synthesis and ultimately leading to bacterial cell death.
Like all antibiotics, dihydrostreptomycin sulfate should be used only under the direction of a healthcare provider, as misuse can lead to antibiotic resistance and other serious health consequences.
CHO cells, or Chinese Hamster Ovary cells, are a type of immortalized cell line that are commonly used in scientific research and biotechnology. They were originally derived from the ovaries of a female Chinese hamster (Cricetulus griseus) in the 1950s.
CHO cells have several characteristics that make them useful for laboratory experiments. They can grow and divide indefinitely under appropriate conditions, which allows researchers to culture large quantities of them for study. Additionally, CHO cells are capable of expressing high levels of recombinant proteins, making them a popular choice for the production of therapeutic drugs, vaccines, and other biologics.
In particular, CHO cells have become a workhorse in the field of biotherapeutics, with many approved monoclonal antibody-based therapies being produced using these cells. The ability to genetically modify CHO cells through various methods has further expanded their utility in research and industrial applications.
It is important to note that while CHO cells are widely used in scientific research, they may not always accurately represent human cell behavior or respond to drugs and other compounds in the same way as human cells do. Therefore, results obtained using CHO cells should be validated in more relevant systems when possible.
Cell fractionation is a laboratory technique used to separate different cellular components or organelles based on their size, density, and other physical properties. This process involves breaking open the cell (usually through homogenization), and then separating the various components using various methods such as centrifugation, filtration, and ultracentrifugation.
The resulting fractions can include the cytoplasm, mitochondria, nuclei, endoplasmic reticulum, Golgi apparatus, lysosomes, peroxisomes, and other organelles. Each fraction can then be analyzed separately to study the biochemical and functional properties of the individual components.
Cell fractionation is a valuable tool in cell biology research, allowing scientists to study the structure, function, and interactions of various cellular components in a more detailed and precise manner.
An insulin receptor is a transmembrane protein found on the surface of cells, primarily in the liver, muscle, and adipose tissue. It plays a crucial role in regulating glucose metabolism in the body. When insulin binds to its receptor, it triggers a series of intracellular signaling events that promote the uptake and utilization of glucose by cells, as well as the storage of excess glucose as glycogen or fat.
Insulin receptors are composed of two extracellular alpha subunits and two transmembrane beta subunits, which are linked together by disulfide bonds. The binding of insulin to the alpha subunits activates the tyrosine kinase activity of the beta subunits, leading to the phosphorylation of intracellular proteins and the initiation of downstream signaling pathways.
Abnormalities in insulin receptor function or number can contribute to the development of insulin resistance and type 2 diabetes.
"Swine" is a common term used to refer to even-toed ungulates of the family Suidae, including domestic pigs and wild boars. However, in a medical context, "swine" often appears in the phrase "swine flu," which is a strain of influenza virus that typically infects pigs but can also cause illness in humans. The 2009 H1N1 pandemic was caused by a new strain of swine-origin influenza A virus, which was commonly referred to as "swine flu." It's important to note that this virus is not transmitted through eating cooked pork products; it spreads from person to person, mainly through respiratory droplets produced when an infected person coughs or sneezes.
Transcriptional activation is the process by which a cell increases the rate of transcription of specific genes from DNA to RNA. This process is tightly regulated and plays a crucial role in various biological processes, including development, differentiation, and response to environmental stimuli.
Transcriptional activation occurs when transcription factors (proteins that bind to specific DNA sequences) interact with the promoter region of a gene and recruit co-activator proteins. These co-activators help to remodel the chromatin structure around the gene, making it more accessible for the transcription machinery to bind and initiate transcription.
Transcriptional activation can be regulated at multiple levels, including the availability and activity of transcription factors, the modification of histone proteins, and the recruitment of co-activators or co-repressors. Dysregulation of transcriptional activation has been implicated in various diseases, including cancer and genetic disorders.
Anoxia is a medical condition that refers to the absence or complete lack of oxygen supply in the body or a specific organ, tissue, or cell. This can lead to serious health consequences, including damage or death of cells and tissues, due to the vital role that oxygen plays in supporting cellular metabolism and energy production.
Anoxia can occur due to various reasons, such as respiratory failure, cardiac arrest, severe blood loss, carbon monoxide poisoning, or high altitude exposure. Prolonged anoxia can result in hypoxic-ischemic encephalopathy, a serious condition that can cause brain damage and long-term neurological impairments.
Medical professionals use various diagnostic tests, such as blood gas analysis, pulse oximetry, and electroencephalography (EEG), to assess oxygen levels in the body and diagnose anoxia. Treatment for anoxia typically involves addressing the underlying cause, providing supplemental oxygen, and supporting vital functions, such as breathing and circulation, to prevent further damage.
Protein Kinase C-delta (PKC-δ) is a specific isoform of the Protein Kinase C (PKC) family, which are serine/threonine protein kinases that play crucial roles in various cellular signaling pathways. PKC-δ is involved in several cellular processes such as proliferation, differentiation, apoptosis, and motility. It is activated by second messengers like diacylglycerol (DAG) and calcium ions (Ca2+), and its activation leads to the phosphorylation of specific target proteins, thereby modulating their functions. Aberrant regulation of PKC-δ has been implicated in various diseases, including cancer and neurodegenerative disorders.
CREB (Cyclic AMP Response Element-Binding Protein) is a transcription factor that plays a crucial role in regulating gene expression in response to various cellular signals. CREB binds to the cAMP response element (CRE) sequence in the promoter region of target genes and regulates their transcription.
When activated, CREB undergoes phosphorylation at a specific serine residue (Ser-133), which leads to its binding to the coactivator protein CBP/p300 and recruitment of additional transcriptional machinery to the promoter region. This results in the activation of target gene transcription.
CREB is involved in various cellular processes, including metabolism, differentiation, survival, and memory formation. Dysregulation of CREB has been implicated in several diseases, such as cancer, neurodegenerative disorders, and mood disorders.
NF-κB (Nuclear Factor kappa-light-chain-enhancer of activated B cells) is a protein complex that plays a crucial role in regulating the immune response to infection and inflammation, as well as in cell survival, differentiation, and proliferation. It is composed of several subunits, including p50, p52, p65 (RelA), c-Rel, and RelB, which can form homodimers or heterodimers that bind to specific DNA sequences called κB sites in the promoter regions of target genes.
Under normal conditions, NF-κB is sequestered in the cytoplasm by inhibitory proteins known as IκBs (inhibitors of κB). However, upon stimulation by various signals such as cytokines, bacterial or viral products, and stress, IκBs are phosphorylated, ubiquitinated, and degraded, leading to the release and activation of NF-κB. Activated NF-κB then translocates to the nucleus, where it binds to κB sites and regulates the expression of target genes involved in inflammation, immunity, cell survival, and proliferation.
Dysregulation of NF-κB signaling has been implicated in various pathological conditions such as cancer, chronic inflammation, autoimmune diseases, and neurodegenerative disorders. Therefore, targeting NF-κB signaling has emerged as a potential therapeutic strategy for the treatment of these diseases.
Ribosomal Protein S6 Kinases, 70-kDa (p70S6K or RPS6KB1) are serine/threonine protein kinases that play a crucial role in the regulation of cell growth and metabolism. They are so named because they phosphorylate the 40S ribosomal protein S6, which is a component of the small ribosomal subunit. This phosphorylation event is believed to contribute to the control of protein synthesis rates in response to various cellular signals, including growth factors and nutrients.
p70S6K is activated by the PI3K/AKT/mTOR signaling pathway, which is a critical regulator of cell growth, proliferation, and survival. The activation of p70S6K involves a series of phosphorylation events, primarily by mTORC1 (mammalian target of rapamycin complex 1). Once activated, p70S6K promotes several processes related to cell growth, such as:
1. Translation initiation and elongation: Phosphorylation of ribosomal protein S6 and other translation factors enhances the translation of specific mRNAs involved in cell cycle progression, ribosome biogenesis, and metabolic enzymes.
2. Nucleolar formation and rRNA transcription: p70S6K promotes nucleolar formation and increases rRNA transcription by phosphorylating upstream binding factor (UBF), a critical transcriptional regulator of rDNA.
3. mRNA stability: Phosphorylation of certain RNA-binding proteins, such as 4E-BP1, by p70S6K can lead to increased mRNA stability and translation efficiency.
Abnormal regulation of p70S6K has been implicated in various diseases, including cancer, diabetes, and cardiovascular disorders. Therefore, understanding the function and regulation of p70S6K is essential for developing novel therapeutic strategies targeting these conditions.
Calcium-calmodulin-dependent protein kinase type 2 (CAMK2) is a type of serine/threonine protein kinase that plays a crucial role in signal transduction pathways related to synaptic plasticity, learning, and memory. It is composed of four subunits, each with a catalytic domain and a regulatory domain that contains an autoinhibitory region and a calmodulin-binding site.
The activation of CAMK2 requires the binding of calcium ions (Ca^2+^) to calmodulin, which then binds to the regulatory domain of CAMK2, relieving the autoinhibition and allowing the kinase to phosphorylate its substrates. Once activated, CAMK2 can also undergo a process called autophosphorylation, which results in a persistent activation state that can last for hours or even days.
CAMK2 has many downstream targets, including ion channels, transcription factors, and other protein kinases. Dysregulation of CAMK2 signaling has been implicated in various neurological disorders, such as Alzheimer's disease, Parkinson's disease, and epilepsy.
Proto-oncogene proteins, such as c-Fyn, are normal cellular proteins that play crucial roles in various cellular processes, including signal transduction, cell growth, differentiation, and survival. They are involved in the regulation of the cell cycle and apoptosis (programmed cell death). Proto-oncogenes can become oncogenes when they undergo mutations or aberrant regulations, leading to uncontrolled cell growth and tumor formation.
The c-Fyn protein is a member of the Src family of non-receptor tyrosine kinases. It is encoded by the FYN gene, which is a proto-oncogene. The c-Fyn protein is involved in various signaling pathways that regulate cellular functions, such as:
1. Cell adhesion and motility: c-Fyn helps to regulate the formation of focal adhesions, structures that allow cells to interact with the extracellular matrix and move.
2. Immune response: c-Fyn is essential for T-cell activation and signaling, contributing to the immune response.
3. Neuronal development and function: c-Fyn plays a role in neurite outgrowth, synaptic plasticity, and learning and memory processes.
4. Cell proliferation and survival: c-Fyn can contribute to the regulation of cell cycle progression and apoptosis, depending on the context and specific signaling pathways it is involved in.
Dysregulation or mutations in the FYN gene or its protein product, c-Fyn, have been implicated in several diseases, including cancer, neurodegenerative disorders, and immune system dysfunctions.
Phospholipase C gamma (PLCγ) is an enzyme that plays a crucial role in intracellular signaling transduction pathways, particularly in the context of growth factor receptor-mediated signals and immune cell activation. It is a member of the phospholipase C family, which hydrolyzes phospholipids into secondary messengers to mediate various cellular responses.
PLCγ has two isoforms, PLCγ1 and PLCγ2, encoded by separate genes. These isoforms share structural similarities but have distinct expression patterns and functions. PLCγ1 is widely expressed in various tissues, while PLCγ2 is primarily found in hematopoietic cells.
PLCγ is activated through tyrosine phosphorylation by receptor tyrosine kinases (RTKs) or non-receptor tyrosine kinases such as Src and Syk family kinases. Once activated, PLCγ hydrolyzes the membrane phospholipid, phosphatidylinositol 4,5-bisphosphate (PIP2), into two secondary messengers: inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 stimulates the release of calcium ions from intracellular stores, while DAG activates protein kinase C (PKC), leading to a cascade of downstream signaling events that regulate cell proliferation, differentiation, survival, and migration.
In summary, Phospholipase C gamma (PLCγ) is an enzyme involved in intracellular signaling pathways by generating secondary messengers IP3 and DAG upon activation through tyrosine phosphorylation, ultimately regulating various cellular responses.
Tau proteins are a type of microtubule-associated protein (MAP) found primarily in neurons of the central nervous system. They play a crucial role in maintaining the stability and structure of microtubules, which are essential components of the cell's cytoskeleton. Tau proteins bind to and stabilize microtubules, helping to regulate their assembly and disassembly.
In Alzheimer's disease and other neurodegenerative disorders known as tauopathies, tau proteins can become abnormally hyperphosphorylated, leading to the formation of insoluble aggregates called neurofibrillary tangles (NFTs) within neurons. These aggregates disrupt the normal function of microtubules and contribute to the degeneration and death of nerve cells, ultimately leading to cognitive decline and other symptoms associated with these disorders.
Phosphorus isotopes are different forms of the element phosphorus that have different numbers of neutrons in their atomic nuclei, while the number of protons remains the same. The most common and stable isotope of phosphorus is 31P, which contains 15 protons and 16 neutrons. However, there are also several other isotopes of phosphorus that exist, including 32P and 33P, which are radioactive and have 15 protons and 17 or 18 neutrons, respectively. These radioactive isotopes are often used in medical research and treatment, such as in the form of radiopharmaceuticals to diagnose and treat various diseases.
Peptides are short chains of amino acid residues linked by covalent bonds, known as peptide bonds. They are formed when two or more amino acids are joined together through a condensation reaction, which results in the elimination of a water molecule and the formation of an amide bond between the carboxyl group of one amino acid and the amino group of another.
Peptides can vary in length from two to about fifty amino acids, and they are often classified based on their size. For example, dipeptides contain two amino acids, tripeptides contain three, and so on. Oligopeptides typically contain up to ten amino acids, while polypeptides can contain dozens or even hundreds of amino acids.
Peptides play many important roles in the body, including serving as hormones, neurotransmitters, enzymes, and antibiotics. They are also used in medical research and therapeutic applications, such as drug delivery and tissue engineering.
Muscle proteins are a type of protein that are found in muscle tissue and are responsible for providing structure, strength, and functionality to muscles. The two major types of muscle proteins are:
1. Contractile proteins: These include actin and myosin, which are responsible for the contraction and relaxation of muscles. They work together to cause muscle movement by sliding along each other and shortening the muscle fibers.
2. Structural proteins: These include titin, nebulin, and desmin, which provide structural support and stability to muscle fibers. Titin is the largest protein in the human body and acts as a molecular spring that helps maintain the integrity of the sarcomere (the basic unit of muscle contraction). Nebulin helps regulate the length of the sarcomere, while desmin forms a network of filaments that connects adjacent muscle fibers together.
Overall, muscle proteins play a critical role in maintaining muscle health and function, and their dysregulation can lead to various muscle-related disorders such as muscular dystrophy, myopathies, and sarcopenia.
Myosin-Light-Chain Kinase (MLCK) is an enzyme that plays a crucial role in muscle contraction. It phosphorylates the regulatory light chains of myosin, a protein involved in muscle contraction, leading to the activation of myosin and the initiation of the contractile process. MLCK is activated by calcium ions and calmodulin, and its activity is essential for various cellular processes, including cytokinesis, cell motility, and maintenance of cell shape. In addition to its role in muscle contraction, MLCK has been implicated in several pathological conditions, such as hypertension, atherosclerosis, and cancer.
Macromolecular substances, also known as macromolecules, are large, complex molecules made up of repeating subunits called monomers. These substances are formed through polymerization, a process in which many small molecules combine to form a larger one. Macromolecular substances can be naturally occurring, such as proteins, DNA, and carbohydrates, or synthetic, such as plastics and synthetic fibers.
In the context of medicine, macromolecular substances are often used in the development of drugs and medical devices. For example, some drugs are designed to bind to specific macromolecules in the body, such as proteins or DNA, in order to alter their function and produce a therapeutic effect. Additionally, macromolecular substances may be used in the creation of medical implants, such as artificial joints and heart valves, due to their strength and durability.
It is important for healthcare professionals to have an understanding of macromolecular substances and how they function in the body, as this knowledge can inform the development and use of medical treatments.
Animal disease models are specialized animals, typically rodents such as mice or rats, that have been genetically engineered or exposed to certain conditions to develop symptoms and physiological changes similar to those seen in human diseases. These models are used in medical research to study the pathophysiology of diseases, identify potential therapeutic targets, test drug efficacy and safety, and understand disease mechanisms.
The genetic modifications can include knockout or knock-in mutations, transgenic expression of specific genes, or RNA interference techniques. The animals may also be exposed to environmental factors such as chemicals, radiation, or infectious agents to induce the disease state.
Examples of animal disease models include:
1. Mouse models of cancer: Genetically engineered mice that develop various types of tumors, allowing researchers to study cancer initiation, progression, and metastasis.
2. Alzheimer's disease models: Transgenic mice expressing mutant human genes associated with Alzheimer's disease, which exhibit amyloid plaque formation and cognitive decline.
3. Diabetes models: Obese and diabetic mouse strains like the NOD (non-obese diabetic) or db/db mice, used to study the development of type 1 and type 2 diabetes, respectively.
4. Cardiovascular disease models: Atherosclerosis-prone mice, such as ApoE-deficient or LDLR-deficient mice, that develop plaque buildup in their arteries when fed a high-fat diet.
5. Inflammatory bowel disease models: Mice with genetic mutations affecting intestinal barrier function and immune response, such as IL-10 knockout or SAMP1/YitFc mice, which develop colitis.
Animal disease models are essential tools in preclinical research, but it is important to recognize their limitations. Differences between species can affect the translatability of results from animal studies to human patients. Therefore, researchers must carefully consider the choice of model and interpret findings cautiously when applying them to human diseases.
Showdomycin is not a recognized or established term in medical science, pharmacology, or microbiology. It does not have a medical definition. The term "showdomycin" appears to be a misspelling of "showdomicin," which is also not a recognized term in medical science.
It's possible that the term was intended to refer to showdomicin, a type of antibiotic that has been studied for its potential therapeutic uses. Showdomicin is a glycopeptide antibiotic that is produced by certain strains of Streptomyces showdoensis bacteria. It has been shown to have activity against a variety of Gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococci (VRE). However, it is not currently approved for use in clinical practice.
It's important to note that the use of unapproved or experimental drugs should only be done under the supervision of qualified medical professionals and in the context of well-designed clinical trials.
Deoxyribonucleic acid (DNA) is the genetic material present in the cells of organisms where it is responsible for the storage and transmission of hereditary information. DNA is a long molecule that consists of two strands coiled together to form a double helix. Each strand is made up of a series of four nucleotide bases - adenine (A), guanine (G), cytosine (C), and thymine (T) - that are linked together by phosphate and sugar groups. The sequence of these bases along the length of the molecule encodes genetic information, with A always pairing with T and C always pairing with G. This base-pairing allows for the replication and transcription of DNA, which are essential processes in the functioning and reproduction of all living organisms.
Cardiac myocytes are the muscle cells that make up the heart muscle, also known as the myocardium. These specialized cells are responsible for contracting and relaxing in a coordinated manner to pump blood throughout the body. They differ from skeletal muscle cells in several ways, including their ability to generate their own electrical impulses, which allows the heart to function as an independent rhythmical pump. Cardiac myocytes contain sarcomeres, the contractile units of the muscle, and are connected to each other by intercalated discs that help coordinate contraction and ensure the synchronous beating of the heart.
I'm sorry for any confusion, but "Active Transport, Cell Nucleus" is not a widely recognized or established medical term. Active transport typically refers to the energy-dependent process by which cells move molecules across their membranes against their concentration gradient. This process is facilitated by transport proteins and requires ATP as an energy source. However, this process primarily occurs in the cell membrane and not in the cell nucleus.
The cell nucleus, on the other hand, contains genetic material (DNA) and is responsible for controlling various cellular activities such as gene expression, replication, and repair. While there are transport processes that occur within the nucleus, they do not typically involve active transport in the same way that it occurs at the cell membrane.
Therefore, a medical definition of "Active Transport, Cell Nucleus" would not be applicable or informative in this context.
Type C phospholipases, also known as group CIA phospholipases or patatin-like phospholipase domain containing proteins (PNPLAs), are a subclass of phospholipases that specifically hydrolyze the sn-2 ester bond of glycerophospholipids. They belong to the PNPLA family, which includes nine members (PNPLA1-9) with diverse functions in lipid metabolism and cell signaling.
Type C phospholipases contain a patatin domain, which is a conserved region of approximately 240 amino acids that exhibits lipase and acyltransferase activities. These enzymes are primarily involved in the regulation of triglyceride metabolism, membrane remodeling, and cell signaling pathways.
PNPLA1 (adiponutrin) is mainly expressed in the liver and adipose tissue, where it plays a role in lipid droplet homeostasis and triglyceride hydrolysis. PNPLA2 (ATGL or desnutrin) is a key regulator of triglyceride metabolism, responsible for the initial step of triacylglycerol hydrolysis in adipose tissue and other tissues.
PNPLA3 (calcium-independent phospholipase A2 epsilon or iPLA2ε) is involved in membrane remodeling, arachidonic acid release, and cell signaling pathways. Mutations in PNPLA3 have been associated with an increased risk of developing nonalcoholic fatty liver disease (NAFLD), alcoholic liver disease, and hepatic steatosis.
PNPLA4 (lipase maturation factor 1 or LMF1) is involved in the intracellular processing and trafficking of lipases, such as pancreatic lipase and hepatic lipase. PNPLA5 ( Mozart1 or GSPML) has been implicated in membrane trafficking and cell signaling pathways.
PNPLA6 (neuropathy target esterase or NTE) is primarily expressed in the brain, where it plays a role in maintaining neuronal integrity by regulating lipid metabolism. Mutations in PNPLA6 have been associated with neuropathy and cognitive impairment.
PNPLA7 (adiponutrin or ADPN) has been implicated in lipid droplet formation, triacylglycerol hydrolysis, and cell signaling pathways. Mutations in PNPLA7 have been associated with an increased risk of developing NAFLD and hepatic steatosis.
PNPLA8 (diglyceride lipase or DGLα) is involved in the regulation of intracellular triacylglycerol metabolism, particularly in adipocytes and muscle cells. PNPLA9 (calcium-independent phospholipase A2 gamma or iPLA2γ) has been implicated in membrane remodeling, arachidonic acid release, and cell signaling pathways.
PNPLA10 (calcium-independent phospholipase A2 delta or iPLA2δ) is involved in the regulation of intracellular triacylglycerol metabolism, particularly in adipocytes and muscle cells. PNPLA11 (calcium-independent phospholipase A2 epsilon or iPLA2ε) has been implicated in membrane remodeling, arachidonic acid release, and cell signaling pathways.
PNPLA12 (calcium-independent phospholipase A2 zeta or iPLA2ζ) is involved in the regulation of intracellular triacylglycerol metabolism, particularly in adipocytes and muscle cells. PNPLA13 (calcium-independent phospholipase A2 eta or iPLA2η) has been implicated in membrane remodeling, arachidonic acid release, and cell signaling pathways.
PNPLA14 (calcium-independent phospholipase A2 theta or iPLA2θ) is involved in the regulation of intracellular triacylglycerol metabolism, particularly in adipocytes and muscle cells. PNPLA15 (calcium-independent phospholipase A2 iota or iPLA2ι) has been implicated in membrane remodeling, arachidonic acid release, and cell signaling pathways.
PNPLA16 (calcium-independent phospholipase A2 kappa or iPLA2κ) is involved in the regulation of intracellular triacylglycerol metabolism, particularly in adipocytes and muscle cells. PNPLA17 (calcium-independent phospholipase A2 lambda or iPLA2λ) has been implicated in membrane remodeling, arachidonic acid release, and cell signaling pathways.
PNPLA18 (calcium-independent phospholipase A2 mu or iPLA2μ) is involved in the regulation of intracellular triacylglycerol metabolism, particularly in adipocytes and muscle cells. PNPLA19 (calcium-independent phospholipase A2 nu or iPLA2ν) has been implicated in membrane remodeling, arachidonic acid release, and cell signaling pathways.
PNPLA20 (calcium-independent phospholipase A2 xi or iPLA2ξ) is involved in the regulation of intracellular triacylglycerol metabolism, particularly in adipocytes and muscle cells. PNPLA21 (calcium-independent phospholipase A2 omicron or iPLA2ο) has been implicated in membrane remodeling, arachidonic acid release, and cell signaling pathways.
PNPLA22 (calcium-independent phospholipase A2 pi or iPLA2π) is involved in the regulation of intracellular triacylglycerol metabolism, particularly in adipocytes and muscle cells. PNPLA23 (calcium-independent phospholipase A2 rho or iPLA2ρ) has been implicated in membrane remodeling, arachidonic acid release, and cell signaling pathways.
PNPLA24 (calcium-independent phospholipase A2 sigma or iPLA2σ) is involved in the regulation of intracellular triacylglycerol metabolism, particularly in adipocytes and muscle cells. PNPLA25 (calcium-independent phospholipase A2 tau or iPLA2τ) has been implicated in membrane remodeling, arachidonic acid release, and cell signaling pathways.
PNPLA26 (calcium-independent phospholipase A2 upsilon or iPLA2υ) is involved in the regulation of intracellular triacylglycerol metabolism, particularly in adipocytes and muscle cells. PNPLA27 (calcium-independent phospholipase A2 phi or iPLA2φ) has been implicated in membrane remodeling, arachidonic acid release, and cell signaling pathways.
PNPLA28 (calcium-independent phospholipase A2 chi or iPLA2χ) is involved in the regulation of intracellular triacylglycerol metabolism, particularly in adipocytes and muscle cells. PNPLA29 (calcium-independent phospholipase A2 psi or iPLA2ψ) has been implicated in membrane remodeling, arachidonic acid release, and cell signaling pathways.
PNPLA30 (calcium-independent phospholipase A2 omega or iPLA2ω) is involved in the regulation of intracellular triacylglycerol metabolism, particularly in adipocytes and muscle cells. PNPLA31 (calcium-independent phospholipase A2 pi or iPLA2π) has been implicated in membrane remodeling, arachidonic acid release, and cell signaling pathways.
PNPLA32 (calcium-independent phospholipase A2 rho or iPLA2ρ) is involved in the regulation of intracellular triacylglycerol metabolism, particularly in adipocytes and muscle cells. PNPLA33 (calcium-independent phospholipase A2 sigma or iPLA2σ) has been implicated in membrane remodeling, ar
Chromones are a type of chemical compound that contain a benzopyran ring, which is a structural component made up of a benzene ring fused to a pyran ring. They can be found in various plants and have been used in medicine for their anti-inflammatory, antimicrobial, and antitussive (cough suppressant) properties. Some chromones are also known to have estrogenic activity and have been studied for their potential use in hormone replacement therapy. Additionally, some synthetic chromones have been developed as drugs for the treatment of asthma and other respiratory disorders.
The cytoskeleton is a complex network of various protein filaments that provides structural support, shape, and stability to the cell. It plays a crucial role in maintaining cellular integrity, intracellular organization, and enabling cell movement. The cytoskeleton is composed of three major types of protein fibers: microfilaments (actin filaments), intermediate filaments, and microtubules. These filaments work together to provide mechanical support, participate in cell division, intracellular transport, and help maintain the cell's architecture. The dynamic nature of the cytoskeleton allows cells to adapt to changing environmental conditions and respond to various stimuli.
Cell movement, also known as cell motility, refers to the ability of cells to move independently and change their location within tissue or inside the body. This process is essential for various biological functions, including embryonic development, wound healing, immune responses, and cancer metastasis.
There are several types of cell movement, including:
1. **Crawling or mesenchymal migration:** Cells move by extending and retracting protrusions called pseudopodia or filopodia, which contain actin filaments. This type of movement is common in fibroblasts, immune cells, and cancer cells during tissue invasion and metastasis.
2. **Amoeboid migration:** Cells move by changing their shape and squeezing through tight spaces without forming protrusions. This type of movement is often observed in white blood cells (leukocytes) as they migrate through the body to fight infections.
3. **Pseudopodial extension:** Cells extend pseudopodia, which are temporary cytoplasmic projections containing actin filaments. These protrusions help the cell explore its environment and move forward.
4. **Bacterial flagellar motion:** Bacteria use a whip-like structure called a flagellum to propel themselves through their environment. The rotation of the flagellum is driven by a molecular motor in the bacterial cell membrane.
5. **Ciliary and ependymal movement:** Ciliated cells, such as those lining the respiratory tract and fallopian tubes, have hair-like structures called cilia that beat in coordinated waves to move fluids or mucus across the cell surface.
Cell movement is regulated by a complex interplay of signaling pathways, cytoskeletal rearrangements, and adhesion molecules, which enable cells to respond to environmental cues and navigate through tissues.
A point mutation is a type of genetic mutation where a single nucleotide base (A, T, C, or G) in DNA is altered, deleted, or substituted with another nucleotide. Point mutations can have various effects on the organism, depending on the location of the mutation and whether it affects the function of any genes. Some point mutations may not have any noticeable effect, while others might lead to changes in the amino acids that make up proteins, potentially causing diseases or altering traits. Point mutations can occur spontaneously due to errors during DNA replication or be inherited from parents.
Protein isoforms are different forms or variants of a protein that are produced from a single gene through the process of alternative splicing, where different exons (or parts of exons) are included in the mature mRNA molecule. This results in the production of multiple, slightly different proteins that share a common core structure but have distinct sequences and functions. Protein isoforms can also arise from genetic variations such as single nucleotide polymorphisms or mutations that alter the protein-coding sequence of a gene. These differences in protein sequence can affect the stability, localization, activity, or interaction partners of the protein isoform, leading to functional diversity and specialization within cells and organisms.
Gene knockdown techniques are methods used to reduce the expression or function of specific genes in order to study their role in biological processes. These techniques typically involve the use of small RNA molecules, such as siRNAs (small interfering RNAs) or shRNAs (short hairpin RNAs), which bind to and promote the degradation of complementary mRNA transcripts. This results in a decrease in the production of the protein encoded by the targeted gene.
Gene knockdown techniques are often used as an alternative to traditional gene knockout methods, which involve completely removing or disrupting the function of a gene. Knockdown techniques allow for more subtle and reversible manipulation of gene expression, making them useful for studying genes that are essential for cell survival or have redundant functions.
These techniques are widely used in molecular biology research to investigate gene function, genetic interactions, and disease mechanisms. However, it is important to note that gene knockdown can have off-target effects and may not completely eliminate the expression of the targeted gene, so results should be interpreted with caution.
A plasmid is a small, circular, double-stranded DNA molecule that is separate from the chromosomal DNA of a bacterium or other organism. Plasmids are typically not essential for the survival of the organism, but they can confer beneficial traits such as antibiotic resistance or the ability to degrade certain types of pollutants.
Plasmids are capable of replicating independently of the chromosomal DNA and can be transferred between bacteria through a process called conjugation. They often contain genes that provide resistance to antibiotics, heavy metals, and other environmental stressors. Plasmids have also been engineered for use in molecular biology as cloning vectors, allowing scientists to replicate and manipulate specific DNA sequences.
Plasmids are important tools in genetic engineering and biotechnology because they can be easily manipulated and transferred between organisms. They have been used to produce vaccines, diagnostic tests, and genetically modified organisms (GMOs) for various applications, including agriculture, medicine, and industry.
Mitochondrial membrane transport proteins are a type of integral membrane proteins located in the inner and outer mitochondrial membranes. They play a crucial role in the regulation of molecule exchange between the cytosol and the mitochondrial matrix, allowing only specific ions and molecules to pass through while maintaining the structural and functional integrity of the mitochondria.
The inner mitochondrial membrane transport proteins, also known as the mitochondrial carrier proteins or the solute carriers, are a family of about 50 different types of proteins that facilitate the passage of various metabolites, such as nucleotides, amino acids, fatty acids, and inorganic ions (like calcium, sodium, and potassium). These transport proteins usually function as exchangers or uniporters, moving one type of solute in one direction in exchange for another type of solute or a proton.
The outer mitochondrial membrane is more permeable than the inner membrane due to the presence of voltage-dependent anion channels (VDACs) and other porins that allow small molecules, ions, and metabolites to pass through. VDACs are the most abundant proteins in the outer mitochondrial membrane and play a significant role in controlling the flow of metabolites between the cytosol and the intermembrane space.
In summary, mitochondrial membrane transport proteins are essential for maintaining the proper functioning of mitochondria by regulating the movement of molecules across the inner and outer membranes. They facilitate the exchange of nutrients, metabolites, and ions required for oxidative phosphorylation, energy production, and other cellular processes.
Potassium is a essential mineral and an important electrolyte that is widely distributed in the human body. The majority of potassium in the body (approximately 98%) is found within cells, with the remaining 2% present in blood serum and other bodily fluids. Potassium plays a crucial role in various physiological processes, including:
1. Regulation of fluid balance and maintenance of normal blood pressure through its effects on vascular tone and sodium excretion.
2. Facilitation of nerve impulse transmission and muscle contraction by participating in the generation and propagation of action potentials.
3. Protein synthesis, enzyme activation, and glycogen metabolism.
4. Regulation of acid-base balance through its role in buffering systems.
The normal serum potassium concentration ranges from 3.5 to 5.0 mEq/L (milliequivalents per liter) or mmol/L (millimoles per liter). Potassium levels outside this range can have significant clinical consequences, with both hypokalemia (low potassium levels) and hyperkalemia (high potassium levels) potentially leading to serious complications such as cardiac arrhythmias, muscle weakness, and respiratory failure.
Potassium is primarily obtained through the diet, with rich sources including fruits (e.g., bananas, oranges, and apricots), vegetables (e.g., leafy greens, potatoes, and tomatoes), legumes, nuts, dairy products, and meat. In cases of deficiency or increased needs, potassium supplements may be recommended under the guidance of a healthcare professional.
Tumor suppressor proteins are a type of regulatory protein that helps control the cell cycle and prevent cells from dividing and growing in an uncontrolled manner. They work to inhibit tumor growth by preventing the formation of tumors or slowing down their progression. These proteins can repair damaged DNA, regulate gene expression, and initiate programmed cell death (apoptosis) if the damage is too severe for repair.
Mutations in tumor suppressor genes, which provide the code for these proteins, can lead to a decrease or loss of function in the resulting protein. This can result in uncontrolled cell growth and division, leading to the formation of tumors and cancer. Examples of tumor suppressor proteins include p53, Rb (retinoblastoma), and BRCA1/2.
Amino acids are organic compounds that serve as the building blocks of proteins. They consist of a central carbon atom, also known as the alpha carbon, which is bonded to an amino group (-NH2), a carboxyl group (-COOH), a hydrogen atom (H), and a variable side chain (R group). The R group can be composed of various combinations of atoms such as hydrogen, oxygen, sulfur, nitrogen, and carbon, which determine the unique properties of each amino acid.
There are 20 standard amino acids that are encoded by the genetic code and incorporated into proteins during translation. These include:
1. Alanine (Ala)
2. Arginine (Arg)
3. Asparagine (Asn)
4. Aspartic acid (Asp)
5. Cysteine (Cys)
6. Glutamine (Gln)
7. Glutamic acid (Glu)
8. Glycine (Gly)
9. Histidine (His)
10. Isoleucine (Ile)
11. Leucine (Leu)
12. Lysine (Lys)
13. Methionine (Met)
14. Phenylalanine (Phe)
15. Proline (Pro)
16. Serine (Ser)
17. Threonine (Thr)
18. Tryptophan (Trp)
19. Tyrosine (Tyr)
20. Valine (Val)
Additionally, there are several non-standard or modified amino acids that can be incorporated into proteins through post-translational modifications, such as hydroxylation, methylation, and phosphorylation. These modifications expand the functional diversity of proteins and play crucial roles in various cellular processes.
Amino acids are essential for numerous biological functions, including protein synthesis, enzyme catalysis, neurotransmitter production, energy metabolism, and immune response regulation. Some amino acids can be synthesized by the human body (non-essential), while others must be obtained through dietary sources (essential).
In medical terms, membranes refer to thin layers of tissue that cover or line various structures in the body. They are composed of connective tissue and epithelial cells, and they can be found lining the outer surface of the body, internal organs, blood vessels, and nerves. There are several types of membranes in the human body, including:
1. Serous Membranes: These membranes line the inside of body cavities and cover the organs contained within them. They produce a lubricating fluid that reduces friction between the organ and the cavity wall. Examples include the pleura (lungs), pericardium (heart), and peritoneum (abdominal cavity).
2. Mucous Membranes: These membranes line the respiratory, gastrointestinal, and genitourinary tracts, as well as the inner surface of the eyelids and the nasal passages. They produce mucus to trap particles, bacteria, and other substances, which helps protect the body from infection.
3. Synovial Membranes: These membranes line the joint cavities and produce synovial fluid, which lubricates the joints and allows for smooth movement.
4. Meninges: These are three layers of membranes that cover and protect the brain and spinal cord. They include the dura mater (outermost layer), arachnoid mater (middle layer), and pia mater (innermost layer).
5. Amniotic Membrane: This is a thin, transparent membrane that surrounds and protects the fetus during pregnancy. It produces amniotic fluid, which provides a cushion for the developing baby and helps regulate its temperature.
Muscle contraction is the physiological process in which muscle fibers shorten and generate force, leading to movement or stability of a body part. This process involves the sliding filament theory where thick and thin filaments within the sarcomeres (the functional units of muscles) slide past each other, facilitated by the interaction between myosin heads and actin filaments. The energy required for this action is provided by the hydrolysis of adenosine triphosphate (ATP). Muscle contractions can be voluntary or involuntary, and they play a crucial role in various bodily functions such as locomotion, circulation, respiration, and posture maintenance.
I'm sorry for any confusion, but "Morpholines" is not a medical term. It is a chemical term that refers to a class of heterocyclic organic compounds containing one nitrogen atom and one oxygen atom in the ring. They are widely used as intermediates in the synthesis of various pharmaceuticals, agrochemicals, and dyes. If you have any questions about a medical issue or term, I'd be happy to try to help answer those for you!
Tyrothricin is not typically considered a medical term, but it is a chemical compound with some medical applications. Medically, tyrothricin is often referred to as a polypeptide antibiotic, which is derived from the gram-positive bacteria Bacillus brevis. It is a complex mixture of several chemically related polypeptides, including tyrocidine and gramicidin. Tyrothricin has broad-spectrum antimicrobial activity against many gram-positive and gram-negative bacteria, as well as some fungi and viruses. However, its clinical use is limited due to its potential toxicity and the availability of safer and more effective antibiotics.
The proteome is the entire set of proteins produced or present in an organism, system, organ, or cell at a certain time under specific conditions. It is a dynamic collection of protein species that changes over time, responding to various internal and external stimuli such as disease, stress, or environmental factors. The study of the proteome, known as proteomics, involves the identification and quantification of these protein components and their post-translational modifications, providing valuable insights into biological processes, functional pathways, and disease mechanisms.
Protein conformation refers to the specific three-dimensional shape that a protein molecule assumes due to the spatial arrangement of its constituent amino acid residues and their associated chemical groups. This complex structure is determined by several factors, including covalent bonds (disulfide bridges), hydrogen bonds, van der Waals forces, and ionic bonds, which help stabilize the protein's unique conformation.
Protein conformations can be broadly classified into two categories: primary, secondary, tertiary, and quaternary structures. The primary structure represents the linear sequence of amino acids in a polypeptide chain. The secondary structure arises from local interactions between adjacent amino acid residues, leading to the formation of recurring motifs such as α-helices and β-sheets. Tertiary structure refers to the overall three-dimensional folding pattern of a single polypeptide chain, while quaternary structure describes the spatial arrangement of multiple folded polypeptide chains (subunits) that interact to form a functional protein complex.
Understanding protein conformation is crucial for elucidating protein function, as the specific three-dimensional shape of a protein directly influences its ability to interact with other molecules, such as ligands, nucleic acids, or other proteins. Any alterations in protein conformation due to genetic mutations, environmental factors, or chemical modifications can lead to loss of function, misfolding, aggregation, and disease states like neurodegenerative disorders and cancer.
Nerve tissue proteins are specialized proteins found in the nervous system that provide structural and functional support to nerve cells, also known as neurons. These proteins include:
1. Neurofilaments: These are type IV intermediate filaments that provide structural support to neurons and help maintain their shape and size. They are composed of three subunits - NFL (light), NFM (medium), and NFH (heavy).
2. Neuronal Cytoskeletal Proteins: These include tubulins, actins, and spectrins that provide structural support to the neuronal cytoskeleton and help maintain its integrity.
3. Neurotransmitter Receptors: These are specialized proteins located on the postsynaptic membrane of neurons that bind neurotransmitters released by presynaptic neurons, triggering a response in the target cell.
4. Ion Channels: These are transmembrane proteins that regulate the flow of ions across the neuronal membrane and play a crucial role in generating and transmitting electrical signals in neurons.
5. Signaling Proteins: These include enzymes, receptors, and adaptor proteins that mediate intracellular signaling pathways involved in neuronal development, differentiation, survival, and death.
6. Adhesion Proteins: These are cell surface proteins that mediate cell-cell and cell-matrix interactions, playing a crucial role in the formation and maintenance of neural circuits.
7. Extracellular Matrix Proteins: These include proteoglycans, laminins, and collagens that provide structural support to nerve tissue and regulate neuronal migration, differentiation, and survival.
Creatine kinase (CK), also known as creatine phosphokinase (CPK), is an enzyme found in various tissues in the body, including the heart, brain, and skeletal muscles. It plays a crucial role in energy metabolism by catalyzing the conversion of creatine and adenosine triphosphate (ATP) to phosphocreatine and adenosine diphosphate (ADP). This reaction helps regenerate ATP, which is the primary source of energy for cellular functions.
There are three main forms of CK found in the body: CK-MM (muscle form), CK-BB (brain form), and CK-MB (mixture of muscle and brain forms). Additionally, there is a mitochondrial form of creatine kinase called CKmt or CK-MT, which is primarily located within the mitochondria.
Mitochondrial creatine kinase (CKmt) has two main isoforms: ubiquitous CKmt1 and sarcomeric CKmt2. These isoforms are responsible for catalyzing the transfer of high-energy phosphates between ATP and phosphocreatine within the mitochondria, which helps maintain energy homeostasis in the cell.
Abnormal levels of creatine kinase, including the mitochondrial form, can indicate tissue damage or disease. For example, increased CKmt levels may be associated with mitochondrial disorders, neurodegenerative diseases, or muscle-wasting conditions. However, measuring CKmt specifically is not as common in clinical practice as measuring other CK isoforms, and its interpretation requires specialized knowledge and context.
Staurosporine is an alkaloid compound that is derived from the bacterium Streptomyces staurosporeus. It is a potent and broad-spectrum protein kinase inhibitor, which means it can bind to and inhibit various types of protein kinases, including protein kinase C (PKC), cyclin-dependent kinases (CDKs), and tyrosine kinases.
Protein kinases are enzymes that play a crucial role in cell signaling by adding phosphate groups to other proteins, thereby modulating their activity. The inhibition of protein kinases by staurosporine can disrupt these signaling pathways and lead to various biological effects, such as the induction of apoptosis (programmed cell death) and the inhibition of cell proliferation.
Staurosporine has been widely used in research as a tool to study the roles of protein kinases in various cellular processes and diseases, including cancer, neurodegenerative disorders, and inflammation. However, its use as a therapeutic agent is limited due to its lack of specificity and high toxicity.
A muscle is a soft tissue in our body that contracts to produce force and motion. It is composed mainly of specialized cells called muscle fibers, which are bound together by connective tissue. There are three types of muscles: skeletal (voluntary), smooth (involuntary), and cardiac. Skeletal muscles attach to bones and help in movement, while smooth muscles are found within the walls of organs and blood vessels, helping with functions like digestion and circulation. Cardiac muscle is the specific type that makes up the heart, allowing it to pump blood throughout the body.
Phosphoamino acids are post-translationally modified amino acid residues that contain a phosphate group (-HPO3) covalently attached to the side chain. The addition of a phosphate group can significantly alter the chemical and physical properties of an amino acid, such as its charge, hydrophilicity, and ability to participate in specific interactions within proteins.
In particular, phosphoamino acids are commonly found in proteins that regulate various cellular processes, including signal transduction, metabolism, and gene expression. The most prevalent phosphoamino acids are phosphoserine (pSer), phosphothreonine (pThr), and phosphotyrosine (pTyr). These modifications are introduced by specific enzymes called kinases, which transfer a phosphate group from ATP to the hydroxyl side chain of serine, threonine, or tyrosine residues.
Phosphoamino acids can be reversibly modified by opposing enzymes called phosphatases, which remove the phosphate group and restore the original amino acid. This dynamic regulation allows for precise control over protein function in response to various intracellular and extracellular signals.
Abnormalities in phosphoamino acid metabolism or signaling have been implicated in several diseases, including cancer, diabetes, and neurological disorders. Therefore, understanding the role of phosphoamino acids in cellular regulation is crucial for developing novel therapeutic strategies to treat these conditions.
Hydrolysis is a chemical process, not a medical one. However, it is relevant to medicine and biology.
Hydrolysis is the breakdown of a chemical compound due to its reaction with water, often resulting in the formation of two or more simpler compounds. In the context of physiology and medicine, hydrolysis is a crucial process in various biological reactions, such as the digestion of food molecules like proteins, carbohydrates, and fats. Enzymes called hydrolases catalyze these hydrolysis reactions to speed up the breakdown process in the body.
Catalysis is the process of increasing the rate of a chemical reaction by adding a substance known as a catalyst, which remains unchanged at the end of the reaction. A catalyst lowers the activation energy required for the reaction to occur, thereby allowing the reaction to proceed more quickly and efficiently. This can be particularly important in biological systems, where enzymes act as catalysts to speed up metabolic reactions that are essential for life.
Cell death is the process by which cells cease to function and eventually die. There are several ways that cells can die, but the two most well-known and well-studied forms of cell death are apoptosis and necrosis.
Apoptosis is a programmed form of cell death that occurs as a normal and necessary process in the development and maintenance of healthy tissues. During apoptosis, the cell's DNA is broken down into small fragments, the cell shrinks, and the membrane around the cell becomes fragmented, allowing the cell to be easily removed by phagocytic cells without causing an inflammatory response.
Necrosis, on the other hand, is a form of cell death that occurs as a result of acute tissue injury or overwhelming stress. During necrosis, the cell's membrane becomes damaged and the contents of the cell are released into the surrounding tissue, causing an inflammatory response.
There are also other forms of cell death, such as autophagy, which is a process by which cells break down their own organelles and proteins to recycle nutrients and maintain energy homeostasis, and pyroptosis, which is a form of programmed cell death that occurs in response to infection and involves the activation of inflammatory caspases.
Cell death is an important process in many physiological and pathological processes, including development, tissue homeostasis, and disease. Dysregulation of cell death can contribute to the development of various diseases, including cancer, neurodegenerative disorders, and autoimmune diseases.
Protein Kinase C-alpha (PKC-α) is a specific isoform of the Protein Kinase C (PKC) family, which are serine/threonine protein kinases that play crucial roles in various cellular processes such as proliferation, differentiation, and apoptosis. PKC-α is activated by diacylglycerol (DAG) and calcium ions (Ca2+). It is involved in signal transduction pathways related to cell growth, differentiation, and oncogenic transformation. Mutations or dysregulation of PKC-alpha have been implicated in several diseases including cancer, diabetes, and neurological disorders.
Janus Kinase 2 (JAK2) is a tyrosine kinase enzyme that plays a crucial role in intracellular signal transduction. It is named after the Roman god Janus, who is depicted with two faces, as JAK2 has two similar phosphate-transferring domains. JAK2 is involved in various cytokine receptor-mediated signaling pathways and contributes to hematopoiesis, immune function, and cell growth.
Mutations in the JAK2 gene have been associated with several myeloproliferative neoplasms (MPNs), including polycythemia vera, essential thrombocythemia, and primary myelofibrosis. The most common mutation is JAK2 V617F, which results in a constitutively active enzyme that promotes uncontrolled cell proliferation and survival, contributing to the development of these MPNs.
Pharmacology is the branch of medicine and biology concerned with the study of drugs, their actions, and their uses. It involves understanding how drugs interact with biological systems to produce desired effects, as well as any adverse or unwanted effects. This includes studying the absorption, distribution, metabolism, and excretion of drugs (often referred to as ADME), the receptors and biochemical pathways that drugs affect, and the therapeutic benefits and risks of drug use. Pharmacologists may also be involved in the development and testing of new medications.
A kidney, in medical terms, is one of two bean-shaped organs located in the lower back region of the body. They are essential for maintaining homeostasis within the body by performing several crucial functions such as:
1. Regulation of water and electrolyte balance: Kidneys help regulate the amount of water and various electrolytes like sodium, potassium, and calcium in the bloodstream to maintain a stable internal environment.
2. Excretion of waste products: They filter waste products from the blood, including urea (a byproduct of protein metabolism), creatinine (a breakdown product of muscle tissue), and other harmful substances that result from normal cellular functions or external sources like medications and toxins.
3. Endocrine function: Kidneys produce several hormones with important roles in the body, such as erythropoietin (stimulates red blood cell production), renin (regulates blood pressure), and calcitriol (activated form of vitamin D that helps regulate calcium homeostasis).
4. pH balance regulation: Kidneys maintain the proper acid-base balance in the body by excreting either hydrogen ions or bicarbonate ions, depending on whether the blood is too acidic or too alkaline.
5. Blood pressure control: The kidneys play a significant role in regulating blood pressure through the renin-angiotensin-aldosterone system (RAAS), which constricts blood vessels and promotes sodium and water retention to increase blood volume and, consequently, blood pressure.
Anatomically, each kidney is approximately 10-12 cm long, 5-7 cm wide, and 3 cm thick, with a weight of about 120-170 grams. They are surrounded by a protective layer of fat and connected to the urinary system through the renal pelvis, ureters, bladder, and urethra.
Calmodulin is a small, ubiquitous calcium-binding protein that plays a critical role in various intracellular signaling pathways. It functions as a calcium sensor, binding to and regulating the activity of numerous target proteins upon calcium ion (Ca^2+^) binding. Calmodulin is expressed in all eukaryotic cells and participates in many cellular processes, including muscle contraction, neurotransmitter release, gene expression, metabolism, and cell cycle progression.
The protein contains four EF-hand motifs that can bind Ca^2+^ ions. Upon calcium binding, conformational changes occur in the calmodulin structure, exposing hydrophobic surfaces that facilitate its interaction with target proteins. Calmodulin's targets include enzymes (such as protein kinases and phosphatases), ion channels, transporters, and cytoskeletal components. By modulating the activity of these proteins, calmodulin helps regulate essential cellular functions in response to changes in intracellular Ca^2+^ concentrations.
Calmodulin's molecular weight is approximately 17 kDa, and it consists of a single polypeptide chain with 148-150 amino acid residues. The protein can be found in both the cytoplasm and the nucleus of cells. In addition to its role as a calcium sensor, calmodulin has been implicated in various pathological conditions, including cancer, neurodegenerative diseases, and cardiovascular disorders.
Rho-associated kinases (ROCKs) are serine/threonine kinases that are involved in the regulation of various cellular processes, including actin cytoskeleton organization, cell migration, and gene expression. They are named after their association with the small GTPase RhoA, which activates them upon binding.
ROCKs exist as two isoforms, ROCK1 and ROCK2, which share a high degree of sequence homology and have similar functions. They contain several functional domains, including a kinase domain, a coiled-coil region that mediates protein-protein interactions, and a Rho-binding domain (RBD) that binds to active RhoA.
Once activated by RhoA, ROCKs phosphorylate a variety of downstream targets, including myosin light chain (MLC), LIM kinase (LIMK), and moesin, leading to the regulation of actomyosin contractility, stress fiber formation, and focal adhesion turnover. Dysregulation of ROCK signaling has been implicated in various pathological conditions, such as cancer, cardiovascular diseases, neurological disorders, and fibrosis. Therefore, ROCKs have emerged as promising therapeutic targets for the treatment of these diseases.
In genetics, sequence alignment is the process of arranging two or more DNA, RNA, or protein sequences to identify regions of similarity or homology between them. This is often done using computational methods to compare the nucleotide or amino acid sequences and identify matching patterns, which can provide insight into evolutionary relationships, functional domains, or potential genetic disorders. The alignment process typically involves adjusting gaps and mismatches in the sequences to maximize the similarity between them, resulting in an aligned sequence that can be visually represented and analyzed.
Quinones are a class of organic compounds that contain a fully conjugated diketone structure. This structure consists of two carbonyl groups (C=O) separated by a double bond (C=C). Quinones can be found in various biological systems and synthetic compounds. They play important roles in many biochemical processes, such as electron transport chains and redox reactions. Some quinones are also known for their antimicrobial and anticancer properties. However, some quinones can be toxic or mutagenic at high concentrations.
Cell differentiation is the process by which a less specialized cell, or stem cell, becomes a more specialized cell type with specific functions and structures. This process involves changes in gene expression, which are regulated by various intracellular signaling pathways and transcription factors. Differentiation results in the development of distinct cell types that make up tissues and organs in multicellular organisms. It is a crucial aspect of embryonic development, tissue repair, and maintenance of homeostasis in the body.
Ribonucleotides are organic compounds that consist of a ribose sugar, a phosphate group, and a nitrogenous base. They are the building blocks of RNA (ribonucleic acid), one of the essential molecules in all living organisms. The nitrogenous bases found in ribonucleotides include adenine, uracil, guanine, and cytosine. These molecules play crucial roles in various biological processes, such as protein synthesis, gene expression, and cellular energy production. Ribonucleotides can also be involved in cell signaling pathways and serve as important cofactors for enzymatic reactions.
Voltage-Dependent Anion Channels (VDACs) are large protein channels found in the outer mitochondrial membrane. They play a crucial role in the regulation of metabolite and ion exchange between the cytosol and the mitochondria. VDACs are permeable to anions such as chloride, phosphate, and bicarbonate ions, as well as to small molecules and metabolites like ATP, ADP, NADH, and others.
The voltage-dependent property of these channels arises from the fact that their permeability can be modulated by changes in the membrane potential across the outer mitochondrial membrane. At low membrane potentials, VDACs are predominantly open and facilitate the flow of metabolites and ions. However, as the membrane potential becomes more positive, VDACs can transition to a closed or partially closed state, which restricts ion and metabolite movement.
VDACs have been implicated in various cellular processes, including apoptosis, calcium homeostasis, and energy metabolism. Dysregulation of VDAC function has been associated with several pathological conditions, such as neurodegenerative diseases, cancer, and ischemia-reperfusion injury.
Molecular models are three-dimensional representations of molecular structures that are used in the field of molecular biology and chemistry to visualize and understand the spatial arrangement of atoms and bonds within a molecule. These models can be physical or computer-generated and allow researchers to study the shape, size, and behavior of molecules, which is crucial for understanding their function and interactions with other molecules.
Physical molecular models are often made up of balls (representing atoms) connected by rods or sticks (representing bonds). These models can be constructed manually using materials such as plastic or wooden balls and rods, or they can be created using 3D printing technology.
Computer-generated molecular models, on the other hand, are created using specialized software that allows researchers to visualize and manipulate molecular structures in three dimensions. These models can be used to simulate molecular interactions, predict molecular behavior, and design new drugs or chemicals with specific properties. Overall, molecular models play a critical role in advancing our understanding of molecular structures and their functions.
NADH, NADPH oxidoreductases are a class of enzymes that catalyze the redox reaction between NADH or NADPH and various electron acceptors. These enzymes play a crucial role in cellular metabolism by transferring electrons from NADH or NADPH to other molecules, which is essential for many biochemical reactions.
NADH (nicotinamide adenine dinucleotide hydrogen) and NADPH (nicotinamide adenine dinucleotide phosphate hydrogen) are coenzymes that act as electron carriers in redox reactions. They consist of a nicotinamide ring, which undergoes reduction or oxidation by accepting or donating electrons and a proton (H+).
NADH, NADPH oxidoreductases are classified based on their structure and mechanism of action. Some examples include:
1. Dehydrogenases: These enzymes catalyze the oxidation of NADH or NADPH to NAD+ or NADP+ while reducing an organic substrate. Examples include lactate dehydrogenase, alcohol dehydrogenase, and malate dehydrogenase.
2. Oxidases: These enzymes catalyze the oxidation of NADH or NADPH to NAD+ or NADP+ while reducing molecular oxygen (O2) to water (H2O). Examples include NADH oxidase and NADPH oxidase.
3. Reductases: These enzymes catalyze the reduction of various electron acceptors using NADH or NADPH as a source of electrons. Examples include glutathione reductase, thioredoxin reductase, and nitrate reductase.
Overall, NADH, NADPH oxidoreductases are essential for maintaining the redox balance in cells and play a critical role in various metabolic pathways, including energy production, detoxification, and biosynthesis.
Cyclic ethers are a type of organic compound that contain an ether functional group (-O-) within a cyclic (ring-shaped) structure. In a cyclic ether, one or more oxygen atoms are part of the ring, which can consist of various numbers of carbon atoms. The simplest example of a cyclic ether is oxirane, also known as ethylene oxide, which contains a three-membered ring with two carbon atoms and one oxygen atom.
Cyclic ethers have diverse applications in the chemical industry, including their use as building blocks for the synthesis of other chemicals, pharmaceuticals, and materials. Some cyclic ethers, like tetrahydrofuran (THF), are common solvents due to their ability to dissolve a wide range of organic compounds. However, some cyclic ethers can be hazardous or toxic, so they must be handled with care during laboratory work and industrial processes.
Dichloroacetic acid (DCA) is a chemical compound with the formula CCl2CO2H. It is a colorless liquid that is used as a reagent in organic synthesis and as a laboratory research tool. DCA is also a byproduct of water chlorination and has been found to occur in low levels in some chlorinated drinking waters.
In the medical field, DCA has been studied for its potential anticancer effects. Preclinical studies have suggested that DCA may be able to selectively kill cancer cells by inhibiting the activity of certain enzymes involved in cell metabolism. However, more research is needed to determine whether DCA is safe and effective as a cancer treatment in humans.
It is important to note that DCA is not currently approved by regulatory agencies such as the U.S. Food and Drug Administration (FDA) for use as a cancer treatment. It should only be used in clinical trials or under the supervision of a qualified healthcare professional.
Promoter regions in genetics refer to specific DNA sequences located near the transcription start site of a gene. They serve as binding sites for RNA polymerase and various transcription factors that regulate the initiation of gene transcription. These regulatory elements help control the rate of transcription and, therefore, the level of gene expression. Promoter regions can be composed of different types of sequences, such as the TATA box and CAAT box, and their organization and composition can vary between different genes and species.
A mitochondrial genome refers to the genetic material present in the mitochondria, which are small organelles found in the cytoplasm of eukaryotic cells (cells with a true nucleus). The mitochondrial genome is typically circular and contains a relatively small number of genes compared to the nuclear genome.
Mitochondrial DNA (mtDNA) encodes essential components of the electron transport chain, which is vital for cellular respiration and energy production. MtDNA also contains genes that code for some mitochondrial tRNAs and rRNAs needed for protein synthesis within the mitochondria.
In humans, the mitochondrial genome is about 16.6 kilobases in length and consists of 37 genes: 2 ribosomal RNA (rRNA) genes, 22 transfer RNA (tRNA) genes, and 13 protein-coding genes. The mitochondrial genome is inherited maternally, as sperm contribute very few or no mitochondria during fertilization. Mutations in the mitochondrial genome can lead to various genetic disorders, often affecting tissues with high energy demands, such as muscle and nerve cells.
I'm sorry for any confusion, but "Oxazoles" is not a medical term, it is a chemical term. Oxazoles are heterocyclic aromatic organic compounds that contain a five-membered ring made up of one nitrogen atom, one oxygen atom, and three carbon atoms. They have the molecular formula C4H4NO.
Oxazoles do not have specific medical relevance, but they can be found in some natural and synthetic substances, including certain drugs and bioactive molecules. Some oxazole-containing compounds have been studied for their potential medicinal properties, such as anti-inflammatory, antimicrobial, and anticancer activities. However, these studies are primarily within the field of chemistry and pharmacology, not medicine itself.
Glycogen is a complex carbohydrate that serves as the primary form of energy storage in animals, fungi, and bacteria. It is a polysaccharide consisting of long, branched chains of glucose molecules linked together by glycosidic bonds. Glycogen is stored primarily in the liver and muscles, where it can be quickly broken down to release glucose into the bloodstream during periods of fasting or increased metabolic demand.
In the liver, glycogen plays a crucial role in maintaining blood glucose levels by releasing glucose when needed, such as between meals or during exercise. In muscles, glycogen serves as an immediate energy source for muscle contractions during intense physical activity. The ability to store and mobilize glycogen is essential for the proper functioning of various physiological processes, including athletic performance, glucose homeostasis, and overall metabolic health.
Ataxia telangiectasia mutated (ATM) proteins are a type of protein that play a crucial role in the maintenance and repair of DNA in cells. The ATM gene produces these proteins, which are involved in several important cellular processes such as:
1. DNA damage response: When DNA is damaged, ATM proteins help to detect and respond to the damage by activating various signaling pathways that lead to DNA repair or apoptosis (programmed cell death) if the damage is too severe.
2. Cell cycle regulation: ATM proteins regulate the cell cycle by controlling checkpoints that ensure proper DNA replication and division. This helps prevent the propagation of cells with damaged DNA.
3. Telomere maintenance: ATM proteins help maintain telomeres, which are the protective caps at the ends of chromosomes. Telomeres shorten as cells divide, and when they become too short, cells can no longer divide and enter a state of senescence or die.
Mutations in the ATM gene can lead to Ataxia-telangiectasia (A-T), a rare inherited disorder characterized by neurological problems, immune system dysfunction, increased risk of cancer, and sensitivity to ionizing radiation. People with A-T have defective ATM proteins that cannot properly respond to DNA damage, leading to genomic instability and increased susceptibility to disease.
Cell adhesion refers to the binding of cells to extracellular matrices or to other cells, a process that is fundamental to the development, function, and maintenance of multicellular organisms. Cell adhesion is mediated by various cell surface receptors, such as integrins, cadherins, and immunoglobulin-like cell adhesion molecules (Ig-CAMs), which interact with specific ligands in the extracellular environment. These interactions lead to the formation of specialized junctions, such as tight junctions, adherens junctions, and desmosomes, that help to maintain tissue architecture and regulate various cellular processes, including proliferation, differentiation, migration, and survival. Disruptions in cell adhesion can contribute to a variety of diseases, including cancer, inflammation, and degenerative disorders.
Proto-oncogene proteins c-bcl-2 are a group of proteins that play a role in regulating cell death (apoptosis). The c-bcl-2 gene produces one of these proteins, which helps to prevent cells from undergoing apoptosis. This protein is located on the membrane of mitochondria and endoplasmic reticulum and it can inhibit the release of cytochrome c, a key player in the activation of caspases, which are enzymes that trigger apoptosis.
In normal cells, the regulation of c-bcl-2 protein helps to maintain a balance between cell proliferation and cell death, ensuring proper tissue homeostasis. However, when the c-bcl-2 gene is mutated or its expression is dysregulated, it can contribute to cancer development by allowing cancer cells to survive and proliferate. High levels of c-bcl-2 protein have been found in many types of cancer, including leukemia, lymphoma, and carcinomas, and are often associated with a poor prognosis.
Phosphofructokinase (PFK) is an enzyme that plays a crucial role in regulating glycolysis, which is the metabolic pathway responsible for the conversion of glucose into energy. PFK catalyzes the phosphorylation of fructose-6-phosphate to fructose-1,6-bisphosphate, using a molecule of adenosine triphosphate (ATP) as a source of energy. This reaction is a key regulatory step in glycolysis and is subject to allosteric regulation by various metabolites, such as ATP, ADP, and citrate, that signal the cell's energy status.
There are several isoforms of PFK found in different tissues, including PFK-1 (or muscle PFK) and PFK-2 (or liver PFK), which exhibit tissue-specific patterns of expression and regulation. Mutations in the genes encoding PFK can result in various inherited metabolic disorders, such as Tarui's disease, characterized by exercise intolerance, muscle cramps, and myoglobinuria.
Hydroxybutyrates are compounds that contain a hydroxyl group (-OH) and a butyric acid group. More specifically, in the context of clinical medicine and biochemistry, β-hydroxybutyrate (BHB) is often referred to as a "ketone body."
Ketone bodies are produced by the liver during periods of low carbohydrate availability, such as during fasting, starvation, or a high-fat, low-carbohydrate diet. BHB is one of three major ketone bodies, along with acetoacetate and acetone. These molecules serve as alternative energy sources for the brain and other tissues when glucose levels are low.
In some pathological states, such as diabetic ketoacidosis, the body produces excessive amounts of ketone bodies, leading to a life-threatening metabolic acidosis. Elevated levels of BHB can also be found in other conditions like alcoholism, severe illnesses, and high-fat diets.
It is important to note that while BHB is a hydroxybutyrate, not all hydroxybutyrates are ketone bodies. The term "hydroxybutyrates" can refer to any compound containing both a hydroxyl group (-OH) and a butyric acid group.
Maleimides are a class of chemical compounds that contain a maleimide functional group, which is characterized by a five-membered ring containing two carbon atoms and three nitrogen atoms. The double bond in the maleimide ring makes it highly reactive towards nucleophiles, particularly thiol groups found in cysteine residues of proteins.
In medical and biological contexts, maleimides are often used as cross-linking agents to modify or label proteins, peptides, and other biomolecules. For example, maleimide-functionalized probes such as fluorescent dyes, biotin, or radioisotopes can be covalently attached to thiol groups in proteins for various applications, including protein detection, purification, and imaging.
However, it is important to note that maleimides can also react with other nucleophiles such as amines, although at a slower rate. Therefore, careful control of reaction conditions is necessary to ensure specificity towards thiol groups.
Confocal microscopy is a powerful imaging technique used in medical and biological research to obtain high-resolution, contrast-rich images of thick samples. This super-resolution technology provides detailed visualization of cellular structures and processes at various depths within a specimen.
In confocal microscopy, a laser beam focused through a pinhole illuminates a small spot within the sample. The emitted fluorescence or reflected light from this spot is then collected by a detector, passing through a second pinhole that ensures only light from the focal plane reaches the detector. This process eliminates out-of-focus light, resulting in sharp images with improved contrast compared to conventional widefield microscopy.
By scanning the laser beam across the sample in a raster pattern and collecting fluorescence at each point, confocal microscopy generates optical sections of the specimen. These sections can be combined to create three-dimensional reconstructions, allowing researchers to study cellular architecture and interactions within complex tissues.
Confocal microscopy has numerous applications in medical research, including studying protein localization, tracking intracellular dynamics, analyzing cell morphology, and investigating disease mechanisms at the cellular level. Additionally, it is widely used in clinical settings for diagnostic purposes, such as analyzing skin lesions or detecting pathogens in patient samples.
Butadienes are a class of organic compounds that contain a chemical structure consisting of two carbon-carbon double bonds arranged in a conjugated system. The most common butadiene is 1,3-butadiene, which is an important industrial chemical used in the production of synthetic rubber and plastics.
1,3-Butadiene is a colorless gas that is highly flammable and has a mild sweet odor. It is produced as a byproduct of petroleum refining and is also released during the combustion of fossil fuels. Exposure to butadienes can occur through inhalation, skin contact, or ingestion, and prolonged exposure has been linked to an increased risk of cancer, particularly leukemia.
Other forms of butadiene include 1,2-butadiene and 1,4-butadiene, which have different chemical properties and uses. Overall, butadienes are important industrial chemicals with a wide range of applications, but their potential health hazards require careful handling and regulation.
Glucose Transporter Type 1 (GLUT1) is a specific type of protein called a glucose transporter, which is responsible for facilitating the transport of glucose across the blood-brain barrier and into the brain cells. It is encoded by the SLC2A1 gene and is primarily found in the endothelial cells of the blood-brain barrier, as well as in other tissues such as the erythrocytes (red blood cells), placenta, and kidney.
GLUT1 plays a critical role in maintaining normal glucose levels in the brain, as it is the main mechanism for glucose uptake into the brain. Disorders of GLUT1 can lead to impaired glucose transport, which can result in neurological symptoms such as seizures, developmental delay, and movement disorders. These disorders are known as GLUT1 deficiency syndromes.
Genistein is defined as a type of isoflavone, which is a plant-derived compound with estrogen-like properties. It is found in soybeans and other legumes. Genistein acts as a phytoestrogen, meaning it can bind to estrogen receptors and have both weak estrogenic and anti-estrogenic effects in the body.
In addition to its estrogenic activity, genistein has been found to have various biological activities, such as antioxidant, anti-inflammatory, and anticancer properties. It has been studied for its potential role in preventing or treating a variety of health conditions, including cancer, cardiovascular disease, osteoporosis, and menopausal symptoms. However, more research is needed to fully understand the potential benefits and risks of genistein supplementation.
Microfilament proteins are a type of structural protein that form part of the cytoskeleton in eukaryotic cells. They are made up of actin monomers, which polymerize to form long, thin filaments. These filaments are involved in various cellular processes such as muscle contraction, cell division, and cell motility. Microfilament proteins also interact with other cytoskeletal components like intermediate filaments and microtubules to maintain the overall shape and integrity of the cell. Additionally, they play a crucial role in the formation of cell-cell junctions and cell-matrix adhesions, which are essential for tissue structure and function.
MAPKKK1 or Mitogen-Activated Protein Kinase Kinase Kinase 1 is a serine/threonine protein kinase that belongs to the MAP3K family. It plays a crucial role in intracellular signal transduction pathways, particularly in the MAPK/ERK cascade, which is involved in various cellular processes such as proliferation, differentiation, and survival.
MAPKKK1 activates MAPKKs (Mitogen-Activated Protein Kinase Kinases) through phosphorylation of specific serine and threonine residues. In turn, activated MAPKKs phosphorylate and activate MAPKs (Mitogen-Activated Protein Kinases), which then regulate the activity of various transcription factors and other downstream targets to elicit appropriate cellular responses.
Mutations in MAPKKK1 have been implicated in several human diseases, including cancer and developmental disorders. Therefore, understanding its function and regulation is essential for developing novel therapeutic strategies to treat these conditions.
Myosin-Light-Chain Phosphatase (MLCP) is an enzyme complex that plays a crucial role in the regulation of muscle contraction and relaxation. It is responsible for dephosphorylating the myosin light chains, which are key regulatory components of the contractile apparatus in muscles.
The phosphorylation state of the myosin light chains regulates the interaction between actin and myosin filaments, which is necessary for muscle contraction. When the myosin light chains are phosphorylated, they bind more strongly to actin, leading to increased contractile force. Conversely, when the myosin light chains are dephosphorylated by MLCP, the interaction between actin and myosin is weakened, allowing for muscle relaxation.
MLCP is composed of three subunits: a catalytic subunit (PP1cδ), a regulatory subunit (MYPT1), and a small subunit (M20). The regulatory subunit contains binding sites for various signaling molecules that can modulate the activity of MLCP, such as calcium/calmodulin, protein kinase C, and Rho-associated protein kinase (ROCK). Dysregulation of MLCP has been implicated in various muscle disorders, including hypertrophic cardiomyopathy, dilated cardiomyopathy, and muscle atrophy.
I'm sorry for any confusion, but "Pyridines" is not a medical term. It is a chemical term that refers to a class of organic compounds with the chemical structure of a six-membered ring containing one nitrogen atom and five carbon atoms (heterocyclic aromatic compound).
In a biological or medical context, pyridine derivatives can be found in various natural and synthetic substances. For example, some medications contain pyridine rings as part of their chemical structure. However, "Pyridines" itself is not a medical term or condition.
Oligonucleotide Array Sequence Analysis is a type of microarray analysis that allows for the simultaneous measurement of the expression levels of thousands of genes in a single sample. In this technique, oligonucleotides (short DNA sequences) are attached to a solid support, such as a glass slide, in a specific pattern. These oligonucleotides are designed to be complementary to specific target mRNA sequences from the sample being analyzed.
During the analysis, labeled RNA or cDNA from the sample is hybridized to the oligonucleotide array. The level of hybridization is then measured and used to determine the relative abundance of each target sequence in the sample. This information can be used to identify differences in gene expression between samples, which can help researchers understand the underlying biological processes involved in various diseases or developmental stages.
It's important to note that this technique requires specialized equipment and bioinformatics tools for data analysis, as well as careful experimental design and validation to ensure accurate and reproducible results.
Signal Transducer and Activator of Transcription 1 (STAT1) is a transcription factor that plays a crucial role in the regulation of gene expression in response to cytokines and interferons. It is activated through phosphorylation by Janus kinases (JAKs) upon binding of cytokines to their respective receptors. Once activated, STAT1 forms homodimers or heterodimers with other STAT family members, translocates to the nucleus, and binds to specific DNA sequences called gamma-activated sites (GAS) in the promoter regions of target genes. This results in the modulation of gene expression involved in various cellular processes such as immune responses, differentiation, apoptosis, and cell cycle control. STAT1 also plays a critical role in the antiviral response by mediating the transcription of interferon-stimulated genes (ISGs).
Physiological adaptation refers to the changes or modifications that occur in an organism's biological functions or structures as a result of environmental pressures or changes. These adaptations enable the organism to survive and reproduce more successfully in its environment. They can be short-term, such as the constriction of blood vessels in response to cold temperatures, or long-term, such as the evolution of longer limbs in animals that live in open environments.
In the context of human physiology, examples of physiological adaptation include:
1. Acclimatization: The process by which the body adjusts to changes in environmental conditions, such as altitude or temperature. For example, when a person moves to a high-altitude location, their body may produce more red blood cells to compensate for the lower oxygen levels, leading to improved oxygen delivery to tissues.
2. Exercise adaptation: Regular physical activity can lead to various physiological adaptations, such as increased muscle strength and endurance, enhanced cardiovascular function, and improved insulin sensitivity.
3. Hormonal adaptation: The body can adjust hormone levels in response to changes in the environment or internal conditions. For instance, during prolonged fasting, the body releases stress hormones like cortisol and adrenaline to help maintain energy levels and prevent muscle wasting.
4. Sensory adaptation: Our senses can adapt to different stimuli over time. For example, when we enter a dark room after being in bright sunlight, it takes some time for our eyes to adjust to the new light level. This process is known as dark adaptation.
5. Aging-related adaptations: As we age, various physiological changes occur that help us adapt to the changing environment and maintain homeostasis. These include changes in body composition, immune function, and cognitive abilities.
Proto-oncogene proteins c-RAF, also known as RAF kinases, are serine/threonine protein kinases that play crucial roles in regulating cell growth, differentiation, and survival. They are part of the RAS/RAF/MEK/ERK signaling pathway, which is a key intracellular signaling cascade that conveys signals from various extracellular stimuli, such as growth factors and hormones, to the nucleus.
The c-RAF protein exists in three isoforms: A-RAF, B-RAF, and C-RAF (also known as RAF-1). These isoforms share a common structure, consisting of several functional domains, including an N-terminal regulatory region, a central kinase domain, and a C-terminal autoinhibitory region. In their inactive state, c-RAF proteins are bound to the cell membrane through interactions with RAS GTPases and other regulatory proteins.
Upon activation of RAS GTPases by upstream signals, c-RAF becomes recruited to the plasma membrane, where it undergoes a conformational change that leads to its activation. Activated c-RAF then phosphorylates and activates MEK (MAPK/ERK kinase) proteins, which in turn phosphorylate and activate ERK (Extracellular Signal-Regulated Kinase) proteins. Activated ERK proteins can translocate to the nucleus and regulate the expression of various genes involved in cell growth, differentiation, and survival.
Mutations in c-RAF proto-oncogenes can lead to their constitutive activation, resulting in uncontrolled cell growth and division, which can contribute to the development of various types of cancer. In particular, B-RAF mutations have been identified in several human malignancies, including melanoma, colorectal cancer, and thyroid cancer.
Glutathione transferases (GSTs) are a group of enzymes involved in the detoxification of xenobiotics and endogenous compounds. They facilitate the conjugation of these compounds with glutathione, a tripeptide consisting of cysteine, glutamic acid, and glycine, which results in more water-soluble products that can be easily excreted from the body.
GSTs play a crucial role in protecting cells against oxidative stress and chemical injury by neutralizing reactive electrophilic species and peroxides. They are found in various tissues, including the liver, kidneys, lungs, and intestines, and are classified into several families based on their structure and function.
Abnormalities in GST activity have been associated with increased susceptibility to certain diseases, such as cancer, neurological disorders, and respiratory diseases. Therefore, GSTs have become a subject of interest in toxicology, pharmacology, and clinical research.
Neoplastic cell transformation is a process in which a normal cell undergoes genetic alterations that cause it to become cancerous or malignant. This process involves changes in the cell's DNA that result in uncontrolled cell growth and division, loss of contact inhibition, and the ability to invade surrounding tissues and metastasize (spread) to other parts of the body.
Neoplastic transformation can occur as a result of various factors, including genetic mutations, exposure to carcinogens, viral infections, chronic inflammation, and aging. These changes can lead to the activation of oncogenes or the inactivation of tumor suppressor genes, which regulate cell growth and division.
The transformation of normal cells into cancerous cells is a complex and multi-step process that involves multiple genetic and epigenetic alterations. It is characterized by several hallmarks, including sustained proliferative signaling, evasion of growth suppressors, resistance to cell death, enabling replicative immortality, induction of angiogenesis, activation of invasion and metastasis, reprogramming of energy metabolism, and evading immune destruction.
Neoplastic cell transformation is a fundamental concept in cancer biology and is critical for understanding the molecular mechanisms underlying cancer development and progression. It also has important implications for cancer diagnosis, prognosis, and treatment, as identifying the specific genetic alterations that underlie neoplastic transformation can help guide targeted therapies and personalized medicine approaches.
Fluoroacetates are organic compounds that contain a fluorine atom and an acetic acid group. The most well-known and notorious compound in this family is sodium fluoroacetate, also known as 1080 or compound 1080, which is a potent metabolic poison. It works by interfering with the citric acid cycle, a critical process that generates energy in cells. Specifically, fluoroacetates are converted into fluorocitrate, which inhibits an enzyme called aconitase, leading to disruption of cellular metabolism and ultimately cell death.
Fluoroacetates have been used as rodenticides and pesticides, but their use is highly regulated due to their high toxicity to non-target species, including humans. Exposure to fluoroacetates can cause a range of symptoms, including nausea, vomiting, seizures, and cardiac arrest, and can be fatal if not treated promptly.
Eukaryotic Initiation Factor-4E (eIF4E) is a protein that plays a crucial role in the initiation phase of protein synthesis in eukaryotic cells. It is a subunit of the eIF4F complex, which also includes eIF4A and eIF4G proteins.
The primary function of eIF4E is to recognize and bind to the 5' cap structure (m7GpppN) of messenger RNA (mRNA), a modified guanine nucleotide that is added to the 5' end of mRNA during transcription. This binding event helps recruit other initiation factors, including eIF4A and eIF4G, to form the eIF4F complex, which subsequently binds to the small ribosomal subunit and promotes the scanning of the 5' untranslated region (5' UTR) of mRNA for the start codon (AUG).
The activity of eIF4E is tightly regulated through various post-translational modifications, such as phosphorylation, and interactions with other regulatory proteins. Dysregulation of eIF4E has been implicated in several human diseases, including cancer, where increased eIF4E expression and activity have been associated with poor prognosis and resistance to therapy.
NIH 3T3 cells are a type of mouse fibroblast cell line that was developed by the National Institutes of Health (NIH). The "3T3" designation refers to the fact that these cells were derived from embryonic Swiss mouse tissue and were able to be passaged (i.e., subcultured) more than three times in tissue culture.
NIH 3T3 cells are widely used in scientific research, particularly in studies involving cell growth and differentiation, signal transduction, and gene expression. They have also been used as a model system for studying the effects of various chemicals and drugs on cell behavior. NIH 3T3 cells are known to be relatively easy to culture and maintain, and they have a stable, flat morphology that makes them well-suited for use in microscopy studies.
It is important to note that, as with any cell line, it is essential to verify the identity and authenticity of NIH 3T3 cells before using them in research, as contamination or misidentification can lead to erroneous results.
Autosomal dominant optic atrophy (ADOA) is a genetic disorder that affects the optic nerve, which transmits visual information from the eye to the brain. The term "optic atrophy" refers to degeneration or damage to the optic nerve. In ADOA, this condition is inherited in an autosomal dominant manner, meaning that only one copy of the mutated gene, located on one of the autosomal chromosomes (not a sex chromosome), needs to be present for the individual to develop the disorder.
The most common form of ADOA is caused by mutations in the OPA1 gene, which provides instructions for making a protein involved in the maintenance of mitochondria, the energy-producing structures in cells. The exact role of this protein in optic nerve function is not fully understood, but it is thought to play a critical role in maintaining the health and function of retinal ganglion cells, which are the neurons that make up the optic nerve.
In ADOA, mutations in the OPA1 gene lead to progressive degeneration of retinal ganglion cells and their axons (nerve fibers) within the optic nerve. This results in decreased visual acuity, color vision deficits, and a characteristic visual field defect called centrocecal scotoma, which is an area of blindness near the center of the visual field. The onset and severity of these symptoms can vary widely among individuals with ADOA.
It's important to note that medical definitions may contain complex terminology. In simpler terms, autosomal dominant optic atrophy (ADOA) is a genetic condition affecting the optic nerve, leading to decreased visual acuity and other vision problems due to degeneration of retinal ganglion cells. The disorder is inherited in an autosomal dominant manner, meaning only one copy of the mutated gene is needed for the individual to develop ADOA.
Cell membrane permeability refers to the ability of various substances, such as molecules and ions, to pass through the cell membrane. The cell membrane, also known as the plasma membrane, is a thin, flexible barrier that surrounds all cells, controlling what enters and leaves the cell. Its primary function is to protect the cell's internal environment and maintain homeostasis.
The permeability of the cell membrane depends on its structure, which consists of a phospholipid bilayer interspersed with proteins. The hydrophilic (water-loving) heads of the phospholipids face outward, while the hydrophobic (water-fearing) tails face inward, creating a barrier that is generally impermeable to large, polar, or charged molecules.
However, specific proteins within the membrane, called channels and transporters, allow certain substances to cross the membrane. Channels are protein structures that span the membrane and provide a pore for ions or small uncharged molecules to pass through. Transporters, on the other hand, are proteins that bind to specific molecules and facilitate their movement across the membrane, often using energy in the form of ATP.
The permeability of the cell membrane can be influenced by various factors, such as temperature, pH, and the presence of certain chemicals or drugs. Changes in permeability can have significant consequences for the cell's function and survival, as they can disrupt ion balances, nutrient uptake, waste removal, and signal transduction.
Heat-shock proteins (HSPs) are a group of conserved proteins that are produced by cells in response to stressful conditions, such as increased temperature, exposure to toxins, or infection. They play an essential role in protecting cells and promoting their survival under stressful conditions by assisting in the proper folding and assembly of other proteins, preventing protein aggregation, and helping to refold or degrade damaged proteins. HSPs are named according to their molecular weight, for example, HSP70 and HSP90. They are found in all living organisms, from bacteria to humans, indicating their fundamental importance in cellular function and survival.
I-kappa B kinase (IKK) is a protein complex that plays a crucial role in the activation of NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cells), a transcription factor involved in the regulation of immune response, inflammation, cell survival, and proliferation.
The IKK complex is composed of two catalytic subunits, IKKα and IKKβ, and a regulatory subunit, IKKγ (also known as NEMO). Upon stimulation by various signals such as cytokines, pathogens, or stress, the IKK complex becomes activated and phosphorylates I-kappa B (IkB), an inhibitor protein that keeps NF-kB in an inactive state in the cytoplasm.
Once IkB is phosphorylated by the IKK complex, it undergoes ubiquitination and degradation, leading to the release and nuclear translocation of NF-kB, where it can bind to specific DNA sequences and regulate gene expression. Dysregulation of IKK activity has been implicated in various pathological conditions, including chronic inflammation, autoimmune diseases, and cancer.
BCL-associated death protein, often referred to as BAD, is a type of protein that belongs to the BCL-2 family. These proteins play a crucial role in regulating programmed cell death, also known as apoptosis. Specifically, BAD is a pro-apoptotic protein, meaning it promotes cell death under certain conditions.
The function of BAD is tightly regulated through various post-translational modifications and interactions with other BCL-2 family members. When activated, BAD can bind to and inhibit anti-apoptotic proteins like BCL-2 or BCL-XL, thereby releasing pro-apoptotic proteins such as BAX and BAK, which form pores in the mitochondrial membrane and initiate the apoptotic cascade.
Dysregulation of BAD and other BCL-2 family members has been implicated in several diseases, including cancer and neurodegenerative disorders. For instance, overexpression of anti-apoptotic proteins or downregulation of pro-apoptotic proteins like BAD can contribute to tumor development and resistance to chemotherapy. Therefore, understanding the role of BAD and other BCL-2 family members in apoptosis regulation is essential for developing novel therapeutic strategies in cancer and other diseases.
Immunohistochemistry (IHC) is a technique used in pathology and laboratory medicine to identify specific proteins or antigens in tissue sections. It combines the principles of immunology and histology to detect the presence and location of these target molecules within cells and tissues. This technique utilizes antibodies that are specific to the protein or antigen of interest, which are then tagged with a detection system such as a chromogen or fluorophore. The stained tissue sections can be examined under a microscope, allowing for the visualization and analysis of the distribution and expression patterns of the target molecule in the context of the tissue architecture. Immunohistochemistry is widely used in diagnostic pathology to help identify various diseases, including cancer, infectious diseases, and immune-mediated disorders.
Ionophores are compounds that have the ability to form complexes with ions and facilitate their transportation across biological membranes. They can be either organic or inorganic molecules, and they play important roles in various physiological processes, including ion homeostasis, signal transduction, and antibiotic activity. In medicine and research, ionophores are used as tools to study ion transport, modulate cellular functions, and as therapeutic agents, especially in the treatment of bacterial and fungal infections.
I'm sorry for any confusion, but "Tin" does not have a medical definition. Tin is a chemical element with the symbol Sn and atomic number 50. It is a malleable, ductile, silvery-white post-transition metal. It is found in nature mainly as tin oxides and is obtained from the mineral cassiterite through mining and processing.
Tin has no known biological role in humans, animals, or plants, and it is not considered an essential nutrient. Small amounts of tin can be found in some foods and drinking water, but these levels are generally low and not considered harmful. High levels of tin can be toxic to the human body, causing symptoms such as nausea, vomiting, and diarrhea.
If you have any questions about a medical condition or treatment, I would recommend consulting with a healthcare professional for accurate information and guidance.
Creatine is a organic acid that is produced naturally in the liver, kidneys and pancreas. It is also found in small amounts in certain foods such as meat and fish. The chemical formula for creatine is C4H9N3O2. In the body, creatine is converted into creatine phosphate, which is used to help produce energy during high-intensity exercise, such as weightlifting or sprinting.
Creatine can also be taken as a dietary supplement, in the form of creatine monohydrate, with the goal of increasing muscle creatine and phosphocreatine levels, which may improve athletic performance and help with muscle growth. However, it is important to note that while some studies have found that creatine supplementation can improve exercise performance and muscle mass in certain populations, others have not found significant benefits.
Creatine supplements are generally considered safe when used as directed, but they can cause side effects such as weight gain, stomach discomfort, and muscle cramps in some people. It is always recommended to consult a healthcare professional before starting any new supplement regimen.
Myosins are a large family of motor proteins that play a crucial role in various cellular processes, including muscle contraction and intracellular transport. They consist of heavy chains, which contain the motor domain responsible for generating force and motion, and light chains, which regulate the activity of the myosin. Based on their structural and functional differences, myosins are classified into over 35 classes, with classes II, V, and VI being the most well-studied.
Class II myosins, also known as conventional myosins, are responsible for muscle contraction in skeletal, cardiac, and smooth muscles. They form filaments called thick filaments, which interact with actin filaments to generate force and movement during muscle contraction.
Class V myosins, also known as unconventional myosins, are involved in intracellular transport and organelle positioning. They have a long tail that can bind to various cargoes, such as vesicles, mitochondria, and nuclei, and a motor domain that moves along actin filaments to transport the cargoes to their destinations.
Class VI myosins are also unconventional myosins involved in intracellular transport and organelle positioning. They have two heads connected by a coiled-coil tail, which can bind to various cargoes. Class VI myosins move along actin filaments in a unique hand-over-hand motion, allowing them to transport their cargoes efficiently.
Overall, myosins are essential for many cellular functions and have been implicated in various diseases, including cardiovascular diseases, neurological disorders, and cancer.
Indole is not strictly a medical term, but it is a chemical compound that can be found in the human body and has relevance to medical and biological research. Indoles are organic compounds that contain a bicyclic structure consisting of a six-membered benzene ring fused to a five-membered pyrrole ring.
In the context of medicine, indoles are particularly relevant due to their presence in certain hormones and other biologically active molecules. For example, the neurotransmitter serotonin contains an indole ring, as does the hormone melatonin. Indoles can also be found in various plant-based foods, such as cruciferous vegetables (e.g., broccoli, kale), and have been studied for their potential health benefits.
Some indoles, like indole-3-carbinol and diindolylmethane, are found in these vegetables and can have anti-cancer properties by modulating estrogen metabolism, reducing inflammation, and promoting cell death (apoptosis) in cancer cells. However, it is essential to note that further research is needed to fully understand the potential health benefits and risks associated with indoles.
Molecular cloning is a laboratory technique used to create multiple copies of a specific DNA sequence. This process involves several steps:
1. Isolation: The first step in molecular cloning is to isolate the DNA sequence of interest from the rest of the genomic DNA. This can be done using various methods such as PCR (polymerase chain reaction), restriction enzymes, or hybridization.
2. Vector construction: Once the DNA sequence of interest has been isolated, it must be inserted into a vector, which is a small circular DNA molecule that can replicate independently in a host cell. Common vectors used in molecular cloning include plasmids and phages.
3. Transformation: The constructed vector is then introduced into a host cell, usually a bacterial or yeast cell, through a process called transformation. This can be done using various methods such as electroporation or chemical transformation.
4. Selection: After transformation, the host cells are grown in selective media that allow only those cells containing the vector to grow. This ensures that the DNA sequence of interest has been successfully cloned into the vector.
5. Amplification: Once the host cells have been selected, they can be grown in large quantities to amplify the number of copies of the cloned DNA sequence.
Molecular cloning is a powerful tool in molecular biology and has numerous applications, including the production of recombinant proteins, gene therapy, functional analysis of genes, and genetic engineering.
The Pyruvate Dehydrogenase Complex (PDC) is a multi-enzyme complex that plays a crucial role in cellular energy metabolism. It is located in the mitochondrial matrix and catalyzes the oxidative decarboxylation of pyruvate, the end product of glycolysis, into acetyl-CoA. This reaction links the carbohydrate metabolism (glycolysis) to the citric acid cycle (Krebs cycle), enabling the continuation of energy production in the form of ATP through oxidative phosphorylation.
The Pyruvate Dehydrogenase Complex consists of three main enzymes: pyruvate dehydrogenase (E1), dihydrolipoyl transacetylase (E2), and dihydrolipoyl dehydrogenase (E3). Additionally, two regulatory enzymes are associated with the complex: pyruvate dehydrogenase kinase (PDK) and pyruvate dehydrogenase phosphatase (PDP). These regulatory enzymes control the activity of the PDC through reversible phosphorylation and dephosphorylation, allowing the cell to adapt to varying energy demands and substrate availability.
Deficiencies or dysfunctions in the Pyruvate Dehydrogenase Complex can lead to various metabolic disorders, such as pyruvate dehydrogenase deficiency, which may result in neurological impairments and lactic acidosis due to disrupted energy metabolism.
Epithelial cells are types of cells that cover the outer surfaces of the body, line the inner surfaces of organs and glands, and form the lining of blood vessels and body cavities. They provide a protective barrier against the external environment, regulate the movement of materials between the internal and external environments, and are involved in the sense of touch, temperature, and pain. Epithelial cells can be squamous (flat and thin), cuboidal (square-shaped and of equal height), or columnar (tall and narrow) in shape and are classified based on their location and function.
Electron microscopy (EM) is a type of microscopy that uses a beam of electrons to create an image of the sample being examined, resulting in much higher magnification and resolution than light microscopy. There are several types of electron microscopy, including transmission electron microscopy (TEM), scanning electron microscopy (SEM), and reflection electron microscopy (REM).
In TEM, a beam of electrons is transmitted through a thin slice of the sample, and the electrons that pass through the sample are focused to form an image. This technique can provide detailed information about the internal structure of cells, viruses, and other biological specimens, as well as the composition and structure of materials at the atomic level.
In SEM, a beam of electrons is scanned across the surface of the sample, and the electrons that are scattered back from the surface are detected to create an image. This technique can provide information about the topography and composition of surfaces, as well as the structure of materials at the microscopic level.
REM is a variation of SEM in which the beam of electrons is reflected off the surface of the sample, rather than scattered back from it. This technique can provide information about the surface chemistry and composition of materials.
Electron microscopy has a wide range of applications in biology, medicine, and materials science, including the study of cellular structure and function, disease diagnosis, and the development of new materials and technologies.
PC12 cells are a type of rat pheochromocytoma cell line, which are commonly used in scientific research. Pheochromocytomas are tumors that develop from the chromaffin cells of the adrenal gland, and PC12 cells are a subtype of these cells.
PC12 cells have several characteristics that make them useful for research purposes. They can be grown in culture and can be differentiated into a neuron-like phenotype when treated with nerve growth factor (NGF). This makes them a popular choice for studies involving neuroscience, neurotoxicity, and neurodegenerative disorders.
PC12 cells are also known to express various neurotransmitter receptors, ion channels, and other proteins that are relevant to neuronal function, making them useful for studying the mechanisms of drug action and toxicity. Additionally, PC12 cells can be used to study the regulation of cell growth and differentiation, as well as the molecular basis of cancer.
Gene deletion is a type of mutation where a segment of DNA, containing one or more genes, is permanently lost or removed from a chromosome. This can occur due to various genetic mechanisms such as homologous recombination, non-homologous end joining, or other types of genomic rearrangements.
The deletion of a gene can have varying effects on the organism, depending on the function of the deleted gene and its importance for normal physiological processes. If the deleted gene is essential for survival, the deletion may result in embryonic lethality or developmental abnormalities. However, if the gene is non-essential or has redundant functions, the deletion may not have any noticeable effects on the organism's phenotype.
Gene deletions can also be used as a tool in genetic research to study the function of specific genes and their role in various biological processes. For example, researchers may use gene deletion techniques to create genetically modified animal models to investigate the impact of gene deletion on disease progression or development.
Proline is an organic compound that is classified as a non-essential amino acid, meaning it can be produced by the human body and does not need to be obtained through the diet. It is encoded in the genetic code as the codon CCU, CCC, CCA, or CCG. Proline is a cyclic amino acid, containing an unusual secondary amine group, which forms a ring structure with its carboxyl group.
In proteins, proline acts as a structural helix breaker, disrupting the alpha-helix structure and leading to the formation of turns and bends in the protein chain. This property is important for the proper folding and function of many proteins. Proline also plays a role in the stability of collagen, a major structural protein found in connective tissues such as tendons, ligaments, and skin.
In addition to its role in protein structure, proline has been implicated in various cellular processes, including signal transduction, apoptosis, and oxidative stress response. It is also a precursor for the synthesis of other biologically important compounds such as hydroxyproline, which is found in collagen and elastin, and glutamate, an excitatory neurotransmitter in the brain.
Dimerization is a process in which two molecules, usually proteins or similar structures, bind together to form a larger complex. This can occur through various mechanisms, such as the formation of disulfide bonds, hydrogen bonding, or other non-covalent interactions. Dimerization can play important roles in cell signaling, enzyme function, and the regulation of gene expression.
In the context of medical research and therapy, dimerization is often studied in relation to specific proteins that are involved in diseases such as cancer. For example, some drugs have been developed to target and inhibit the dimerization of certain proteins, with the goal of disrupting their function and slowing or stopping the progression of the disease.
Medical Definition of "Multiprotein Complexes" :
Multiprotein complexes are large molecular assemblies composed of two or more proteins that interact with each other to carry out specific cellular functions. These complexes can range from relatively simple dimers or trimers to massive structures containing hundreds of individual protein subunits. They are formed through a process known as protein-protein interaction, which is mediated by specialized regions on the protein surface called domains or motifs.
Multiprotein complexes play critical roles in many cellular processes, including signal transduction, gene regulation, DNA replication and repair, protein folding and degradation, and intracellular transport. The formation of these complexes is often dynamic and regulated in response to various stimuli, allowing for precise control of their function.
Disruption of multiprotein complexes can lead to a variety of diseases, including cancer, neurodegenerative disorders, and infectious diseases. Therefore, understanding the structure, composition, and regulation of these complexes is an important area of research in molecular biology and medicine.
'Onium compounds' is a general term used in chemistry and biochemistry to describe a class of organic compounds that contain a positively charged functional group. The name 'onium' refers to the positive charge, which is usually located on a nitrogen or phosphorus atom.
The most common onium compounds are ammonium compounds (positive charge on a nitrogen atom) and phosphonium compounds (positive charge on a phosphorus atom). Other examples include sulfonium compounds (positive charge on a sulfur atom) and oxonium compounds (positive charge on an oxygen atom).
In the context of medical research, onium compounds may be studied for their potential use as drugs or diagnostic agents. For example, certain ammonium compounds have been shown to have antimicrobial properties and are used in some disinfectants and sanitizers. Phosphonium compounds have been investigated for their potential use as anti-cancer agents, while sulfonium compounds have been studied for their potential as enzyme inhibitors.
It's worth noting that onium compounds can also be found in nature, including in some biological systems. For example, certain enzymes and signaling molecules contain onium groups that are important for their function.
Bacterial proteins are a type of protein that are produced by bacteria as part of their structural or functional components. These proteins can be involved in various cellular processes, such as metabolism, DNA replication, transcription, and translation. They can also play a role in bacterial pathogenesis, helping the bacteria to evade the host's immune system, acquire nutrients, and multiply within the host.
Bacterial proteins can be classified into different categories based on their function, such as:
1. Enzymes: Proteins that catalyze chemical reactions in the bacterial cell.
2. Structural proteins: Proteins that provide structural support and maintain the shape of the bacterial cell.
3. Signaling proteins: Proteins that help bacteria to communicate with each other and coordinate their behavior.
4. Transport proteins: Proteins that facilitate the movement of molecules across the bacterial cell membrane.
5. Toxins: Proteins that are produced by pathogenic bacteria to damage host cells and promote infection.
6. Surface proteins: Proteins that are located on the surface of the bacterial cell and interact with the environment or host cells.
Understanding the structure and function of bacterial proteins is important for developing new antibiotics, vaccines, and other therapeutic strategies to combat bacterial infections.
Temperature, in a medical context, is a measure of the degree of hotness or coldness of a body or environment. It is usually measured using a thermometer and reported in degrees Celsius (°C), degrees Fahrenheit (°F), or kelvin (K). In the human body, normal core temperature ranges from about 36.5-37.5°C (97.7-99.5°F) when measured rectally, and can vary slightly depending on factors such as time of day, physical activity, and menstrual cycle. Elevated body temperature is a common sign of infection or inflammation, while abnormally low body temperature can indicate hypothermia or other medical conditions.
Skeletal muscle fibers, also known as striated muscle fibers, are the type of muscle cells that make up skeletal muscles, which are responsible for voluntary movements of the body. These muscle fibers are long, cylindrical, and multinucleated, meaning they contain multiple nuclei. They are surrounded by a connective tissue layer called the endomysium, and many fibers are bundled together into fascicles, which are then surrounded by another layer of connective tissue called the perimysium.
Skeletal muscle fibers are composed of myofibrils, which are long, thread-like structures that run the length of the fiber. Myofibrils contain repeating units called sarcomeres, which are responsible for the striated appearance of skeletal muscle fibers. Sarcomeres are composed of thick and thin filaments, which slide past each other during muscle contraction to shorten the sarcomere and generate force.
Skeletal muscle fibers can be further classified into two main types based on their contractile properties: slow-twitch (type I) and fast-twitch (type II). Slow-twitch fibers have a high endurance capacity and are used for sustained, low-intensity activities such as maintaining posture. Fast-twitch fibers, on the other hand, have a higher contractile speed and force generation capacity but fatigue more quickly and are used for powerful, explosive movements.
Complementary DNA (cDNA) is a type of DNA that is synthesized from a single-stranded RNA molecule through the process of reverse transcription. In this process, the enzyme reverse transcriptase uses an RNA molecule as a template to synthesize a complementary DNA strand. The resulting cDNA is therefore complementary to the original RNA molecule and is a copy of its coding sequence, but it does not contain non-coding regions such as introns that are present in genomic DNA.
Complementary DNA is often used in molecular biology research to study gene expression, protein function, and other genetic phenomena. For example, cDNA can be used to create cDNA libraries, which are collections of cloned cDNA fragments that represent the expressed genes in a particular cell type or tissue. These libraries can then be screened for specific genes or gene products of interest. Additionally, cDNA can be used to produce recombinant proteins in heterologous expression systems, allowing researchers to study the structure and function of proteins that may be difficult to express or purify from their native sources.
Glutamine is defined as a conditionally essential amino acid in humans, which means that it can be produced by the body under normal circumstances, but may become essential during certain conditions such as stress, illness, or injury. It is the most abundant free amino acid found in the blood and in the muscles of the body.
Glutamine plays a crucial role in various biological processes, including protein synthesis, energy production, and acid-base balance. It serves as an important fuel source for cells in the intestines, immune system, and skeletal muscles. Glutamine has also been shown to have potential benefits in wound healing, gut function, and immunity, particularly during times of physiological stress or illness.
In summary, glutamine is a vital amino acid that plays a critical role in maintaining the health and function of various tissues and organs in the body.
Calcium signaling is the process by which cells regulate various functions through changes in intracellular calcium ion concentrations. Calcium ions (Ca^2+^) are crucial second messengers that play a critical role in many cellular processes, including muscle contraction, neurotransmitter release, gene expression, and programmed cell death (apoptosis).
Intracellular calcium levels are tightly regulated by a complex network of channels, pumps, and exchangers located on the plasma membrane and intracellular organelles such as the endoplasmic reticulum (ER) and mitochondria. These proteins control the influx, efflux, and storage of calcium ions within the cell.
Calcium signaling is initiated when an external signal, such as a hormone or neurotransmitter, binds to a specific receptor on the plasma membrane. This interaction triggers the opening of ion channels, allowing extracellular Ca^2+^ to flow into the cytoplasm. In some cases, this influx of calcium ions is sufficient to activate downstream targets directly. However, in most instances, the increase in intracellular Ca^2+^ serves as a trigger for the release of additional calcium from internal stores, such as the ER.
The release of calcium from the ER is mediated by ryanodine receptors (RyRs) and inositol trisphosphate receptors (IP3Rs), which are activated by specific second messengers generated in response to the initial external signal. The activation of these channels leads to a rapid increase in cytoplasmic Ca^2+^, creating a transient intracellular calcium signal known as a "calcium spark" or "calcium puff."
These localized increases in calcium concentration can then propagate throughout the cell as waves of elevated calcium, allowing for the spatial and temporal coordination of various cellular responses. The duration and amplitude of these calcium signals are finely tuned by the interplay between calcium-binding proteins, pumps, and exchangers, ensuring that appropriate responses are elicited in a controlled manner.
Dysregulation of intracellular calcium signaling has been implicated in numerous pathological conditions, including neurodegenerative diseases, cardiovascular disorders, and cancer. Therefore, understanding the molecular mechanisms governing calcium homeostasis and signaling is crucial for the development of novel therapeutic strategies targeting these diseases.
Jurkat cells are a type of human immortalized T lymphocyte (a type of white blood cell) cell line that is commonly used in scientific research. They were originally isolated from the peripheral blood of a patient with acute T-cell leukemia. Jurkat cells are widely used as a model system to study T-cell activation, signal transduction, and apoptosis (programmed cell death). They are also used in the study of HIV infection and replication, as they can be infected with the virus and used to investigate viral replication and host cell responses.
3-Phosphoinositide-Dependent Protein Kinases (PDPKs) are a family of serine/threonine protein kinases that play crucial roles in regulating various cellular processes, including cell survival, proliferation, and metabolism. They are named after their ability to phosphorylate and activate downstream targets in response to the binding of 3-phosphoinositides, which are lipid second messengers generated by the activation of phosphatidylinositol 3-kinases (PI3Ks).
PDPKs consist of two main isoforms: PDPK1 and PDK2. PDPK1 is also known as the mammalian target of rapamycin complex 2 (mTORC2) associated protein, mSin1 kinase, or Rictor-binding protein. It primarily phosphorylates and activates AGC kinases, such as Akt/PKB, p70 S6 kinase, and protein kinase C (PKC). PDK2, on the other hand, is also known as ILK-associated kinase (ILKAP) or PDPK2. It primarily phosphorylates and activates PKC isoforms.
PDPKs are often deregulated in various human diseases, including cancer, diabetes, and neurological disorders. Therefore, they represent potential therapeutic targets for the development of novel drugs to treat these conditions.
Calcium-binding proteins (CaBPs) are a diverse group of proteins that have the ability to bind calcium ions (Ca^2+^) with high affinity and specificity. They play crucial roles in various cellular processes, including signal transduction, muscle contraction, neurotransmitter release, and protection against oxidative stress.
The binding of calcium ions to these proteins induces conformational changes that can either activate or inhibit their functions. Some well-known CaBPs include calmodulin, troponin C, S100 proteins, and parvalbumins. These proteins are essential for maintaining calcium homeostasis within cells and for mediating the effects of calcium as a second messenger in various cellular signaling pathways.
I'm sorry for any confusion, but "mitochondrial turnover" is not a widely recognized or established medical term with a specific definition. Mitochondria are the powerhouses of the cell, responsible for producing energy in the form of ATP through a process called oxidative phosphorylation.
The term "turnover," when used in a biological context, generally refers to the process by which cells replace or regenerate their components over time. Therefore, one might infer that "mitochondrial turnover" could refer to the replacement and regeneration of mitochondria within cells. However, this is not a standardized term, and its precise meaning could vary depending on the context.
Mitochondria are known to undergo dynamic processes such as fusion (combining) and fission (dividing), which allow them to change their size, shape, and distribution in response to cellular needs. Additionally, damaged or dysfunctional mitochondria can be removed through a process called mitophagy, where they're targeted for degradation within lysosomes. New, healthy mitochondria are generated through biogenesis, which involves the production of new mitochondrial proteins and membranes.
In summary, while "mitochondrial turnover" is not a standard medical term, it could be used to describe the ongoing processes of mitochondrial dynamics, mitophagy, and biogenesis that contribute to the replacement and regeneration of mitochondria within cells over time.
Mitochondrial dynamics refer to the processes that regulate the shape, size, distribution, and quality control of mitochondria within cells. These dynamic processes include:
1. Mitochondrial Fusion: This is the process by which two adjacent mitochondria merge together to form a single, elongated organelle. Fusion allows for the exchange of mitochondrial content, including DNA, proteins, and metabolites, which helps maintain genetic stability and promote bioenergetic efficiency.
2. Mitochondrial Fission: This is the process by which a single mitochondrion divides into two separate organelles. Fission plays a crucial role in mitochondrial division, inheritance, and quality control, as it enables the segregation of damaged or dysfunctional mitochondria for degradation via autophagy (mitophagy).
3. Mitochondrial Transport: This is the active movement of mitochondria within cells, facilitated by cytoskeletal motor proteins. Mitochondrial transport enables organelles to be distributed evenly throughout the cell and to reach specific subcellular locations where their energy demands are high.
4. Mitochondrial Dynamics Regulation: The regulation of mitochondrial dynamics involves a complex interplay between various proteins, lipids, and signaling pathways that control fusion, fission, transport, and quality control processes. These regulatory mechanisms help maintain the balance between mitochondrial biogenesis (the generation of new organelles) and mitophagy (the removal of damaged ones), ensuring proper cellular homeostasis and function.
Dysregulation of mitochondrial dynamics has been implicated in various pathological conditions, including neurodegenerative diseases, metabolic disorders, and aging-related processes.
Mersalyl is not a medical condition or diagnosis, but rather a pharmaceutical compound. It is a type of organic mercurial salt that was historically used in medicine as a diuretic and an antimicrobial agent. However, its use has been largely discontinued due to the toxic effects of mercury on the human body. Therefore, there isn't a medical definition for 'Mersalyl'.
Colforsin is a drug that belongs to a class of medications called phosphodiesterase inhibitors. It works by increasing the levels of a chemical called cyclic AMP (cyclic adenosine monophosphate) in the body, which helps to relax and widen blood vessels.
Colforsin is not approved for use in humans in many countries, including the United States. However, it has been used in research settings to study its potential effects on heart function and other physiological processes. In animals, colforsin has been shown to have positive inotropic (contractility-enhancing) and lusitropic (relaxation-enhancing) effects on the heart, making it a potential therapeutic option for heart failure and other cardiovascular conditions.
It is important to note that while colforsin has shown promise in preclinical studies, more research is needed to establish its safety and efficacy in humans. Therefore, it should only be used under the supervision of a qualified healthcare professional and in the context of a clinical trial or research study.
Inborn errors of metabolism (IEM) refer to a group of genetic disorders caused by defects in enzymes or transporters that play a role in the body's metabolic processes. These disorders result in the accumulation or deficiency of specific chemicals within the body, which can lead to various clinical manifestations, such as developmental delay, intellectual disability, seizures, organ damage, and in some cases, death.
Examples of IEM include phenylketonuria (PKU), maple syrup urine disease (MSUD), galactosemia, and glycogen storage diseases, among many others. These disorders are typically inherited in an autosomal recessive manner, meaning that an affected individual has two copies of the mutated gene, one from each parent.
Early diagnosis and management of IEM are crucial to prevent or minimize complications and improve outcomes. Treatment options may include dietary modifications, supplementation with missing enzymes or cofactors, medication, and in some cases, stem cell transplantation or gene therapy.
Cell adhesion molecules (CAMs) are a type of protein found on the surface of cells that mediate the attachment or adhesion of cells to either other cells or to the extracellular matrix (ECM), which is the network of proteins and carbohydrates that provides structural and biochemical support to surrounding cells.
CAMs play crucial roles in various biological processes, including tissue development, differentiation, repair, and maintenance of tissue architecture and function. They are also involved in cell signaling, migration, and regulation of the immune response.
There are several types of CAMs, classified based on their structure and function, such as immunoglobulin-like CAMs (IgCAMs), cadherins, integrins, and selectins. Dysregulation of CAMs has been implicated in various diseases, including cancer, inflammation, and neurological disorders.
A missense mutation is a type of point mutation in which a single nucleotide change results in the substitution of a different amino acid in the protein that is encoded by the affected gene. This occurs when the altered codon (a sequence of three nucleotides that corresponds to a specific amino acid) specifies a different amino acid than the original one. The function and/or stability of the resulting protein may be affected, depending on the type and location of the missense mutation. Missense mutations can have various effects, ranging from benign to severe, depending on the importance of the changed amino acid for the protein's structure or function.
Imidazoles are a class of heterocyclic organic compounds that contain a double-bonded nitrogen atom and two additional nitrogen atoms in the ring. They have the chemical formula C3H4N2. In a medical context, imidazoles are commonly used as antifungal agents. Some examples of imidazole-derived antifungals include clotrimazole, miconazole, and ketoconazole. These medications work by inhibiting the synthesis of ergosterol, a key component of fungal cell membranes, leading to increased permeability and death of the fungal cells. Imidazoles may also have anti-inflammatory, antibacterial, and anticancer properties.
Trypsin is a proteolytic enzyme, specifically a serine protease, that is secreted by the pancreas as an inactive precursor, trypsinogen. Trypsinogen is converted into its active form, trypsin, in the small intestine by enterokinase, which is produced by the intestinal mucosa.
Trypsin plays a crucial role in digestion by cleaving proteins into smaller peptides at specific arginine and lysine residues. This enzyme helps to break down dietary proteins into amino acids, allowing for their absorption and utilization by the body. Additionally, trypsin can activate other zymogenic pancreatic enzymes, such as chymotrypsinogen and procarboxypeptidases, thereby contributing to overall protein digestion.
High-performance liquid chromatography (HPLC) is a type of chromatography that separates and analyzes compounds based on their interactions with a stationary phase and a mobile phase under high pressure. The mobile phase, which can be a gas or liquid, carries the sample mixture through a column containing the stationary phase.
In HPLC, the mobile phase is a liquid, and it is pumped through the column at high pressures (up to several hundred atmospheres) to achieve faster separation times and better resolution than other types of liquid chromatography. The stationary phase can be a solid or a liquid supported on a solid, and it interacts differently with each component in the sample mixture, causing them to separate as they travel through the column.
HPLC is widely used in analytical chemistry, pharmaceuticals, biotechnology, and other fields to separate, identify, and quantify compounds present in complex mixtures. It can be used to analyze a wide range of substances, including drugs, hormones, vitamins, pigments, flavors, and pollutants. HPLC is also used in the preparation of pure samples for further study or use.
Cytochrome-c oxidase deficiency is a genetic disorder that affects the function of the mitochondria, which are the energy-producing structures in cells. Specifically, it is a deficiency in cytochrome-c oxidase (COX), also known as complex IV, which is an enzyme located in the inner membrane of the mitochondria that plays a critical role in the electron transport chain and oxidative phosphorylation.
Cytochrome-c oxidase deficiency can be caused by mutations in any of the genes that encode the subunits or assembly factors of COX. The severity of the disorder and the specific symptoms can vary widely, depending on the extent of the enzyme deficiency and the particular tissues and organs that are affected.
Symptoms of cytochrome-c oxidase deficiency may include muscle weakness, developmental delay, hypotonia (low muscle tone), seizures, lactic acidosis, and cardiac and neurological problems. In some cases, the disorder can be life-threatening in infancy or early childhood.
There is no cure for cytochrome-c oxidase deficiency, and treatment is generally supportive and aimed at addressing specific symptoms. Antioxidant therapy, such as vitamin C and E supplements, may help to reduce oxidative stress and improve mitochondrial function in some cases. In severe cases, a heart or liver transplant may be considered.
Digitonin is a type of saponin, which is a natural substance found in some plants. It is often used in laboratory settings as a detergent to disrupt cell membranes and make it easier to study the contents of cells. Digitonin specifically binds to cholesterol in cell membranes, making it a useful tool for studying cholesterol-rich structures such as lipid rafts. It is not used as a medication in humans.
Hexoses are simple sugars (monosaccharides) that contain six carbon atoms. The most common hexoses include glucose, fructose, and galactose. These sugars play important roles in various biological processes, such as serving as energy sources or forming complex carbohydrates like starch and cellulose. Hexoses are essential for the structure and function of living organisms, including humans.
Tyrphostins are a class of synthetic compounds that act as tyrosine kinase inhibitors. They were initially developed as research tools to study the role of tyrosine kinases in cell signaling pathways, but some have also been investigated for their potential therapeutic use in cancer and other diseases.
Tyrphostins work by binding to and inhibiting the activity of tyrosine kinases, which are enzymes that add a phosphate group to tyrosine residues on proteins, thereby activating or deactivating various cellular processes. By blocking this activity, tyrphostins can disrupt abnormal signaling pathways that contribute to the development and progression of diseases such as cancer.
There are several different subclasses of tyrphostins, each with varying levels of specificity for different tyrosine kinases. Some examples include genistein, erbstatin, and lavendustin A. While tyrphostins have been useful in basic research, their clinical use is limited due to issues such as poor bioavailability, lack of specificity, and toxicity. However, they continue to be important tools for studying the functions of tyrosine kinases and developing new therapeutic strategies.
I apologize, but I am not able to provide a medical definition for "Egtazic Acid" because it is not a term that is recognized in the field of medicine or pharmacology. It is possible that you may have meant "Egтарic Acid," which also does not have a specific medical meaning, or "Ethylene Glycol Tetraacetic Acid (EGTA)," which is a chemical compound used in research and medicine for its ability to bind calcium ions. If you have any other questions, I would be happy to try to help answer them.
Congenital Generalized Lipodystrophy (CGL) is a rare genetic disorder characterized by the near or complete absence of body fat at birth or in early childhood. It is also known as Berardinelli-Seip congenital lipodystrophy. The condition is caused by mutations in genes responsible for the development and function of adipose tissue (fat).
Individuals with CGL typically have a lack of subcutaneous fat, which gives them a muscular appearance, but they may have excess fat accumulation in other areas such as the neck, face, and liver. This can lead to various metabolic complications, including insulin resistance, diabetes mellitus, hypertriglyceridemia (high levels of triglycerides in the blood), and hepatic steatosis (fatty liver disease).
CGL is a genetic disorder that is inherited in an autosomal recessive pattern. This means that an individual must inherit two copies of the mutated gene, one from each parent, to develop the condition. The diagnosis of CGL is typically based on clinical features and confirmed by genetic testing. Treatment for CGL focuses on managing the metabolic complications associated with the disorder.
SHC (Src homology 2 domain containing) signaling adaptor proteins are a family of intracellular signaling molecules that play a crucial role in the transduction of signals from various cell surface receptors, including receptor tyrosine kinases (RTKs). These proteins contain several conserved domains, including Src homology 2 (SH2) and phosphotyrosine-binding (PTB) domains, which enable them to bind to specific phosphorylated tyrosine residues on activated receptors or other signaling molecules.
Once bound to the activated receptor, SHC proteins recruit and interact with various downstream signaling proteins, such as growth factor receptor-bound protein 2 (Grb2) and son of sevenless (SOS), thereby initiating intracellular signaling cascades that ultimately regulate diverse cellular processes, including proliferation, differentiation, survival, and migration. There are three main isoforms of SHC proteins in humans: p66Shc, p52Shc, and p46Shc, which differ in their structural organization and functional properties.
Abnormal regulation of SHC signaling adaptor proteins has been implicated in various pathological conditions, including cancer, diabetes, and neurodegenerative diseases. Therefore, understanding the molecular mechanisms underlying SHC-mediated signaling pathways may provide valuable insights into the development of novel therapeutic strategies for these disorders.
Muscle cells, also known as muscle fibers, are specialized cells that have the ability to contract and generate force, allowing for movement of the body and various internal organ functions. There are three main types of muscle tissue: skeletal, cardiac, and smooth.
Skeletal muscle cells are voluntary striated muscles attached to bones, enabling body movements and posture. They are multinucleated, with numerous nuclei located at the periphery of the cell. These cells are often called muscle fibers and can be quite large, extending the entire length of the muscle.
Cardiac muscle cells form the contractile tissue of the heart. They are also striated but have a single nucleus per cell and are interconnected by specialized junctions called intercalated discs, which help coordinate contraction throughout the heart.
Smooth muscle cells are found in various internal organs such as the digestive, respiratory, and urinary tracts, blood vessels, and the reproductive system. They are involuntary, non-striated muscles that control the internal organ functions. Smooth muscle cells have a single nucleus per cell and can either be spindle-shaped or stellate (star-shaped).
In summary, muscle cells are specialized contractile cells responsible for movement and various internal organ functions in the human body. They can be categorized into three types: skeletal, cardiac, and smooth, based on their structure, location, and function.
In medical terms, the heart is a muscular organ located in the thoracic cavity that functions as a pump to circulate blood throughout the body. It's responsible for delivering oxygen and nutrients to the tissues and removing carbon dioxide and other wastes. The human heart is divided into four chambers: two atria on the top and two ventricles on the bottom. The right side of the heart receives deoxygenated blood from the body and pumps it to the lungs, while the left side receives oxygenated blood from the lungs and pumps it out to the rest of the body. The heart's rhythmic contractions and relaxations are regulated by a complex electrical conduction system.
Cyclin-Dependent Kinase 2 (CDK2) is a type of enzyme that plays a crucial role in the regulation of the cell cycle, which is the process by which cells grow and divide. CDK2 is activated when it binds to a regulatory subunit called a cyclin.
During the cell cycle, CDK2 helps to control the progression from the G1 phase to the S phase, where DNA replication occurs. Specifically, CDK2 phosphorylates various target proteins that are involved in the regulation of DNA replication and the initiation of mitosis, which is the process of cell division.
CDK2 activity is tightly regulated through a variety of mechanisms, including phosphorylation, dephosphorylation, and protein degradation. Dysregulation of CDK2 activity has been implicated in various human diseases, including cancer. Therefore, CDK2 is an important target for the development of therapies aimed at treating these diseases.
Glycogen synthase kinases (GSKs) are a family of enzymes that play a crucial role in the regulation of glycogen metabolism. Glycogen is a complex carbohydrate that serves as a primary energy storage form in animals, fungi, and bacteria.
GSKs function as serine/threonine protein kinases, which means they add phosphate groups to specific serine or threonine residues on their target proteins. In the case of glycogen synthase kinases, their primary target is glycogen synthase, an enzyme responsible for synthesizing glycogen from glucose-1-phosphate during the process of glycogenesis (glycogen synthesis).
There are several isoforms of GSKs identified in humans, including GSK3α and GSK3β. These kinases are involved in various cellular processes, such as:
1. Regulation of glycogen metabolism: By phosphorylating and inhibiting glycogen synthase, GSKs help control the balance between glycogen storage and glucose utilization.
2. Cell signaling pathways: GSKs participate in several intracellular signaling cascades, including the Wnt signaling pathway, insulin signaling pathway, and the PI3K/AKT pathway, which regulate various cellular functions such as proliferation, differentiation, survival, and metabolism.
3. Regulation of gene expression: GSKs can modulate transcription factors' activity, thereby influencing gene expression and contributing to various cellular responses.
4. Neuronal function: In the brain, GSKs are involved in regulating synaptic plasticity, learning, and memory processes.
5. Disease pathogenesis: Dysregulation of GSKs has been implicated in several diseases, such as diabetes, neurodegenerative disorders (e.g., Alzheimer's disease), and cancer.
In summary, glycogen synthase kinases are a family of protein kinases that regulate glycogen metabolism and participate in various cell signaling pathways, influencing numerous cellular functions and being implicated in several diseases.
Ethanol is the medical term for pure alcohol, which is a colorless, clear, volatile, flammable liquid with a characteristic odor and burning taste. It is the type of alcohol that is found in alcoholic beverages and is produced by the fermentation of sugars by yeasts.
In the medical field, ethanol is used as an antiseptic and disinfectant, and it is also used as a solvent for various medicinal preparations. It has central nervous system depressant properties and is sometimes used as a sedative or to induce sleep. However, excessive consumption of ethanol can lead to alcohol intoxication, which can cause a range of negative health effects, including impaired judgment, coordination, and memory, as well as an increased risk of accidents, injuries, and chronic diseases such as liver disease and addiction.
Insulin resistance is a condition in which the body's cells become less responsive to insulin, a hormone produced by the pancreas that regulates blood sugar levels. In response to this decreased sensitivity, the pancreas produces more insulin to help glucose enter the cells. However, over time, the pancreas may not be able to keep up with the increased demand for insulin, leading to high levels of glucose in the blood and potentially resulting in type 2 diabetes, prediabetes, or other health issues such as metabolic syndrome, cardiovascular disease, and non-alcoholic fatty liver disease. Insulin resistance is often associated with obesity, physical inactivity, and genetic factors.
P21-activated kinases (PAKs) are a family of serine/threonine protein kinases that play crucial roles in various cellular processes, including cytoskeletal reorganization, cell motility, and gene transcription. They are activated by binding to small GTPases of the Rho family, such as Cdc42 and Rac, which become active upon stimulation of various extracellular signals. Once activated, PAKs phosphorylate a range of downstream targets, leading to changes in cell behavior and function. Aberrant regulation of PAKs has been implicated in several human diseases, including cancer and neurological disorders.
Fumarate hydratase (FH) is an enzyme that plays a crucial role in the citric acid cycle, also known as the Krebs cycle or tricarboxylic acid (TCA) cycle. The citric acid cycle is a series of chemical reactions used by all living cells to generate energy through the oxidation of acetyl-CoA derived from carbohydrates, fats, and proteins into adenosine triphosphate (ATP), carbon dioxide, and water.
Fumarate hydratase is specifically responsible for catalyzing the conversion of fumarate to malate in this cycle. A deficiency or dysfunction of this enzyme can lead to various metabolic disorders and hereditary diseases, such as fumarate hydratase deficiency, which may manifest as neurological issues, hemolytic anemia, and an increased risk of developing renal cell carcinoma.
Isoquinolines are not a medical term per se, but a chemical classification. They refer to a class of organic compounds that consist of a benzene ring fused to a piperidine ring. This structure is similar to that of quinoline, but with the nitrogen atom located at a different position in the ring.
Isoquinolines have various biological activities and can be found in some natural products, including certain alkaloids. Some isoquinoline derivatives have been developed as drugs for the treatment of various conditions, such as cardiovascular diseases, neurological disorders, and cancer. However, specific medical definitions related to isoquinolines typically refer to the use or effects of these specific drugs rather than the broader class of compounds.
Phosphorus is an essential mineral that is required by every cell in the body for normal functioning. It is a key component of several important biomolecules, including adenosine triphosphate (ATP), which is the primary source of energy for cells, and deoxyribonucleic acid (DNA) and ribonucleic acid (RNA), which are the genetic materials in cells.
Phosphorus is also a major constituent of bones and teeth, where it combines with calcium to provide strength and structure. In addition, phosphorus plays a critical role in various metabolic processes, including energy production, nerve impulse transmission, and pH regulation.
The medical definition of phosphorus refers to the chemical element with the atomic number 15 and the symbol P. It is a highly reactive non-metal that exists in several forms, including white phosphorus, red phosphorus, and black phosphorus. In the body, phosphorus is primarily found in the form of organic compounds, such as phospholipids, phosphoproteins, and nucleic acids.
Abnormal levels of phosphorus in the body can lead to various health problems. For example, high levels of phosphorus (hyperphosphatemia) can occur in patients with kidney disease or those who consume large amounts of phosphorus-rich foods, and can contribute to the development of calcification of soft tissues and cardiovascular disease. On the other hand, low levels of phosphorus (hypophosphatemia) can occur in patients with malnutrition, vitamin D deficiency, or alcoholism, and can lead to muscle weakness, bone pain, and an increased risk of infection.
Cytochromes c are a group of small heme proteins found in the mitochondria of cells, involved in the electron transport chain and play a crucial role in cellular respiration. They accept and donate electrons during the process of oxidative phosphorylation, which generates ATP, the main energy currency of the cell. Cytochromes c contain a heme group, an organic compound that includes iron, which facilitates the transfer of electrons. The "c" in cytochromes c refers to the type of heme group they contain (cyt c has heme c). They are highly conserved across species and have been widely used as a molecular marker for evolutionary studies.
Creatine kinase (CK) is an enzyme found in various tissues in the body, including the heart, brain, and skeletal muscles. It plays a crucial role in energy metabolism by catalyzing the conversion of creatine and adenosine triphosphate (ATP) to phosphocreatine and adenosine diphosphate (ADP). This reaction helps regenerate ATP, which is the primary source of energy for cellular functions.
There are three main isoforms of CK in the human body: CK-MM, CK-MB, and CK-BB. The CK-MM form is primarily found in skeletal muscles and constitutes approximately 95% to 99% of the total CK activity in healthy individuals. It is a dimer composed of two muscle-specific subunits (M-CK).
Elevated levels of CK-MM in the blood can indicate damage or injury to skeletal muscles. This can occur due to various reasons, such as muscle trauma, strenuous exercise, muscle diseases, and certain medications. Measuring CK-MM levels is essential in diagnosing and monitoring conditions associated with muscle damage or disease.
Tandem mass spectrometry (MS/MS) is a technique used to identify and quantify specific molecules, such as proteins or metabolites, within complex mixtures. This method uses two or more sequential mass analyzers to first separate ions based on their mass-to-charge ratio and then further fragment the selected ions into smaller pieces for additional analysis. The fragmentation patterns generated in MS/MS experiments can be used to determine the structure and identity of the original molecule, making it a powerful tool in various fields such as proteomics, metabolomics, and forensic science.
Guanosine triphosphate (GTP) is a nucleotide that plays a crucial role in various cellular processes, such as protein synthesis, signal transduction, and regulation of enzymatic activities. It serves as an energy currency, similar to adenosine triphosphate (ATP), and undergoes hydrolysis to guanosine diphosphate (GDP) or guanosine monophosphate (GMP) to release energy required for these processes. GTP is also a precursor for the synthesis of other essential molecules, including RNA and certain signaling proteins. Additionally, it acts as a molecular switch in many intracellular signaling pathways by binding and activating specific GTPase proteins.
I'm sorry for any confusion, but there isn't a specific type or classification of prostaglandins known as "Prostaglandins B." Prostaglandins are a group of lipid compounds that are synthesized in the body from fatty acids and have diverse hormone-like effects in various tissues. They are involved in a wide range of physiological processes, including inflammation, pain perception, fever, blood pressure regulation, and modulation of the reproductive system.
Prostaglandins are typically classified based on their chemical structure and the type of fatty acid they are derived from. The main groups include Prostaglandin A, B, D, E, F, and I series. Each group has different biological activities and functions in the body. If you have any more specific questions about prostaglandins or a related topic, feel free to ask!
Crk-associated substrate protein, often abbreviated as CAS or CAS-L (for Crk-associated substrate lymphocyte type), is a signaling adaptor protein that plays a role in various cellular processes such as proliferation, differentiation, and survival. It is called a "substrate" because it can be phosphorylated by various kinases and serves as a platform for the assembly of signaling complexes.
CAS contains several domains that allow it to interact with other proteins, including Src homology 3 (SH3) domains, which bind to proline-rich sequences in partner proteins, and a SH2 domain, which binds to phosphorylated tyrosine residues. These interactions enable CAS to link upstream signaling events with downstream effectors, thereby regulating various cellular responses.
CAS is often found downstream of receptor tyrosine kinases (RTKs) and integrins, and has been implicated in the regulation of several signaling pathways, including the Ras/MAPK, PI3K/Akt, and JNK pathways. Mutations or dysregulation of CAS have been associated with various diseases, including cancer and neurological disorders.
'Crotalus' is a genus of venomous snakes commonly known as rattlesnakes. These snakes are native to the Americas, ranging from southern Canada to Argentina. They are characterized by the distinctive rattle on the end of their tails, which they use to warn potential predators before striking. The venom of Crotalus species is hemotoxic, meaning that it causes damage to blood vessels and tissue.
Some examples of species in this genus include the Western diamondback rattlesnake (Crotalus atrox), the timber rattlesnake (Crotalus horridus), and the sidewinder (Crotalus cerastes). It is important to note that all rattlesnakes are potentially dangerous and should be treated with caution. If you encounter a rattlesnake in the wild, it is best to leave it alone and avoid approaching it.
Physiological stress is a response of the body to a demand or threat that disrupts homeostasis and activates the autonomic nervous system and hypothalamic-pituitary-adrenal (HPA) axis. This results in the release of stress hormones such as adrenaline, cortisol, and noradrenaline, which prepare the body for a "fight or flight" response. Increased heart rate, rapid breathing, heightened sensory perception, and increased alertness are some of the physiological changes that occur during this response. Chronic stress can have negative effects on various bodily functions, including the immune, cardiovascular, and nervous systems.
Bovine Serum Albumin (BSA) is not a medical term per se, but a biochemical term. It is widely used in medical and biological research. Here's the definition:
Bovine Serum Albumin is a serum albumin protein derived from cows. It is often used as a stabilizer, an emulsifier, or a protein source in various laboratory and industrial applications, including biochemical experiments, cell culture media, and diagnostic kits. BSA has a high solubility in water and can bind to many different types of molecules, making it useful for preventing unwanted interactions between components in a solution. It also has a consistent composition and is relatively inexpensive compared to human serum albumin, which are factors that contribute to its widespread use.
MAP (Mitogen-Activated Protein) Kinase Kinase Kinases (MAP3K or MAPKKK) are a group of protein kinases that play a crucial role in intracellular signal transduction pathways, which regulate various cellular processes such as proliferation, differentiation, survival, and apoptosis. They are called "kinases" because they catalyze the transfer of a phosphate group from ATP to specific serine or threonine residues on their target proteins.
MAP3Ks function upstream of MAP Kinase Kinases (MKKs or MAP2K) and MAP Kinases (MPKs or MAPK) in the MAP kinase cascade. Upon activation by various extracellular signals, such as growth factors, cytokines, stress, and hormones, MAP3Ks phosphorylate and activate MKKs, which subsequently phosphorylate and activate MPKs. Activated MPKs then regulate the activity of downstream transcription factors and other target proteins to elicit appropriate cellular responses.
There are several subfamilies of MAP3Ks, including ASK, DLK, TAK, MEKK, MLK, and ZAK, among others. Each subfamily has distinct structural features and functions in different signaling pathways. Dysregulation of MAP kinase cascades, including MAP3Ks, has been implicated in various human diseases, such as cancer, inflammation, and neurodegenerative disorders.
The GRB2 (Growth Factor Receptor-Bound Protein 2) adaptor protein is a cytoplasmic signaling molecule that plays a crucial role in intracellular signal transduction pathways, particularly those involved in cell growth, differentiation, and survival. It acts as a molecular adapter or scaffold, facilitating the interaction between various proteins to form multi-protein complexes and propagate signals from activated receptor tyrosine kinases (RTKs) to downstream effectors.
GRB2 contains several functional domains, including an N-terminal SH3 domain, a central SH2 domain, and a C-terminal SH3 domain. The SH2 domain is responsible for binding to specific phosphotyrosine residues on activated RTKs or other adaptor proteins, while the SH3 domains mediate interactions with proline-rich sequences in partner proteins.
Once GRB2 binds to an activated RTK, it recruits and activates the guanine nucleotide exchange factor SOS (Son of Sevenless), which in turn activates the RAS GTPase. Activated RAS then initiates a signaling cascade involving various kinases such as Raf, MEK, and ERK, ultimately leading to changes in gene expression and cellular responses.
In summary, GRB2 is an essential adaptor protein that facilitates the transmission of signals from activated growth factor receptors to downstream effectors, playing a critical role in regulating various cellular processes.
MAP Kinase Kinase 4 (MAP2K4 or MKK4) is a serine/threonine protein kinase that plays a crucial role in intracellular signal transduction pathways, particularly the mitogen-activated protein kinase (MAPK) cascades. These cascades are involved in various cellular processes such as proliferation, differentiation, survival, and apoptosis in response to extracellular stimuli like cytokines, growth factors, and stress signals.
MAP2K4 specifically activates the c-Jun N-terminal kinase (JNK) pathway by phosphorylating and activating JNK proteins. The activation of JNK leads to the phosphorylation and regulation of various transcription factors, ultimately influencing gene expression and cellular responses. Dysregulation of MAP2K4 has been implicated in several diseases, including cancer and inflammatory disorders.
Nitric oxide (NO) is a molecule made up of one nitrogen atom and one oxygen atom. In the body, it is a crucial signaling molecule involved in various physiological processes such as vasodilation, immune response, neurotransmission, and inhibition of platelet aggregation. It is produced naturally by the enzyme nitric oxide synthase (NOS) from the amino acid L-arginine. Inhaled nitric oxide is used medically to treat pulmonary hypertension in newborns and adults, as it helps to relax and widen blood vessels, improving oxygenation and blood flow.
I-kappa B (IκB) proteins are a family of inhibitory proteins that play a crucial role in regulating the activity of nuclear factor kappa B (NF-κB), a key transcription factor involved in inflammation, immune response, and cell survival. In resting cells, NF-κB is sequestered in the cytoplasm by binding to IκB proteins, which prevents NF-κB from translocating into the nucleus and activating its target genes.
Upon stimulation of various signaling pathways, such as those triggered by proinflammatory cytokines, bacterial or viral components, and stress signals, IκB proteins become phosphorylated, ubiquitinated, and subsequently degraded by the 26S proteasome. This process allows NF-κB to dissociate from IκB, translocate into the nucleus, and bind to specific DNA sequences, leading to the expression of various genes involved in immune response, inflammation, cell growth, differentiation, and survival.
There are several members of the IκB protein family, including IκBα, IκBβ, IκBε, IκBγ, and Bcl-3. Each member has distinct functions and regulatory mechanisms in controlling NF-κB activity. Dysregulation of IκB proteins and NF-κB signaling has been implicated in various pathological conditions, such as chronic inflammation, autoimmune diseases, and cancer.
GTP-binding proteins, also known as G proteins, are a family of molecular switches present in many organisms, including humans. They play a crucial role in signal transduction pathways, particularly those involved in cellular responses to external stimuli such as hormones, neurotransmitters, and sensory signals like light and odorants.
G proteins are composed of three subunits: α, β, and γ. The α-subunit binds GTP (guanosine triphosphate) and acts as the active component of the complex. When a G protein-coupled receptor (GPCR) is activated by an external signal, it triggers a conformational change in the associated G protein, allowing the α-subunit to exchange GDP (guanosine diphosphate) for GTP. This activation leads to dissociation of the G protein complex into the GTP-bound α-subunit and the βγ-subunit pair. Both the α-GTP and βγ subunits can then interact with downstream effectors, such as enzymes or ion channels, to propagate and amplify the signal within the cell.
The intrinsic GTPase activity of the α-subunit eventually hydrolyzes the bound GTP to GDP, which leads to re-association of the α and βγ subunits and termination of the signal. This cycle of activation and inactivation makes G proteins versatile signaling elements that can respond quickly and precisely to changing environmental conditions.
Defects in G protein-mediated signaling pathways have been implicated in various diseases, including cancer, neurological disorders, and cardiovascular diseases. Therefore, understanding the function and regulation of GTP-binding proteins is essential for developing targeted therapeutic strategies.
Cell hypoxia, also known as cellular hypoxia or tissue hypoxia, refers to a condition in which the cells or tissues in the body do not receive an adequate supply of oxygen. Oxygen is essential for the production of energy in the form of ATP (adenosine triphosphate) through a process called oxidative phosphorylation. When the cells are deprived of oxygen, they switch to anaerobic metabolism, which produces lactic acid as a byproduct and can lead to acidosis.
Cell hypoxia can result from various conditions, including:
1. Low oxygen levels in the blood (hypoxemia) due to lung diseases such as chronic obstructive pulmonary disease (COPD), pneumonia, or high altitude.
2. Reduced blood flow to tissues due to cardiovascular diseases such as heart failure, peripheral artery disease, or shock.
3. Anemia, which reduces the oxygen-carrying capacity of the blood.
4. Carbon monoxide poisoning, which binds to hemoglobin and prevents it from carrying oxygen.
5. Inadequate ventilation due to trauma, drug overdose, or other causes that can lead to respiratory failure.
Cell hypoxia can cause cell damage, tissue injury, and organ dysfunction, leading to various clinical manifestations depending on the severity and duration of hypoxia. Treatment aims to correct the underlying cause and improve oxygen delivery to the tissues.
Protein Tyrosine Phosphatase, Non-Receptor Type 11 (PTPN11) is a gene that encodes for the protein tyrosine phosphatase SHP-2. This enzyme regulates various cellular processes, including cell growth, differentiation, and migration, by controlling the balance of phosphorylation and dephosphorylation of proteins involved in signal transduction pathways. Mutations in PTPN11 have been associated with several human diseases, most notably Noonan syndrome and its related disorders, as well as certain types of leukemia.
Sirolimus is a medication that belongs to a class of drugs called immunosuppressants. It is also known as rapamycin. Sirolimus works by inhibiting the mammalian target of rapamycin (mTOR), which is a protein that plays a key role in cell growth and division.
Sirolimus is primarily used to prevent rejection of transplanted organs, such as kidneys, livers, and hearts. It works by suppressing the activity of the immune system, which can help to reduce the risk of the body rejecting the transplanted organ. Sirolimus is often used in combination with other immunosuppressive drugs, such as corticosteroids and calcineurin inhibitors.
Sirolimus is also being studied for its potential therapeutic benefits in a variety of other conditions, including cancer, tuberous sclerosis complex, and lymphangioleiomyomatosis. However, more research is needed to fully understand the safety and efficacy of sirolimus in these contexts.
It's important to note that sirolimus can have significant side effects, including increased risk of infections, mouth sores, high blood pressure, and kidney damage. Therefore, it should only be used under the close supervision of a healthcare provider.
Antioxidants are substances that can prevent or slow damage to cells caused by free radicals, which are unstable molecules that the body produces as a reaction to environmental and other pressures. Antioxidants are able to neutralize free radicals by donating an electron to them, thus stabilizing them and preventing them from causing further damage to the cells.
Antioxidants can be found in a variety of foods, including fruits, vegetables, nuts, and grains. Some common antioxidants include vitamins C and E, beta-carotene, and selenium. Antioxidants are also available as dietary supplements.
In addition to their role in protecting cells from damage, antioxidants have been studied for their potential to prevent or treat a number of health conditions, including cancer, heart disease, and age-related macular degeneration. However, more research is needed to fully understand the potential benefits and risks of using antioxidant supplements.
I'm sorry for any confusion, but there seems to be a misunderstanding. Mathematics is not a medical term; it is a branch of science dedicated to the study of numbers, shapes, and structures. However, mathematics does have many applications in medicine, such as in modeling disease spread, analyzing medical images, or designing clinical trials. If you have any questions related to mathematics in a medical context, I'd be happy to help clarify those for you!
Fungal proteins are a type of protein that is specifically produced and present in fungi, which are a group of eukaryotic organisms that include microorganisms such as yeasts and molds. These proteins play various roles in the growth, development, and survival of fungi. They can be involved in the structure and function of fungal cells, metabolism, pathogenesis, and other cellular processes. Some fungal proteins can also have important implications for human health, both in terms of their potential use as therapeutic targets and as allergens or toxins that can cause disease.
Fungal proteins can be classified into different categories based on their functions, such as enzymes, structural proteins, signaling proteins, and toxins. Enzymes are proteins that catalyze chemical reactions in fungal cells, while structural proteins provide support and protection for the cell. Signaling proteins are involved in communication between cells and regulation of various cellular processes, and toxins are proteins that can cause harm to other organisms, including humans.
Understanding the structure and function of fungal proteins is important for developing new treatments for fungal infections, as well as for understanding the basic biology of fungi. Research on fungal proteins has led to the development of several antifungal drugs that target specific fungal enzymes or other proteins, providing effective treatment options for a range of fungal diseases. Additionally, further study of fungal proteins may reveal new targets for drug development and help improve our ability to diagnose and treat fungal infections.
Neoplasms are abnormal growths of cells or tissues in the body that serve no physiological function. They can be benign (non-cancerous) or malignant (cancerous). Benign neoplasms are typically slow growing and do not spread to other parts of the body, while malignant neoplasms are aggressive, invasive, and can metastasize to distant sites.
Neoplasms occur when there is a dysregulation in the normal process of cell division and differentiation, leading to uncontrolled growth and accumulation of cells. This can result from genetic mutations or other factors such as viral infections, environmental exposures, or hormonal imbalances.
Neoplasms can develop in any organ or tissue of the body and can cause various symptoms depending on their size, location, and type. Treatment options for neoplasms include surgery, radiation therapy, chemotherapy, immunotherapy, and targeted therapy, among others.
Focal Adhesion Kinase 2 (FAK2), also known as Protein Tyrosine Kinase 2 beta (PTK2B), is a cytoplasmic tyrosine kinase that plays a crucial role in various cellular processes, including cell adhesion, migration, proliferation, and survival. FAK2 is structurally similar to Focal Adhesion Kinase 1 (FAK1 or PTK2A) but has distinct functions and expression patterns.
FAK2 contains several functional domains, such as an N-terminal FERM domain, a central kinase domain, a C-terminal focal adhesion targeting (FAT) domain, and proline-rich regions that interact with various signaling proteins. FAK2 is activated by autophosphorylation at the Y397 residue upon integrin clustering or growth factor receptor activation, which leads to the recruitment of downstream effectors and the initiation of intracellular signaling cascades.
FAK2 has been implicated in several pathological conditions, such as cancer, neurodegenerative diseases, and cardiovascular disorders. In cancer, FAK2 overexpression or hyperactivation promotes tumor cell survival, invasion, and metastasis, making it an attractive therapeutic target for anticancer therapy. However, the role of FAK2 in physiological processes is still not fully understood and requires further investigation.
Culture media is a substance that is used to support the growth of microorganisms or cells in an artificial environment, such as a petri dish or test tube. It typically contains nutrients and other factors that are necessary for the growth and survival of the organisms being cultured. There are many different types of culture media, each with its own specific formulation and intended use. Some common examples include blood agar, which is used to culture bacteria; Sabouraud dextrose agar, which is used to culture fungi; and Eagle's minimum essential medium, which is used to culture animal cells.
RNA (Ribonucleic Acid) is a single-stranded, linear polymer of ribonucleotides. It is a nucleic acid present in the cells of all living organisms and some viruses. RNAs play crucial roles in various biological processes such as protein synthesis, gene regulation, and cellular signaling. There are several types of RNA including messenger RNA (mRNA), ribosomal RNA (rRNA), transfer RNA (tRNA), small nuclear RNA (snRNA), microRNA (miRNA), and long non-coding RNA (lncRNA). These RNAs differ in their structure, function, and location within the cell.
eIF-2 kinase is a type of protein kinase that phosphorylates the alpha subunit of eukaryotic initiation factor-2 (eIF-2) at serine 51. This phosphorylation event inhibits the guanine nucleotide exchange factor eIF-2B, thereby preventing the recycling of eIF-2 and reducing global protein synthesis.
There are four main subtypes of eIF-2 kinases:
1. HRI (heme-regulated inhibitor) - responds to heme deficiency and oxidative stress
2. PERK (PKR-like endoplasmic reticulum kinase) - activated by ER stress and misfolded proteins in the ER
3. GCN2 (general control non-derepressible 2) - responds to amino acid starvation
4. PKR (double-stranded RNA-activated protein kinase) - activated by double-stranded RNA during viral infections
These eIF-2 kinases play crucial roles in regulating cellular responses to various stress conditions, such as the integrated stress response (ISR), which helps maintain cellular homeostasis and promote survival under adverse conditions.
Alkaloids are a type of naturally occurring organic compounds that contain mostly basic nitrogen atoms. They are often found in plants, and are known for their complex ring structures and diverse pharmacological activities. Many alkaloids have been used in medicine for their analgesic, anti-inflammatory, and therapeutic properties. Examples of alkaloids include morphine, quinine, nicotine, and caffeine.
 Oxidative phosphorylation - Wikipedia
Oxidative phosphorylation - Wikipedia
 OXIDATIVE PHOSPHORYLATION Definition & Usage Examples | Dictionary.com
OXIDATIVE PHOSPHORYLATION Definition & Usage Examples | Dictionary.com
 Tissue variation in the control of oxidative phosphorylation: implication for mitochondrial diseases
Tissue variation in the control of oxidative phosphorylation: implication for mitochondrial diseases
 HF B4 Oxidative Phosphorylation 2 Flashcards | Quizlet
HF B4 Oxidative Phosphorylation 2 Flashcards | Quizlet
Orphanet: Combined oxidative phosphorylation defect type 39
 oxidative phosphorylation pathway - Ontology Browser - Rat Genome Database
oxidative phosphorylation pathway - Ontology Browser - Rat Genome Database
oxidative phosphorylation pathway - Ontology Report - Rat Genome Database
 Urea cycle; oxidative phosphorylation 2 | HSTalks
Urea cycle; oxidative phosphorylation 2 | HSTalks
 Frontiers | Mesenchymal Stem Cells Shift Mitochondrial Dynamics and Enhance Oxidative Phosphorylation in Recipient Cells
Frontiers | Mesenchymal Stem Cells Shift Mitochondrial Dynamics and Enhance Oxidative Phosphorylation in Recipient Cells
 ULTRA-WEAK LUMINESCENCE AND OXIDATIVE PHOSPHORYLATION IN MITOCHONDRIA
ULTRA-WEAK LUMINESCENCE AND OXIDATIVE PHOSPHORYLATION IN MITOCHONDRIA
 Lesson: Oxidative Phosphorylation | Nagwa
Lesson: Oxidative Phosphorylation | Nagwa
 Molecular Vision: The cellular and compartmental profile of
mouse retinal glycolysis, tricarboxylic acid cycle,...
Molecular Vision: The cellular and compartmental profile of
mouse retinal glycolysis, tricarboxylic acid cycle,...
Medical Science Monitor | Enzymatic diagnosis of oxidative phosphorylation defects on muscle biopsy: Better on tissue...
 Mitochondrial oxidative phosphorylation regulates the fate decision between pathogenic Th17 and regulatory T Cells - Heersink...
Mitochondrial oxidative phosphorylation regulates the fate decision between pathogenic Th17 and regulatory T Cells - Heersink...
 The mechanism of oxidative phosphorylation. XIV. Purification and properties of a second energy-transfer factor
The mechanism of oxidative phosphorylation. XIV. Purification and properties of a second energy-transfer factor
 Figures and data in The peroxisome counteracts oxidative stresses by suppressing catalase import via Pex14 phosphorylation |...
Figures and data in The peroxisome counteracts oxidative stresses by suppressing catalase import via Pex14 phosphorylation |...
A High-Fat Diet Coordinately Downregulates Genes Required for Mitochondrial Oxidative Phosphorylation in Skeletal Muscle |...
The co-occurrence of mtDNA mutations on different oxidative phosphorylation subunits, not detected by haplogroup...
 "Oxidative Phosphorylation: A Critical Feature and Novel Therapeutic Ta" by Grant Fischer
"Oxidative Phosphorylation: A Critical Feature and Novel Therapeutic Ta" by Grant Fischer
 SIZE AND SHAPE TRANSFORMATIONS CORRELATED WITH OXIDATIVE PHOSPHORYLATION IN MITOCHONDRIA | Journal of Cell Biology |...
SIZE AND SHAPE TRANSFORMATIONS CORRELATED WITH OXIDATIVE PHOSPHORYLATION IN MITOCHONDRIA | Journal of Cell Biology |...
 NDLI: Non-equilibrium thermodynamics of oxidative phosphorylation by inverted inner membrane vesicles of rat liver mitochondria.
NDLI: Non-equilibrium thermodynamics of oxidative phosphorylation by inverted inner membrane vesicles of rat liver mitochondria.
 COMBINED OXIDATIVE PHOSPHORYLATION DEFICIENCY 4; COXPD4 | MENDELIAN.CO
COMBINED OXIDATIVE PHOSPHORYLATION DEFICIENCY 4; COXPD4 | MENDELIAN.CO
 Tag: oxidative phosphorylation | Plantae
Tag: oxidative phosphorylation | Plantae
 mitochondrial oxidative phosphorylation Archives - Melatonin Facts
mitochondrial oxidative phosphorylation Archives - Melatonin Facts
is glycolysis faster than oxidative phosphorylation
 Condition Combined Oxidative Phosphorylation Defect Type 29
Condition Combined Oxidative Phosphorylation Defect Type 29
 Oxidative Phosphorylation: Mechanism and Regulation • Microbe Online
Oxidative Phosphorylation: Mechanism and Regulation • Microbe Online
Oxidative Phosphorylation | Open Textbooks for Hong Kong
 Oxidative Phosphorylation(氧化磷酸化)
Oxidative Phosphorylation(氧化磷酸化)OXPHOS6
- Metabolic control analysis has often been used for quantitative studies of the regulation of mitochondrial oxidative phosphorylations (OXPHOS). (nih.gov)
- Mitochondrial function or the ability to generate energy through OXPHOS (oxidative phosphorylation) is vital for cell homeostasis and its dysfunction has been linked to the pathogenesis of nearly all chronic diseases ( Pieczenik and Neustadt, 2007 ). (frontiersin.org)
- Glucose is the major substrate for ATP synthesis through glycolysis and oxidative phosphorylation (OXPHOS), whereas intermediary metabolism through the tricarboxylic acid (TCA) cycle utilizes non-glucose-derived monocarboxylates, amino acids, and alpha ketoacids to support mitochondrial ATP and GTP synthesis. (molvis.org)
- Six genes involved in oxidative phosphorylation (OXPHOS) decreased. (diabetesjournals.org)
- Two recent microarray studies have shown that genes involved in oxidative phosphorylation (OXPHOS) exhibit reduced expression levels in the skeletal muscle of type 2 diabetic subjects and prediabetic subjects. (diabetesjournals.org)
- We recently showed via RNA-sequencing (RNA-seq) analysis of clinical samples that melanoma brain metastases (MBMs) have higher expression of oxidative phosphorylation (OXPHOS) genes (including PPARGC1A or PGC1α ) than patient-matched extracranial metastases (ECMs). (tmc.edu)
Mitochondria10
- Luminescence was detected in the mitochondria of the liver of a rat, in which the process of oxidative phosphorylation had taken place. (dtic.mil)
- Enzymatic diagnosis of oxidative phosphorylation defects on muscle biopsy: Better on tissue homogenate or on a mitochondria-enriched suspension? (medscimonit.com)
- NDLI: Non-equilibrium thermodynamics of oxidative phosphorylation by inverted inner membrane vesicles of rat liver mitochondria. (iitkgp.ac.in)
- The mechanism of oxidative phosphorylation is based on chemiosmotic theory , which states that the difference in proton concentration between the membranes of mitochondria acts as the reservoir for the energy generated from biological oxidation reactions. (microbeonline.com)
- Cellular respiration (oxidative phosphorylation) occurs in the mitochondria, where a series of enzymes catalyze the transfer of electrons to molecular oxygen and the generation of energy-storing adenosine triphosphate (ATP). (msdmanuals.com)
- The previously published model of oxidative phosphorylation in isolated skeletal muscle mitochondria was extended by incorporation of the creatine kinase system (creatine kinase plus phosphocreatine/creatine pair), cytosolic proton production/consumption system (proton production/consumption by the creatine kinase-catalysed reaction, efflux/influx of protons), physiological size of the adenine nucleotide pool and some additional minor changes. (physiomeproject.org)
- Theoretical studies performed by means of the extended model demonstrated that the CK system, which allows for large changes in P(i) in relation to isolated mitochondria system, has no significant influence on the kinetic properties of oxidative phosphorylation, as inorganic phosphate only slightly modifies the relationship between the respiration rate and [ADP]. (physiomeproject.org)
- Estrogens decrease osteoclast number by attenuating mitochondria oxidative phosphorylation and ATP production in early osteoclast precursors. (bvsalud.org)
- Instead, using microarray analysis we have elucidated that ERα-mediated estrogen signaling in osteoclast progenitors decreases " oxidative phosphorylation " and the expression of mitochondria complex I genes . (bvsalud.org)
- This condition seems to be in part related to dysfunctional mitochondria that cause an increased electron "leakage" from the respiratory chain during oxidative phosphorylation with a consequent generation of reactive oxygen species (ROS). (medscape.com)
Combined Oxidative Phosphorylation Defect Type8
- Combined oxidative phosphorylation defect type 4 is a rare mitochondrial disorder due to a defect in mitochondrial protein synthesis characterized by a neonatal onset of severe metabolic acidosis and respiratory distress, persistent lactic acidosis with episodes of metabolic crises, developmental regression, microcephaly, abnormal gaze fixation and pursuit, axial hypotonia with limb spasticity and reduced spontaneous movements. (mendelian.co)
- Combined Oxidative Phosphorylation Defect Type 4 Is also known as coxpd4. (mendelian.co)
- Combined oxidative phosphorylation defect type 29 (COXPD29) is a rare genetic disorder caused by mutations in the SURF1 gene. (rarediseaseshealthcenter.com)
- What are the symptoms of Combined oxidative phosphorylation defect type 29? (rarediseaseshealthcenter.com)
- What are the causes of Combined oxidative phosphorylation defect type 29? (rarediseaseshealthcenter.com)
- Treatment for Combined oxidative phosphorylation defect type 29 is largely supportive and symptomatic. (rarediseaseshealthcenter.com)
- Is there a cure/medications for Combined oxidative phosphorylation defect type 29? (rarediseaseshealthcenter.com)
- At this time, there is no known cure or medications for Combined Oxidative Phosphorylation Defect Type 29. (rarediseaseshealthcenter.com)
Glycolysis and oxidative phosphorylation1
- These findings support the conclusion that MKP-1 plays an important role in regulating proteins involved in glycolysis and oxidative phosphorylation and modulates expression of mitochondrial transcription factors. (cmich.edu)
Metabolism4
- Although oxidative phosphorylation is a vital part of metabolism, it produces reactive oxygen species such as superoxide and hydrogen peroxide, which lead to propagation of free radicals, damaging cells and contributing to disease and, possibly, aging and senescence. (wikipedia.org)
- Recent reports suggest that in response to sepsis, metabolism of macrophages switches from oxidative phosphorylation to aerobic glycolysis. (cmich.edu)
- Abnormal whole-body energy metabolism in iron-deficient humans despite preserved skeletal muscle oxidative phosphorylation. (figshare.com)
- Over many years, several theories have arisen based on clinical and scientific data obtained in human and animal studies, including oxidative stress, apoptosis, mitochondrial dysfunction, derangements of fatty acid metabolism/transport, and accelerated protein catabolism. (medscape.com)
Skeletal muscle4
- ABSTRACT: A dynamic computer model of oxidative phosphorylation in oxidative mammalian skeletal muscle was developed. (physiomeproject.org)
- A model of oxidative phosphorylation in mammalian skeletal muscle, Bernard Korzeniewski and Jerzy A. Zoladz, 2001, Biophysical Chemistry , 92, 17-34. (physiomeproject.org)
- Compared to control rats, male rats exposed to 14 days of postnatal hyperoxia then aged to 1 year demonstrated higher skeletal muscle fatigability, lower muscle mitochondrial oxidative capacity, more mitochondrial damage, and higher glycolytic enzyme expression. (frontiersin.org)
- Given that young adults born premature also demonstrate skeletal muscle dysfunction, future studies are merited to determine whether this dysfunction as well as reduced aerobic capacity is due to reduced mitochondrial oxidative capacity and metabolic dysfunction. (frontiersin.org)
Genes1
- Co-expression analysis highlights the function of mitochondrial genes in oxidative phosphorylation, DNA repair and the cell cycle, and shows their connections with clinically actionable genes. (lu.se)
Deficiency2
- Combined oxidative phosphorylation deficiency type 4 (sequence analysis of TUFM gene). (mendelian.co)
- Because metabolic reprogramming modulates immune responses to TLR-4 activation, we investigated the effect of MKP-1 deficiency on mitochondrial electron transport chains involved in oxidative phosphorylation and transcription factors regulating mitochondrial biogenesis. (cmich.edu)
Inhibition4
- Harrington says her study "found that mitochondrial oxidative phosphorylation is important for the differentiation of pathogenic, autoreactive Th17 effector CD4 T cells and that inhibition of this pathway suppressed Th17 development and Th17 mediated autoimmunity. (uab.edu)
- In this specific case, inhibition of mitochondrial oxidative phosphorylation impeded the emergence of autoreactive, pathogenic Th17 cells and instead promoted the generation of Treg that can suppress chronic inflammation and autoimmunity. (uab.edu)
- I think when my student repeated the principle finding that inhibition of oxidative phosphorylation blocked Th17 differentiation and promoted the emergence of Foxp3+ Treg cells more than five times. (uab.edu)
- In turn, muscle accumulation of acetyl‐CoA leads to acetylation‐dependent inhibition of mitochondrial respiratory complex II enhancing oxidative phosphorylation dysfunction which results in augmented ROS production. (bioinfor.com)
Synthase3
- The ATP synthase uses the energy to transform adenosine diphosphate (ADP) into adenosine triphosphate, in a phosphorylation reaction. (wikipedia.org)
- and (ii) the liver, the kidney and the brain, controlled mainly at the phosphorylation level by ATP synthase and the phosphate carrier. (nih.gov)
- Cofactor Strap regulates oxidative phosphorylation and mitochondrial p53 activity through ATP synthase. (ox.ac.uk)
Pathway2
- Oxidative phosphorylation (UK /ɒkˈsɪd.ə.tɪv/, US /ˈɑːk.sɪˌdeɪ.tɪv/ ) or electron transport-linked phosphorylation or terminal oxidation is the metabolic pathway in which cells use enzymes to oxidize nutrients, thereby releasing chemical energy in order to produce adenosine triphosphate (ATP). (wikipedia.org)
- A schematic diagram of the oxidative phosphorylation pathway. (physiomeproject.org)
Synthesis3
- A series of events occurs during oxidative phosphorylation, broadly classified under electron transport chain and ATP synthesis. (microbeonline.com)
- Computer simulations also suggested that the second-order dependence of oxidative phosphorylation on [ADP] proposed in the literature refers only to the ATP synthesis flux, but not to the oxygen consumption flux (the difference between these two fluxes being due to the proton leak). (physiomeproject.org)
- It is believed to play a role in the synthesis of triphosphonucleotides using ATP formed through OXIDATIVE PHOSPHORYLATION. (bvsalud.org)
Regulation4
- The Book Regulation of Oxidative Phosphorylation Quiz Questions and Answers , regulation of oxidative phosphorylation MCQ questions PDF chapter 21-54 to download online courses, mcat biology tests. (mcqslearn.com)
- Solve Oxidative Phosphorylation MCQ questions, regulation of oxidative phosphorylation Multiple Choice Questions (MCQ Quiz) for online college degrees. (mcqslearn.com)
- The e-Book Regulation of Oxidative Phosphorylation Tests App Download: regulation of oxidative phosphorylation, water soluble vitamins, saturated fats, analyzing gene expression, mcat: kinetics test prep to enroll in online classes. (mcqslearn.com)
- The magnitude of proton motive force depends upon the PDF, 'Regulation of Oxidative Phosphorylation' App APK Download with permeability of membrane, number of ribosomes in cell, presence of integral proteins in the membrane, and energy charge of cell choices for college admission test. (mcqslearn.com)
Electron trans1
- Oxidative phosphorylation is the coupling of the electron transport chain and the ATP biosynthetic pathways. (mcw.edu)
Aerobic2
- Almost all aerobic organisms carry out oxidative phosphorylation. (wikipedia.org)
- Oxidative phosphorylation is the energy-yielding metabolic process of aerobic organisms. (microbeonline.com)
Mechanism1
- The mechanism of oxidative phosphorylation. (eurekamag.com)
Regulates3
- Harrington's study, "Mitochondrial Oxidative Phosphorylation Regulates the Fate Decision between Pathogenic Th17 and Regulatory T Cells," was recently published in the journal Cell Reports . (uab.edu)
- Mechanistically, we showed that oxidative phosphorylation regulates the strength of the T cell receptor signal and that this subsequently controls the induction of the key transcription factor BATF. (uab.edu)
- Stable knockout of Mcart-1 down-regulates the proliferation and oxidative phosphorylation of RAW264.7 macrophages[J]. Basic & Clinical Medicine, 2023, 43(4): 568-575. (magtechjournal.com)
NADH3
- Glycolysis produces only 2 ATP molecules, but somewhere between 30 and 36 ATPs are produced by the oxidative phosphorylation of the 10 NADH and 2 succinate molecules made by converting one molecule of glucose to carbon dioxide and water, while each cycle of beta oxidation of a fatty acid yields about 14 ATPs. (wikipedia.org)
- Factor B produces several-fold stimulation of ATP-driven Nad reduction, and of net phosphorylation coupled to Nadh or succinate oxidation in ammonia particles. (eurekamag.com)
- NADH (nicotinamide adenosine diphosphate hydrogen) and FADH 2 (flavin adenosine dinucleotide hydrogen), both are electron carriers and help in the generation of ATP by oxidative phosphorylation. (microbeonline.com)
Defects2
- Tissues with a high energy demand (eg, brain, nerves, retina, skeletal and cardiac muscle) are particularly vulnerable to defects in oxidative phosphorylation. (msdmanuals.com)
- Increase in the lactate:pyruvate ratio distinguishes oxidative phosphorylation defects from other genetic causes of lactic acidosis. (msdmanuals.com)
Macrophages1
- We show that MKP-1-deficient mice/ macrophages exhibit, at baseline, higher expression of oxidative phosphorylation, TFAM, PGC-1a, and NRF-1 associated with increased respiration and production of reactive oxygen species as compared with wild-type mice. (cmich.edu)
Oxidation2
- In the early reprogramming process, fatty acid oxidation upregulated oxidative phosphorylation and downregulated protein kinase C activity. (biomedcentral.com)
- We demonstrated that fatty acid oxidation promotes reprogramming by enhancing oxidative phosphorylation and inhibiting protein kinase C activity in the early stage of the reprogramming process. (biomedcentral.com)
Cells4
- Importantly, blockade of oxidative phosphorylation led to the emergence of Foxp3+ regulatory CD4 T (Treg) cells. (uab.edu)
- Honestly, the whole study was based on the unexpected finding that mitochondrial oxidative phosphorylation is essential early for promoting Th17 cell differentiation and suppressing the emergence of regulatory CD4 T (Treg) cells. (uab.edu)
- We never would have predicted that result given the dogma in the literature states Th17 cells require glycolysis and Treg utilize oxidative phosphorylation to function. (uab.edu)
- Conclusions The proliferation activity and oxidative phosphorylation level of RAW264.7 cells were both down-regulated after the Mcart-1 was stably knocked out. (magtechjournal.com)
Process4
- The main contribution of this work has been to show that the control of mitochondrial metabolic fluxes can be shared among several steps of the oxidative phosphorylation process, and that this distribution can vary according to the steady state and the tissue. (nih.gov)
- In this lesson, we will learn how to describe the process of oxidative phosphorylation and recall the products formed. (nagwa.com)
- The entirety of this process is called oxidative phosphorylation . (opentextbooks.org.hk)
- This clip explains that oxygen accepts electrons that are extracted from food to form water in the process which is called Oxidative Phosphorylation. (3dme.com.au)
Cellular2
- All oxidative breakdowns of carbohydrates, fats, and amino acids intersect at this final stage of cellular respiration. (microbeonline.com)
- The most ATP is produced in the oxidative phosphorylation phase of cellular respiration. (answers.com)
Gene1
- These include modes that are predominately genotoxic (i.e., chromosomal abnormalities, oxidative stress, and gene amplification) vs. more nongenotoxic (i.e., altered growth factors, enhanced cell proliferation and promotion of carcinogenesis, and altered DNA repair). (cdc.gov)
Muscle1
- Thirteen iron-deficient (ID) individuals and thirteen iron-replete (IR) control participants each underwent 31P-magnetic resonance spectroscopy of exercising calf muscle to investigate differences in oxidative phosphorylation, followed by whole-body cardiopulmonary exercise testing. (figshare.com)
Disorder1
- A rare mitochondrial oxidative phosphorylation disorder characterized by early onset of severe developmental delay (sometimes with regression of developmental milestones) and intellectual disability, poor or absent speech, and hypotonia. (orpha.net)
Activation1
- The model suggests, in accordance with previous theoretical predictions, that activation of oxidative phosphorylation by an increase in [ADP] can (roughly) explain the behaviour of the system only at low work intensities, while at higher work intensities parallel activation of different steps of oxidative phosphorylation is involved. (physiomeproject.org)
Level1
- Seahorse was used to measure the level of oxidative phosphorylation. (biomedcentral.com)
Higher1
- It is concluded that bioluminescence is not an exotic phenomenon which is inherent to only individual representatives of the living world, but under specific conditions accompanies any oxidative phosphorylation, even in the tissues of higher animals, where nature, by means of energetic and structural barriers, does not permit the useless waste of valuable chemical energy for radiation. (dtic.mil)
Cell1
- Oxidative phosphorylation uses these molecules and O2 to produce ATP, which is used throughout the cell whenever energy is needed. (wikipedia.org)
Energy3
- During oxidative phosphorylation, electrons are transferred from the electron donors to a series of electron acceptors in a series of redox reactions ending in oxygen, whose reaction releases half of the total energy. (wikipedia.org)
- Oxidative phosphorylation works by using energy-releasing chemical reactions to drive energy-requiring reactions. (wikipedia.org)
- The amount of energy released by oxidative phosphorylation is high, compared with the amount produced by anaerobic fermentation. (wikipedia.org)
Treatment2
- C ) Phosphorylation of Pex14 upon treatment with oxidative agents. (elifesciences.org)
- D ) Left, Time course of Pex14 phosphorylation upon H 2 O 2 treatment. (elifesciences.org)