Niemann-Pick Disease, Type A
Pick Disease of the Brain
Niemann-Pick Disease, Type C
Niemann-Pick Diseases
1-Deoxynojirimycin
Cholesterol
Glycogen Storage Disease Type I
tau Proteins
Filipin
Dementia
Tauopathies
Supranuclear Palsy, Progressive
Charcot-Marie-Tooth Disease
Carrier Proteins
Alzheimer Disease
Glycogen Storage Disease Type II
Lysosomes
Glycogen Storage Disease Type III
Neurofibrillary Tangles
Endogenous neuroprotective factors: neurosteroids. (1/138)
Neurosteroids are a group of steroid hormones synthesized by the brain in the presence of steroidogenic enzymes. Specific neurosteroids modulate function of several receptors, and also regulate growth of neurons, myelinization and synaptogenesis in the central nervous system. Some neurosteroids have been shown to display neuroprotective properties, which may have important implications for their potential use in the treatment of various neuropathologies such as: age-dependent dementia, stroke, epilepsy, spinal cord injury, Alzheimer's disease (AD), Parkinson's disease (PD) and Niemann-Pick type C disease (NP-C). This paper focuses on neuroprotection afforded by neurosteroids. (+info)The adult form of Niemann-Pick disease type C. (2/138)
Niemann-Pick disease type C (NPC) is a fatal neurovisceral lipid storage disease of autosomal inheritance resulting from mutations in either the NPC1 (95% of families) or NPC2 gene. The encoded proteins appear to be involved in lysosomal/late endosomal transport of cholesterol, glycolipids and other molecules but their exact function is still unknown. The clinical spectrum of the disease ranges from a neonatal rapidly fatal disorder to an adult-onset chronic neurodegenerative disease. Based upon a comprehensive study of 13 unrelated adult patients diagnosed in France over the past 20 years as well as the analysis of the 55 other cases published since 1969, we have attempted to delineate the major clinical, radiological, biochemical and genotypic characteristics of adult NPC. Overall, mean age at onset (+/-SD) of neuropsychiatric symptoms was 25 +/- 9.7 years. The diagnosis of NPC was established after a mean delay of 6.2 +/- 6.4 years and the mean age at death (calculated from 20 cases) was 38 +/- 10.2 years. Major clinical features included cerebellar ataxia (76%), vertical supranuclear ophthalmoplegia (VSO, 75%), dysarthria, (63%), cognitive troubles (61%), movement disorders (58%), splenomegaly (54%), psychiatric disorders (45%) and dysphagia (37%). Less frequent signs were epilepsy and cataplexy. During the course of the disease, clinical features could be subdivided into (i) visceral signs (hepatomegaly or splenomegaly), (ii) cortical signs (psychiatric cognitive disorders and epilepsy); and (iii) deep brain signs (VSO, ataxia, movement disorders, dysarthria, dysphagia, cataplexy) which exhibited different evolution patterns. Asymptomatic and non-evolutive visceral signs were often noticed since early childhood (38.5% of our patients), followed by mild cortical signs in childhood (learning difficulties) and early adulthood (62% of cases among which 38% were psychiatric disorders). Deep brain signs were observed in 96% of patients and were usually responsible for death. In general, there was a good correlation between clinical signs and the localization of brain atrophy on MRI. The 'variant' biochemical phenotype characterized by mild abnormalities of the cellular trafficking of endocytosed cholesterol was over-represented in the adult form of NPC and seemed associated with less frequent splenomegaly in childhood and lesser psychiatric signs. Involvement of the NPC1 gene was shown in 33 families and of the NPC2 gene in one. Improving the knowledge of the disease among psychiatrists and neurologists appears essential since emerging treatments should be more efficient at the visceral or cognitive/psychiatric stages of the disease, before the occurrence of widespread deep brain neurological lesions. (+info)Lipid homeostasis and lipoprotein secretion in Niemann-Pick C1-deficient hepatocytes. (3/138)
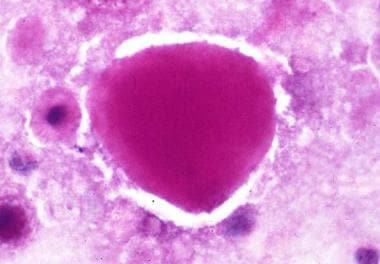
Niemann-Pick C (NPC) disease is a fatal inherited disorder characterized by an accumulation of cholesterol and other lipids in late endosomes/lysosomes. Although this disease is considered to be primarily a neurodegenerative disorder, many NPC patients suffer from liver disease. We have investigated alterations that occur in hepatic lipid homeostasis using primary hepatocytes isolated from NPC1-deficient mice. The cholesterol content of Npc1(-/-) hepatocytes was 5-fold higher than that of Npc1(+/+) hepatocytes; phospholipids and cholesteryl esters also accumulated. In contrast, the triacylglycerol content of Npc1(-/-) hepatocytes was 50% lower than of Npc1(+/+) hepatocytes. We hypothesized that the cholesterol sequestration induced by NPC1 deficiency might inhibit very low density lipoprotein secretion. However, this process was enhanced by NPC1 deficiency and the secreted particles were enriched in cholesteryl esters. We investigated the mechanisms responsible for these changes. The synthesis of phosphatidylcholine, cholesteryl esters, and cholesterol in hepatocytes was increased by NPC1 deficiency and the amount of the mature form of sterol response element-binding protein-1 was also increased. These observations indicate that the enhanced secretion of lipoproteins from NPC1-deficient hepatocytes is due, at least in part, to increased lipid synthesis. (+info)Cholesterol-dependent balance between evoked and spontaneous synaptic vesicle recycling. (4/138)
Cholesterol is a prominent component of nerve terminals. To examine cholesterol's role in central neurotransmission, we treated hippocampal cultures with methyl-beta-cyclodextrin, which reversibly binds cholesterol, or mevastatin, an inhibitor of cholesterol biosynthesis, to deplete cholesterol. We also used hippocampal cultures from Niemann-Pick type C1-deficient mice defective in intracellular cholesterol trafficking. These conditions revealed an augmentation in spontaneous neurotransmission detected electrically and an increase in spontaneous vesicle endocytosis judged by horseradish peroxidase uptake after cholesterol depletion by methyl-beta-cyclodextrin. In contrast, responses evoked by action potentials and hypertonicity were severely impaired after the same treatments. The increase in spontaneous vesicle recycling and the decrease in evoked neurotransmission were reversible upon cholesterol addition. Cholesterol removal did not impact on the low level of evoked neurotransmission seen in the absence of synaptic vesicle SNARE protein synaptobrevin-2 whereas the increase in spontaneous fusion remained. These results suggest that synaptic cholesterol balances evoked and spontaneous neurotransmission by hindering spontaneous synaptic vesicle turnover and sustaining evoked exo-endocytosis. (+info)Clues to neuro-degeneration in Niemann-Pick type C disease from global gene expression profiling. (5/138)
BACKGROUND: Niemann-Pick Type C (NPC) disease is a neurodegenerative disease that is characterized by the accumulation of cholesterol and glycosphingolipids in the late endocytic pathway. The majority of NPC cases are due to mutations in the NPC1 gene. The precise function of this gene is not yet known. METHODOLOGY/PRINCIPAL FINDINGS: Using cDNA microarrays, we analyzed the genome-wide expression patterns of human fibroblasts homozygous for the I1061T NPC1 mutation that is characterized by a severe defect in the intracellular processing of low density lipoprotein-derived cholesterol. A distinct gene expression profile was identified in NPC fibroblasts from different individuals when compared with fibroblasts isolated from normal subjects. As expected, NPC1 mutant cells displayed an inappropriate homeostatic response to accumulated intracellular cholesterol. In addition, a number of striking parallels were observed between NPC disease and Alzheimer's disease. CONCLUSIONS/SIGNIFICANCE: Many genes involved in the trafficking and processing of amyloid precursor protein and the microtubule binding protein, tau, were more highly expressed. Numerous genes important for membrane traffic and the cellular regulation of calcium, metals and other ions were upregulated. Finally, NPC fibroblasts exhibited a gene expression profile indicative of oxidative stress. These changes are likely contributors to the pathophysiology of Niemann-Pick Type C disease. (+info)Defect in fatty acid esterification of dolichol in Niemann-Pick type C1 mouse livers in vivo. (6/138)
Fatty acid esterification of dolichol and cholesterol in Niemann-Pick type C1 mouse (Balb/c NIH npc1(-/-)) livers was investigated in response to treatment with peroxisomal proliferators. These inducers have hypolipidemic properties and influence the mevalonate pathway and the intracellular transport of the final products of this biosynthetic route. Such inducers are consequently interesting to use in a disease model with defective intracellular transport of lipids. In wild-type mice, the levels of dolichol and cholesterol found as free alcohols were not changed to any great extent upon treatment with the peroxisomal inducers dehydroepiandrosterone, clofibrate and diethylhexylphtalate. In contrast, the amounts of dolichyl esters increased whereas cholesteryl esters decreased by the same treatments. The rate of enzymatic esterification of dolichol in isolated microsomes was accordingly elevated after 5- to 7-day treatments with the efficient peroxisomal proliferators DEHP and PFOA, while the corresponding esterification of cholesterol was decreased. Upon peroxisomal induction in npc1(-/-) mice, the enzymatic dolichol esterification in vitro increased whereas the low concentration of dolichyl esters remained unchanged. The results thus demonstrate that the induction of fatty acid esterification of dolichol in vivo is impaired in npc1(-/-) mouse liver. It is therefore proposed that the intracellular lipid transport defect in npc1(-/-) mouse liver disables either dolichol and/or the fatty acid from reaching the site of esterification in vivo. This proposal was strengthened by the finding that the amount of dolichol was decreased in an isolated Golgi fraction from npc1(-/-) mice. (+info)Endosomal accumulation of Toll-like receptor 4 causes constitutive secretion of cytokines and activation of signal transducers and activators of transcription in Niemann-Pick disease type C (NPC) fibroblasts: a potential basis for glial cell activation in the NPC brain. (7/138)
Niemann-Pick disease type C (NPC) is an inherited lipid storage disorder caused by mutations in NPC1 or NPC2 genes. Loss of function of either protein results in the endosomal accumulation of cholesterol and other lipids, progressive neurodegeneration, and robust glial cell activation. Here, we report that cultured human NPC fibroblasts secrete interferon-beta, interleukin-6 (IL-6), and IL-8, and contain increased levels of signal transducers and activators of transcription (STATs). These cells also contained increased levels of Toll-like receptor 4 (TLR4) that accumulated in cholesterol-enriched endosomes/lysosomes, and small interfering RNA knockdown of this receptor reduced cytokine secretion. In the NPC1-/- mouse brain, glial cells expressed TLR4 and IL-6, whereas both glial and neuronal cells expressed STATs. Genetic deletion of TLR4 in NPC1-/- mice reduced IL-6 secretion by cultured fibroblasts but failed to alter STAT levels or glial cell activation in the brain. In contrast, genetic deletion of IL-6 normalized STAT levels and suppressed glial cell activation. These findings indicate that constitutive cytokine secretion leads to activation of STATs in NPC fibroblasts and that this secretion is partly caused by an endosomal accumulation of TLR4. These results also suggest that similar signaling events may underlie glial cell activation in the NPC1-/- mouse brain. (+info)Monocyte cholesterol homeostasis correlates with the presence of detergent resistant membrane microdomains. (8/138)
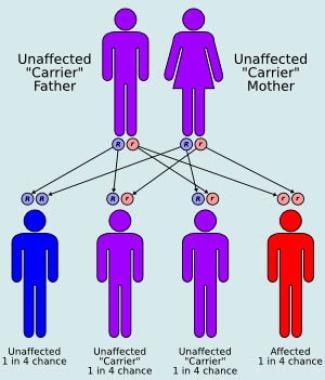
BACKGROUND: Lipid membrane microdomains are involved in the regulation of biological functions of monocyte membrane proteins. These microdomains show a relative resistance to non-ionic detergents providing an easy analytical tool to study them. METHODS: Here, we applied a rapid detergent-based flow cytometric assay to investigate microdomain association of proteins on monocytes from whole blood samples. The association of known surface antigens with detergent resistant fraction of membranes (DRMs) was compared using monocytes from healthy blood donors, patients with genetic disorders affecting cellular cholesterol traffic and patients with systemic inflammatory response. RESULTS: All investigated surface antigens of Niemann-Pick type C (NPC)-mutant monocytes with impaired cholesterol influx and defective late endosome cholesterol trafficking, presented a strongly increased DRM-association. Though, membrane antigens of ATP binding cassette transporter A1 (ABCA1)-mutant monocytes with impaired cholesterol efflux did not show alterations in DRM-association. Differential CD14-dependent receptor clustering within microdomains was also investigated in response to in vivo lipopolysaccharide (LPS) and/or atherogenic lipoprotein activation. Increased DRM-association of the GPI-anchored proteins CD14, CD55, the Fcgamma receptor CD64, the scavenger receptors CD36, CD91 and CD163, the integrin CD11a, and complement receptor 3 complex CD11b/CD18 were observed from patients with systemic inflammatory response syndrome (SIRS)/sepsis or coronary artery disease (CAD)/myocardial infarction. Interestingly, the tetraspanin CD81 presented increased DRM-association in SIRS/sepsis patients, but not in CAD patients. Moreover, the pentaspanin CD47 and the Fcgamma RIII CD16 showed an increased DRM partition in CAD patients but disassembled from DRMs in SIRS/sepsis patients. CONCLUSIONS: Our results demonstrate that flow cytometric analysis of short time in situ detergent extraction provides a powerful tool for rapid screening of blood monocyte DRMs to preselect patients with potential raft/microdomain abnormalities for more detailed analysis. (+info)Niemann-Pick Disease, Type A (NPD A) is a rare inherited metabolic disorder caused by a deficiency of the enzyme acid sphingomyelinase (ASM). This enzyme defect results in the accumulation of lipids, particularly sphingomyelin and cholesterol, within various cells of the body, including brain cells, liver cells, and white blood cells.
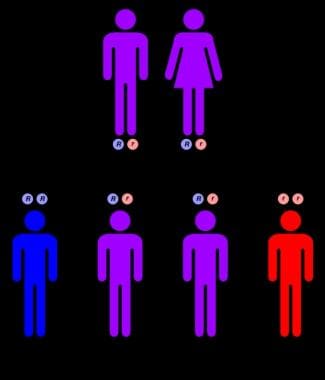
The accumulation of these lipids leads to progressive damage to these organs, causing a range of symptoms such as an enlarged liver (hepatomegaly), anemia, jaundice, and neurological problems like developmental delay, seizures, loss of muscle tone, and difficulty with swallowing. NPD A is typically diagnosed in infancy or early childhood and is often fatal by around two to three years of age due to severe neurological complications. It is an autosomal recessive disorder, meaning that an individual must inherit two copies of the defective gene (one from each parent) to develop the condition.
Pick's disease, also known as Frontotemporal dementia (FTD), is a rare form of degenerative brain disorder that affects the frontal and temporal lobes of the brain. It is characterized by progressive shrinkage (atrophy) of these regions, resulting in a decline in cognitive abilities, behavioral changes, and language difficulties.
The medical definition of Pick's disease includes the following key features:
1. Progressive deterioration of cognitive functions, including memory, judgment, and problem-solving skills.
2. Changes in personality, emotional blunting, and loss of social inhibitions.
3. Language difficulties, such as difficulty with word finding, grammar, and comprehension.
4. Presence of abnormal protein deposits called Pick bodies or Pick cells in the affected brain regions.
5. Exclusion of other causes of dementia, such as Alzheimer's disease, vascular dementia, or Lewy body dementia.
Pick's disease typically affects people between the ages of 40 and 60, and it tends to progress more rapidly than other forms of dementia. Currently, there is no cure for Pick's disease, and treatment focuses on managing symptoms and improving quality of life.
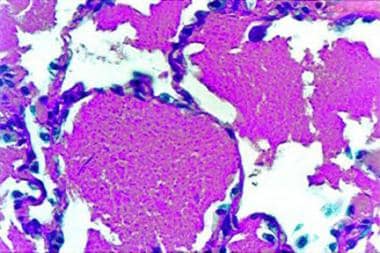
Niemann-Pick Disease, Type C (NPC) is a rare, progressive, and fatal neurovisceral lipid storage disorder caused by mutations in the NPC1 or NPC2 genes. These genetic defects result in impaired intracellular transport of cholesterol and other lipids, leading to excessive accumulation within lysosomes of various tissues, particularly in the brain, liver, spleen, and lungs.
The disease primarily affects children, although late-onset forms have been reported in adults. The symptoms and severity can vary widely among patients but often include neurological manifestations such as ataxia, dysarthria, dysphagia, cognitive decline, seizures, and vertical supranuclear gaze palsy (VSGP). Other features may involve visceral involvement like hepatosplenomegaly, jaundice, or pulmonary complications.
There is currently no cure for NPC, but treatments aim to manage symptoms, slow disease progression, and improve quality of life. Miglustat and cyclodextrin (HPβCD) are two FDA-approved therapeutic options that have shown some promise in stabilizing or delaying neurological decline in NPC patients. Early diagnosis and intervention are crucial for optimizing outcomes and providing appropriate supportive care.
Niemann-Pick diseases are a group of inherited metabolic disorders characterized by the accumulation of lipids, particularly sphingomyelin and cholesterol, within cells due to deficiencies in certain enzymes. These diseases are caused by mutations in the SMPD1, NPC1, or NPC2 genes, among others. There are four main types of Niemann-Pick disease (Types A, B, C, and D), each with varying severity and symptoms.
Type A and Type B diseases, also known as Acid Sphingomyelinase Deficiency or ASMD, result from mutations in the SMPD1 gene leading to a deficiency of acid sphingomyelinase enzyme. This causes excessive accumulation of sphingomyelin in various tissues, particularly in the liver, spleen, lungs, and brain.
Type A is the most severe form, typically presenting in infancy with symptoms such as developmental delay, feeding difficulties, enlarged liver and spleen, lung infection, and progressive neurological degeneration, which often leads to early death, usually before age 3.
Type B has a broader range of severity and onset, from infancy to adulthood. Symptoms may include enlarged liver and spleen, lung disease, poor growth, and varying degrees of neurological impairment. Type B patients can survive into adolescence or adulthood, depending on the severity of their symptoms.
Type C and Type D diseases, also known as Niemann-Pick Type C Disease (NPC), are caused by mutations in either the NPC1 or NPC2 genes, leading to defective intracellular lipid transport. This results in excessive accumulation of cholesterol and other lipids within cells, particularly in the brain, liver, spleen, and lungs.
Type C typically presents in childhood but can also manifest in adolescence or adulthood. Symptoms include progressive neurological degeneration, ataxia, seizures, dementia, problems with speech and swallowing, and yellowish skin (jaundice) at birth or during infancy due to liver involvement. Type C patients usually have a shorter life expectancy, often surviving into their teens, twenties, or thirties.
Type D is a subtype of NPC that affects people of Nova Scotian descent and has similar symptoms to Type C but with an earlier onset and faster progression.
1-Deoxynojirimycin (DNJ) is an antagonist of the enzyme alpha-glucosidase, which is involved in the digestion of carbohydrates. DNJ is a naturally occurring compound found in some plants, including mulberry leaves and the roots of the African plant Moringa oleifera. It works by binding to the active site of alpha-glucosidase and inhibiting its activity, which can help to slow down the digestion and absorption of carbohydrates in the small intestine. This can help to reduce postprandial glucose levels (the spike in blood sugar that occurs after a meal) and may have potential benefits for the management of diabetes and other metabolic disorders. DNJ is also being studied for its potential anti-cancer effects.
Cholesterol is a type of lipid (fat) molecule that is an essential component of cell membranes and is also used to make certain hormones and vitamins in the body. It is produced by the liver and is also obtained from animal-derived foods such as meat, dairy products, and eggs.
Cholesterol does not mix with blood, so it is transported through the bloodstream by lipoproteins, which are particles made up of both lipids and proteins. There are two main types of lipoproteins that carry cholesterol: low-density lipoproteins (LDL), also known as "bad" cholesterol, and high-density lipoproteins (HDL), also known as "good" cholesterol.
High levels of LDL cholesterol in the blood can lead to a buildup of cholesterol in the walls of the arteries, increasing the risk of heart disease and stroke. On the other hand, high levels of HDL cholesterol are associated with a lower risk of these conditions because HDL helps remove LDL cholesterol from the bloodstream and transport it back to the liver for disposal.
It is important to maintain healthy levels of cholesterol through a balanced diet, regular exercise, and sometimes medication if necessary. Regular screening is also recommended to monitor cholesterol levels and prevent health complications.
Glycogen Storage Disease Type I (GSD I) is a rare inherited metabolic disorder caused by deficiency of the enzyme glucose-6-phosphatase, which is necessary for the liver to release glucose into the bloodstream. This leads to an accumulation of glycogen in the liver and abnormally low levels of glucose in the blood (hypoglycemia).
There are two main subtypes of GSD I: Type Ia and Type Ib. In Type Ia, there is a deficiency of both glucose-6-phosphatase enzyme activity in the liver, kidney, and intestine, leading to hepatomegaly (enlarged liver), hypoglycemia, lactic acidosis, hyperlipidemia, and growth retardation. Type Ib is characterized by a deficiency of glucose-6-phosphatase enzyme activity only in the neutrophils, leading to recurrent bacterial infections.
GSD I requires lifelong management with frequent feedings, high-carbohydrate diet, and avoidance of fasting to prevent hypoglycemia. In some cases, treatment with continuous cornstarch infusions or liver transplantation may be necessary.
Tau proteins are a type of microtubule-associated protein (MAP) found primarily in neurons of the central nervous system. They play a crucial role in maintaining the stability and structure of microtubules, which are essential components of the cell's cytoskeleton. Tau proteins bind to and stabilize microtubules, helping to regulate their assembly and disassembly.
In Alzheimer's disease and other neurodegenerative disorders known as tauopathies, tau proteins can become abnormally hyperphosphorylated, leading to the formation of insoluble aggregates called neurofibrillary tangles (NFTs) within neurons. These aggregates disrupt the normal function of microtubules and contribute to the degeneration and death of nerve cells, ultimately leading to cognitive decline and other symptoms associated with these disorders.
Filipin is not a medical term itself, but it is the name given to a group of compounds that are used in medicine and research. Medically, Filipin is often referred to as Filipin III or Filipin stain, which is a fluorescent polyene antibiotic used in the study of lipids, particularly in diagnosing certain types of lipid storage diseases such as Niemann-Pick disease type C. The Filipin stain binds to unesterified cholesterol and forms complexes that exhibit blue fluorescence under ultraviolet light. This property is used to detect the accumulation of free cholesterol in various tissues and cells, which can be indicative of certain diseases or conditions.
Dementia is a broad term that describes a decline in cognitive functioning, including memory, language, problem-solving, and judgment, severe enough to interfere with daily life. It is not a specific disease but rather a group of symptoms that may be caused by various underlying diseases or conditions. Alzheimer's disease is the most common cause of dementia, accounting for 60-80% of cases. Other causes include vascular dementia, Lewy body dementia, frontotemporal dementia, and Huntington's disease.
The symptoms of dementia can vary widely depending on the cause and the specific areas of the brain that are affected. However, common early signs of dementia may include:
* Memory loss that affects daily life
* Difficulty with familiar tasks
* Problems with language or communication
* Difficulty with visual and spatial abilities
* Misplacing things and unable to retrace steps
* Decreased or poor judgment
* Withdrawal from work or social activities
* Changes in mood or behavior
Dementia is a progressive condition, meaning that symptoms will gradually worsen over time. While there is currently no cure for dementia, early diagnosis and treatment can help slow the progression of the disease and improve quality of life for those affected.
Tauopathies are a group of neurodegenerative disorders that are characterized by the abnormal accumulation and aggregation of the microtubule-associated protein Tau in neurons and glial cells. These misfolded Tau proteins form insoluble inclusions, such as neurofibrillary tangles (NFTs) and neuropil threads, which are associated with the degeneration and loss of neurons in specific regions of the brain.
Tauopathies include several well-known diseases, such as Alzheimer's disease (AD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and frontotemporal dementia with Parkinsonism-17 (FTDP-17). The exact cause of Tauopathies remains unclear, but genetic mutations, environmental factors, or a combination of both may contribute to the development and progression of these disorders.
The accumulation of abnormal Tau aggregates is believed to play a central role in the neurodegenerative process, leading to cognitive decline, motor impairment, and other neurological symptoms associated with Tauopathies. The diagnosis of Tauopathies typically involves clinical evaluation, imaging studies, and sometimes postmortem examination of brain tissue. Currently, there are no effective disease-modifying treatments for Tauopathies, but ongoing research is focused on developing therapies that target Tau aggregation and clearance to slow down or halt the progression of these debilitating disorders.
Progressive Supranuclear Palsy (PSP) is a rare neurological disorder characterized by the progressive degeneration of brain cells that regulate movement, thoughts, behavior, and eye movements. The term "supranuclear" refers to the location of the damage in the brain, specifically above the level of the "nuclei" which are clusters of nerve cells that control voluntary movements.
The most common early symptom of PSP is a loss of balance and difficulty coordinating eye movements, particularly vertical gaze. Other symptoms may include stiffness or rigidity of muscles, slowness of movement, difficulty swallowing, changes in speech and writing, and cognitive decline leading to dementia.
PSP typically affects people over the age of 60, and its progression can vary from person to person. Currently, there is no cure for PSP, and treatment is focused on managing symptoms and maintaining quality of life.
Charcot-Marie-Tooth disease (CMT) is a group of inherited disorders that cause nerve damage, primarily affecting the peripheral nerves. These are the nerves that transmit signals between the brain and spinal cord to the rest of the body. CMT affects both motor and sensory nerves, leading to muscle weakness and atrophy, as well as numbness or tingling in the hands and feet.
The disease is named after the three physicians who first described it: Jean-Martin Charcot, Pierre Marie, and Howard Henry Tooth. CMT is characterized by its progressive nature, meaning symptoms typically worsen over time, although the rate of progression can vary significantly among individuals.
There are several types of CMT, classified based on their genetic causes and patterns of inheritance. The two most common forms are CMT1 and CMT2:
1. CMT1: This form is caused by mutations in the genes responsible for the myelin sheath, which insulates peripheral nerves and allows for efficient signal transmission. As a result, demyelination occurs, slowing down nerve impulses and causing muscle weakness, particularly in the lower limbs. Symptoms usually begin in childhood or adolescence and include foot drop, high arches, and hammertoes.
2. CMT2: This form is caused by mutations in the genes responsible for the axons, the nerve fibers that transmit signals within peripheral nerves. As a result, axonal degeneration occurs, leading to muscle weakness and atrophy. Symptoms usually begin in early adulthood and progress more slowly than CMT1. They primarily affect the lower limbs but can also involve the hands and arms.
Diagnosis of CMT typically involves a combination of clinical evaluation, family history, nerve conduction studies, and genetic testing. While there is no cure for CMT, treatment focuses on managing symptoms and maintaining mobility and function through physical therapy, bracing, orthopedic surgery, and pain management.
Carrier proteins, also known as transport proteins, are a type of protein that facilitates the movement of molecules across cell membranes. They are responsible for the selective and active transport of ions, sugars, amino acids, and other molecules from one side of the membrane to the other, against their concentration gradient. This process requires energy, usually in the form of ATP (adenosine triphosphate).
Carrier proteins have a specific binding site for the molecule they transport, and undergo conformational changes upon binding, which allows them to move the molecule across the membrane. Once the molecule has been transported, the carrier protein returns to its original conformation, ready to bind and transport another molecule.
Carrier proteins play a crucial role in maintaining the balance of ions and other molecules inside and outside of cells, and are essential for many physiological processes, including nerve impulse transmission, muscle contraction, and nutrient uptake.
Alzheimer's disease is a progressive disorder that causes brain cells to waste away (degenerate) and die. It's the most common cause of dementia — a continuous decline in thinking, behavioral and social skills that disrupts a person's ability to function independently.
The early signs of the disease include forgetting recent events or conversations. As the disease progresses, a person with Alzheimer's disease will develop severe memory impairment and lose the ability to carry out everyday tasks.
Currently, there's no cure for Alzheimer's disease. However, treatments can temporarily slow the worsening of dementia symptoms and improve quality of life.
Glycogen Storage Disease Type II, also known as Pompe Disease, is a genetic disorder caused by a deficiency of the enzyme acid alpha-glucosidase (GAA). This enzyme is responsible for breaking down glycogen, a complex sugar that serves as energy storage, within lysosomes. When GAA is deficient, glycogen accumulates in various tissues, particularly in muscle cells, leading to their dysfunction and damage.
The severity of Pompe Disease can vary significantly, depending on the amount of functional enzyme activity remaining. The classic infantile-onset form presents within the first few months of life with severe muscle weakness, hypotonia, feeding difficulties, and respiratory insufficiency. This form is often fatal by 1-2 years of age if left untreated.
A later-onset form, which can present in childhood, adolescence, or adulthood, has a more variable clinical course. Affected individuals may experience progressive muscle weakness, respiratory insufficiency, and cardiomyopathy, although the severity and rate of progression are generally less pronounced than in the infantile-onset form.
Enzyme replacement therapy with recombinant human GAA is available for the treatment of Pompe Disease and has been shown to improve survival and motor function in affected individuals.
Lysosomes are membrane-bound organelles found in the cytoplasm of eukaryotic cells. They are responsible for breaking down and recycling various materials, such as waste products, foreign substances, and damaged cellular components, through a process called autophagy or phagocytosis. Lysosomes contain hydrolytic enzymes that can break down biomolecules like proteins, nucleic acids, lipids, and carbohydrates into their basic building blocks, which can then be reused by the cell. They play a crucial role in maintaining cellular homeostasis and are often referred to as the "garbage disposal system" of the cell.
Glycogen Storage Disease Type III, also known as Cori or Forbes disease, is a rare inherited metabolic disorder caused by deficiency of the debranching enzyme amylo-1,6-glucosidase, which is responsible for breaking down glycogen in the liver and muscles. This results in an abnormal accumulation of glycogen in these organs leading to its associated symptoms.
There are two main types: Type IIIa affects both the liver and muscles, while Type IIIb affects only the liver. Symptoms can include hepatomegaly (enlarged liver), hypoglycemia (low blood sugar), hyperlipidemia (high levels of fats in the blood), and growth retardation. In Type IIIa, muscle weakness and cardiac problems may also occur.
The diagnosis is usually made through biochemical tests and genetic analysis. Treatment often involves dietary management with frequent meals to prevent hypoglycemia, and in some cases, enzyme replacement therapy. However, there is no cure for this condition and life expectancy can be reduced depending on the severity of the symptoms.
Neurofibrillary tangles are a pathological hallmark of several neurodegenerative disorders, most notably Alzheimer's disease. They are intracellular inclusions composed of abnormally phosphorylated and aggregated tau protein, which forms paired helical filaments. These tangles accumulate within the neurons, leading to their dysfunction and eventual death. The presence and density of neurofibrillary tangles are strongly associated with cognitive decline and disease progression in Alzheimer's disease and other related dementias.