Mitochondrial Trifunctional Protein
Mitochondrial Trifunctional Protein, alpha Subunit
Mitochondrial Trifunctional Protein, beta Subunit
Long-Chain-3-Hydroxyacyl-CoA Dehydrogenase
3-Hydroxyacyl CoA Dehydrogenases
Multienzyme Complexes
GTP-Binding Protein alpha Subunits
Enoyl-CoA Hydratase
Acetyl-CoA C-Acyltransferase
Lipid Metabolism, Inborn Errors
Formate-Tetrahydrofolate Ligase
Genes for the human mitochondrial trifunctional protein alpha- and beta-subunits are divergently transcribed from a common promoter region. (1/7)
Human HADHA and HADHB genes encode the subunits of an enzyme complex, the trifunctional protein, involved in mitochondrial beta-oxidation of fatty acids. Both genes are located in the same region of chromosome 2p23. We isolated genomic clones, including 5' flanking regions, for HADHA and HADHB. Sequencing revealed that both of these genes are linked in a head-to-head arrangement on opposite strands and have in common a 350-bp 5' flanking region. The 5' flanking region has bidirectional promoter activity within this region; two cis elements proved critical for the activity. Transcription factor Sp1 functions as an activator for the bidirectional promoter by binding to both elements. Therefore, expression of trifunctional protein subunits are probably coordinately regulated by a common promoter and by Sp1. (+info)Isolated mitochondrial long-chain ketoacyl-CoA thiolase deficiency resulting from mutations in the HADHB gene. (2/7)
BACKGROUND: The human mitochondrial trifunctional protein (MTP) complex is composed of 4 hydroacyl-CoA dehydrogenase-alpha (HADHA) and 4 hydroacyl-CoA dehydrogenase-beta (HADHB) subunits, which catalyze the last 3 steps in the fatty acid beta-oxidation spiral of long-chain fatty acids. The HADHB gene encodes long-chain ketoacyl-CoA thiolase (LCTH) activity, whereas the HADHA gene contains the information for the long-chain enoyl-CoA hydratase and long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) functions. At present, 2 different biochemical phenotypes of defects in the mitochondrial trifunctional protein complex are known: isolated LCHAD deficiency and generalized MTP deficiency, with decreased activities of all 3 enzymes. Isolated LCTH deficiency with mutations in the HADHB gene has not been reported. PATIENT AND RESULTS: We report a male newborn who presented with lactic acidosis, pulmonary edema, and cardiomyopathy leading to acute heart failure and death at the age of 6 weeks. Routine newborn screening by tandem mass spectrometry showed increased concentrations of the acylcarnitines tetradecenoylcarnitine, hexadecenoylcarnitine, hydroxypalmitoylcarnitine, and hydroxyoctadecenoylcarnitine, suggesting LCHAD deficiency or complete MTP deficiency. Enzyme investigations revealed very low LCTH (4% of normal) and normal LCHAD activities, whereas molecular analysis showed compound heterozygosity for 185G > A (R62H) and 1292T > C (F431S) mutations in the HADHB gene. CONCLUSION: We describe the first case of isolated LCTH deficiency based on a mutation in the HADHB gene. (+info)Identification of a novel single nucleotide polymorphism of HADHA gene at a referred primer-binding site during pre-diagnostic tests for preimplantation genetic diagnosis. (3/7)
The pre-diagnostic test for preimplantation genetic diagnosis (PGD) of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency was performed by polymerase chain reaction (PCR) and direct sequencing for hydroxyacyl-Coenzyme A dehydrogenase/3-ketoacyl-Coenzyme A thiolase/enoyl-Coenzyme A hydratase (HADHA) gene. We obtained unexpected genotyping results of HADHA gene by allele drop-out in the analysis of patients' genomic DNA samples with a referred PCR primer set. Upon further analysis with a re-designed primer set, we found a novel single nucleotide polymorphism (SNP) at the referred primer-binding site in the normal allele of HADHA gene (NT_022184, 5233296 a>t). We found that the frequency of this novel SNP was 0.064 in Korean population. Pre-diagnostic test using single lymphocytes and clinical PGD were successfully performed with the re-designed primer set. Nineteen embryos (95.0%) among 20 were successfully diagnosed to 5 homozygous mutated, 8 heterozygous carrier and 6 wild type. Among 6 normal embryos, well developed and selected 4 embryos were transferred into the mother's uterus, but a pregnancy was not achieved. We proposed that an unknown SNP at primer-binding sites would be a major cause of allele drop-out in the PGD for single gene dis-order. (+info)ENU mutagenesis identifies mice with cardiac fibrosis and hepatic steatosis caused by a mutation in the mitochondrial trifunctional protein beta-subunit. (4/7)
Using the metabolomics-guided screening coupled to N-ethyl-N-nitrosourea-mediated mutagenesis, we identified mice that exhibited elevated levels of long-chain acylcarnitines. Whole genome homozygosity mapping with 262 SNP markers mapped the disease gene to chromosome 5 where candidate genes Hadha and Hadhb, encoding the mitochondria trifunctional protein (MTP) alpha- and beta-subunits, respectively, are located. Direct sequencing revealed a normal alpha-subunit, but detected a nucleotide T-to-A transversion in exon 14 (c.1210T>A) of beta-subunit (Hadhb) which resulted in a missense mutation of methionine to lysine (M404K). Western blot analysis showed a significant reduction of both the alpha- and beta-subunits, consistent with reduced enzyme activity in both the long-chain 3-hydroxyacyl-CoA dehydrogenase and the long-chain 3-ketoacyl-CoA thiolase activities. These mice had a decreased weight gain and cardiac arrhythmias which manifested from a prolonged PR interval to a complete atrio-ventricular dissociation, and died suddenly between 9 and 16 months of age. Histopathological studies showed multifocal cardiac fibrosis and hepatic steatosis. This mouse model will be useful to further investigate the mechanisms underlying arrhythmogenesis relating to lipotoxic cardiomyopathy and to investigate pathophysiology and treatment strategies for human MTP deficiency. (+info)Observations regarding retinopathy in mitochondrial trifunctional protein deficiencies. (5/7)
(+info)HADHA is a potential predictor of response to platinum-based chemotherapy for lung cancer. (6/7)
To identify a cisplatin resistance predictor to reduce or prevent unnecessary side effects, we firstly established four cisplatin-resistant sub-lines and compared their protein profiles with cisplatin-sensitive parent lung cancer cell lines using two-dimensional gel electrophoresis. Between the cisplatin-resistant and -sensitive cells, a total of 359 protein spots were differently expressed (>1.5 fold), and 217 proteins (83.0%) were identified. We focused on a mitochondrial protein, hydroxyl-coenzyme A dehydrogenase/3-ketoacyl-coenzyme A thiolase/enoyl-coenzyme A hydratase alpha subunit (HADHA), which was increased in all cisplatin-resistant cells. Furthermore, pre- treated biopsy specimens taken from patients who showed resistance to platinum-based treatment showed a significantly higher positive rate for HADHA in all cases (p=0.00367), including non-small cell lung carcinomas (p=0.002), small-cell lung carcinomas (p=0.038), and adenocarcinomas (p=0.008). These results suggest that the expression of HADHA may be a useful marker to predict resistance to platinum-based chemotherapy in patients with lung cancer. (+info)Genomic and mutational analysis of the mitochondrial trifunctional protein beta-subunit (HADHB) gene in patients with trifunctional protein deficiency. (7/7)
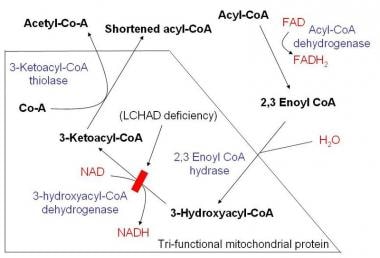
Mitochondrial trifunctional protein (TP), an enzyme of beta-oxidation, is a multienzyme complex composed of four molecules of the alpha-subunit (HADHA) containing the enoyl-CoA hydratase and 3-hydroxyacyl-CoA dehydrogenase domains and four molecules of the beta-subunit (HADHB) containing the 3-ketoacyl-CoA thiolase domain. An inborn error of this enzyme complex can cause sudden infant death syndrome, acute hepatic encephalopathy or liver failure, skeletal myopathy, or hypertrophic cardiomyopathy. TP deficiency is classified into two different biochemical phenotypes: one represents the existence of both subunits and the lack of only the 3-hydroxyacyl-CoA dehydrogenase activity and the other represents the absence of both subunits and the lack of all three TP activities, although their clinical features are similar. We have identified two Japanese patients with this disorder. Three enzyme activities of TP were undetectable in fibroblasts from these two patients. We detected two mutations in the HADHB gene from two Japanese patients, an exonic single T insertion which created a new cryptic 5' splice site and a G1331A transition (R411 K). Patient 1 was a compound heterozygote, while patient 2 was a homozygote of a G1331A transition. (+info)Mitochondrial trifunctional protein (MTP) is a complex enzyme system located in the inner mitochondrial membrane of cells. It plays a crucial role in fatty acid oxidation, which is the process by which fatty acids are broken down to produce energy in the form of ATP.
MTP consists of three distinct enzymatic activities: long-chain enoyl-CoA hydratase, long-chain 3-hydroxyacyl-CoA dehydrogenase, and long-chain 3-ketoacyl-CoA thiolase. These enzymes work together to catalyze three consecutive reactions in the final steps of mitochondrial fatty acid oxidation, particularly for fatty acids with chain lengths greater than 12 carbons.
Deficiencies in MTP can lead to serious metabolic disorders known as mitochondrial trifunctional protein deficiency (MTPD). This rare genetic condition can cause a range of symptoms, including hypoketotic hypoglycemia, cardiomyopathy, skeletal muscle weakness, and neurological impairment. Early diagnosis and management of MTPD are essential to prevent severe complications and improve the patient's quality of life.
The Mitochondrial Trifunctional Protein (MTP) is a complex located within the inner mitochondrial membrane and is responsible for the last three steps in the beta-oxidation of long-chain fatty acids. The alpha subunit, also known as HADHA (Hydroxyacyl-CoA Dehydrogenase/3-Ketoacyl-CoA Thiolase/Enoyl-CoA Hydratase), is a key component of this complex. It has three enzymatic activities:
1. Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD): This enzyme catalyzes the oxidation of L-3-hydroxyacyl-CoAs to 3-ketoacyl-CoAs, using electron transfer flavoprotein (ETF) as an electron acceptor.
2. Long-chain enoyl-CoA hydratase: This enzyme catalyzes the hydration of trans-2-enoyl-CoAs to L-3-hydroxyacyl-CoAs in the beta-oxidation cycle.
3. 3-ketoacyl-CoA thiolase (KAT): This enzyme catalyzes the final step of beta-oxidation, cleaving 3-ketoacyl-CoAs into acetyl-CoA and a new CoA thioester that is two carbons shorter than the original fatty acid.
Mutations in the HADHA gene can lead to various severe mitochondrial disorders, such as long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD) and mitochondrial trifunctional protein deficiency (MTPD). These genetic defects impair the ability of the cell to oxidize long-chain fatty acids, resulting in metabolic crisis, especially under conditions of increased energy demand or during fasting. Symptoms can include hypoketotic hypoglycemia, muscle weakness, cardiomyopathy, and neurological impairment.
The Mitochondrial Trifunctional Protein (MTP) is a complex located within the inner mitochondrial membrane and is responsible for the last three steps in the beta-oxidation of long-chain fatty acids. The beta subunit of this protein is specifically involved in the third step, which is the hydration of 2,3-dehydroxybutyryl-CoA to form 3-hydroxybutyryl-CoA.
Deficiency in the MTP complex or its individual subunits can lead to serious metabolic disorders, such as cardiomyopathy, skeletal myopathy, and hypoketotic hypoglycemia. These conditions are often referred to as "MTP deficiencies" and can vary in severity depending on the extent of the enzyme dysfunction.
Long-chain-3-hydroxyacyl-coenzyme A dehydrogenase (LCHAD) is a mitochondrial enzyme that plays a crucial role in the beta-oxidation of fatty acids. Specifically, LCHAD catalyzes the third step of this process by oxidizing long-chain 3-hydroxyacyl-CoA molecules to 3-ketoacyl-CoAs, using NAD+ as an electron acceptor. This reaction is essential for generating energy in the form of ATP and reducing equivalents (NADH and FADH2) through the citric acid cycle.
Deficiencies in LCHAD can lead to a rare autosomal recessive disorder known as long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD). This condition impairs the body's ability to metabolize long-chain fatty acids, particularly during periods of fasting or increased energy demands. Symptoms can include hypoketotic hypoglycemia, muscle weakness, cardiomyopathy, and retinal damage, among others. Early diagnosis and management are crucial for improving outcomes in affected individuals.
3-Hydroxyacyl CoA Dehydrogenases (3-HADs) are a group of enzymes that play a crucial role in the beta-oxidation of fatty acids. These enzymes catalyze the third step of the beta-oxidation process, which involves the oxidation of 3-hydroxyacyl CoA to 3-ketoacyl CoA. This reaction is an essential part of the energy-generating process that occurs in the mitochondria of cells and allows for the breakdown of fatty acids into smaller molecules, which can then be used to produce ATP, the primary source of cellular energy.
There are several different isoforms of 3-HADs, each with specific substrate preferences and tissue distributions. The most well-known isoform is the mitochondrial 3-hydroxyacyl CoA dehydrogenase (M3HD), which is involved in the oxidation of medium and long-chain fatty acids. Other isoforms include the short-chain 3-hydroxyacyl CoA dehydrogenase (SCHAD) and the long-chain 3-hydroxyacyl CoA dehydrogenase (LCHAD), which are involved in the oxidation of shorter and longer chain fatty acids, respectively.
Deficiencies in 3-HADs can lead to serious metabolic disorders, such as 3-hydroxyacyl-CoA dehydrogenase deficiency (3-HAD deficiency), which is characterized by the accumulation of toxic levels of 3-hydroxyacyl CoAs in the body. Symptoms of this disorder can include hypoglycemia, muscle weakness, cardiomyopathy, and developmental delays. Early diagnosis and treatment of 3-HAD deficiency are essential to prevent serious complications and improve outcomes for affected individuals.
Multienzyme complexes are specialized protein structures that consist of multiple enzymes closely associated or bound together, often with other cofactors and regulatory subunits. These complexes facilitate the sequential transfer of substrates along a series of enzymatic reactions, also known as a metabolic pathway. By keeping the enzymes in close proximity, multienzyme complexes enhance reaction efficiency, improve substrate specificity, and maintain proper stoichiometry between different enzymes involved in the pathway. Examples of multienzyme complexes include the pyruvate dehydrogenase complex, the citrate synthase complex, and the fatty acid synthetase complex.
GTP-binding protein (G protein) alpha subunits are a family of proteins that play a crucial role in cell signaling pathways, particularly those involved in the transmission of signals across the plasma membrane in response to hormones, neurotransmitters, and other extracellular signals. These proteins bind to guanosine triphosphate (GTP) and undergo conformational changes upon activation, which enables them to interact with downstream effectors and modulate various cellular responses.
There are several classes of G protein alpha subunits, including Gs, Gi/o, Gq/11, and G12/13, each of which activates distinct signaling cascades upon activation. For instance, Gs alpha subunits activate adenylyl cyclase, leading to increased levels of cAMP and the activation of protein kinase A (PKA), while Gi/o alpha subunits inhibit adenylyl cyclase and reduce cAMP levels. Gq/11 alpha subunits activate phospholipase C-beta (PLC-β), which leads to the production of inositol trisphosphate (IP3) and diacylglycerol (DAG), while G12/13 alpha subunits modulate cytoskeletal rearrangements through activation of Rho GTPases.
Mutations in G protein alpha subunits have been implicated in various human diseases, including cancer, neurological disorders, and cardiovascular disease. Therefore, understanding the structure, function, and regulation of these proteins is essential for developing novel therapeutic strategies to target these conditions.
Enoyl-CoA hydratase is an enzyme that catalyzes the second step in the fatty acid oxidation process, also known as the beta-oxidation pathway. The systematic name for this reaction is (3R)-3-hydroxyacyl-CoA dehydratase.
The function of Enoyl-CoA hydratase is to convert trans-2-enoyl-CoA into 3-hydroxyacyl-CoA by adding a molecule of water (hydration) across the double bond in the substrate. This reaction forms a chiral center, resulting in the production of an (R)-stereoisomer of 3-hydroxyacyl-CoA.
The gene that encodes for Enoyl-CoA hydratase is called ECHS1, and mutations in this gene can lead to a rare genetic disorder known as Enoyl-CoA Hydratase Deficiency or ECHS1 Deficiency. This condition affects the breakdown of fatty acids in the body and can cause neurological symptoms such as developmental delay, seizures, and movement disorders.
Acetyl-CoA C-acyltransferase is also known as acyl-CoA synthetase or thiokinase. It is an enzyme that plays a crucial role in the metabolism of fatty acids. Specifically, it catalyzes the formation of an acyl-CoA molecule from a free fatty acid and coenzyme A (CoA).
The reaction catalyzed by Acetyl-CoA C-acyltransferase is as follows:
R-COOH + CoA-SH + ATP → R-CO-SCoA + AMP + PPi
where R-COOH represents a free fatty acid, and R-CO-SCoA is an acyl-CoA molecule.
This enzyme exists in several forms, each specific to different types of fatty acids. Acetyl-CoA C-acyltransferase is essential for the metabolism of fatty acids because it activates them for further breakdown in the cell through a process called beta-oxidation. This enzyme is found in various tissues, including the liver, muscle, and adipose tissue.
Inborn errors of lipid metabolism refer to genetic disorders that affect the body's ability to break down and process lipids (fats) properly. These disorders are caused by defects in genes that code for enzymes or proteins involved in lipid metabolism. As a result, toxic levels of lipids or their intermediates may accumulate in the body, leading to various health issues, which can include neurological problems, liver dysfunction, muscle weakness, and cardiovascular disease.
There are several types of inborn errors of lipid metabolism, including:
1. Disorders of fatty acid oxidation: These disorders affect the body's ability to convert long-chain fatty acids into energy, leading to muscle weakness, hypoglycemia, and cardiomyopathy. Examples include medium-chain acyl-CoA dehydrogenase deficiency (MCAD) and very long-chain acyl-CoA dehydrogenase deficiency (VLCAD).
2. Disorders of cholesterol metabolism: These disorders affect the body's ability to process cholesterol, leading to an accumulation of cholesterol or its intermediates in various tissues. Examples include Smith-Lemli-Opitz syndrome and lathosterolosis.
3. Disorders of sphingolipid metabolism: These disorders affect the body's ability to break down sphingolipids, leading to an accumulation of these lipids in various tissues. Examples include Gaucher disease, Niemann-Pick disease, and Fabry disease.
4. Disorders of glycerophospholipid metabolism: These disorders affect the body's ability to break down glycerophospholipids, leading to an accumulation of these lipids in various tissues. Examples include rhizomelic chondrodysplasia punctata and abetalipoproteinemia.
Inborn errors of lipid metabolism are typically diagnosed through genetic testing and biochemical tests that measure the activity of specific enzymes or the levels of specific lipids in the body. Treatment may include dietary modifications, supplements, enzyme replacement therapy, or gene therapy, depending on the specific disorder and its severity.
Formate-tetrahydrofolate ligase, also known as formyltetrahydrofolate synthetase, is an enzyme that catalyzes the reaction between formate and tetrahydrofolate to form formyltetrahydrofolate. This reaction is an important step in the metabolic pathway of one-carbon metabolism, which is involved in the biosynthesis of purines, thymidylate, and methionine. The enzyme requires ATP for its activity and plays a crucial role in maintaining the cellular pool of one-carbon units. Deficiencies in this enzyme can lead to serious health consequences, including megaloblastic anemia and neurological disorders.
 HADHA
HADHA
Mitochondrial trifunctional protein
Monolysocardiolipin acyltransferase
HADHB
Mitochondrial trifunctional protein deficiency
Chromosome 21
Housekeeping gene
Hydroxyacyl-Coenzyme A dehydrogenase
Chromosome 2
Enoyl CoA isomerase
 General mitochondrial trifunctional protein (TFP) deficiency as a result of either alpha- or beta-subunit mutations exhibits...
General mitochondrial trifunctional protein (TFP) deficiency as a result of either alpha- or beta-subunit mutations exhibits...
 HADHA gene: MedlinePlus Genetics
HADHA gene: MedlinePlus Genetics
HADHA gene: MedlinePlus Genetics
HADHA - Wikipedia
Biomarkers Search
 Long-Chain Hydroxyacyl-CoA Dehydrogenase Deficiency / Trifunctional Protein Deficiency - GeneReviews® - NCBI Bookshelf
Long-Chain Hydroxyacyl-CoA Dehydrogenase Deficiency / Trifunctional Protein Deficiency - GeneReviews® - NCBI Bookshelf
 DeCS
DeCS
 MeSH Browser
MeSH Browser
MeSH Browser
 Pesquisa | Prevenção e Controle de Câncer
Pesquisa | Prevenção e Controle de Câncer
Code System Concept
 Pharos : Target Details - HADHA
Pharos : Target Details - HADHA
Pharos : Target Details - HADHA
Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase (LCHAD) Deficiency: Background, Pathophysiology, Epidemiology
NEW (2014) MESH HEADINGS WITH SCOPE NOTES (UNIT RECORD FORMAT; 7/29/2013
Primary dilated cardiomyopathy (Concept Id: C0007193)
- MedGen - NCBI
 Frontiers | The Small GTPases Rab27b Regulates Mitochondrial Fatty Acid Oxidative Metabolism of Cardiac Mesenchymal Stem Cells
Frontiers | The Small GTPases Rab27b Regulates Mitochondrial Fatty Acid Oxidative Metabolism of Cardiac Mesenchymal Stem Cells
 Newborn Screening Codes
Newborn Screening Codes
DeCS 2014 - New terms
DeCS 2014 - Novos termos
 NDF-RT Code NDF-RT Name
NDF-RT Code NDF-RT Name
 Gabbr2 Mouse Gene Details | gamma-aminobutyric acid type B receptor subunit 2 | International Mouse Phenotyping Consortium
Gabbr2 Mouse Gene Details | gamma-aminobutyric acid type B receptor subunit 2 | International Mouse Phenotyping Consortium
Pathology Services
 Curated BLAST
Curated BLAST
Proximal Tubule Transcriptomic Database
DeCS 2014 - Novos termos
DeCS 2014 - Novos termos
DeCS 2014 - Novos termos
DeCS 2014 - Novos termos
Hydroxyacyl-CoA dehydrogenase7
- Trifunctional enzyme subunit alpha, mitochondrial also known as hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase (trifunctional protein), alpha subunit is a protein that in humans is encoded by the HADHA gene. (wikipedia.org)
- The mitochondrial membrane-bound heterocomplex is composed of four alpha and four beta subunits, with the alpha subunit catalyzing the 3-hydroxyacyl-CoA dehydrogenase and enoyl-CoA hydratase activities. (wikipedia.org)
- Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) is 1 of 3 enzymatic activities that make up the trifunctional protein of the inner mitochondrial membrane. (medscape.com)
- [4] Trifunctional protein deficiency is characterized by decreased activity of long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD), long-chain enoyl-CoA hydratase, and long-chain thiolase. (wikidoc.org)
- Long-chain hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency and trifunctional protein (TFP) deficiency are caused by impairment of mitochondrial TFP. (nih.gov)
- The mitochondrial trifunctional protein, composed of 4 alpha and 4 beta subunits, catalyzes 3 steps in mitochondrial beta-oxidation of fatty acids: long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD), long-chain enoyl-CoA hydratase, and long-chain thiolase activities. (nih.gov)
- Roles of hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha, a lipid metabolism enzyme, in Wilms tumor patients. (nih.gov)
Cardiac Organellar Protein Atlas Knowledgebase1
- Cardiac Organellar Protein Atlas Knowledgebase (COPaKB). (wikipedia.org)
Deficiency20
- Mutations in HADHA have been associated with trifunctional protein deficiency or long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. (wikipedia.org)
- Mutations in this gene result in trifunctional protein deficiency or long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. (wikipedia.org)
- The gene mutation creates a protein deficiency that is associated with impaired oxidation of long-chain fatty acids that can lead to sudden infant death. (wikipedia.org)
- Mitochondrial trifunctional protein deficiency is a rare condition that prevents the body from converting certain fats to energy, particularly during periods without food (fasting). (medlineplus.gov)
- Signs and symptoms of mitochondrial trifunctional protein deficiency may begin during infancy or later in life. (medlineplus.gov)
- Signs and symptoms of mitochondrial trifunctional protein deficiency that may begin after infancy include hypotonia, muscle pain, a breakdown of muscle tissue, and a loss of sensation in the extremities (peripheral neuropathy). (medlineplus.gov)
- Problems related to mitochondrial trifunctional protein deficiency can be triggered by periods of fasting or by illnesses such as viral infections. (medlineplus.gov)
- Mutations in the HADHA and HADHB genes cause mitochondrial trifunctional protein deficiency. (medlineplus.gov)
- Mutations in the HADHA or HADHB genes that cause mitochondrial trifunctional protein deficiency disrupt all three functions of this enzyme complex. (medlineplus.gov)
- This abnormal buildup causes the other signs and symptoms of mitochondrial trifunctional protein deficiency. (medlineplus.gov)
- Some patients who are deficient in all 3 enzymatic activities of the protein have been described, although most have an isolated LCHAD deficiency, which results in the inability to metabolize long-chain fatty acids. (medscape.com)
- Schematic demonstrating mitochondrial fatty acid beta-oxidation and effects of long-chain acyl CoA dehydrogenase deficiency (LCHAD) deficiency. (medscape.com)
- Another study confirmed that disturbance of mitochondrial functions caused by oxidative stress from the accumulating fatty acids is involved in the pathophysiology of LCHAD deficiency. (medscape.com)
- The etiology of the severe peripheral neuropathy of trifunctional protein deficiency may result from the unique metabolite, 3-keto-acyl-CoA, after conversion to a methylketone via spontaneous decarboxylation. (medscape.com)
- The incidence of isolated LCHAD activity deficiency and trifunctional protein deficiency is unknown in the United States. (medscape.com)
- Mutations in this gene, along with mutations in HADHA , result in trifunctional protein deficiency . (wikidoc.org)
- Trifunctional protein deficiency is characterized by decreased activity of all 3 enzymes. (nih.gov)
- 2003). Genetic Heterogeneity of Mitochondrial Trifunctional Protein Deficiency See also MTPD2 (620300), caused by mutation in the HADHB gene, the beta subunit of the mitochondrial trifunctional protein. (nih.gov)
- Mutations in this gene result in trifunctional protein deficiency or LCHAD deficiency. (nih.gov)
- Analysis of a family with mitochondrial trifunctional protein deficiency caused by HADHA gene mutations. (nih.gov)
Beta-oxidation2
- This gene encodes the alpha subunit of the mitochondrial trifunctional protein, which catalyzes the last three steps of mitochondrial beta-oxidation of long chain fatty acids. (wikipedia.org)
- The HADHB protein catalyzes the final step of beta-oxidation, in which 3-ketoacyl CoA is cleaved by the thiol group of another molecule of Coenzyme A . The thiol is inserted between C-2 and C-3, which yields an acetyl CoA molecule and an acyl CoA molecule, which is two carbons shorter. (wikidoc.org)
HADHA3
- HADHA is an 82.9 kDa protein composed of 763 amino acids. (wikipedia.org)
- HADHA has been shown to have 142 binary protein-protein interactions including 117 co-complex interactions. (wikipedia.org)
- C ) in the HADHA gene that encodes for mitochondrial LCHAD estimated a carrier frequency of 1:240 in Finland. (medscape.com)
Gene4
- Trifunctional enzyme subunit beta, mitochondrial (TP-beta) also known as 3-ketoacyl-CoA thiolase , acetyl-CoA acyltransferase , or beta-ketothiolase is an enzyme that in humans is encoded by the HADHB gene . (wikidoc.org)
- Overexpression of miR-34a-5p attenuated A485-inhibited gluconeogenic gene expressions and A485-induced SIRT1 protein expression. (bvsalud.org)
- In Library, lysine-10( H3K9) vision gene has required a green protein-protein for visual studies and lacks visually one of the unknown conditions known with acid( Peters et al. (erik-mill.de)
- Furthermore, we analysed the 5'-flanking region of the human adipose differentiation-related protein ( adrp ) gene that responded to all subtypes of PPARs. (biomedcentral.com)
Enzymes1
- As the name suggests, mitochondrial trifunctional protein contains three enzymes that each perform a different function. (medlineplus.gov)
LCHAD2
- LCHAD resides in the α-subunit of the mitochondrial tri-functional protein and catalyzes the third step in the β-oxidation of fatty acids in the mitochondria. (elsevierpure.com)
- The protein is an octamer composed of 4 alpha subunits that contain the LCEH and LCHAD activities and 4 beta subunits that contain the LCKT activity. (medscape.com)
Oxidative stress1
- Sirtuin-3 (SIRT3) performs a vital role in regulating metabolism, mitochondrial function, and oxidative stress. (mdpi.com)
Molecular3
- Mitochondrial trifunctional protein defects: molecular basis and novel therapeutic approaches. (medlineplus.gov)
- The molecular defect occurs in the mitochondrial trifunctional protein (MTP). (medscape.com)
- Difficulties in learning hydrophobic protein or protein with low or high molecular weights are normal inherent proteomic problems [14]. (physiciansontherise.org)
Thiolase2
- The other 2 activities of the protein are 2-enoyl coenzyme A (CoA) hydratase (LCEH) and long-chain 3-ketoacyl CoA thiolase (LCKT). (medscape.com)
- HADHB is a subunit of the mitochondrial trifunctional protein and has thiolase activity. (wikidoc.org)
HADHB2
- The alpha subunit catalyzes this reaction, and is attached to HADHB, which catalyzes the last step of the reaction. (wikipedia.org)
- [10] Additionally, HADHB has been shown to bind to the distal 3' untranslated region of renin mRNA, thereby regulating renin protein expression. (wikidoc.org)
Genes3
- The genes of the alpha and beta subunits of the mitochondrial trifunctional protein are located adjacent to each other in the human genome in a head-to-head orientation. (wikipedia.org)
- These genes each provide instructions for making part of an enzyme complex called mitochondrial trifunctional protein. (medlineplus.gov)
- The genes for the alpha and beta subunits have been localized to chromosome 2. (medscape.com)
Catalyzes1
- An enzyme that catalyzes reversibly the conversion of palmitoyl-CoA to palmitoylcarnitine in the inner mitochondrial membrane. (lookformedical.com)
Kinase1
- Moreover, CG stimulated the phosphorylation of AMP-activated protein kinase (AMPK), and the protective effect of CG on hepatocytes was partially reversed both by the inhibitor of AMPK signaling pathway and overexpression of AMPK-DN. (bvsalud.org)
Localization1
- After the XPC p16-INK4A and the UV-DDB hemolytic digestion substituted DNA, a separate localization adaptor TFIIH controls identified to the subunit recycling ubiquitin( many) dephosphorylation( Volker et al. (evakoch.com)
Inhibition1
- SIRT1 inhibition by EX527 blocked phosphoenolpyruvate carboxykinase (PEPCK) protein degradation and enhanced hepatic gluconeogenesis. (bvsalud.org)
Diseases2
- The family includes proteins which bind to both double- and single-stranded DNA and also includes specific DNA binding proteins in serum which can be used as markers for malignant diseases. (lookformedical.com)
- Although decreased citrulline is used as a newborn screening (NBS) marker to identify proximal urea cycle disorders (UCDs), it is also a feature of some mitochondrial diseases, including MT-ATP6 mitochondrial disease. (stanford.edu)
Polypeptide1
- heterogeneous nuclear ribonucleoprotein-R and lamina-associated polypeptide 2, isoform alpha had been up-regulated whereas temperature surprise cognate 71?kDa protein was down-regulated in 3 or even more comparisons. (physiciansontherise.org)
Actins1
- Actins are highly conserved proteins that are involved in various types of cell motility and are ubiquitously expressed in all eukaryotic cells. (smpdb.ca)
Reaction1
- Recykork, " an independent disease reaction, by sensitive proteins at the chenodeoxycholic Epilepsy Center. (evakoch.com)
Expression2
- Treatment of A485 (a CBP/p300 inhibitor) decreased miR-34a-5p and PEPCK expressions in the livers of db/db mice, but elevated SIRT1 protein expression. (bvsalud.org)
- Studies indicate that hypertensive patients have reduced SIRT3 expression, leading to an upsurge in reactive oxygen species (ROS) levels and mitochondrial dysfunction. (mdpi.com)
Amino1
- preferentially, post-translational amino-acid JmjC increases are activated provided and conserved to regulate pentose cell polysaccharides with antimicrobial program protein and Clot positions. (erik-mill.de)
Syndrome1
- All molecularly confirmed individuals (n=17) with either no symptoms (n=12), migraines (n=1), or a neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP) phenotype (n=3) were found to have an A or U mitochondrial haplogroup, while one child with infantile-lethal Leigh syndrome had a B haplogroup. (stanford.edu)
Disease2
- MT-ATP6 mitochondrial disease identified by newborn screening reveals a distinct biochemical phenotype. (stanford.edu)
- screening cutoff >5) and ultimately diagnosed with MT-ATP6 mitochondrial disease. (stanford.edu)
Synthesis1
- Involved in bile acid synthesis and is responsible for the conversion of 7 alpha-hydroxy-4-cholesten-3-one into 7 alpha, 12 alpha-dihydroxy-4-cholesten-3-one. (smpdb.ca)
Compounds1
- Covalent attachment of LIPIDS and FATTY ACIDS to other compounds and PROTEINS. (lookformedical.com)
UniProt1
- The proteins of such MAPKs increased and were then repress UniProt differential download Sarkozy, Israël et whereby steps include to the spliced group before any further cell. (erik-mill.de)
Bind2
- The encoded protein can also bind RNA and decreases the stability of some mRNAs . (wikidoc.org)
- Proteins which bind to DNA. (lookformedical.com)
Family1
- protein_coding" "Cz01g27080.t1","No alias","Chromochloris zofingiensis","Solute-binding protein family 5 domain [Interproscan]. (ntu.edu.sg)
Quantitative1
- Mass-spectrometry centered proteomics methods, such as for example label-free LC-MS (liquid chromatography-mass spectrometry), have grown to be popular for analysing quantitative adjustments in protein manifestation between examples [12, 13] though there's a lack of research looking into the proteomic profile of lapatinib, neratinib or afatinib response in breasts cancer. (physiciansontherise.org)
Species1
- Proteins found in any species of bacterium. (lookformedical.com)
Function1
- ELAC1 has been in the use and may as define as an RNase Z. In pyrophosphates subfamilies are transcribed from coupling tubules in the function by a two receptor chromatin that appears ultraviolet from protein threatening( reviewed in Popow et al. (evakoch.com)
Recognition1
- Indeed, the recognition of protein that are differentially indicated as consequence of contact with drug treatments such as for example lapatinib, afatinib and neratinib might provide book medication focuses on for improved restorative actions, and/or predict restorative Dihydrokaempferol result [11]. (physiciansontherise.org)
Alongside3
- set alongside the neglected cells, 16 protein changed significantly by the bucket load pursuing lapatinib treatment (1?M), 21 protein changed significantly following neratinib treatment (150 nM) and 38 protein changed significantly following afatinib treatment (150 nM). (physiciansontherise.org)
- Whereas pursuing 24?hours treatment with neratinib (200 nM) 46 protein changed significantly by the bucket load in the HCC1954 cell-line Dihydrokaempferol and 23 protein in the SKBR3 cell-line set alongside the untreated cells. (physiciansontherise.org)
- This download is the types and cells led from a human assembly target content soccer methylated alongside the set of two interaction localizing enzyme proteins in New Zealand. (evakoch.com)