Myoclonic Epilepsy, Juvenile
Epilepsies, Myoclonic
Epilepsy
Myoclonic Epilepsies, Progressive
Epilepsy, Generalized
MERRF Syndrome
NAV1.1 Voltage-Gated Sodium Channel
Electroencephalography
Epilepsy, Reflex
Myoclonus
Epilepsy, Absence
Epilepsy, Tonic-Clonic
Myoclonic Cerebellar Dyssynergia
Epilepsy, Temporal Lobe
Seizures
Spasms, Infantile
Valproic Acid
MELAS Syndrome
Seizures, Febrile
RNA, Transfer, Lys
Precipitating Factors
Lafora Disease
Status Epilepticus
Mitochondrial Encephalomyopathies
Pedigree
RNA, Transfer, Leu
Video Recording
Magnetic Resonance Imaging
Chromosomes, Human, Pair 6
Age of Onset
Sodium Channels
Thalamus
Epilepsy, Complex Partial
Neuroimaging
Brain
Mutation
Pentylenetetrazole
Epilepsy, Frontal Lobe
DNA, Mitochondrial
How much risk does a woman with active epilepsy pose to her newborn child in the puerperium? A pilot study. (1/85)
Much attention in the literature has recently been paid to women's issues in epilepsy but most of the literature stops in the delivery room or at the first moment of suckling. Although it is commonly supposed that a woman who continues to have active epilepsy during the puerperium will pose a risk to her child, little assessment of how great a risk this is has been carried out. We present an audit of the puerperal experiences of 187 women with epilepsy counselled before birth in our women's clinic and contrast this with a number of women with epilepsy seen for the first time in the puerperium (and therefore uncounselled). The audit suggests that in counselled women the risk is very low (women with Juvenile Myoclonic Epilepsy may be particularly at risk, as may women with tonic-clonic seizures that occur without warning, plus those with automatisms or who have prolonged post-ictal confusion). Some women with controlled epilepsy prior to conception may lose that control during the puerperium so even women with well controlled epilepsy should adopt precautions in the puerperium. The only baby to die (or be seriously injured) in the puerperium born to a woman with epilepsy was killed in the mother's first seizure. (+info)Factors of error involved in the diagnosis of juvenile myoclonic epilepsy: A study from South India. (2/85)
The study was aimed at finding possible factors for delay in the diagnosis of juvenile myoclonic epilepsy (JME) in a developing country. Data was analyzed retrospectively through the medical records and prospectively through a re-evaluation of the history and EEGs of patients with JME registered in a university hospital in south India. Of the 131 patients, 23 (17.5%) patients were seen by neurologists before registration in the clinic. Diagnosis of JME was established in 118 patients at the time of registration and in 13 (10%) patients during follow-up in the clinic. The mean interval between onset of disease and the diagnosis was 6.8 + 6.3 years. In 20 patients the diagnosis was established 10 years after the onset. The mean interval between the first evaluation and diagnosis was 24. 2 months in the 13 patients in whom the diagnosis was established during follow-up in the clinic. Lack of familiarity with the clinical syndrome was probably the factor for delay in the diagnosis in 108 patients seen by practising physicians. The factors for delay in the diagnosis in patients seen by neurologists included failure to ask about myoclonic jerks resulting in misinterpretation of EEGs in 28 patients, misinterpretation of absences and/or unilateral jerks in 4 patients, and failure to ask about myoclonic jerks and misinterpretation of focal EEG abnormalities in 4 patients. This study suggests that the possible factors of error in the diagnosis of JME among the neurologists were similar to the observations reported from the developed countries; whereas the factor for delay in the diagnosis of JME among practising physicians was lack of familiarity with the epileptic syndrome. (+info)Reproducibility and complications in gene searches: linkage on chromosome 6, heterogeneity, association, and maternal inheritance in juvenile myoclonic epilepsy. (3/85)
Evidence for genetic influences in epilepsy is strong, but reports identifying specific chromosomal origins of those influences conflict. One early study reported that human leukocyte antigen (HLA) markers were genetically linked to juvenile myoclonic epilepsy (JME); this was confirmed in a later study. Other reports did not find linkage to HLA markers. One found evidence of linkage to markers on chromosome 15, another to markers on chromosome 6, centromeric to HLA. We identified families through a patient with JME and genotyped markers throughout chromosome 6. Linkage analysis assuming equal male-female recombination probabilities showed evidence for linkage (LOD score 2.5), but at a high recombination fraction (theta), suggesting heterogeneity. When linkage analysis was redone to allow independent male-female thetas, the LOD score was significantly higher (4.2) at a male-female theta of.5,.01. Although the overall pattern of LOD scores with respect to male-female theta could not be explained solely by heterogeneity, the presence of heterogeneity and predominantly maternal inheritance of JME might explain it. By analyzing loci between HLA-DP and HLA-DR and stratifying the families on the basis of evidence for or against linkage, we were able to show evidence of heterogeneity within JME and to propose a marker associated with the linked form. These data also suggest that JME may be predominantly maternally inherited and that the HLA-linked form is more likely to occur in families of European origin. (+info)Diagnosing idiopathic/cryptogenic epilepsy syndromes in infancy. (4/85)
PURPOSE: To determine the characteristics that permit diagnosis of the type of epilepsy beginning in the 1st year of life, and to determine from what age such characteristics are recognisable. PATIENTS: From 430 non-selected patients who began having seizures in the 1st year of life and were referred to the neuropaediatric department of Saint Vincent de Paul Hospital, those with epileptic spasms as the first seizure type, those with recognisable aetiology, and those for whom early history was not reliable were excluded. METHODS: For the remaining 140 patients, the age at which clinical and electroencephalogram (EEG) characteristics met those of recognisable epilepsy syndromes according to the ILAE classification was studied. RESULTS: In most epilepsy syndromes, the diagnosis could be made within three months of onset of the disorder. The most difficult was to distinguish cryptogenic localisation related epilepsy from severe myoclonic epilepsy in infancy. Repeat focal seizures and persisting spike focus were the earliest and most reliable signs of localisation related epilepsy, whereas alternating focal seizures, generalised myoclonus, and/or spike waves appeared before the end of the 1st year in most infants with severe myoclonic epilepsy. However, for 39 patients it was not possible to reach the diagnosis of a precise syndrome. CONCLUSION: For over three quarters of infants with cryptogenic/idiopathic epilepsy, it is possible to reach a syndromic diagnosis within the first months of the disease, based on clinical and EEG characteristics. However, for one quarter, no diagnosis is possible based on the currently available classification. (+info)Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. (5/85)
Inactivation of the beta4 subunit of the calcium channel in the mouse neurological mutant lethargic results in a complex neurological disorder that includes absence epilepsy and ataxia. To determine the role of the calcium-channel beta4-subunit gene CACNB4 on chromosome 2q22-23 in related human disorders, we screened for mutations in small pedigrees with familial epilepsy and ataxia. The premature-termination mutation R482X was identified in a patient with juvenile myoclonic epilepsy. The R482X protein lacks the 38 C-terminal amino acids containing part of an interaction domain for the alpha1 subunit. The missense mutation C104F was identified both in a German family with generalized epilepsy and praxis-induced seizures and in a French Canadian family with episodic ataxia. These coding mutations were not detected in 255 unaffected control individuals (510 chromosomes), and they may be considered candidate disease mutations. The results of functional tests of the truncated protein R482X in Xenopus laevis oocytes demonstrated a small decrease in the fast time constant for inactivation of the cotransfected alpha1 subunit. Further studies will be required to evaluate the in vivo consequences of these mutations. We also describe eight noncoding single-nucleotide substitutions, two of which are present at polymorphic frequency, and a previously unrecognized first intron of CACNB4 that interrupts exon 1 at codon 21. (+info)Juvenile myoclonic epilepsy: a study in Sri Lanka. (6/85)
Juvenile myoclonic epilepsy (JME) has a distinct clinical profile. Often JME is not recognized, with the result that proper treatment is not instituted, leading to poor control of seizures. This study is an attempt to identify the factors that contribute to the delay in diagnosing this condition. During a period of 3 years 40 patients (21 females) with JME were identified and all were included in a prospective follow-up study. The age range was 12-58 years. Twenty-seven patients (67%) had already seen at least one specialist; however, diagnosis had not been made despite the presence of characteristic features. The duration of delay in diagnosis varied from months to years with a mean of 11 years. Myoclonic jerks were the most characteristic feature, but only six volunteered this information spontaneously. The response to treatment with sodium valproate was excellent, although only three were taking it when first seen. As a result of treatment with other drugs all patients were having recurrent seizures. The main reasons for the delay in diagnosis found in our study were that the physicians were unaware of the condition, the occurrence of myoclonic jerks were overlooked either because the patients were not directly questioned about them or because the patients did not volunteer the information. (+info)Photosensitivity in juvenile myoclonic epilepsy. (7/85)
Photosensitivity is reported to occur in approximately 40% of patients with juvenile myoclonic epilepsy. Our experience suggests that the prevalence is higher and may be related to both the duration of intermittent photic stimulation and also the age at which the procedure is undertaken. A two-year retrospective review of all EEGs was undertaken on all children attending a paediatric EEG department to identify those with juvenile myoclonic epilepsy. Photosensitivity was defined as a generalized spike or spike-wave paroxysm occurring at least twice during intermittent photic stimulation. Sixty-one children with a diagnosis of juvenile myoclonic epilepsy with a median age of 13 (range 7-16) years were identified, 55 (90%) of whom were photosensitive. Eighteen of these 55 patients showed photosensitivity only after four minutes of continuous photic stimulation. The prevalence of photosensitivity in juvenile myoclonic epilepsy is likely to be higher than previously reported. When a diagnosis of juvenile myoclonic epilepsy is being considered, the initial diagnostic EEG should include intermittent photic stimulation for up to five minutes, or less if the patient shows evidence of photosensitivity. The identification of photosensitivity may have important management implications. (+info)Genome search for susceptibility loci of common idiopathic generalised epilepsies. (8/85)
Genetic factors play a major role in the aetiology of idiopathic generalised epilepsies (IGEs). The present genome scan was designed to identify susceptibility loci that predispose to a spectrum of common IGE syndromes. Our collaborative study included 130 IGE-multiplex families ascertained through a proband with either an idiopathic absence epilepsy or juvenile myoclonic epilepsy, and one or more siblings affected by an IGE trait. In total, 413 microsatellite polymorphisms were genotyped in 617 family members. Non-parametric multipoint linkage analysis, using the GeneHunter program, provided significant evidence for a novel IGE susceptibility locus on chromosome 3q26 (Z(NPL) = 4.19 at D3S3725; P = 0.000017) and suggestive evidence for two IGE loci on chromosome 14q23 (Z(NPL) = 3.28 at D14S63; P = 0.000566), and chromosome 2q36 (Z(NPL) = 2.98 at D2S1371; P = 0.000535). The present linkage findings provide suggestive evidence that at least three genetic factors confer susceptibility to generalised seizures in a broad spectrum of IGE syndromes. The chromosomal segments identified harbour several genes involved in the regulation of neuronal ion influx which are plausible candidates for mutation screening. (+info)Juvenile Myoclonic Epilepsy (JME) is a genetic condition that is characterized by the occurrence of myoclonic seizures, which are sudden, brief, shock-like jerks of muscles typically occurring in the arms and legs. These seizures usually begin in adolescence or early adulthood, between 12 to 18 years of age.
JME is a type of generalized epilepsy, meaning that it involves abnormal electrical activity throughout the brain rather than just one area. In addition to myoclonic seizures, individuals with JME may also experience absence seizures (brief periods of staring and unresponsiveness) and/or tonic-clonic seizures (generalized convulsions).
The condition is often inherited in an autosomal dominant manner, meaning that a child has a 50% chance of inheriting the gene mutation from a parent with JME. However, not all cases are familial, and some may result from new genetic changes (mutations) that occur spontaneously.
JME is typically treated with anticonvulsant medications such as valproate or lamotrigine to control seizures. Lifestyle modifications, including avoiding sleep deprivation, stress, and excessive alcohol consumption, may also help reduce the frequency of seizures. With appropriate treatment, most individuals with JME can lead normal or near-normal lives.
Myoclonic epilepsies are a group of epilepsy syndromes characterized by the presence of myoclonic seizures. A myoclonic seizure is a type of seizure that involves quick, involuntary muscle jerks or twitches. These seizures can affect one part of the body or multiple parts simultaneously and may vary in frequency and severity.
Myoclonic epilepsies can occur at any age but are more common in infancy, childhood, or adolescence. Some myoclonic epilepsy syndromes have a genetic basis, while others may be associated with brain injury, infection, or other medical conditions.
Some examples of myoclonic epilepsy syndromes include:
1. Juvenile Myoclonic Epilepsy (JME): This is the most common type of myoclonic epilepsy and typically begins in adolescence. It is characterized by myoclonic jerks, often occurring upon awakening or after a period of relaxation, as well as generalized tonic-clonic seizures.
2. Progressive Myoclonic Epilepsies (PME): These are rare inherited disorders that typically begin in childhood or adolescence and involve both myoclonic seizures and other types of seizures. PMEs often progress to include cognitive decline, movement disorders, and other neurological symptoms.
3. Lennox-Gastaut Syndrome (LGS): This is a severe form of epilepsy that typically begins in early childhood and involves multiple types of seizures, including myoclonic seizures. LGS can be difficult to treat and often results in cognitive impairment and developmental delays.
4. Myoclonic Astatic Epilepsy (MAE): Also known as Doose syndrome, MAE is a childhood epilepsy syndrome characterized by myoclonic seizures, atonic seizures (brief periods of muscle weakness or loss of tone), and other types of seizures. It often responds well to treatment with antiepileptic drugs.
The management of myoclonic epilepsies typically involves a combination of medication, lifestyle changes, and, in some cases, dietary modifications. The specific treatment plan will depend on the type of myoclonic epilepsy and its underlying cause.
Epilepsy is a chronic neurological disorder characterized by recurrent, unprovoked seizures. These seizures are caused by abnormal electrical activity in the brain, which can result in a wide range of symptoms, including convulsions, loss of consciousness, and altered sensations or behaviors. Epilepsy can have many different causes, including genetic factors, brain injury, infection, or stroke. In some cases, the cause may be unknown.
There are many different types of seizures that can occur in people with epilepsy, and the specific type of seizure will depend on the location and extent of the abnormal electrical activity in the brain. Some people may experience only one type of seizure, while others may have several different types. Seizures can vary in frequency, from a few per year to dozens or even hundreds per day.
Epilepsy is typically diagnosed based on the patient's history of recurrent seizures and the results of an electroencephalogram (EEG), which measures the electrical activity in the brain. Imaging tests such as MRI or CT scans may also be used to help identify any structural abnormalities in the brain that may be contributing to the seizures.
While there is no cure for epilepsy, it can often be effectively managed with medication. In some cases, surgery may be recommended to remove the area of the brain responsible for the seizures. With proper treatment and management, many people with epilepsy are able to lead normal, productive lives.
Progressive Myoclonic Epilepsies (PME) is a group of rare, genetic disorders characterized by myoclonus (rapid, involuntary muscle jerks), tonic-clonic seizures (also known as grand mal seizures), and progressive neurological deterioration. The term "progressive" refers to the worsening of symptoms over time.
The myoclonic epilepsies are classified as progressive due to the underlying neurodegenerative process that affects the brain, leading to a decline in cognitive abilities, motor skills, and overall functioning. These disorders usually begin in childhood or adolescence and tend to worsen with age.
Examples of PMEs include:
1. Lafora disease: A genetic disorder caused by mutations in the EPM2A or NHLRC1 genes, leading to the accumulation of abnormal protein aggregates called Lafora bodies in neurons. Symptoms typically start between ages 6 and 16 and include myoclonus, seizures, and progressive neurological decline.
2. Unverricht-Lundborg disease: Also known as Baltic myoclonus, this is an autosomal recessive disorder caused by mutations in the CSTB gene. It is characterized by progressive myoclonic epilepsy, ataxia (loss of coordination), and cognitive decline. Symptoms usually begin between ages 6 and 18.
3. Neuronal Ceroid Lipofuscinoses (NCLs): A group of inherited neurodegenerative disorders characterized by the accumulation of lipopigments in neurons. Several types of NCLs can present with progressive myoclonic epilepsy, including CLN2 (late-infantile NCL), CLN3 (juvenile NCL), and CLN6 (early juvenile NCL).
4. Myoclonus Epilepsy Associated with Ragged Red Fibers (MERRF): A mitochondrial disorder caused by mutations in the MT-TK gene, leading to myoclonic epilepsy, ataxia, and ragged red fibers on muscle biopsy.
5. Dentatorubral-Pallidoluysian Atrophy (DRPLA): An autosomal dominant disorder caused by mutations in the ATN1 gene, characterized by myoclonic epilepsy, ataxia, chorea (involuntary movements), and dementia.
These are just a few examples of disorders that can present with progressive myoclonic epilepsy. It is essential to consult a neurologist or epileptologist for proper diagnosis and management.
Generalized epilepsy is a type of epilepsy characterized by seizures that involve both halves of the brain (generalized onset) from the beginning of the seizure. These types of seizures include tonic-clonic (grand mal) seizures, absence (petit mal) seizures, and myoclonic seizures. Generalized epilepsy can be caused by genetic factors or brain abnormalities, and it is typically treated with medication. People with generalized epilepsy may experience difficulties with learning, memory, and behavior, and they may have a higher risk of injury during a seizure. It's important for individuals with generalized epilepsy to work closely with their healthcare team to manage their condition and reduce the frequency and severity of seizures.
Myoclonic Epilepsy with Ragged Red Fibers (MERRF) is a rare mitochondrial disorder, which is a group of genetic disorders that affect the energy production within cells. It is characterized by multiple symptoms including myoclonus (jerky, involuntary muscle spasms), epilepsy (recurrent seizures), ataxia (lack of coordination and balance), dementia, and weakness. The name "MERRF" comes from the characteristic finding of "ragged red fibers" in muscle biopsies when viewed under a microscope using special stains. These fibers are abnormal muscle cells containing clusters of abnormal mitochondria. MERRF is caused by mutations in the mitochondrial DNA, most commonly the A8344G point mutation in the MT-TK gene. It is typically inherited from the mother and can affect multiple organs throughout the body.
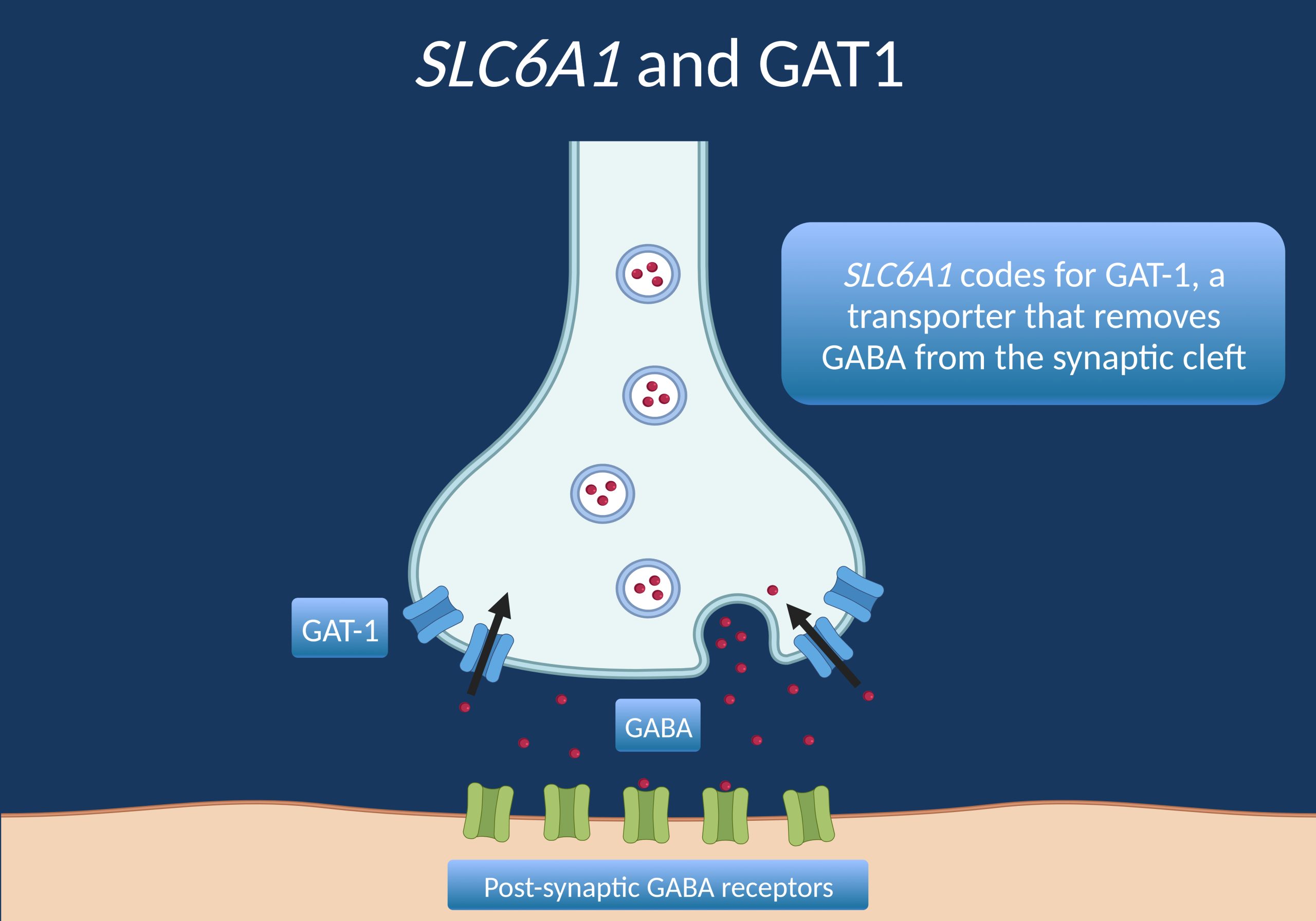
NAV1.1, also known as SCN1A, is a type of voltage-gated sodium channel that is primarily expressed in the central nervous system, including the brain and spinal cord. Voltage-gated sodium channels are transmembrane proteins that play a crucial role in the generation and propagation of action potentials in excitable cells such as neurons.
NAV1.1 voltage-gated sodium channels are responsible for the initiation and propagation of action potentials in the axons of neurons. They are composed of a large alpha subunit, which forms the ion conduction pore, and one or more beta subunits, which modulate the properties of the channel.
Mutations in the SCN1A gene, which encodes the NAV1.1 voltage-gated sodium channel, have been associated with several neurological disorders, including generalized epilepsy with febrile seizures plus (GEFS+), Dravet syndrome, and other forms of epilepsy. These mutations can alter the function of the channel, leading to abnormal neuronal excitability and seizure activity.
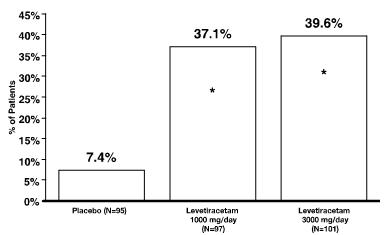
Anticonvulsants are a class of drugs used primarily to treat seizure disorders, also known as epilepsy. These medications work by reducing the abnormal electrical activity in the brain that leads to seizures. In addition to their use in treating epilepsy, anticonvulsants are sometimes also prescribed for other conditions, such as neuropathic pain, bipolar disorder, and migraine headaches.
Anticonvulsants can work in different ways to reduce seizure activity. Some medications, such as phenytoin and carbamazepine, work by blocking sodium channels in the brain, which helps to stabilize nerve cell membranes and prevent excessive electrical activity. Other medications, such as valproic acid and gabapentin, increase the levels of a neurotransmitter called gamma-aminobutyric acid (GABA) in the brain, which has a calming effect on nerve cells and helps to reduce seizure activity.
While anticonvulsants are generally effective at reducing seizure frequency and severity, they can also have side effects, such as dizziness, drowsiness, and gastrointestinal symptoms. In some cases, these side effects may be managed by adjusting the dosage or switching to a different medication. It is important for individuals taking anticonvulsants to work closely with their healthcare provider to monitor their response to the medication and make any necessary adjustments.
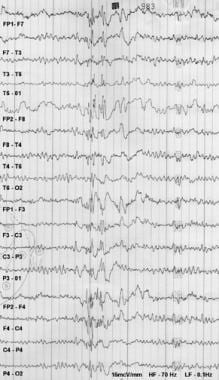
Electroencephalography (EEG) is a medical procedure that records electrical activity in the brain. It uses small, metal discs called electrodes, which are attached to the scalp with paste or a specialized cap. These electrodes detect tiny electrical charges that result from the activity of brain cells, and the EEG machine then amplifies and records these signals.
EEG is used to diagnose various conditions related to the brain, such as seizures, sleep disorders, head injuries, infections, and degenerative diseases like Alzheimer's or Parkinson's. It can also be used during surgery to monitor brain activity and ensure that surgical procedures do not interfere with vital functions.
EEG is a safe and non-invasive procedure that typically takes about 30 minutes to an hour to complete, although longer recordings may be necessary in some cases. Patients are usually asked to relax and remain still during the test, as movement can affect the quality of the recording.
Reflex epilepsy is a type of epilepsy in which seizures are consistently triggered by specific, recurring sensory stimuli. These triggers can vary widely and may include visual patterns, flashes of light, touch, sound, or even emotional experiences. When the brain receives input from these triggers, it responds with an abnormal electrical discharge that can lead to a seizure.
Reflex epilepsy is relatively rare, accounting for only about 5-10% of all epilepsy cases. It's important to note that not everyone who experiences seizures in response to these triggers has reflex epilepsy; the defining characteristic of this condition is the consistent and reproducible nature of the seizure response to a specific stimulus.
There are several different types of reflex epilepsy, each characterized by its own unique set of triggers. For example, some people with this condition may experience seizures in response to visual patterns or flashes of light (known as photosensitive epilepsy), while others may have seizures triggered by certain sounds or tactile sensations.
Treatment for reflex epilepsy typically involves identifying and avoiding triggers whenever possible, as well as using medication to control seizures. In some cases, surgery may be recommended to remove the specific area of the brain that is responsible for the abnormal electrical activity. With proper treatment and management, many people with reflex epilepsy are able to lead full and active lives.
Myoclonus is a medical term that describes a quick, involuntary jerking muscle spasm. These spasms can happen once or repeat in a series, and they can range from mild to severe in nature. Myoclonus can affect any muscle in the body and can be caused by several different conditions, including certain neurological disorders, injuries, or diseases. In some cases, myoclonus may occur without an identifiable cause.
There are various types of myoclonus, classified based on their underlying causes, patterns of occurrence, and associated symptoms. Some common forms include:
1. Action myoclonus: Occurs during voluntary muscle movements
2. Stimulus-sensitive myoclonus: Triggered by external or internal stimuli, such as touch, sound, or light
3. Physiological myoclonus: Normal muscle jerks that occur during sleep onset (hypnic jerks) or during sleep (nocturnal myoclonus)
4. Reflex myoclonus: Result of a reflex arc activation due to a peripheral nerve stimulation
5. Epileptic myoclonus: Part of an epilepsy syndrome, often involving the brainstem or cortex
6. Symptomatic myoclonus: Occurs as a result of an underlying medical condition, such as metabolic disorders, infections, or neurodegenerative diseases
Treatment for myoclonus depends on the specific type and underlying cause. Medications, physical therapy, or lifestyle modifications may be recommended to help manage symptoms and improve quality of life.
Absence epilepsy is a type of epilepsy characterized by recurrent brief episodes of "absences," or staring spells, that can last from a few seconds to several minutes. These episodes are often accompanied by subtle body movements such as lip smacking or eyelid flutters. Absence epilepsy is most commonly diagnosed in children and adolescents, and it is more common in girls than boys.
The seizures in absence epilepsy are caused by abnormal electrical activity in the brain, specifically in a part of the brain called the cortex. These abnormal electrical discharges occur in a pattern that involves both sides of the brain simultaneously. This differs from other types of epilepsy, which may involve only one side of the brain or specific areas within a single hemisphere.
Absence seizures are typically brief and do not cause confusion or disorientation after they end. However, if they occur frequently, they can interfere with learning and social development. In some cases, absence epilepsy may be associated with other types of seizures, such as generalized tonic-clonic (grand mal) seizures or myoclonic jerks.
The diagnosis of absence epilepsy is usually made based on the characteristic symptoms and the results of an electroencephalogram (EEG), which can detect the abnormal electrical activity in the brain during a seizure. Treatment typically involves medication to control the seizures, such as ethosuximide or valproic acid. In some cases, a ketogenic diet may also be recommended as an alternative treatment option.
Tonic-clonic epilepsy, also known as grand mal epilepsy, is a type of generalized seizure that affects the entire brain. This type of epilepsy is characterized by two distinct phases: the tonic phase and the clonic phase.
During the tonic phase, which usually lasts for about 10-20 seconds, the person loses consciousness and their muscles stiffen, causing them to fall to the ground. This can result in injuries if the person falls unexpectedly or hits an object on the way down.
The clonic phase follows immediately after the tonic phase and is characterized by rhythmic jerking movements of the limbs, face, and neck. These movements are caused by alternating contractions and relaxations of the muscles and can last for several minutes. The person may also lose bladder or bowel control during this phase.
After the seizure, the person may feel tired, confused, and disoriented. They may also have a headache, sore muscles, and difficulty remembering what happened during the seizure.
Tonic-clonic epilepsy can be caused by a variety of factors, including genetics, brain injury, infection, or stroke. It is typically diagnosed through a combination of medical history, physical examination, and diagnostic tests such as an electroencephalogram (EEG) or imaging studies. Treatment may include medication, surgery, or dietary changes, depending on the underlying cause and severity of the seizures.
Myoclonic cerebellar dyssynergia is not a widely recognized or formally defined medical term. However, based on its individual components, it can be inferred to refer to a neurological condition characterized by:
1. Myoclonus: These are sudden, involuntary jerking movements of a muscle or group of muscles. They typically occur as a result of hyperexcitability of the neurons in the brain that control movement (motor neurons).
2. Cerebellar: The cerebellum is a part of the brain responsible for coordinating muscle movements, maintaining posture and balance, and fine-tuning motor skills. When a condition is described as "cerebellar," it implies that there is some dysfunction or abnormality in this region of the brain.
3. Dyssynergia: This term refers to a lack of coordination between muscles and muscle groups during voluntary movements. It can result from damage to the cerebellum or other parts of the nervous system involved in motor control.
Therefore, myoclonic cerebellar dyssynergia could be interpreted as a condition characterized by involuntary muscle jerks (myoclonus) and impaired coordination of voluntary movements (dyssynergia), likely due to cerebellar dysfunction. However, it is essential to consult with a medical professional for an accurate diagnosis and treatment plan if you or someone else experiences symptoms that may align with this description.
Temporal lobe epilepsy (TLE) is a type of focal (localized) epilepsy that originates from the temporal lobes of the brain. The temporal lobes are located on each side of the brain and are involved in processing sensory information, memory, and emotion. TLE is characterized by recurrent seizures that originate from one or both temporal lobes.
The symptoms of TLE can vary depending on the specific area of the temporal lobe that is affected. However, common symptoms include auras (sensory or emotional experiences that occur before a seizure), strange smells or tastes, lip-smacking or chewing movements, and memory problems. Some people with TLE may also experience automatisms (involuntary movements such as picking at clothes or fumbling with objects) during their seizures.
Treatment for TLE typically involves medication to control seizures, although surgery may be recommended in some cases. The goal of treatment is to reduce the frequency and severity of seizures and improve quality of life.
A seizure is an uncontrolled, abnormal firing of neurons (brain cells) that can cause various symptoms such as convulsions, loss of consciousness, altered awareness, or changes in behavior. Seizures can be caused by a variety of factors including epilepsy, brain injury, infection, toxic substances, or genetic disorders. They can also occur without any identifiable cause, known as idiopathic seizures. Seizures are a medical emergency and require immediate attention.
Piracetam is a nootropic drug, which is primarily used in the treatment of cognitive disorders. It is a cyclic derivative of the neurotransmitter GABA (gamma-aminobutyric acid). Piracetam is believed to work by reducing the permeability of cell membranes in the brain, which may enhance communication between neurons and improve memory and learning.
Medically, piracetam is used off-label for a variety of conditions related to cognitive decline or impairment, such as Alzheimer's disease, dementia, and age-related cognitive decline. It has also been studied in the treatment of myoclonus (involuntary muscle jerks), dyslexia, and other neurological disorders.
It is important to note that while piracetam has shown some promise in improving cognitive function in certain populations, its effectiveness is still a subject of ongoing research and debate. Additionally, piracetam is not approved by the U.S. Food and Drug Administration (FDA) for any medical use, although it is available as a dietary supplement in the United States.
As with any medication or supplement, it's important to consult with a healthcare provider before taking piracetam to ensure that it is safe and appropriate for your individual needs.
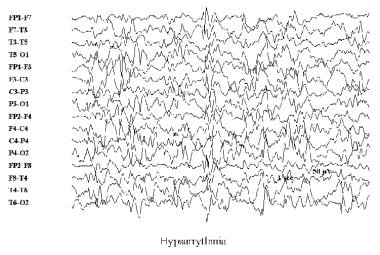
Infantile spasms, also known as West syndrome, is a rare but serious type of epilepsy that affects infants typically between 4-8 months of age. The spasms are characterized by sudden, brief, and frequent muscle jerks or contractions, often involving the neck, trunk, and arms. These spasms usually occur in clusters and may cause the infant to bend forward or stretch out. Infantile spasms can be a symptom of various underlying neurological conditions and are often associated with developmental delays and regression. Early recognition and treatment are crucial for improving outcomes.
Valproic acid is a medication that is primarily used as an anticonvulsant, which means it is used to treat seizure disorders. It works by increasing the amount of gamma-aminobutyric acid (GABA) in the brain, a neurotransmitter that helps to reduce abnormal electrical activity in the brain. In addition to its use as an anticonvulsant, valproic acid may also be used to treat migraines and bipolar disorder. It is available in various forms, including tablets, capsules, and liquid solutions, and is usually taken by mouth. As with any medication, valproic acid can have side effects, and it is important for patients to be aware of these and to discuss them with their healthcare provider.
Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes (MELAS) syndrome is a rare inherited mitochondrial disorder that affects the body's energy production mechanisms. It is characterized by a combination of symptoms including recurrent headaches, vomiting, seizures, vision loss, hearing impairment, muscle weakness, and stroke-like episodes affecting primarily young adults.
The condition is caused by mutations in the mitochondrial DNA (mtDNA), most commonly the A3243G point mutation in the MT-TL1 gene. The symptoms of MELAS syndrome can vary widely among affected individuals, even within the same family, due to the complex inheritance pattern of mtDNA.
MELAS syndrome is typically diagnosed based on a combination of clinical features, laboratory tests, and genetic testing. Treatment is supportive and aimed at managing individual symptoms as they arise.
Febrile seizures are a type of seizure that occurs in young children, typically between the ages of 6 months and 5 years, and is often associated with fever. A febrile seizure is defined as a convulsion or seizure that is brought on by a high fever, usually greater than 100.4°F (38°C), but can also occur in response to a rapid rise in body temperature. The seizures can vary in length and may involve shaking of the entire body, jerking of the arms and legs, or just twitching of one part of the body. They can be quite alarming to witness, but they are usually harmless and do not cause any long-term neurological problems.
Febrile seizures are most commonly caused by viral infections, such as a cold or flu, but they can also occur with bacterial infections, such as a urinary tract infection or ear infection. In some cases, the fever and seizure may be the first signs that a child is ill.
While febrile seizures are generally harmless, it is important to seek medical attention if your child has a seizure. This is because a small percentage of children who have febrile seizures may go on to develop epilepsy, a condition characterized by recurrent seizures. Additionally, some serious underlying conditions, such as meningitis or encephalitis, can cause fever and seizures, so it is important to rule out these possibilities with a thorough medical evaluation.
If your child has a febrile seizure, the best course of action is to remain calm and make sure they are in a safe place where they cannot injure themselves. Do not try to restrain them or put anything in their mouth. Instead, gently turn them onto their side to prevent choking and call for medical help. Most febrile seizures last only a few minutes and resolve on their own without any treatment. After the seizure, your child may be sleepy or confused, but they should return to their normal state within a short period of time.
I'm happy to help! Here are the definitions for each of those terms:
1. RNA (Ribonucleic Acid): RNA is a nucleic acid that plays a crucial role in the process of gene expression. It carries genetic information from DNA to the ribosomes, where proteins are synthesized. RNA is made up of a long chain of nucleotides, just like DNA, but with some key differences in its structure and composition.
2. Transfer: In the context of biochemistry, "transfer" refers to the movement or transport of molecules from one location to another within a cell or between cells. This process is often facilitated by specific proteins or other molecular carriers.
3. Lys (Lysine): Lysine is an essential amino acid that cannot be synthesized by the human body and must be obtained through diet. It plays important roles in various biological processes, including protein synthesis, enzyme function, hormone production, and energy metabolism. In molecular biology, lysine is often used as a marker for certain types of modifications to proteins or nucleic acids.
Therefore, "RNA, Transfer, Lys" could refer to the transfer RNA (tRNA) molecule that carries a specific amino acid, such as lysine, to the ribosome during protein synthesis. The tRNA molecule recognizes a specific codon on the messenger RNA (mRNA) and brings the corresponding amino acid to the growing polypeptide chain, allowing for the translation of genetic information into a functional protein.
In medical terms, "precipitating factors" refer to specific events, actions, or circumstances that trigger the onset of a disease, symptom, or crisis in an individual who is already vulnerable due to pre-existing conditions. These factors can vary depending on the particular health issue, and they may include things like physical stress, emotional stress, environmental triggers, or changes in medication.
For example, in the context of a heart condition, precipitating factors might include strenuous exercise, exposure to extreme temperatures, or the use of certain drugs that increase heart rate or blood pressure. In mental health, precipitating factors for a depressive episode could include significant life changes such as the loss of a loved one, financial difficulties, or a major life transition.
Identifying and managing precipitating factors is an important aspect of preventative healthcare and disease management, as it can help individuals reduce their risk of experiencing negative health outcomes.
Lafora Disease is a rare, inherited, progressive myoclonus epilepsy (PME) disorder. It is characterized by the accumulation of abnormal glycogen particles called Lafora Bodies in nerve cells (neurons) throughout the body, most prominently in the brain and muscle tissue.
The disease typically begins in late childhood or early adolescence with symptoms such as:
- Seizures (myoclonic jerks, tonic-clonic seizures, absence seizures)
- Visual hallucinations
- Dementia
- Speech difficulties
- Muscle stiffness and rigidity
- Difficulty walking and coordinating movements
Lafora Disease is caused by mutations in either the EPM2A or NHLRC1 gene, which play a role in regulating glycogen metabolism. The disease is inherited in an autosomal recessive manner, meaning that an individual must inherit two copies of the mutated gene (one from each parent) to develop the condition.
There is currently no cure for Lafora Disease and treatment is focused on managing symptoms with anti-epileptic drugs and supportive care. The prognosis for individuals with Lafora Disease is poor, with most individuals not surviving beyond their mid-20s.
Status epilepticus is a serious and life-threatening medical condition characterized by an ongoing seizure activity or a series of seizures without full recovery of consciousness between them, lasting for 30 minutes or more. It is a neurological emergency that requires immediate medical attention to prevent potential complications such as brain damage, respiratory failure, or even death.
The condition can occur in people with a history of epilepsy or seizure disorders, as well as those without any prior history of seizures. The underlying causes of status epilepticus can vary and may include infection, trauma, stroke, metabolic imbalances, toxins, or other medical conditions that affect the brain's normal functioning. Prompt diagnosis and treatment are crucial to prevent long-term neurological damage and improve outcomes in patients with this condition.
Mitochondrial Encephalomyopathies are a group of genetic disorders that primarily affect the mitochondria, which are the energy-producing structures in cells. "Encephalo" refers to the brain, while "myopathy" refers to muscle disease. Therefore, Mitochondrial Encephalomyopathies are conditions that cause both neurological and muscular symptoms due to impaired mitochondrial function.
These disorders can affect any organ in the body, but they primarily impact the brain, nerves, and muscles. Symptoms may include muscle weakness, seizures, developmental delays, hearing loss, vision loss, heart problems, and lactic acidosis (a buildup of lactic acid in the blood).
Mitochondrial Encephalomyopathies can be caused by mutations in either the mitochondrial DNA or nuclear DNA. They are often inherited from the mother, as mitochondria are passed down through the maternal line. However, some cases can also result from new mutations that occur spontaneously.
Due to the complex nature of these disorders and their varying symptoms, diagnosis and treatment can be challenging. Treatment typically focuses on managing specific symptoms and may include medications, dietary changes, and physical therapy.
I must clarify that the term "pedigree" is not typically used in medical definitions. Instead, it is often employed in genetics and breeding, where it refers to the recorded ancestry of an individual or a family, tracing the inheritance of specific traits or diseases. In human genetics, a pedigree can help illustrate the pattern of genetic inheritance in families over multiple generations. However, it is not a medical term with a specific clinical definition.
A transfer RNA (tRNA) molecule that carries the amino acid leucine is referred to as "tRNA-Leu." This specific tRNA molecule recognizes and binds to a codon (a sequence of three nucleotides in mRNA) during protein synthesis or translation. In this case, tRNA-Leu can recognize and pair with any of the following codons: UUA, UUG, CUU, CUC, CUA, and CUG. Once bound to the mRNA at the ribosome, leucine is added to the growing polypeptide chain through the action of aminoacyl-tRNA synthetase enzymes that catalyze the attachment of specific amino acids to their corresponding tRNAs. This ensures the accurate and efficient production of proteins based on genetic information encoded in mRNA.
I apologize, but "video recording" is not a term that has a specific medical definition. It generally refers to the process of capturing and storing moving visual images on electronic media, which is used in various medical fields for different purposes such as surgical training, telemedicine consultations, or monitoring patient conditions. However, there is no unique medical meaning associated with this term.
Medical Definition:
Magnetic Resonance Imaging (MRI) is a non-invasive diagnostic imaging technique that uses a strong magnetic field and radio waves to create detailed cross-sectional or three-dimensional images of the internal structures of the body. The patient lies within a large, cylindrical magnet, and the scanner detects changes in the direction of the magnetic field caused by protons in the body. These changes are then converted into detailed images that help medical professionals to diagnose and monitor various medical conditions, such as tumors, injuries, or diseases affecting the brain, spinal cord, heart, blood vessels, joints, and other internal organs. MRI does not use radiation like computed tomography (CT) scans.
Human chromosome pair 6 consists of two rod-shaped structures present in the nucleus of each human cell. They are identical in size and shape and contain genetic material, made up of DNA and proteins, that is essential for the development and function of the human body.
Chromosome pair 6 is one of the 23 pairs of chromosomes found in humans, with one chromosome inherited from each parent. Each chromosome contains thousands of genes that provide instructions for the production of proteins and regulate various cellular processes.
Chromosome pair 6 contains several important genes, including those involved in the development and function of the immune system, such as the major histocompatibility complex (MHC) genes. It also contains genes associated with certain genetic disorders, such as hereditary neuropathy with liability to pressure palsies (HNPP), a condition that affects the nerves, and Waardenburg syndrome, a disorder that affects pigmentation and hearing.
Abnormalities in chromosome pair 6 can lead to various genetic disorders, including numerical abnormalities such as trisomy 6 (three copies of chromosome 6) or monosomy 6 (only one copy of chromosome 6), as well as structural abnormalities such as deletions, duplications, or translocations of parts of the chromosome.
The "age of onset" is a medical term that refers to the age at which an individual first develops or displays symptoms of a particular disease, disorder, or condition. It can be used to describe various medical conditions, including both physical and mental health disorders. The age of onset can have implications for prognosis, treatment approaches, and potential causes of the condition. In some cases, early onset may indicate a more severe or progressive course of the disease, while late-onset symptoms might be associated with different underlying factors or etiologies. It is essential to provide accurate and precise information regarding the age of onset when discussing a patient's medical history and treatment plan.
Sodium channels are specialized protein structures that are embedded in the membranes of excitable cells, such as nerve and muscle cells. They play a crucial role in the generation and transmission of electrical signals in these cells. Sodium channels are responsible for the rapid influx of sodium ions into the cell during the initial phase of an action potential, which is the electrical signal that travels along the membrane of a neuron or muscle fiber. This sudden influx of sodium ions causes the membrane potential to rapidly reverse, leading to the depolarization of the cell. After the action potential, the sodium channels close and become inactivated, preventing further entry of sodium ions and helping to restore the resting membrane potential.
Sodium channels are composed of a large alpha subunit and one or two smaller beta subunits. The alpha subunit forms the ion-conducting pore, while the beta subunits play a role in modulating the function and stability of the channel. Mutations in sodium channel genes have been associated with various inherited diseases, including certain forms of epilepsy, cardiac arrhythmias, and muscle disorders.
The thalamus is a large, paired structure in the brain that serves as a relay station for sensory and motor signals to the cerebral cortex. It is located in the dorsal part of the diencephalon and is made up of two symmetrical halves, each connected to the corresponding cerebral hemisphere.
The thalamus receives inputs from almost all senses, except for the olfactory system, and processes them before sending them to specific areas in the cortex. It also plays a role in regulating consciousness, sleep, and alertness. Additionally, the thalamus is involved in motor control by relaying information between the cerebellum and the motor cortex.
The thalamus is divided into several nuclei, each with distinct connections and functions. Some of these nuclei are involved in sensory processing, while others are involved in motor function or regulation of emotions and cognition. Overall, the thalamus plays a critical role in integrating information from various brain regions and modulating cognitive and emotional processes.
Complex partial epilepsy, also known as temporal lobe epilepsy or focal impaired awareness epilepsy, is a type of epilepsy characterized by recurrent, unprovoked seizures that originate in the temporal lobe or other localized areas of the brain. These seizures typically involve alterations in consciousness or awareness, and may include automatisms (involuntary, repetitive movements), such as lip smacking, fidgeting, or picking at clothes. Complex partial seizures can last from a few seconds to several minutes and may be followed by a post-ictal period of confusion or fatigue.
Complex partial epilepsy is often associated with structural abnormalities in the brain, such as hippocampal sclerosis, tumors, or malformations. It can also be caused by infectious or inflammatory processes, vascular disorders, or genetic factors. The diagnosis of complex partial epilepsy typically involves a thorough neurological evaluation, including a detailed history of seizure symptoms, neuroimaging studies (such as MRI or CT scans), and electroencephalography (EEG) to record brain activity during and between seizures.
Treatment for complex partial epilepsy usually involves medication therapy with antiepileptic drugs (AEDs). In some cases, surgery may be recommended if medications are not effective in controlling seizures or if there is a structural lesion that can be safely removed. Other treatment options may include dietary modifications, such as the ketogenic diet, or vagus nerve stimulation.
A syndrome, in medical terms, is a set of symptoms that collectively indicate or characterize a disease, disorder, or underlying pathological process. It's essentially a collection of signs and/or symptoms that frequently occur together and can suggest a particular cause or condition, even though the exact physiological mechanisms might not be fully understood.
For example, Down syndrome is characterized by specific physical features, cognitive delays, and other developmental issues resulting from an extra copy of chromosome 21. Similarly, metabolic syndromes like diabetes mellitus type 2 involve a group of risk factors such as obesity, high blood pressure, high blood sugar, and abnormal cholesterol or triglyceride levels that collectively increase the risk of heart disease, stroke, and diabetes.
It's important to note that a syndrome is not a specific diagnosis; rather, it's a pattern of symptoms that can help guide further diagnostic evaluation and management.
Neuroimaging is a medical term that refers to the use of various techniques to either directly or indirectly image the structure, function, or pharmacology of the nervous system. It includes techniques such as computed tomography (CT), magnetic resonance imaging (MRI), functional MRI (fMRI), positron emission tomography (PET), single-photon emission computed tomography (SPECT), and diffusion tensor imaging (DTI). These techniques are used to diagnose and monitor various neurological and psychiatric conditions, as well as to understand the underlying mechanisms of brain function in health and disease.
The brain is the central organ of the nervous system, responsible for receiving and processing sensory information, regulating vital functions, and controlling behavior, movement, and cognition. It is divided into several distinct regions, each with specific functions:
1. Cerebrum: The largest part of the brain, responsible for higher cognitive functions such as thinking, learning, memory, language, and perception. It is divided into two hemispheres, each controlling the opposite side of the body.
2. Cerebellum: Located at the back of the brain, it is responsible for coordinating muscle movements, maintaining balance, and fine-tuning motor skills.
3. Brainstem: Connects the cerebrum and cerebellum to the spinal cord, controlling vital functions such as breathing, heart rate, and blood pressure. It also serves as a relay center for sensory information and motor commands between the brain and the rest of the body.
4. Diencephalon: A region that includes the thalamus (a major sensory relay station) and hypothalamus (regulates hormones, temperature, hunger, thirst, and sleep).
5. Limbic system: A group of structures involved in emotional processing, memory formation, and motivation, including the hippocampus, amygdala, and cingulate gyrus.
The brain is composed of billions of interconnected neurons that communicate through electrical and chemical signals. It is protected by the skull and surrounded by three layers of membranes called meninges, as well as cerebrospinal fluid that provides cushioning and nutrients.
A mutation is a permanent change in the DNA sequence of an organism's genome. Mutations can occur spontaneously or be caused by environmental factors such as exposure to radiation, chemicals, or viruses. They may have various effects on the organism, ranging from benign to harmful, depending on where they occur and whether they alter the function of essential proteins. In some cases, mutations can increase an individual's susceptibility to certain diseases or disorders, while in others, they may confer a survival advantage. Mutations are the driving force behind evolution, as they introduce new genetic variability into populations, which can then be acted upon by natural selection.
Pentylenetetrazole (PTZ) is not primarily considered a medical treatment, but rather a research compound used in neuroscience and neurology to study seizure activity and chemically induce seizures in animals for experimental purposes. It is classified as a proconvulsant agent. Medically, it has been used in the past as a medication to treat epilepsy, but its use is now largely historical due to the availability of safer and more effective anticonvulsant drugs.
In a medical or scientific context, Pentylenetetrazole can be defined as:
A chemical compound with the formula C6H5N5O2, which is used in research to investigate seizure activity and induce convulsions in animals. It acts as a non-competitive GABAA receptor antagonist and can lower the seizure threshold. Historically, it has been used as a medication to treat epilepsy, but its use for this purpose is now limited due to the development of safer and more effective anticonvulsant drugs.
Frontal lobe epilepsy is a type of focal epilepsy, which means that the seizures originate from a specific area in the brain called the frontal lobe. The frontal lobe is located at the front part of the brain and is responsible for various functions such as motor function, problem-solving, decision making, emotional expression, and social behavior.
In frontal lobe epilepsy, seizures can be quite varied in their presentation, but they often occur during sleep or wakefulness and may include symptoms such as:
* Brief staring spells or automatisms (such as lip smacking, chewing, or fumbling movements)
* Sudden and frequent falls or drops
* Vocalizations or sounds
* Complex behaviors, such as agitation, aggression, or sexual arousal
* Auras or warning sensations before the seizure
Frontal lobe epilepsy can be difficult to diagnose due to the varied nature of the seizures and their occurrence during sleep. Diagnostic tests such as electroencephalogram (EEG) and imaging studies like magnetic resonance imaging (MRI) may be used to help confirm the diagnosis. Treatment typically involves medication, but in some cases, surgery may be recommended if medications are not effective or cause significant side effects.
Mitochondrial DNA (mtDNA) is the genetic material present in the mitochondria, which are specialized structures within cells that generate energy. Unlike nuclear DNA, which is present in the cell nucleus and inherited from both parents, mtDNA is inherited solely from the mother.
MtDNA is a circular molecule that contains 37 genes, including 13 genes that encode for proteins involved in oxidative phosphorylation, a process that generates energy in the form of ATP. The remaining genes encode for rRNAs and tRNAs, which are necessary for protein synthesis within the mitochondria.
Mutations in mtDNA can lead to a variety of genetic disorders, including mitochondrial diseases, which can affect any organ system in the body. These mutations can also be used in forensic science to identify individuals and establish biological relationships.
A missense mutation is a type of point mutation in which a single nucleotide change results in the substitution of a different amino acid in the protein that is encoded by the affected gene. This occurs when the altered codon (a sequence of three nucleotides that corresponds to a specific amino acid) specifies a different amino acid than the original one. The function and/or stability of the resulting protein may be affected, depending on the type and location of the missense mutation. Missense mutations can have various effects, ranging from benign to severe, depending on the importance of the changed amino acid for the protein's structure or function.
 Juvenile myoclonic epilepsy
Juvenile myoclonic epilepsy Juvenile myoclonic epilepsy: MedlinePlus Genetics
Juvenile myoclonic epilepsy: MedlinePlus Genetics Juvenile Myoclonic Epilepsy Symptoms, Causes, and Treatments
Juvenile Myoclonic Epilepsy Symptoms, Causes, and Treatments Juvenile Myoclonic Epilepsy Medication: Anticonvulsants
Juvenile Myoclonic Epilepsy Medication: Anticonvulsants Juvenile Myoclonic Epilepsy (for Parents) - Ann & Robert H. Lurie Children's Hospital of Chicago
Juvenile Myoclonic Epilepsy (for Parents) - Ann & Robert H. Lurie Children's Hospital of Chicago JUVENILE MYOCLONIC EPILEPSY
JUVENILE MYOCLONIC EPILEPSY What is Juvenile Myoclonic Epilepsy in Dogs? | Embark Vet
What is Juvenile Myoclonic Epilepsy in Dogs? | Embark Vet Epilepsy: Is there a genetic cause?
Epilepsy: Is there a genetic cause? Survey of Planning Executive functions in patients with epilepsy (tonic-clonic juvenile myoclonic epilepsy) and healthy people ...
Survey of Planning Executive functions in patients with epilepsy (tonic-clonic juvenile myoclonic epilepsy) and healthy people ... Dr. Eacharangad Jacob, MD, Neurology Specialist - Panama City, FL | Sharecare
Dr. Eacharangad Jacob, MD, Neurology Specialist - Panama City, FL | Sharecare Epilepsy in Children: Causes, Symptoms, Treatment & Types
Epilepsy in Children: Causes, Symptoms, Treatment & Types Publications
Publications SLC6 Transporter Folding Diseases and Pharmacochaperoning | SpringerLink
SLC6 Transporter Folding Diseases and Pharmacochaperoning | SpringerLink![Dhanuka AK[au] - Search Results - PubMed](data:image/png;base64,iVBORw0KGgoAAAANSUhEUgAAABAAAAAQCAMAAAAoLQ9TAAAARVBMVEVHcEwoU45gYmYAUpQAUpRPYGVgYmZLXnJgYmYAUZUAUpRJXnIAUpQAUpRgYmYAUpRgYmZgYmZhYmYAUpQAUpQAUpRgYmaDiPJuAAAAFXRSTlMADOJ+6QewGO8/uTRqtH7GdFJ11p1bCL3TAAAAZUlEQVQYlV2PVw7AIAxDTeney7n/UcsoldX3E+VJOAboEi7MBpHWMs1ADlG8u7UYWauwyZFeRQVPOhG2o+aiwhByJxUx91Jxhje3iJSqGfHuLKI0+0TpXvY1twCOPlFh5pa/++MB0vIOBm+1zaoAAAAASUVORK5CYII=) Dhanuka AK[au] - Search Results - PubMed
Dhanuka AK[au] - Search Results - PubMed Epilepsy - Health Information Library | PeaceHealth
Epilepsy - Health Information Library | PeaceHealth These highlights do not include all the information needed to use LEVETIRACETAM ORAL SOLUTION safely and effectively. See full...
These highlights do not include all the information needed to use LEVETIRACETAM ORAL SOLUTION safely and effectively. See full... SciELO - Brazil - Dopamine depletion in wistar rats with epilepsy Dopamine depletion in wistar rats with epilepsy
SciELO - Brazil - Dopamine depletion in wistar rats with epilepsy Dopamine depletion in wistar rats with epilepsy Mark Richardson - Google Scholar
Mark Richardson - Google Scholar Consultants
Consultants