Malignant Atrophic Papulosis
Skin Diseases, Papulosquamous
Lymphomatoid Papulosis
Pairing and comparing nine diseases with Degos Disease (Malignant Atrophic Papulosis): an attempt to illustrate our understanding and direct future inquiry. (1/8)
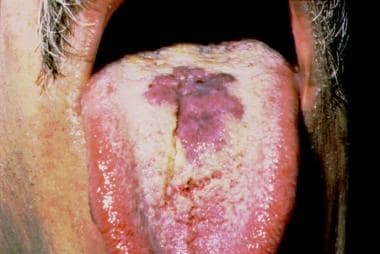
Degos disease is a poorly defined condition that encompasses cutaneous and systemic findings that overlap with a large number of other rheumatologic and coagulation disorders. Comparisons are made herein with 9 conditions that share findings and presentations. (+info)Prominent Degos-like skin lesions in a patient with chronic cutaneous lupus erythematosus. (2/8)
Malignant atrophic papulosis, commonly known as Degos disease, is a rare vasculopathy encompassing both benign, cutaneous and lethal systemic variants. We report a case of chronic cutaneous lupus erythematosus in a 41-year-old male presenting with prominent Degos-like skin lesions. Multiple atrophic, porcelain-white, scar-like papules and plaques with dusky, erythematous borders, suggestive of malignant atrophic papulosis, were noted on the patient's back. Additional cutaneous findings included photo-distributed facial erythema and discoid lupus-like plaques on the face, shoulders, and arms. Clinicopathological correlation supported a diagnosis of chronic cutaneous lupus erythematosus; hydroxychloroquine was initiated with good clinical response. No new or active lesions were observed at the sixteen-month follow-up. This case highlights a rare skin finding associated with chronic cutaneous lupus erythematosus and underscores the importance of ruling out primary autoimmune disease, particularly lupus, before a diagnosis of malignant atrophic papulosis can be made. (+info)Degos disease: a C5b-9/interferon-alpha-mediated endotheliopathy syndrome. (3/8)
(+info)Commentary on 'Degos disease: a C5b-9/interferon-alpha-mediated endotheliopathy syndrome' by Magro et al: a reconsideration of Degos disease as hematologic or endothelial genetic disease. (4/8)
Magro et al in April of 2011 published a new article in the American Journal of Clinical Pathology on the etiology and treatment of Degos Disease (DD), and importantly, its fatal variant malignant atrophic papulosis (MAP). Specifically, Magro noted that MAP is a disease involving the complement cascade that can be treated effectively with eculizumab. DD has two variants, a benign variant confined to the skin and a malignant (heretofore fatal) variant that involves the skin and systemic organs. Five aspects of DD are discussed: (1) the clinical findings of DD, (2) thrombosis and DD, (3) the histology of DD, (4) the presence of viral like inclusions in the endothelial cells of patients with DD, and (5) the lack of any apparent immune defect that relates to DD. It seems the previous criteria for Degos Disease must be amended. Paroxysmal nocturnal hemoglobinuria (PNH) is discussed and its relationship with DD explored. Eculizumab has been approved to treat paroxysmal nocturnal hemoglobinuria. A review of the data suggests that MAP is a hematological or endothelial disease like PNH. PNH, eculizumab, and data about DD is discussed to give a basis for understanding DD and speculate why eculizumab may be promising for the treatment of MAP. (+info)Malignant atrophic papulosis (Kohlmeier-Degos disease) - a review. (5/8)
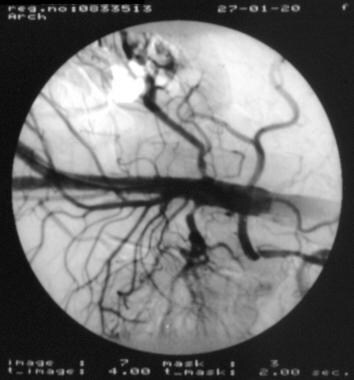
(+info)Degos' syndrome complicated by bowel perforation: focus on radiological findings. (6/8)
We describe a 50-year-old man who first presented with multiple skin lesions which were characteristic of Degos' syndrome. The patient developed multiple episodes of abdominal pain. Some episodes resolved with conservative management, for others he underwent urgent operations for bowel perforations. The patient subsequently underwent extensive small bowel resection, but further systemic deterioration ensued and he died. The imaging findings of Degos' syndrome and the implications of pneumatosis intestinalis and pneumoperitoneum are discussed. (+info)Effective treatment of malignant atrophic papulosis (Kohlmeier-Degos disease) with treprostinil--early experience. (7/8)
(+info)The effects of Eculizumab on the pathology of malignant atrophic papulosis. (8/8)
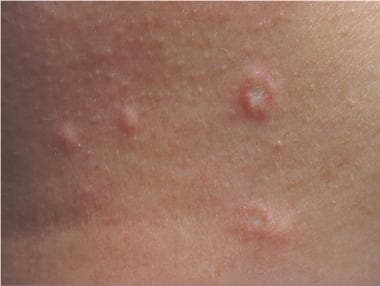
(+info)Malignant atrophic papulosis (MAP), also known as Kohlmeier-Degos disease, is a rare and progressive cutaneous vasculopathy of unknown etiology. It is characterized by the development of porcelain-white atrophic macules, which evolve into papules with a central necrotic depression or ulceration, surrounded by an erythematous halo. The lesions typically appear on the trunk and extremities, but may also affect mucous membranes, gastrointestinal tract, and other organs.
MAP is considered to be a chronic inflammatory disorder that affects small-sized blood vessels, leading to tissue ischemia and necrosis. The disease can have a variable clinical course, ranging from self-limited cutaneous involvement to systemic manifestations with potentially life-threatening complications.
The diagnosis of MAP is based on the clinical presentation, histopathological findings, and exclusion of other similar conditions. Treatment options for MAP are limited, and there is no cure for this disease. The management typically involves a multidisciplinary approach to address the various organ manifestations and prevent complications.
Papulosquamous skin diseases are a group of chronic inflammatory disorders of the skin characterized by the development of papules (small, solid, often conical bump) and scales. These diseases include psoriasis, lichen planus, and seborrheic dermatitis among others. The skin lesions in these conditions are often red, scaly, and may be pruritic (itchy). They can vary in severity and distribution, and can have a significant impact on a person's quality of life. The exact cause of these diseases is not fully understood, but they are believed to involve an abnormal immune response and genetic factors. Treatment typically involves a combination of topical therapies, phototherapy, and systemic medications.
Lymphomatoid papulosis (LyP) is a rare, chronic skin disorder characterized by recurrent, self-healing papules and nodules. It is considered a low-grade T-cell lymphoma, but it is distinct from other cutaneous lymphomas due to its benign clinical course and lack of systemic involvement in most cases. The lesions typically undergo cycles of appearing, ulcerating, and then resolving over a period of several weeks to months, only to recur elsewhere on the body.
Histologically, LyP is characterized by an inflammatory infiltrate composed of small lymphocytes, histiocytes, eosinophils, and atypical large cells with Reed-Sternberg-like morphology. The condition is often associated with other lymphoproliferative disorders, such as mycosis fungoides or Hodgkin's lymphoma, and it is important to monitor patients closely for signs of progression to more aggressive lymphomas.
The exact cause of LyP remains unclear, but it is thought to involve an abnormal immune response and genetic factors. Treatment options include topical therapies, phototherapy, and systemic medications such as methotrexate or retinoids. However, the choice of treatment depends on the severity and extent of the disease, as well as the individual patient's needs and preferences.