Hexosaminidase A
Hexosaminidase B
Hexosaminidases
beta-N-Acetylhexosaminidases
Tay-Sachs Disease
Gangliosidoses
Lipidoses
G(M2) Ganglioside
Sandhoff Disease
G(M2) Activator Protein
Acetylglucosaminidase
Chromosomes, Human, 4-5
Sphingolipid Activator Proteins
Hymecromone
Gangliosidoses, GM2
Isoenzymes
Saposins
Chromosomes, Human, 13-15
Electrophoresis, Starch Gel
Lysosomes
Isoelectric Focusing
Gangliosides
Hexosamines
Imines
Hybrid Cells
Chromatography, DEAE-Cellulose
Carbohydrate Epimerases
Amniotic Fluid
Immunoelectrophoresis
Heterozygote
Chromatography, Ion Exchange
Leukocytes
Prenatal Diagnosis
Fibroblasts
Liver
Substrate Specificity
Pyruvate Kinase
Glycoside Hydrolases
Mannose
Immunodiffusion
Placenta
Hydrogen-Ion Concentration
Chromatography, Gel
Cross Reactions
Immune Sera
Pedigree
Conversion of brain-specific complex type sugar chains by N-acetyl-beta-D-hexosaminidase B. (1/105)
The N-linked sugar chains, GlcNAcbeta1-2Manalpha1-6(GlcNAcbeta1-4)(Manalpha1++ +-3)Manbeta1-4GlcNAcb eta1-4(Fucalpha1-6)GlcNAc (BA-1) and GlcNAcbeta1-2Manalpha1-6(GlcNAcbeta1-4)(GlcNAcbeta1 -2Manalpha1-3)Manb eta1-4GlcNAcbeta1-4(Fucalpha1-6)GlcNAc (BA-2), were recently found to be linked to membrane proteins of mouse brain in a development-dependent manner [S. Nakakita, S. Natsuka, K. Ikenaka, and S. Hase, J. Biochem. 123, 1164-1168 (1998)]. The GlcNAc residue linked to the Manalpha1-3 branch of BA-2 is lacking in BA-1 and the removal of this GlcNAc residue is not part of the usual biosynthetic pathway for N-linked sugar chains, suggesting the existence of an N-acetyl-beta-D-hexosaminidase. Using pyridylaminated BA-2 (BA-2-PA) as a substrate the activity of this enzyme was found in all four subcellular fractions obtained. The activity was much greater in the cerebrum than in the cerebellum. To further identify the N-acetyl-beta-D-hexosaminidase, BA-1 and BA-2 in brain tissues of Hex gene-disrupted mutant mice were detected and quantified. PA-sugar chains were liberated from the cerebrum and cerebellum of the mutant mice by hydrazinolysis-N-acetylation followed by pyridylamination. PA-sugar chains were separated by anion-exchange HPLC, size-fractionation, and reversed-phase HPLC. Each peak was quantified by measuring the peaks at the elution positions of authentic BA-1-PA and BA-2-PA. BA-2-PA was detected in all the PA-sugar chain fractions prepared from Hexa, Hexb, and both Hexa and Hexb (double knockout) gene-disrupted mice, but BA-1 was not found in the fractions from Hexb gene-disrupted and double knockout mice. These results indicate that N-acetyl-beta-D-hexosaminidase B encoded by the Hexb gene hydrolyzed BA-2 to BA-1. (+info)Adenoviral gene therapy of the Tay-Sachs disease in hexosaminidase A-deficient knock-out mice. (2/105)
The severe neurodegenerative disorder, Tays-Sachs disease, is caused by a beta-hexosaminidase alpha-subunit deficiency which prevents the formation of lysosomal heterodimeric alpha-beta enzyme, hexosaminidase A (HexA). No treatment is available for this fatal disease; however, gene therapy could represent a therapeutic approach. We previously have constructed and characterized, in vitro, adenoviral and retroviral vectors coding for alpha- and beta-subunits of the human beta-hexosaminidases. Here, we have determined the in vivo strategy which leads to the highest HexA activity in the maximum number of tissues in hexA -deficient knock-out mice. We demonstrated that intravenous co-administration of adenoviral vectors coding for both alpha- and beta-subunits, resulting in preferential liver transduction, was essential to obtain the most successful results. Only the supply of both subunits allowed for HexA overexpression leading to massive secretion of the enzyme in serum, and full or partial enzymatic activity restoration in all peripheral tissues tested. The enzymatic correction was likely to be due to direct cellular transduction by adenoviral vectors and/or uptake of secreted HexA by different organs. These results confirmed that the liver was the preferential target organ to deliver a large amount of secreted proteins. In addition, the need to overexpress both subunits of heterodimeric proteins in order to obtain a high level of secretion in animals defective in only one subunit is emphasized. The endogenous non-defective subunit is otherwise limiting. (+info)Biochemical consequences of mutations causing the GM2 gangliosidoses. (3/105)
The hydrolysis of GM2-ganglioside is unusual in its requirements for the correct synthesis, processing, and ultimate combination of three gene products. Whereas two of these proteins are the alpha- (HEXA gene) and beta- (HEXB) subunits of beta-hexosaminidase A, the third is a small glycolipid transport protein, the GM2 activator protein (GM2A), which acts as a substrate specific co-factor for the enzyme. A deficiency of any one of these proteins leads to storage of the ganglioside, primarily in the lysosomes of neuronal cells, and one of the three forms of GM2-gangliosidosis, Tay-Sachs disease, Sandhoff disease or the AB-variant form. Studies of the biochemical impact of naturally occurring mutations associated with the GM2 gangliosidoses on mRNA splicing and stability, and on the intracellular transport and stability of the affected protein have provided some general insights into these complex cellular mechanisms. However, such studies have revealed little in the way of structure-function information on the proteins. It appears that the detrimental effect of most mutations is not specifically on functional elements of the protein, but rather on the proteins' overall folding and/or intracellular transport. The few exceptions to this generalization are missense mutations at two codons in HEXA, causing the unique biochemical phenotype known as the B1-variant, and one codon in both the HEXB and GM2A genes. Biochemical characterization of these mutations has led to the localization of functional residues and/or domains within each of the encoded proteins. (+info)Primer system for single cell detection of double mutation for Tay-Sachs disease. (4/105)
PURPOSE: Nearly 100% of infantile Tay-Sachs disease is produced by two mutations occurring in the alpha chain of the lysosomal enzyme beta-N-acetylhexosaminidase (HEXA) in the Ashkenazi Jewish population. Although others have described primer systems used to amplify both sites simultaneously, few discuss the allele dropout problems inherent in this test. Our goal was to construct a more robust test enabling stronger signal generation for single cell preimplantation genetic diagnosis and to investigate the occurrence of allele dropout. METHODS: New nested primers were designed to optimize detection of both major Tay-Sachs mutations. Four hundred fifty-seven single cells, including normal cells and those carrying mutations of either the 4bp insertion exon 11 or splice-site intron 12 defects, were used to screen a new primer system. RESULTS: Based on PCR amplified product analysis, total efficiency of amplification was 85.3%, (390/457). The allele dropout rate for the 4bp insertion mutation in exon 11 and splice-site mutation in intron 12 was 4.8% and 5.8%, respectively. CONCLUSIONS: Multiple mutation detection and analysis within the Tay-Sachs disease gene (HEXA) is possible using single cells for clinical preimplantation genetic diagnosis. Alternative PCR primers and conditions offer various methods for developing systems compatible to specific program requirements. (+info)Effect of collection, transport, processing and storage of blood specimens on the activity of lysosomal enzymes in plasma and leukocytes. (5/105)
This study was designed to evaluate the effect of different conditions of collection, transport and storage on the quality of blood samples from normal individuals in terms of the activity of the enzymes ss-glucuronidase, total hexosaminidase, hexosaminidase A, arylsulfatase A and ss-galactosidase. The enzyme activities were not affected by the different materials used for collection (plastic syringes or vacuum glass tubes). In the evaluation of different heparin concentrations (10% heparin, 5% heparin, and heparinized syringe) in the syringes, it was observed that higher doses resulted in an increase of at least 1-fold in the activities of ss-galactosidase, total hexosaminidase and hexosaminidase A in leukocytes, and ss-glucuronidase in plasma. When the effects of time and means of transportation were studied, samples that had been kept at room temperature showed higher deterioration with time (72 and 96 h) before processing, and in this case it was impossible to isolate leukocytes from most samples. Comparison of heparin and acid citrate-dextrose (ACD) as anticoagulants revealed that ss-glucuronidase and hexosaminidase activities in plasma reached levels near the lower normal limits when ACD was used. In conclusion, we observed that heparin should be used as the preferable anticoagulant when measuring these lysosomal enzyme activities, and we recommend that, when transport time is more than 24 h, samples should be shipped by air in a styrofoam box containing wet ice. (+info)Degradation of membrane-bound ganglioside GM2 by beta -hexosaminidase A. Stimulation by GM2 activator protein and lysosomal lipids. (6/105)
According to a recent hypothesis, glycosphingolipids originating from the plasma membrane are degraded in the acidic compartments of the cell as components of intraendosomal and intralysosomal vesicles and structures. Since most previous in vitro investigations used micellar ganglioside GM2 as substrate, we studied the degradation of membrane-bound ganglioside GM2 by water-soluble beta-hexosaminidase A in the presence of the GM2 activator protein in a detergent-free, liposomal assay system. Our results show that anionic lipids such as the lysosomal components bis(monoacylglycero)phosphate or phosphatidylinositol stimulate the degradation of GM2 by beta-hexosaminidase A up to 180-fold in the presence of GM2 activator protein. In contrast, the degradation rate of GM2 incorporated into liposomes composed of neutral lysosomal lipids such as dolichol, cholesterol, or phosphatidylcholine was significantly lower than in negatively charged liposomes. This demonstrates that both, the GM2 activator protein and anionic lysosomal phospholipids, are needed to achieve a significant degradation of membrane-bound GM2 under physiological conditions. The interaction of GM2 activator protein with immobilized membranes was studied with surface plasmon resonance spectroscopy at an acidic pH value as it occurs in the lysosomes. Increasing the concentration of bis(monoacylglycero)phosphate in immobilized liposomes led to a significant drop of the resonance signal in the presence of GM2 activator protein. This suggests that in the presence of bis(monoacylglycero)phosphate, which has been shown to occur in inner membranes of the acidic compartment, GM2 activator protein is able to solubilize lipids from the surface of immobilized membrane structures. (+info)Physiological substrates for human lysosomal beta -hexosaminidase S. (7/105)
Human lysosomal beta-hexosaminidases remove terminal beta-glycosidically bound N-acetylhexosamine residues from a number of glycoconjugates. Three different isozymes composed of two noncovalently linked subunits alpha and beta exist: Hex A (alphabeta), Hex B (betabeta), and Hex S (alphaalpha). While the role of Hex A and B for the degradation of several anionic and neutral glycoconjugates has been well established, the physiological significance of labile Hex S has remained unclear. However, the striking accumulation of anionic oligosaccharides in double knockout mice totally deficient in hexosaminidase activity but not in mice expressing Hex S (Sango, K., McDonald, M. P., Crawley, J. N., Mack, M. L., Tifft, C.J., Skop, E., Starr, C. M., Hoffmann, A., Sandhoff, K., Suzuki, K., and Proia, R. L., (1996) Nat. Genet. 14, 348-352) prompted us to reinvestigate the substrate specificity of Hex S. To identify physiological substrates of Hex S, anionic and neutral oligosaccharides excreted in the urine of the double knockout mice were isolated and analyzed. Using ESI-MS/MS and glycosidase digestion the anionic glycans were identified as products of incomplete dermatan sulfate degradation whereas the neutral storage oligosaccharides were found to be fragments of N-glycan degradation. In vitro, recombinant Hex S was highly active on water-soluble and amphiphilic glycoconjugates including artificial substrates, sulfated GAG fragments, and the sulfated glycosphingolipid SM2. Hydrolysis of membrane-bound SM2 by the recombinant Hex S was synergistically stimulated by the GM2 activator protein and the lysosomal anionic phospholipid bis(monoacylglycero)phosphate. (+info)A single site in human beta-hexosaminidase A binds both 6-sulfate-groups on hexosamines and the sialic acid moiety of GM2 ganglioside. (8/105)
Human beta-hexosaminidase A (Hex A) (alphabeta) is composed of two subunits whose primary structures are approximately 60% identical. Deficiency of either subunit results in severe neurological disease due to the storage of GM2 ganglioside; Tay-Sachs disease, alpha deficiency, and Sandhoff disease, beta deficiency. Whereas both subunits contain active sites only the alpha-site can efficiently bind negatively charged 6-sulfated hexosamine substrates and GM2 ganglioside. We have recently identified the alphaArg(424) as playing a critical role in the binding of 6-sulfate-containing substrates, and betaAsp(452) as actively inhibiting their binding. To determine if these same residues affect the binding of the sialic acid moiety of GM2 ganglioside, an alphaArg(424)Gln form of Hex A was expressed and its kinetics analyzed using the GM2 activator protein:[3H]-GM2 ganglioside complex as a substrate. The mutant showed a approximately 3-fold increase in its K(m) for the complex. Next a form of Hex B (betabeta) containing a double mutation, betaAspLeu(453)AsnArg (duplicating the alpha-aligning sequences), was expressed. As compared to the wild type (WT), the mutant exhibited a >30-fold increase in its ability to hydrolyze a 6-sulfated substrate and was now able to hydrolyze GM2 ganglioside when the GM2 activator protein was replaced by sodium taurocholate. Thus, this alpha-site is critical for binding both types of negatively charge substrates. (+info)Hexosaminidase A is an enzyme that is responsible for breaking down certain complex molecules in the body, specifically gangliosides. This enzyme is composed of two subunits, alpha and beta, which are encoded by the genes HEXA and HEXB, respectively.
Deficiency or mutation in the HEXA gene can lead to a genetic disorder called Tay-Sachs disease, which is characterized by an accumulation of gangliosides in the nerve cells, leading to progressive neurological degeneration. The function of hexosaminidase A is to break down these gangliosides into simpler molecules that can be eliminated from the body. Without sufficient levels of this enzyme, the gangliosides build up and cause damage to the nervous system.
Hexosaminidase B is a type of enzyme that is involved in the breakdown of complex lipids called gangliosides in the body. These enzymes are found in lysosomes, which are structures inside cells that break down and recycle various materials.
Hexosaminidase B specifically helps to break down a particular type of ganglioside called GM2 ganglioside, which is abundant in the nervous system. Mutations in the gene that provides instructions for making this enzyme can lead to a condition called Tay-Sachs disease, which is characterized by the accumulation of GM2 gangliosides in the nerve cells, leading to progressive neurological deterioration.
In summary, Hexosaminidase B is an essential enzyme for breaking down certain types of lipids in the body, and its deficiency can lead to serious health consequences.
Hexosaminidases are a group of enzymes that play a crucial role in the breakdown of complex carbohydrates, specifically glycoproteins and glycolipids, in the human body. These enzymes are responsible for cleaving the terminal N-acetyl-D-glucosamine (GlcNAc) residues from these molecules during the process of glycosidase digestion.
There are several types of hexosaminidases, including Hexosaminidase A and Hexosaminidase B, which are encoded by different genes and have distinct functions. Deficiencies in these enzymes can lead to serious genetic disorders, such as Tay-Sachs disease and Sandhoff disease, respectively. These conditions are characterized by the accumulation of undigested glycolipids and glycoproteins in various tissues, leading to progressive neurological deterioration and other symptoms.
Beta-N-Acetylhexosaminidases are a group of enzymes that play a role in the breakdown and recycling of complex carbohydrates in the body. Specifically, they help to break down gangliosides, which are a type of molecule found in cell membranes.
There are several different isoforms of beta-N-Acetylhexosaminidases, including A, B, and S. These isoforms are formed by different combinations of subunits, which can affect their activity and substrate specificity.
Mutations in the genes that encode for these enzymes can lead to a variety of genetic disorders, including Tay-Sachs disease and Sandhoff disease. These conditions are characterized by an accumulation of gangliosides in the brain, which can cause progressive neurological deterioration and death.
Treatment for these conditions typically involves managing symptoms and providing supportive care, as there is currently no cure. Enzyme replacement therapy has been explored as a potential treatment option, but its effectiveness varies depending on the specific disorder and the age of the patient.
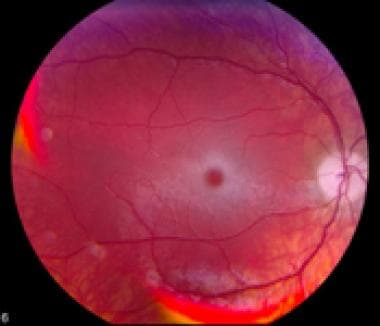
Tay-Sachs Disease is a rare, inherited autosomal recessive disorder that affects the nervous system's functioning. It results from the deficiency of an enzyme called hexosaminidase A (Hex-A), which is necessary for breaking down gangliosides, a type of fatty substance found in nerve cells. When Hex-A is absent or insufficient, gangliosides accumulate abnormally in the nerve cells, leading to their progressive destruction and severe neurological deterioration.
The classic infantile form of Tay-Sachs Disease manifests within the first six months of life with symptoms such as loss of motor skills, seizures, paralysis, dementia, blindness, and eventually death, usually by age four. Late-onset forms of the disease also exist, which may present in childhood or adulthood with milder symptoms.
Tay-Sachs Disease is more prevalent among individuals of Ashkenazi Jewish, French Canadian, and Cajun descent. Genetic counseling and prenatal testing are recommended for couples at risk of passing on the disease.
Gangliosidoses are a group of inherited metabolic disorders caused by the accumulation of certain complex lipids called gangliosides in the brain and nervous system. This buildup is due to a deficiency of specific enzymes needed to break down these substances. The three main types of gangliosidoses are:
1. Type 1 - Infantile Neurovisceral or Tay-Sachs Disease: Characterized by the absence of the enzyme hexosaminidase A, leading to severe neurological symptoms such as muscle weakness, blindness, and developmental delay in early infancy, with rapid progression and death usually occurring before age 4.
2. Type 2 - Juvenile or Subacute GM1 Gangliosidosis: Caused by a deficiency of the enzyme beta-galactosidase, resulting in progressive neurological symptoms such as motor and cognitive decline, beginning between ages 6 months and 2 years. Affected individuals may survive into adolescence or early adulthood.
3. Type 3 - Adult or Chronic GM1 Gangliosidosis: Characterized by a deficiency of beta-galactosidase, leading to milder neurological symptoms that appear in late childhood, adolescence, or even adulthood. The progression is slower compared to the other types, and life expectancy varies widely.
Gangliosidoses are autosomal recessive disorders, meaning an individual must inherit two copies of the defective gene (one from each parent) to develop the condition.
Lipidoses are a group of genetic disorders characterized by abnormal accumulation of lipids (fats or fat-like substances) in various tissues and cells of the body due to defects in lipid metabolism. These disorders include conditions such as Gaucher's disease, Tay-Sachs disease, Niemann-Pick disease, Fabry disease, and Wolman disease, among others. The accumulation of lipids can lead to progressive damage in multiple organs, resulting in a range of symptoms and health complications. Early diagnosis and management are essential for improving the quality of life and prognosis of affected individuals.
Sandhoff disease is a rare inherited disorder that affects the nervous system. It's a type of GM2 gangliosidosis, which is a group of conditions characterized by the body's inability to break down certain fats (lipids) called gangliosides.
In Sandhoff disease, deficiencies in the enzymes hexosaminidase A and B lead to an accumulation of GM2 ganglioside in various cells, particularly in nerve cells of the brain. This accumulation results in progressive damage to the nervous system.
The symptoms of Sandhoff disease typically appear between 6 months and 2 years of age and can include developmental delay, seizures, an exaggerated startle response, muscle weakness, loss of motor skills, and vision and hearing loss. The condition is often fatal by around age 3. It's caused by mutations in the HEXB gene, and it's inherited in an autosomal recessive manner, meaning an individual must inherit two copies of the mutated gene (one from each parent) to develop the disease.
Acetylglucosaminidase (ACG) is an enzyme that catalyzes the hydrolysis of N-acetyl-beta-D-glucosaminides, which are found in glycoproteins and glycolipids. This enzyme plays a crucial role in the degradation and recycling of these complex carbohydrates within the body.
Deficiency or malfunction of Acetylglucosaminidase can lead to various genetic disorders, such as mucolipidosis II (I-cell disease) and mucolipidosis III (pseudo-Hurler polydystrophy), which are characterized by the accumulation of glycoproteins and glycolipids in lysosomes, resulting in cellular dysfunction and progressive damage to multiple organs.
Chromosomes are thread-like structures located in the nucleus of cells that carry genetic information in the form of genes. In humans, there are 23 pairs of chromosomes for a total of 46 chromosomes in every cell of the body, except for the sperm and egg cells which contain only 23 chromosomes.
Human chromosomes are numbered from 1 to 22, based on their size, with chromosome 1 being the largest and chromosome 22 being the smallest. The last two pairs of human chromosomes are known as the sex chromosomes because they determine a person's biological sex. These are labeled X and Y, with females having two X chromosomes (44+XX) and males having one X and one Y chromosome (44+XY).
Therefore, "Chromosomes, Human, 4-5" refers to the fourth and fifth pairs of human chromosomes. Chromosome 4 is an acrocentric chromosome, meaning its centromere is located near one end, resulting in a short arm (p) and a long arm (q). It contains about 190 million base pairs and encodes approximately 700 genes.
Chromosome 5 is a submetacentric chromosome, with the centromere located closer to the middle, creating two arms of roughly equal length: the short arm (p) and the long arm (q). It contains about 182 million base pairs and encodes approximately 900 genes.
Both chromosomes 4 and 5 are involved in various genetic disorders when abnormalities occur, such as deletions, duplications, or translocations. Some of the well-known genetic conditions associated with these chromosomes include:
* Chromosome 4: Wolf-Hirschhorn syndrome (deletion), Charcot-Marie-Tooth disease type 1A (duplication)
* Chromosome 5: Cri du Chat syndrome (deletion), Duchenne muscular dystrophy (deletion or mutation in a gene located on chromosome 5)
Sphingolipid activator proteins (SAPs), also known as saposins, are a group of small proteins that play a crucial role in the metabolism of sphingolipids, a class of lipids found in cell membranes. These proteins are produced by the cleavage of a precursor protein called prosaposin.
SAPs facilitate the hydrolysis of sphingolipids by activating specific lysosomal hydrolases, enzymes that break down these lipids into simpler molecules. Each SAP has a unique structure and function, and they are named SapA, SapB, SapC, and SapD.
SapA and SapB activate the enzyme glucocerebrosidase, which breaks down glucosylceramide into glucose and ceramide. SapC activates the enzyme galactocerebrosidase, which breaks down galactosylceramide into galactose and ceramide. SapD has multiple functions, including activating the enzyme acid sphingomyelinase, which breaks down sphingomyelin into ceramide and phosphorylcholine.
Deficiencies in SAPs can lead to lysosomal storage disorders, such as Gaucher disease (caused by a deficiency in glucocerebrosidase) and Krabbe disease (caused by a deficiency in galactocerebrosidase). These disorders are characterized by the accumulation of undigested sphingolipids in various tissues, leading to cell dysfunction and tissue damage.
Hymecromone, also known as fladrafinic acid, is an antispasmodic and anti-inflammatory medication that is primarily used to treat biliary tract spasms and cholestasis (a condition in which the flow of bile from the liver is reduced or blocked). It works by relaxing the smooth muscles in the bile ducts, thereby reducing spasms and allowing for improved bile flow. Hymecromone has also been studied for its potential use in treating other conditions such as liver disease and cancer, but more research is needed to confirm its effectiveness in these areas. It's important to note that this medication should only be used under the supervision of a healthcare professional, as it can have side effects and interactions with other medications.
GM2 gangliosidoses are a group of inherited metabolic disorders caused by the accumulation of harmful amounts of GM2 gangliosides in the body's cells, particularly in the nerve cells of the brain. There are three main types of GM2 gangliosidoses: Tay-Sachs disease, Sandhoff disease, and AB variant of GM2 gangliosidosis. These conditions are characterized by progressive neurological degeneration, which can lead to severe physical and mental disabilities, and ultimately death in childhood or early adulthood.
The underlying cause of GM2 gangliosides is a deficiency in the enzyme hexosaminidase A (Tay-Sachs and AB variant) or both hexosaminidase A and B (Sandhoff disease), which are responsible for breaking down GM2 gangliosides. Without sufficient enzyme activity, GM2 gangliosides accumulate in the lysosomes of cells, leading to cell dysfunction and death.
Symptoms of GM2 gangliosidoses can vary depending on the specific type and severity of the disorder, but often include developmental delay, muscle weakness, loss of motor skills, seizures, blindness, and dementia. There is currently no cure for GM2 gangliosidoses, and treatment is focused on managing symptoms and improving quality of life.
Isoenzymes, also known as isoforms, are multiple forms of an enzyme that catalyze the same chemical reaction but differ in their amino acid sequence, structure, and/or kinetic properties. They are encoded by different genes or alternative splicing of the same gene. Isoenzymes can be found in various tissues and organs, and they play a crucial role in biological processes such as metabolism, detoxification, and cell signaling. Measurement of isoenzyme levels in body fluids (such as blood) can provide valuable diagnostic information for certain medical conditions, including tissue damage, inflammation, and various diseases.
Saposins are a group of naturally occurring lipid-binding proteins that play an essential role in the metabolism of lipids within cells. They are named after a skin disease called "Niemann-Pick disease," where defects in saposin function lead to an accumulation of lipids in various tissues, including the brain.
There are four types of saposins (SapA, SapB, SapC, and SapD) that are produced by the cleavage of a larger precursor protein called prosaposin. These proteins help to facilitate the breakdown of lipids in lysosomes, which are specialized organelles within cells that break down and recycle various materials.
Saposins play an important role in activating certain enzymes that are involved in breaking down lipids, such as sphingolipids and gangliosides. They do this by binding to these enzymes and presenting them with their lipid substrates in a way that allows the enzymes to efficiently break them down.
Defects in saposin function can lead to a variety of diseases, including Niemann-Pick disease, Gaucher disease, and Krabbe disease, which are characterized by an accumulation of lipids in various tissues and neurological symptoms.
Human chromosomes 13-15 are part of a set of 23 pairs of chromosomes found in the cells of the human body. Chromosomes are thread-like structures that contain genetic material, or DNA, that is inherited from each parent. They are responsible for the development and function of all the body's organs and systems.
Chromosome 13 is a medium-sized chromosome and contains an estimated 114 million base pairs of DNA. It is associated with several genetic disorders, including cri du chat syndrome, which is caused by a deletion on the short arm of the chromosome. Chromosome 13 also contains several important genes, such as those involved in the production of enzymes and proteins that help regulate growth and development.
Chromosome 14 is a medium-sized chromosome and contains an estimated 107 million base pairs of DNA. It is known to contain many genes that are important for the normal functioning of the brain and nervous system, as well as genes involved in the production of immune system proteins. Chromosome 14 is also associated with a number of genetic disorders, including Wolf-Hirschhorn syndrome, which is caused by a deletion on the short arm of the chromosome.
Chromosome 15 is a medium-sized chromosome and contains an estimated 102 million base pairs of DNA. It is associated with several genetic disorders, including Prader-Willi syndrome and Angelman syndrome, which are caused by abnormalities in the expression of genes on the chromosome. Chromosome 15 also contains important genes involved in the regulation of growth and development, as well as genes that play a role in the production of neurotransmitters, the chemical messengers of the brain.
It is worth noting that while chromosomes 13-15 are important for normal human development and function, abnormalities in these chromosomes can lead to a variety of genetic disorders and developmental issues.
Electrophoresis, starch gel is a type of electrophoretic technique used in laboratory settings for the separation and analysis of large biomolecules such as DNA, RNA, and proteins. In this method, a gel made from cooked starch is used as the supporting matrix for the molecules being separated.
The sample containing the mixture of biomolecules is loaded onto the gel and an electric field is applied, causing the negatively charged molecules to migrate towards the positive electrode. The starch gel acts as a molecular sieve, with smaller molecules moving more quickly through the gel than larger ones. This results in the separation of the mixture into individual components based on their size and charge.
Once the separation is complete, the gel can be stained to visualize the separated bands. Different staining techniques are used depending on the type of biomolecule being analyzed. For example, proteins can be stained with dyes such as Coomassie Brilliant Blue or silver nitrate, while nucleic acids can be stained with dyes such as ethidium bromide.
Starch gel electrophoresis is a relatively simple and inexpensive technique that has been widely used in molecular biology research and diagnostic applications. However, it has largely been replaced by other electrophoretic techniques, such as polyacrylamide gel electrophoresis (PAGE), which offer higher resolution and can be automated for high-throughput analysis.
Acetylglucosamine is a type of sugar that is commonly found in the body and plays a crucial role in various biological processes. It is a key component of glycoproteins and proteoglycans, which are complex molecules made up of protein and carbohydrate components.
More specifically, acetylglucosamine is an amino sugar that is formed by the addition of an acetyl group to glucosamine. It can be further modified in the body through a process called acetylation, which involves the addition of additional acetyl groups.
Acetylglucosamine is important for maintaining the structure and function of various tissues in the body, including cartilage, tendons, and ligaments. It also plays a role in the immune system and has been studied as a potential therapeutic target for various diseases, including cancer and inflammatory conditions.
In summary, acetylglucosamine is a type of sugar that is involved in many important biological processes in the body, and has potential therapeutic applications in various diseases.
Lysosomes are membrane-bound organelles found in the cytoplasm of eukaryotic cells. They are responsible for breaking down and recycling various materials, such as waste products, foreign substances, and damaged cellular components, through a process called autophagy or phagocytosis. Lysosomes contain hydrolytic enzymes that can break down biomolecules like proteins, nucleic acids, lipids, and carbohydrates into their basic building blocks, which can then be reused by the cell. They play a crucial role in maintaining cellular homeostasis and are often referred to as the "garbage disposal system" of the cell.
Heterozygote detection is a method used in genetics to identify individuals who carry one normal and one mutated copy of a gene. These individuals are known as heterozygotes and they do not typically show symptoms of the genetic disorder associated with the mutation, but they can pass the mutated gene on to their offspring, who may then be affected.
Heterozygote detection is often used in genetic counseling and screening programs for recessive disorders such as cystic fibrosis or sickle cell anemia. By identifying heterozygotes, individuals can be informed of their carrier status and the potential risks to their offspring. This information can help them make informed decisions about family planning and reproductive options.
Various methods can be used for heterozygote detection, including polymerase chain reaction (PCR) based tests, DNA sequencing, and genetic linkage analysis. The choice of method depends on the specific gene or mutation being tested, as well as the availability and cost of the testing technology.
Isoelectric focusing (IEF) is a technique used in electrophoresis, which is a method for separating proteins or other molecules based on their electrical charges. In IEF, a mixture of ampholytes (molecules that can carry both positive and negative charges) is used to create a pH gradient within a gel matrix. When an electric field is applied, the proteins or molecules migrate through the gel until they reach the point in the gradient where their net charge is zero, known as their isoelectric point (pI). At this point, they focus into a sharp band and stop moving, resulting in a highly resolved separation of the different components based on their pI. This technique is widely used in protein research for applications such as protein identification, characterization, and purification.
Gangliosides are a type of complex lipid molecule known as sialic acid-containing glycosphingolipids. They are predominantly found in the outer leaflet of the cell membrane, particularly in the nervous system. Gangliosides play crucial roles in various biological processes, including cell recognition, signal transduction, and cell adhesion. They are especially abundant in the ganglia (nerve cell clusters) of the peripheral and central nervous systems, hence their name.
Gangliosides consist of a hydrophobic ceramide portion and a hydrophilic oligosaccharide chain that contains one or more sialic acid residues. The composition and structure of these oligosaccharide chains can vary significantly among different gangliosides, leading to the classification of various subtypes, such as GM1, GD1a, GD1b, GT1b, and GQ1b.
Abnormalities in ganglioside metabolism or expression have been implicated in several neurological disorders, including Parkinson's disease, Alzheimer's disease, and various lysosomal storage diseases like Tay-Sachs and Gaucher's diseases. Additionally, certain bacterial toxins, such as botulinum neurotoxin and tetanus toxin, target gangliosides to gain entry into neuronal cells, causing their toxic effects.
Hexosamines are amino sugars that are formed by the substitution of an amino group (-NH2) for a hydroxyl group (-OH) in a hexose sugar. The most common hexosamine is N-acetylglucosamine (GlcNAc), which is derived from glucose. Other hexosamines include galactosamine, mannosamine, and fucosamine.
Hexosamines play important roles in various biological processes, including the formation of glycosaminoglycans, proteoglycans, and glycoproteins. These molecules are involved in many cellular functions, such as cell signaling, cell adhesion, and protein folding. Abnormalities in hexosamine metabolism have been implicated in several diseases, including diabetes, cancer, and neurodegenerative disorders.
In the field of organic chemistry, imines are a class of compounds that contain a functional group with the general structure =CR-NR', where C=R and R' can be either alkyl or aryl groups. Imines are also commonly referred to as Schiff bases. They are formed by the condensation of an aldehyde or ketone with a primary amine, resulting in the loss of a molecule of water.
It is important to note that imines do not have a direct medical application, but they can be used as intermediates in the synthesis of various pharmaceuticals and bioactive compounds. Additionally, some imines have been found to exhibit biological activity, such as antimicrobial or anticancer properties. However, these are areas of ongoing research and development.
In a medical context, "hot temperature" is not a standard medical term with a specific definition. However, it is often used in relation to fever, which is a common symptom of illness. A fever is typically defined as a body temperature that is higher than normal, usually above 38°C (100.4°F) for adults and above 37.5-38°C (99.5-101.3°F) for children, depending on the source.
Therefore, when a medical professional talks about "hot temperature," they may be referring to a body temperature that is higher than normal due to fever or other causes. It's important to note that a high environmental temperature can also contribute to an elevated body temperature, so it's essential to consider both the body temperature and the environmental temperature when assessing a patient's condition.
I'm sorry for any confusion, but "hybrid cells" is not a standard medical term with a widely accepted or specific definition in the field of medicine. The term "hybrid" is used in various scientific and medical contexts to describe combinations or mixtures of different elements, such as hybridoma cells (a type of fusion cell used in research, created by combining a B cell and a tumor cell) or hybridization (in genetics, the process of combining DNA from two different sources).
Without more specific context, it's difficult to provide an accurate medical definition for "hybrid cells." If you could provide more information about the context in which this term was used, I would be happy to help you further!
DEAE-cellulose chromatography is a method of purification and separation of biological molecules such as proteins, nucleic acids, and enzymes. DEAE stands for diethylaminoethyl, which is a type of charged functional group that is covalently bound to cellulose, creating a matrix with positive charges.
In this method, the mixture of biological molecules is applied to a column packed with DEAE-cellulose. The positively charged DEAE groups attract and bind negatively charged molecules in the mixture, such as nucleic acids and proteins, while allowing uncharged or neutrally charged molecules to pass through.
By adjusting the pH, ionic strength, or concentration of salt in the buffer solution used to elute the bound molecules from the column, it is possible to selectively elute specific molecules based on their charge and binding affinity to the DEAE-cellulose matrix. This makes DEAE-cellulose chromatography a powerful tool for purifying and separating biological molecules with high resolution and efficiency.
Carbohydrate epimerases are a group of enzymes that catalyze the interconversion of specific stereoisomers (epimers) of carbohydrates by the reversible oxidation and reduction of carbon atoms, usually at the fourth or fifth position. These enzymes play important roles in the biosynthesis and modification of various carbohydrate-containing molecules, such as glycoproteins, proteoglycans, and glycolipids, which are involved in numerous biological processes including cell recognition, signaling, and adhesion.
The reaction catalyzed by carbohydrate epimerases involves the transfer of a hydrogen atom and a proton between two adjacent carbon atoms, leading to the formation of new stereochemical configurations at these positions. This process can result in the conversion of one epimer into another, thereby expanding the structural diversity of carbohydrates and their derivatives.
Carbohydrate epimerases are classified based on the type of substrate they act upon and the specific stereochemical changes they induce. Some examples include UDP-glucose 4-epimerase, which interconverts UDP-glucose and UDP-galactose; UDP-N-acetylglucosamine 2-epimerase, which converts UDP-N-acetylglucosamine to UDP-N-acetylmannosamine; and GDP-fucose synthase, which catalyzes the conversion of GDP-mannose to GDP-fucose.
Understanding the function and regulation of carbohydrate epimerases is crucial for elucidating their roles in various biological processes and developing strategies for targeting them in therapeutic interventions.
Amniotic fluid is a clear, slightly yellowish liquid that surrounds and protects the developing baby in the uterus. It is enclosed within the amniotic sac, which is a thin-walled sac that forms around the embryo during early pregnancy. The fluid is composed of fetal urine, lung secretions, and fluids that cross over from the mother's bloodstream through the placenta.
Amniotic fluid plays several important roles in pregnancy:
1. It provides a shock-absorbing cushion for the developing baby, protecting it from injury caused by movement or external forces.
2. It helps to maintain a constant temperature around the fetus, keeping it warm and comfortable.
3. It allows the developing baby to move freely within the uterus, promoting normal growth and development of the muscles and bones.
4. It provides a source of nutrients and hydration for the fetus, helping to support its growth and development.
5. It helps to prevent infection by providing a barrier between the fetus and the outside world.
Throughout pregnancy, the volume of amniotic fluid increases as the fetus grows. The amount of fluid typically peaks around 34-36 weeks of gestation, after which it begins to gradually decrease. Abnormalities in the volume of amniotic fluid can indicate problems with the developing baby or the pregnancy itself, and may require medical intervention.
Immunoelectrophoresis (IEP) is a laboratory technique used in the field of clinical pathology and immunology. It is a method for separating and identifying proteins, particularly immunoglobulins or antibodies, in a sample. This technique combines the principles of electrophoresis, which separates proteins based on their electric charge and size, with immunological reactions, which detect specific proteins using antigen-antibody interactions.
In IEP, a protein sample is first separated by electrophoresis in an agarose or agar gel matrix on a glass slide or in a test tube. After separation, an antibody specific to the protein of interest is layered on top of the gel and allowed to diffuse towards the separated proteins. This creates a reaction between the antigen (protein) and the antibody, forming a visible precipitate at the point where they meet. The precipitate line's position and intensity can then be analyzed to identify and quantify the protein of interest.
Immunoelectrophoresis is particularly useful in diagnosing various medical conditions, such as immunodeficiency disorders, monoclonal gammopathies (like multiple myeloma), and other plasma cell dyscrasias. It can help detect abnormal protein patterns, quantify specific immunoglobulins, and identify the presence of M-proteins or Bence Jones proteins, which are indicative of monoclonal gammopathies.
A heterozygote is an individual who has inherited two different alleles (versions) of a particular gene, one from each parent. This means that the individual's genotype for that gene contains both a dominant and a recessive allele. The dominant allele will be expressed phenotypically (outwardly visible), while the recessive allele may or may not have any effect on the individual's observable traits, depending on the specific gene and its function. Heterozygotes are often represented as 'Aa', where 'A' is the dominant allele and 'a' is the recessive allele.
Consanguinity is a medical and genetic term that refers to the degree of genetic relationship between two individuals who share common ancestors. Consanguineous relationships exist when people are related by blood, through a common ancestor or siblings who have children together. The closer the relationship between the two individuals, the higher the degree of consanguinity.
The degree of consanguinity is typically expressed as a percentage or fraction, with higher values indicating a closer genetic relationship. For example, first-degree relatives, such as parents and children or full siblings, share approximately 50% of their genes and have a consanguinity coefficient of 0.25 (or 25%).
Consanguinity can increase the risk of certain genetic disorders and birth defects in offspring due to the increased likelihood of sharing harmful recessive genes. The risks depend on the degree of consanguinity, with closer relationships carrying higher risks. It is important for individuals who are planning to have children and have a history of consanguinity to consider genetic counseling and testing to assess their risk of passing on genetic disorders.
Ion exchange chromatography is a type of chromatography technique used to separate and analyze charged molecules (ions) based on their ability to exchange bound ions in a solid resin or gel with ions of similar charge in the mobile phase. The stationary phase, often called an ion exchanger, contains fixed ated functional groups that can attract counter-ions of opposite charge from the sample mixture.
In this technique, the sample is loaded onto an ion exchange column containing the charged resin or gel. As the sample moves through the column, ions in the sample compete for binding sites on the stationary phase with ions already present in the column. The ions that bind most strongly to the stationary phase will elute (come off) slower than those that bind more weakly.
Ion exchange chromatography can be performed using either cation exchangers, which exchange positive ions (cations), or anion exchangers, which exchange negative ions (anions). The pH and ionic strength of the mobile phase can be adjusted to control the binding and elution of specific ions.
Ion exchange chromatography is widely used in various applications such as water treatment, protein purification, and chemical analysis.
Leukocytes, also known as white blood cells (WBCs), are a crucial component of the human immune system. They are responsible for protecting the body against infections and foreign substances. Leukocytes are produced in the bone marrow and circulate throughout the body in the bloodstream and lymphatic system.
There are several types of leukocytes, including:
1. Neutrophils - These are the most abundant type of leukocyte and are primarily responsible for fighting bacterial infections. They contain enzymes that can destroy bacteria.
2. Lymphocytes - These are responsible for producing antibodies and destroying virus-infected cells, as well as cancer cells. There are two main types of lymphocytes: B-lymphocytes and T-lymphocytes.
3. Monocytes - These are the largest type of leukocyte and help to break down and remove dead or damaged tissues, as well as microorganisms.
4. Eosinophils - These play a role in fighting parasitic infections and are also involved in allergic reactions and inflammation.
5. Basophils - These release histamine and other chemicals that cause inflammation in response to allergens or irritants.
An abnormal increase or decrease in the number of leukocytes can indicate an underlying medical condition, such as an infection, inflammation, or a blood disorder.
Prenatal diagnosis is the medical testing of fetuses, embryos, or pregnant women to detect the presence or absence of certain genetic disorders or birth defects. These tests can be performed through various methods such as chorionic villus sampling (CVS), amniocentesis, or ultrasound. The goal of prenatal diagnosis is to provide early information about the health of the fetus so that parents and healthcare providers can make informed decisions about pregnancy management and newborn care. It allows for early intervention, treatment, or planning for the child's needs after birth.
Fibroblasts are specialized cells that play a critical role in the body's immune response and wound healing process. They are responsible for producing and maintaining the extracellular matrix (ECM), which is the non-cellular component present within all tissues and organs, providing structural support and biochemical signals for surrounding cells.
Fibroblasts produce various ECM proteins such as collagens, elastin, fibronectin, and laminins, forming a complex network of fibers that give tissues their strength and flexibility. They also help in the regulation of tissue homeostasis by controlling the turnover of ECM components through the process of remodeling.
In response to injury or infection, fibroblasts become activated and start to proliferate rapidly, migrating towards the site of damage. Here, they participate in the inflammatory response, releasing cytokines and chemokines that attract immune cells to the area. Additionally, they deposit new ECM components to help repair the damaged tissue and restore its functionality.
Dysregulation of fibroblast activity has been implicated in several pathological conditions, including fibrosis (excessive scarring), cancer (where they can contribute to tumor growth and progression), and autoimmune diseases (such as rheumatoid arthritis).
The liver is a large, solid organ located in the upper right portion of the abdomen, beneath the diaphragm and above the stomach. It plays a vital role in several bodily functions, including:
1. Metabolism: The liver helps to metabolize carbohydrates, fats, and proteins from the food we eat into energy and nutrients that our bodies can use.
2. Detoxification: The liver detoxifies harmful substances in the body by breaking them down into less toxic forms or excreting them through bile.
3. Synthesis: The liver synthesizes important proteins, such as albumin and clotting factors, that are necessary for proper bodily function.
4. Storage: The liver stores glucose, vitamins, and minerals that can be released when the body needs them.
5. Bile production: The liver produces bile, a digestive juice that helps to break down fats in the small intestine.
6. Immune function: The liver plays a role in the immune system by filtering out bacteria and other harmful substances from the blood.
Overall, the liver is an essential organ that plays a critical role in maintaining overall health and well-being.
Substrate specificity in the context of medical biochemistry and enzymology refers to the ability of an enzyme to selectively bind and catalyze a chemical reaction with a particular substrate (or a group of similar substrates) while discriminating against other molecules that are not substrates. This specificity arises from the three-dimensional structure of the enzyme, which has evolved to match the shape, charge distribution, and functional groups of its physiological substrate(s).
Substrate specificity is a fundamental property of enzymes that enables them to carry out highly selective chemical transformations in the complex cellular environment. The active site of an enzyme, where the catalysis takes place, has a unique conformation that complements the shape and charge distribution of its substrate(s). This ensures efficient recognition, binding, and conversion of the substrate into the desired product while minimizing unwanted side reactions with other molecules.
Substrate specificity can be categorized as:
1. Absolute specificity: An enzyme that can only act on a single substrate or a very narrow group of structurally related substrates, showing no activity towards any other molecule.
2. Group specificity: An enzyme that prefers to act on a particular functional group or class of compounds but can still accommodate minor structural variations within the substrate.
3. Broad or promiscuous specificity: An enzyme that can act on a wide range of structurally diverse substrates, albeit with varying catalytic efficiencies.
Understanding substrate specificity is crucial for elucidating enzymatic mechanisms, designing drugs that target specific enzymes or pathways, and developing biotechnological applications that rely on the controlled manipulation of enzyme activities.
Pyruvate kinase is an enzyme that plays a crucial role in the final step of glycolysis, a process by which glucose is broken down to produce energy in the form of ATP (adenosine triphosphate). Specifically, pyruvate kinase catalyzes the transfer of a phosphate group from phosphoenolpyruvate (PEP) to adenosine diphosphate (ADP), resulting in the formation of pyruvate and ATP.
There are several isoforms of pyruvate kinase found in different tissues, including the liver, muscle, and brain. The type found in red blood cells is known as PK-RBC or PK-M2. Deficiencies in pyruvate kinase can lead to a genetic disorder called pyruvate kinase deficiency, which can result in hemolytic anemia due to the premature destruction of red blood cells.
Glycoside hydrolases are a class of enzymes that catalyze the hydrolysis of glycosidic bonds found in various substrates such as polysaccharides, oligosaccharides, and glycoproteins. These enzymes break down complex carbohydrates into simpler sugars by cleaving the glycosidic linkages that connect monosaccharide units.
Glycoside hydrolases are classified based on their mechanism of action and the type of glycosidic bond they hydrolyze. The classification system is maintained by the International Union of Biochemistry and Molecular Biology (IUBMB). Each enzyme in this class is assigned a unique Enzyme Commission (EC) number, which reflects its specificity towards the substrate and the type of reaction it catalyzes.
These enzymes have various applications in different industries, including food processing, biofuel production, pulp and paper manufacturing, and biomedical research. In medicine, glycoside hydrolases are used to diagnose and monitor certain medical conditions, such as carbohydrate-deficient glycoprotein syndrome, a rare inherited disorder affecting the structure of glycoproteins.
Mannose is a simple sugar (monosaccharide) that is similar in structure to glucose. It is a hexose, meaning it contains six carbon atoms. Mannose is a stereoisomer of glucose, meaning it has the same chemical formula but a different structural arrangement of its atoms.
Mannose is not as commonly found in foods as other simple sugars, but it can be found in some fruits, such as cranberries, blueberries, and peaches, as well as in certain vegetables, like sweet potatoes and turnips. It is also found in some dietary fibers, such as those found in beans and whole grains.
In the body, mannose can be metabolized and used for energy, but it is also an important component of various glycoproteins and glycolipids, which are molecules that play critical roles in many biological processes, including cell recognition, signaling, and adhesion.
Mannose has been studied as a potential therapeutic agent for various medical conditions, including urinary tract infections (UTIs), because it can inhibit the attachment of certain bacteria to the cells lining the urinary tract. Additionally, mannose-binding lectins have been investigated for their potential role in the immune response to viral and bacterial infections.
In the context of medicine and pharmacology, "kinetics" refers to the study of how a drug moves throughout the body, including its absorption, distribution, metabolism, and excretion (often abbreviated as ADME). This field is called "pharmacokinetics."
1. Absorption: This is the process of a drug moving from its site of administration into the bloodstream. Factors such as the route of administration (e.g., oral, intravenous, etc.), formulation, and individual physiological differences can affect absorption.
2. Distribution: Once a drug is in the bloodstream, it gets distributed throughout the body to various tissues and organs. This process is influenced by factors like blood flow, protein binding, and lipid solubility of the drug.
3. Metabolism: Drugs are often chemically modified in the body, typically in the liver, through processes known as metabolism. These changes can lead to the formation of active or inactive metabolites, which may then be further distributed, excreted, or undergo additional metabolic transformations.
4. Excretion: This is the process by which drugs and their metabolites are eliminated from the body, primarily through the kidneys (urine) and the liver (bile).
Understanding the kinetics of a drug is crucial for determining its optimal dosing regimen, potential interactions with other medications or foods, and any necessary adjustments for special populations like pediatric or geriatric patients, or those with impaired renal or hepatic function.
Molecular weight, also known as molecular mass, is the mass of a molecule. It is expressed in units of atomic mass units (amu) or daltons (Da). Molecular weight is calculated by adding up the atomic weights of each atom in a molecule. It is a useful property in chemistry and biology, as it can be used to determine the concentration of a substance in a solution, or to calculate the amount of a substance that will react with another in a chemical reaction.
Immunodiffusion is a laboratory technique used in immunology to detect and measure the presence of specific antibodies or antigens in a sample. It is based on the principle of diffusion, where molecules move from an area of high concentration to an area of low concentration until they reach equilibrium. In this technique, a sample containing an unknown quantity of antigen or antibody is placed in a gel or agar medium that contains a known quantity of antibody or antigen, respectively.
The two substances then diffuse towards each other and form a visible precipitate at the point where they meet and reach equivalence, which indicates the presence and quantity of the specific antigen or antibody in the sample. There are several types of immunodiffusion techniques, including radial immunodiffusion (RID) and double immunodiffusion (Ouchterlony technique). These techniques are widely used in diagnostic laboratories to identify and measure various antigens and antibodies, such as those found in infectious diseases, autoimmune disorders, and allergic reactions.
The placenta is an organ that develops in the uterus during pregnancy and provides oxygen and nutrients to the growing baby through the umbilical cord. It also removes waste products from the baby's blood. The placenta attaches to the wall of the uterus, and the baby's side of the placenta contains many tiny blood vessels that connect to the baby's circulatory system. This allows for the exchange of oxygen, nutrients, and waste between the mother's and baby's blood. After the baby is born, the placenta is usually expelled from the uterus in a process called afterbirth.
Hydrogen-ion concentration, also known as pH, is a measure of the acidity or basicity of a solution. It is defined as the negative logarithm (to the base 10) of the hydrogen ion activity in a solution. The standard unit of measurement is the pH unit. A pH of 7 is neutral, less than 7 is acidic, and greater than 7 is basic.
In medical terms, hydrogen-ion concentration is important for maintaining homeostasis within the body. For example, in the stomach, a high hydrogen-ion concentration (low pH) is necessary for the digestion of food. However, in other parts of the body such as blood, a high hydrogen-ion concentration can be harmful and lead to acidosis. Conversely, a low hydrogen-ion concentration (high pH) in the blood can lead to alkalosis. Both acidosis and alkalosis can have serious consequences on various organ systems if not corrected.
Gel chromatography is a type of liquid chromatography that separates molecules based on their size or molecular weight. It uses a stationary phase that consists of a gel matrix made up of cross-linked polymers, such as dextran, agarose, or polyacrylamide. The gel matrix contains pores of various sizes, which allow smaller molecules to penetrate deeper into the matrix while larger molecules are excluded.
In gel chromatography, a mixture of molecules is loaded onto the top of the gel column and eluted with a solvent that moves down the column by gravity or pressure. As the sample components move down the column, they interact with the gel matrix and get separated based on their size. Smaller molecules can enter the pores of the gel and take longer to elute, while larger molecules are excluded from the pores and elute more quickly.
Gel chromatography is commonly used to separate and purify proteins, nucleic acids, and other biomolecules based on their size and molecular weight. It is also used in the analysis of polymers, colloids, and other materials with a wide range of applications in chemistry, biology, and medicine.
Enzyme precursors are typically referred to as zymogens or proenzymes. These are inactive forms of enzymes that can be activated under specific conditions. When the need for the enzyme's function arises, the proenzyme is converted into its active form through a process called proteolysis, where it is cleaved by another enzyme. This mechanism helps control and regulate the activation of certain enzymes in the body, preventing unwanted or premature reactions. A well-known example of an enzyme precursor is trypsinogen, which is converted into its active form, trypsin, in the digestive system.
Cross reactions, in the context of medical diagnostics and immunology, refer to a situation where an antibody or a immune response directed against one antigen also reacts with a different antigen due to similarities in their molecular structure. This can occur in allergy testing, where a person who is allergic to a particular substance may have a positive test result for a different but related substance because of cross-reactivity between them. For example, some individuals who are allergic to birch pollen may also have symptoms when eating certain fruits, such as apples, due to cross-reactive proteins present in both.
Genetic variation refers to the differences in DNA sequences among individuals and populations. These variations can result from mutations, genetic recombination, or gene flow between populations. Genetic variation is essential for evolution by providing the raw material upon which natural selection acts. It can occur within a single gene, between different genes, or at larger scales, such as differences in the number of chromosomes or entire sets of chromosomes. The study of genetic variation is crucial in understanding the genetic basis of diseases and traits, as well as the evolutionary history and relationships among species.
'Immune sera' refers to the serum fraction of blood that contains antibodies produced in response to an antigenic stimulus, such as a vaccine or an infection. These antibodies are proteins known as immunoglobulins, which are secreted by B cells (a type of white blood cell) and can recognize and bind to specific antigens. Immune sera can be collected from an immunized individual and used as a source of passive immunity to protect against infection or disease. It is often used in research and diagnostic settings to identify or measure the presence of specific antigens or antibodies.
I must clarify that the term "pedigree" is not typically used in medical definitions. Instead, it is often employed in genetics and breeding, where it refers to the recorded ancestry of an individual or a family, tracing the inheritance of specific traits or diseases. In human genetics, a pedigree can help illustrate the pattern of genetic inheritance in families over multiple generations. However, it is not a medical term with a specific clinical definition.
Pregnancy is a physiological state or condition where a fertilized egg (zygote) successfully implants and grows in the uterus of a woman, leading to the development of an embryo and finally a fetus. This process typically spans approximately 40 weeks, divided into three trimesters, and culminates in childbirth. Throughout this period, numerous hormonal and physical changes occur to support the growing offspring, including uterine enlargement, breast development, and various maternal adaptations to ensure the fetus's optimal growth and well-being.
 Hexosaminidase
Hexosaminidase
Tay-Sachs disease
GM2 gangliosidoses
Glycoside hydrolase family 20
Prevention of Tay-Sachs disease
Peptidoglycan β-N-acetylmuramidase
GM2-gangliosidosis, AB variant
Protein O-GlcNAcase
HEXA
Dispersin B
Sandhoff disease
Paucimannosylation
Biofilm
History of Tay-Sachs disease
Pyrimethamine
HEXB
Pseudodeficiency alleles
GM2A
Granule (cell biology)
CHB HEX N-terminal domain
Cathepsin D
Asthma-related microbes
Gaucher's disease
Joanne Lemieux
Compound heterozygosity
Point mutation
N-acetyl-β-d-glucosaminidase
Jacob sheep
Enzyme
Maclyn McCarty
Deficiency11
- There are numerous mutations that lead to hexosaminidase deficiency including gene deletions, nonsense mutations, and missense mutations. (wikipedia.org)
- These diseases result from a deficiency of lysosomal enzyme ß- hexosaminidase A (HexA), which is responsible for GM2 ganglioside degradation. (bvsalud.org)
- Hexosaminidase activator deficiency is caused by absence or defects of the hexosaminidase activator. (medscape.com)
- Type AB G M2 gangliosidosis is also known as hexosaminidase activator deficiency. (medscape.com)
- Kaback MM. Hexosaminidase A Deficiency. (epnet.com)
- 10. Late-onset hexosaminidase A and hexosaminidase A and B deficiency: family study and review. (nih.gov)
- inherited disorders of metabolism, caused by hexosaminidase deficiency that causes severe neurologic symptoms and early death. (msdmanuals.com)
- Deficiency of hexosaminidase A results in accumulation of GM2 in the brain. (msdmanuals.com)
- There is a combined hexosaminidase A and B deficiency. (msdmanuals.com)
- The documented enzyme deficiency should support a genetically confirmed diagnosis of GM2-gangliosidosis caused by beta-hexosaminidase deficiency resulting from mutations in the HEXA or HEXB genes. (nih.gov)
- The accumulation of GM 2 (due to a deficiency in beta-hexosaminidase) has characterized Tay-Sachs disease (due to a mutation in the gene HEXA) and Sandhoff disease (due to a mutation in the gene HEXB). (matreya.com)
HEXA4
- The HEXA gene provides instructions for making one part (subunit) of an enzyme called beta-hexosaminidase A. Specifically, the protein produced from the HEXA gene forms the alpha subunit of this enzyme. (medlineplus.gov)
- The HEXA gene variants that cause Tay-Sachs disease eliminate or severely reduce the activity of the enzyme beta-hexosaminidase A. This lack of enzyme activity prevents the enzyme from breaking down GM2 ganglioside. (medlineplus.gov)
- Most of the known HEXA gene variants result in a completely nonfunctional version of beta-hexosaminidase A. These variants cause a severe form of Tay-Sachs disease, known as infantile Tay-Sachs disease, which appears in infancy. (medlineplus.gov)
- Dersh D, Iwamoto Y, Argon Y. Tay-Sachs disease mutations in HEXA target the alpha chain of hexosaminidase A to endoplasmic reticulum-associated degradation. (medlineplus.gov)
Beta-hexosaminidases4
- Human lysosomal beta-hexosaminidases remove terminal beta-glycosidically bound N-acetylhexosamine residues from a number of glycoconjugates. (nih.gov)
- 18. Serum beta-hexosaminidases in pregnancy. (nih.gov)
- This domain represents the N terminal domain in chitobiases and beta-hexosaminidases EC:3.2.1.52. (embl-heidelberg.de)
- This domain is found in the N terminus of chitobiases and beta-hexosaminidases. (embl-heidelberg.de)
HEXB3
- Description: This is Double-antibody Sandwich Enzyme-linked immunosorbent assay for detection of Mouse Hexosaminidase B Beta (HEXb) in serum, plasma, tissue homogenates and other biological fluids. (srbiosystem.com)
- Description: A sandwich quantitative ELISA assay kit for detection of Rat Hexosaminidase B Beta (HEXb) in samples from serum, plasma, tissue homogenates or other biological fluids. (srbiosystem.com)
- One alpha subunit joins with one beta subunit (produced from the HEXB gene) to form a functioning beta-hexosaminidase A enzyme. (medlineplus.gov)
Sandhoff7
- Hereditary inability to form functional hexosaminidase enzymes are the cause of lipid storage disorders Tay-Sachs disease and Sandhoff disease. (wikipedia.org)
- The technique is applied here to PGD of Sandhoff disease caused by 16-kb deletion of the hexosaminidase B gene for a couple with a religious objection to discarding embryos irrespective of embryo genotype. (nih.gov)
- 3. [Reconstruction of hexosaminidase isoenzymes during hybridization of fibroblasts from Tay-Sachs and Sandhoff diseases]. (nih.gov)
- 11. Evidence for a hybrid hexosaminidase isoenzyme in heterozygotes for Sandhoff disease. (nih.gov)
- 12. Crystal structure of human beta-hexosaminidase B: understanding the molecular basis of Sandhoff and Tay-Sachs disease. (nih.gov)
- The structure allows us to model the catalytic domain ofthe homologous hexosaminidases to give a structural rationale topathogenic mutations that underlie Tay-Sachs and Sandhoff disease. (embl-heidelberg.de)
- Disease/enzymes combinations selected for study are, in order of priority, MPS I/ alpha-L-iduronidase, MPS III B/ Alpha-N-acetylglucosaminidase, MPS VII/ beta-glucuronidase and Sandhoff disease/beta-hexosaminidase. (nih.gov)
Lysosomal hexosaminidase3
- Even though the α and β subunits of lysosomal hexosaminidase can both cleave GalNAc residues, only the α subunit is able to hydrolyze GM2 gangliosides because of a key residue, Arg-424, and a loop structure that forms from the amino acid sequence in the alpha subunit. (wikipedia.org)
- NCOAT is also known as hexosaminidase C and has distinct substrate specificities compared to lysosomal hexosaminidase A. A single-nucleotide polymorphism in the human O-GlcNAcase gene is linked to diabetes mellitus type 2. (wikipedia.org)
- 19. The lysosomal hexosaminidase isozymes. (nih.gov)
Mutations1
- Crystallographic structure of human beta-hexosaminidase A: interpretation of Tay-Sachs mutations and loss of GM2 ganglioside hydrolysis. (medlineplus.gov)
Serum1
- Description: A sandwich ELISA kit for detection of Hexosaminidase B Beta from Mouse in samples from blood, serum, plasma, cell culture fluid and other biological fluids. (srbiosystem.com)
Lysosomes1
- Within lysosomes, beta-hexosaminidase A forms part of a complex that breaks down a fatty substance called GM2 ganglioside found in cell membranes. (medlineplus.gov)
Genes1
- Assignment of genes encoding dihydrofolate reductase and hexosaminidase B to Mus musculus chromosome 13. (nih.gov)
Genetics1
- 2. The biochemical genetics of the hexosaminidase system in man. (nih.gov)
Activator2
- β-Hexosaminidase and the cofactor GM2 activator protein catalyze the degradation of the GM2 gangliosides and other molecules containing terminal N-acetyl hexosamines. (wikipedia.org)
- The GM2 activator protein transports GM2 gangliosides and presents the lipids to hexosaminidase, so a functional hexosaminidase enzyme is able to hydrolyze GM2 gangliosides into GM3 gangliosides by removing the N-acetylgalactosamine (GalNAc) residue from GM2 gangliosides. (wikipedia.org)
Subunits1
- The two subunits of hexosaminidase A are shown below: The bifunctional protein NCOAT (nuclear cytoplasmic O-GlcNAcase and acetyltransferase) that is encoded by the MGEA5 gene possesses both hexosaminidase and histone acetyltransferase activities. (wikipedia.org)
Histamine1
- AEDK inhibited the release of histamine and β-hexosaminidase from mast cells by modulating cAMP and intracellular calcium levels. (spandidos-publications.com)
Disease3
- Tay-Sachs disease occurs when hexosaminidase A loses its ability to function. (wikipedia.org)
- Other variants severely reduce but do not eliminate the activity of beta-hexosaminidase A. These genetic changes are responsible for less severe forms of Tay-Sachs disease, known as the juvenile and late-onset forms, which appear later in life. (medlineplus.gov)
- 4. A new form of residual hexosaminidase activity in infantile Tay Sachs disease fibroblasts. (nih.gov)
Proteins1
- MS revealed that phycocyanin and the core-membrane linker peptide are the responsible allergens, and MC(-) extracts containing these proteins induced β-hexosaminidase release in rat basophil leukemia cells. (nih.gov)
Catalytic1
- The GH20 hexosaminidases are thought to act via a catalytic mechanism in which the catalytic nucleophile is not provided by solvent or the enzyme, but by the substrate itself. (unl.edu)
Genetically1
- Increasing ß-hexosaminidase A activity using genetically modified mesenchymal stem cells. (bvsalud.org)
Dimer1
- β-Hexosaminidase B (Hex B) is a dimer of beta chains. (medscape.com)
Functional1
- Functional lysosomal β-hexosaminidase enzymes are dimeric in structure. (wikipedia.org)
Release1
- hexosaminidase release assay to examine the inhibitory effects of fCD23. (ncl.edu.tw)
Human3
- 1. The expression of hex A and hex B isozymes of hexosaminidase in parental and experimental human fibroblast cells and their components. (nih.gov)
- 13. beta-hexosaminidase in cultured normal and mutant human fibroblasts: an immunohistochemical and biochemical investigation. (nih.gov)
- 17. Human hexosaminidase isozymes: chromatographic separation as an aid to heterozygote identification. (nih.gov)
Blood1
- Elevated levels of hexosaminidase in blood and/or urine have been proposed as a biomarker of relapse in the treatment of alcoholism. (wikipedia.org)
Brain1
- Beta-hexosaminidase A plays a critical role in the brain and spinal cord (central nervous system). (medlineplus.gov)