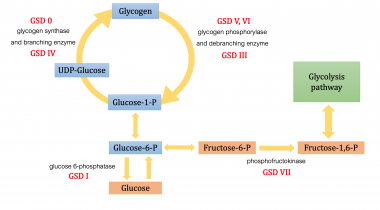
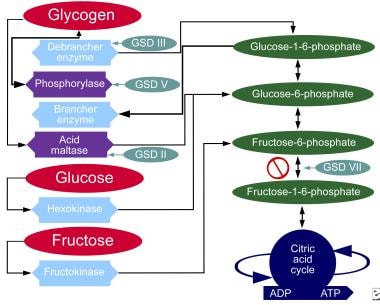
Glycogen Storage Disease Type VII
Glycogen Storage Disease Type I
Glycogen Storage Disease Type III
Glycogen Storage Disease
Glycogen Storage Disease Type IV
Glycogen Storage Disease Type II
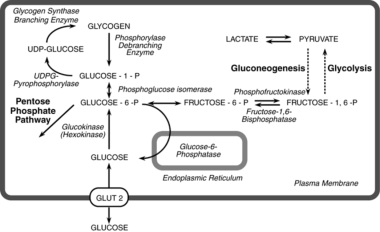
Glucose-6-Phosphatase
Glycogen Storage Disease Type VI
Glycogen
alpha-Glucosidases
Glycogen Debranching Enzyme System
Glycogen Storage Disease Type V
Glycogen Storage Disease Type VIII
Glucan 1,4-alpha-Glucosidase
Antiporters
Glucose-6-Phosphate
Collagen Type VII
1,4-alpha-Glucan Branching Enzyme
Glycogen Storage Disease Type IIb
Fructose-1,6-Diphosphatase Deficiency
The contribution of Ca+ calmodulin activation of human erythrocyte AMP deaminase (isoform E) to the erythrocyte metabolic dysregulation of familial phosphofructokinase deficiency. (1/18)
Erythrocyte membrane leakage of Ca2+ in familial phosphofructokinase deficiency results in a compensatory increase of Ca2+-ATPase activity that depletes ATP and leads to diminished erythrocyte deformability and a higher rate of hemolysis. Lowered ATP levels in circulating erythrocytes are accompanied by increased IMP, indicating that activated AMP deaminase plays a role in this metabolic dysregulation. Exposure to a calmodulin antagonist significantly slows IMP accumulation during experimental energy imbalance in patients' cells to levels that are similar to those in untreated controls, implying that Ca2+-calmodulin is involved in erythrocyte AMP deaminase activation in familial phosphofructokinase deficiency. Therapies directed against activated isoform E may be beneficial in this compensated anemia. (+info)Tissue-dependent loss of phosphofructokinase-M in mice with interrupted activity of the distal promoter: impairment in insulin secretion. (2/18)
Phosphofructokinase is a key enzyme of glycolysis that exists as homo- and heterotetramers of three subunit isoforms: muscle, liver, and C type. Mice with a disrupting tag inserted near the distal promoter of the phosphofructokinase-M gene showed tissue-dependent differences in loss of that isoform: 99% in brain and 95-98% in islets, but only 50-75% in skeletal muscle and little if any loss in heart. This correlated with the continued presence of proximal transcripts specifically in muscle tissues. These data strongly support the proposed two-promoter system of the gene, with ubiquitous use of the distal promoter and additional use of the proximal promoter selectively in muscle. Interestingly, the mice were glucose intolerant and had somewhat elevated fasting and fed blood glucose levels; however, they did not have an abnormal insulin tolerance test, consistent with the less pronounced loss of phosphofructokinase-M in muscle. Isolated perifused islets showed about 50% decreased glucose-stimulated insulin secretion and reduced amplitude and regularity of secretory oscillations. Oscillations in cytoplasmic free Ca(2+) and the rise in the ATP/ADP ratio appeared normal. Secretory oscillations still occurred in the presence of diazoxide and high KCl, indicating an oscillation mechanism not requiring dynamic Ca(2+) changes. The results suggest the importance of phosphofructokinase-M for insulin secretion, although glucokinase is the overall rate-limiting glucose sensor. Whether the Ca(2+) oscillations and residual insulin oscillations in this mouse model are due to the residual 2-5% phosphofructokinase-M or to other phosphofructokinase isoforms present in islets or involve another metabolic oscillator remains to be determined. (+info)Tarui disease and distal glycogenoses: clinical and genetic update. (3/18)
Phosphofructokinase deficiency (Tarui disease) was the first disorder recognized to directly affect glycolysis. Since the discovery of the disease, in 1965, a wide range of biochemical, physiological and molecular studies have greatly contributed to our knowledge concerning not only phosphofructokinase function in normal muscle but also on the general control of glycolysis and glycogen metabolism. Studies on phosphofructokinase deficiency vastly enriched the field of glycogen storage diseases, making a relevant improvement also in the molecular genetic area. So far, more than one hundred patients have been described with prominent clinical symptoms characterized by muscle cramps, exercise intolerance, rhabdomyolysis and myoglobinuria, often associated with haemolytic anaemia and hyperuricaemia. The muscle phosphofructokinase gene is located on chromosome 12 and about 20 mutations have been described. Other glycogenoses have been recognised in the distal part of the glycolytic pathway: these are infrequent but some may induce muscle cramps, exercise intolerance and rhabdomyolysis. Phosphoglycerate Kinase, Phosphoglycerate Mutase, Lactate Dehydrogenase, beta-Enolase and Aldolase A deficiencies have been described as distal glycogenoses. From the molecular point of view, the majority of these enzyme deficiencies are sustained by "private" mutations. (+info)Phosphofructo-1-kinase deficiency leads to a severe cardiac and hematological disorder in addition to skeletal muscle glycogenosis. (4/18)
(+info)Polysaccharide storage myopathy in canine phosphofructokinase deficiency (type VII glycogen storage disease). (5/18)
A severe, progressive myopathy developed in an 11-year-old, phosphofructokinase (PFK)-deficient, male, English Springer Spaniel dog. Results from a routine neurological examination were normal. Examination of histologic sections of skeletal muscle revealed large accumulations of material in some myofibers. These deposits were pale, basophilic, somewhat flocculent, and slightly granular with hematoxylin and eosin stain. Most fascicles examined in sections of limb and trunk muscles were affected to some degree, with up to 10% of muscle fibers being involved. Deposits stained strongly with periodic acid-Schiff and were resistant to digestion by alpha amylase but were removed by incubation with gamma amylase. Deposits were faintly positive with Gomori's methenamine silver technique and alcian blue (pH 2.5) and were brown-gray with Lugol's iodine solution but were negative with other stains. Based on staining characteristics, the deposits seemed to consist primarily of an amylopectin-like polysaccharide(s). Alcian blue staining (pH 2.5) was removed by treatment with neuraminidase but not with hyaluronidase, indicating that some sialic acid residues were also present. Electron microscopically, the deposits were composed of short granular filaments, small granules and amorphous material. They were not membrane bound. The morphologic appearance and staining characteristics of the deposits were remarkably similar to deposits previously described in human PFK-deficient myopathy. As expected, total PFK activities were markedly reduced when assayed in skeletal muscles of this dog. In contrast with other PFK-deficient dogs, muscle glycogen in this animal was not increased above that of normal dogs. (+info)Genetic defect in muscle phosphofructokinase deficiency. Abnormal splicing of the muscle phosphofructokinase gene due to a point mutation at the 5'-splice site. (6/18)
The genetic defect in muscle phosphofructokinase deficiency (type VII glycogenosis, Tarui disease) was investigated. Six cDNAs for muscle phosphofructokinase, including a full-length clone, were isolated from a non-amplified library of muscle from a patient. By sequence analysis of these clones, a 75-base in-frame deletion was identified. The rest of the sequence was identical to that of the normal cDNA, except for a silent base transition at position 516 (ACT (Thr) to ACC (Thr]. The deletion was located in the 3'-terminal region of exon 13 (numbered with reference to the rabbit muscle phosphofructokinase gene (Lee, C.-P., Kao, M.-C., French, B.A., Putney, S.D., and Chang, S.H. (1987) J. Biol. Chem. 262, 4195-4199]. Genomic DNA of the patient was amplified by polymerase chain reaction. Sequence analysis of the amplified DNA revealed a point mutation from G to T at the 5'-end of intron 13. This mutation changed the normal 5'-splice site of CAG:GTATGG to CAG:TTATGG. A cryptic splice site of ACT:GTGAGG located 75 bases upstream from the normal splice site was recognized and spliced in the patient. (+info)Missense mutation in PFKM associated with muscle-type phosphofructokinase deficiency in the Wachtelhund dog. (7/18)
(+info)Characterization of the enzymatic defect in late-onset muscle phosphofructokinase deficiency. New subtype of glycogen storage disease type VII. (8/18)
Human phosphofructokinase (PFK) exists in tetrameric isozymic forms, at least in vitro. Muscle and liver contain homotetramers M4 and L4, respectively, whereas red cells contain five isozymes composed of M (muscle) and L (liver) type subunits, i.e., M4, M3L, M2L2, and ML3, and L4. Homozygous deficiency of muscle PFK results in the classic glycogen storage disease type VII characterized by exertional myopathy and hemolytic syndrome beginning in early childhood. The genetic lesion results in a total and partial loss of muscle and red cell PFK, respectively. Characteristically, the residual red cell PFK from the patients consists of isolated L4 isozyme; the M-containing hybrid isozymes are completely absent. In this study, we investigated an 80-yr-old man who presented with a 10-yr history of progressive weakness of the lower limbs as the only symptom. The residual red cell PFK showed the presence of a few M-containing isozymes in addition to the predominant L4 species, indicating that the genetic lesion is a "leaky" mutation of the gene coding for the M subunit. The presence of a small amount of enzyme activity in the muscle may account for the atypical myopathy in this patient. (+info)Glycogen Storage Disease Type VII, also known as Tarui's disease, is a rare inherited metabolic disorder caused by a deficiency of the enzyme phosphofructokinase (PFK), which is required for glycogenolysis – the breakdown of glycogen to glucose-1-phosphate and ultimately into glucose. This enzyme deficiency results in the accumulation of glycogen, particularly in muscle and red blood cells, leading to symptoms such as exercise-induced muscle cramps, myoglobinuria (the presence of myoglobin in the urine), and hemolytic anemia. The disease can also cause muscle weakness, fatigue, and dark-colored urine after strenuous exercise. It is inherited in an autosomal recessive manner, meaning that an individual must inherit two copies of the mutated gene (one from each parent) to develop the condition.
Glycogen Storage Disease Type I (GSD I) is a rare inherited metabolic disorder caused by deficiency of the enzyme glucose-6-phosphatase, which is necessary for the liver to release glucose into the bloodstream. This leads to an accumulation of glycogen in the liver and abnormally low levels of glucose in the blood (hypoglycemia).
There are two main subtypes of GSD I: Type Ia and Type Ib. In Type Ia, there is a deficiency of both glucose-6-phosphatase enzyme activity in the liver, kidney, and intestine, leading to hepatomegaly (enlarged liver), hypoglycemia, lactic acidosis, hyperlipidemia, and growth retardation. Type Ib is characterized by a deficiency of glucose-6-phosphatase enzyme activity only in the neutrophils, leading to recurrent bacterial infections.
GSD I requires lifelong management with frequent feedings, high-carbohydrate diet, and avoidance of fasting to prevent hypoglycemia. In some cases, treatment with continuous cornstarch infusions or liver transplantation may be necessary.
Glycogen Storage Disease Type III, also known as Cori or Forbes disease, is a rare inherited metabolic disorder caused by deficiency of the debranching enzyme amylo-1,6-glucosidase, which is responsible for breaking down glycogen in the liver and muscles. This results in an abnormal accumulation of glycogen in these organs leading to its associated symptoms.
There are two main types: Type IIIa affects both the liver and muscles, while Type IIIb affects only the liver. Symptoms can include hepatomegaly (enlarged liver), hypoglycemia (low blood sugar), hyperlipidemia (high levels of fats in the blood), and growth retardation. In Type IIIa, muscle weakness and cardiac problems may also occur.
The diagnosis is usually made through biochemical tests and genetic analysis. Treatment often involves dietary management with frequent meals to prevent hypoglycemia, and in some cases, enzyme replacement therapy. However, there is no cure for this condition and life expectancy can be reduced depending on the severity of the symptoms.
Glycogen storage disease (GSD) is a group of rare inherited metabolic disorders that affect the body's ability to break down and store glycogen, a complex carbohydrate that serves as the primary form of energy storage in the body. These diseases are caused by deficiencies or dysfunction in enzymes involved in the synthesis, degradation, or transport of glycogen within cells.
There are several types of GSDs, each with distinct clinical presentations and affected organs. The most common type is von Gierke disease (GSD I), which primarily affects the liver and kidneys. Other types include Pompe disease (GSD II), McArdle disease (GSD V), Cori disease (GSD III), Andersen disease (GSD IV), and others.
Symptoms of GSDs can vary widely depending on the specific type, but may include:
* Hypoglycemia (low blood sugar)
* Growth retardation
* Hepatomegaly (enlarged liver)
* Muscle weakness and cramping
* Cardiomyopathy (heart muscle disease)
* Respiratory distress
* Developmental delays
Treatment for GSDs typically involves dietary management, such as frequent feedings or a high-protein, low-carbohydrate diet. In some cases, enzyme replacement therapy may be used to manage symptoms. The prognosis for individuals with GSDs depends on the specific type and severity of the disorder.
Glycogen Storage Disease Type IV (GSD IV), also known as Andersen's disease, is a rare inherited metabolic disorder that affects the body's ability to break down glycogen, a complex carbohydrate that serves as a source of energy for the body.
In GSD IV, there is a deficiency in the enzyme called glycogen branching enzyme (GBE), which is responsible for adding branches to the glycogen molecule during its synthesis. This results in an abnormal form of glycogen that accumulates in various organs and tissues, particularly in the liver, heart, and muscles.
The accumulation of this abnormal glycogen can lead to progressive damage and failure of these organs, resulting in a variety of symptoms such as muscle weakness, hypotonia, hepatomegaly (enlarged liver), cardiomyopathy (heart muscle disease), and developmental delay. The severity of the disease can vary widely, with some individuals experiencing milder symptoms while others may have a more severe and rapidly progressing form of the disorder.
Currently, there is no cure for GSD IV, and treatment is focused on managing the symptoms and slowing down the progression of the disease. This may include providing nutritional support, addressing specific organ dysfunction, and preventing complications.
Glycogen Storage Disease Type II, also known as Pompe Disease, is a genetic disorder caused by a deficiency of the enzyme acid alpha-glucosidase (GAA). This enzyme is responsible for breaking down glycogen, a complex sugar that serves as energy storage, within lysosomes. When GAA is deficient, glycogen accumulates in various tissues, particularly in muscle cells, leading to their dysfunction and damage.
The severity of Pompe Disease can vary significantly, depending on the amount of functional enzyme activity remaining. The classic infantile-onset form presents within the first few months of life with severe muscle weakness, hypotonia, feeding difficulties, and respiratory insufficiency. This form is often fatal by 1-2 years of age if left untreated.
A later-onset form, which can present in childhood, adolescence, or adulthood, has a more variable clinical course. Affected individuals may experience progressive muscle weakness, respiratory insufficiency, and cardiomyopathy, although the severity and rate of progression are generally less pronounced than in the infantile-onset form.
Enzyme replacement therapy with recombinant human GAA is available for the treatment of Pompe Disease and has been shown to improve survival and motor function in affected individuals.
Glucose-6-phosphatase is an enzyme that plays a crucial role in the regulation of glucose metabolism. It is primarily located in the endoplasmic reticulum of cells in liver, kidney, and intestinal mucosa. The main function of this enzyme is to remove the phosphate group from glucose-6-phosphate (G6P), converting it into free glucose, which can then be released into the bloodstream and used as a source of energy by cells throughout the body.
The reaction catalyzed by glucose-6-phosphatase is as follows:
Glucose-6-phosphate + H2O → Glucose + Pi (inorganic phosphate)
This enzyme is essential for maintaining normal blood glucose levels, particularly during periods of fasting or starvation. In these situations, the body needs to break down stored glycogen in the liver and convert it into glucose to supply energy to the brain and other vital organs. Glucose-6-phosphatase is a key enzyme in this process, allowing for the release of free glucose into the bloodstream.
Deficiencies or mutations in the gene encoding glucose-6-phosphatase can lead to several metabolic disorders, such as glycogen storage disease type I (von Gierke's disease) and other related conditions. These disorders are characterized by an accumulation of glycogen and/or fat in various organs, leading to impaired glucose metabolism, growth retardation, and increased risk of infection and liver dysfunction.
Glycogen Storage Disease Type VI, also known as Hers disease, is a rare inherited metabolic disorder caused by deficiency of the liver enzyme called glycogen phosphorylase. This enzyme is responsible for breaking down glycogen, which is a stored form of glucose, into glucose-1-phosphate during the process of glycogenolysis.
In GSD Type VI, the lack of this enzyme leads to an abnormal accumulation of glycogen in the liver, causing hepatomegaly (enlarged liver) and elevated liver enzymes. The symptoms of this condition are usually milder compared to other types of GSD, and may include fatigue, weakness, and hypoglycemia (low blood sugar), especially after prolonged fasting or physical exertion.
The diagnosis of GSD Type VI is typically made through biochemical tests that measure the activity of the glycogen phosphorylase enzyme in liver tissue, as well as genetic testing to identify mutations in the gene responsible for the enzyme's production. Treatment may involve dietary management, such as frequent feeding and avoidance of prolonged fasting, to prevent hypoglycemia. In some cases, medication may be necessary to manage symptoms and prevent complications.
Glycogen is a complex carbohydrate that serves as the primary form of energy storage in animals, fungi, and bacteria. It is a polysaccharide consisting of long, branched chains of glucose molecules linked together by glycosidic bonds. Glycogen is stored primarily in the liver and muscles, where it can be quickly broken down to release glucose into the bloodstream during periods of fasting or increased metabolic demand.
In the liver, glycogen plays a crucial role in maintaining blood glucose levels by releasing glucose when needed, such as between meals or during exercise. In muscles, glycogen serves as an immediate energy source for muscle contractions during intense physical activity. The ability to store and mobilize glycogen is essential for the proper functioning of various physiological processes, including athletic performance, glucose homeostasis, and overall metabolic health.
Alpha-glucosidases are a group of enzymes that break down complex carbohydrates into simpler sugars, such as glucose, by hydrolyzing the alpha-1,4 and alpha-1,6 glycosidic bonds in oligosaccharides, disaccharides, and polysaccharides. These enzymes are located on the brush border of the small intestine and play a crucial role in carbohydrate digestion and absorption.
Inhibitors of alpha-glucosidases, such as acarbose and miglitol, are used in the treatment of type 2 diabetes to slow down the digestion and absorption of carbohydrates, which helps to reduce postprandial glucose levels and improve glycemic control.
The Glycogen Debranching Enzyme System, also known as glycogen debranching enzyme or Amy-1, is a crucial enzyme complex in human biochemistry. It plays an essential role in the metabolism of glycogen, which is a large, branched polymer of glucose that serves as the primary form of energy storage in animals and fungi.
The Glycogen Debranching Enzyme System consists of two enzymatic activities: a transferase and an exo-glucosidase. The transferase activity transfers a segment of a branched glucose chain to another part of the same or another glycogen molecule, while the exo-glucosidase activity cleaves the remaining single glucose units from the outer branches of the glycogen molecule.
This enzyme system is responsible for removing the branched structures of glycogen, allowing the linear chains to be further degraded by other enzymes into glucose molecules that can be used for energy production or stored for later use. Defects in this enzyme complex can lead to several genetic disorders, such as Glycogen Storage Disease Type III (Cori's disease) and Type IV (Andersen's disease), which are characterized by the accumulation of abnormal glycogen molecules in various tissues.
Glycogen Storage Disease Type V, also known as McArdle's disease, is a genetic disorder that affects the body's ability to break down glycogen, a complex carbohydrate stored in muscles, into glucose, which provides energy for muscle contraction.
This condition results from a deficiency of the enzyme myophosphorylase, which is responsible for breaking down glycogen into glucose-1-phosphate within the muscle fibers. Without sufficient myophosphorylase activity, muscles become easily fatigued and may cramp or become rigid during exercise due to a lack of available energy.
Symptoms typically appear in childhood or adolescence and can include muscle weakness, stiffness, cramps, and myoglobinuria (the presence of myoglobin, a protein found in muscle cells, in the urine) following exercise. Diagnosis is usually confirmed through genetic testing and enzyme assays. Treatment typically involves avoiding strenuous exercise and ensuring adequate hydration and rest before and after physical activity. In some cases, dietary modifications such as high-protein or high-carbohydrate intake may be recommended to help manage symptoms.
Glycogen Storage Disease Type VIII, also known as Phosphorylase Kinase Deficiency, is a rare genetic metabolic disorder that affects the production and breakdown of glycogen in the body. Glycogen is a complex carbohydrate that serves as the primary form of energy storage in the body.
In this condition, there is a deficiency or dysfunction of the enzyme phosphorylase kinase (PhK), which plays a crucial role in activating glycogen phosphorylase, an enzyme responsible for breaking down glycogen into glucose-1-phosphate during periods of increased energy demand.
The deficiency or dysfunction of PhK leads to the abnormal accumulation of glycogen in various tissues, particularly in the liver and muscles. This accumulation can result in hepatomegaly (enlarged liver), hypoglycemia (low blood sugar levels), growth retardation, and muscle weakness.
Glycogen Storage Disease Type VIII is inherited in an autosomal recessive manner, meaning that an individual must inherit two defective copies of the gene, one from each parent, to develop the condition. There are four subtypes of GSD Type VIII, classified based on the specific genetic mutation and the severity of symptoms.
Treatment for Glycogen Storage Disease Type VIII typically involves managing the symptoms and complications associated with the disorder, such as providing a high-carbohydrate diet to prevent hypoglycemia and addressing any liver or muscle dysfunction. Regular monitoring by a healthcare team experienced in metabolic disorders is essential for optimizing treatment and ensuring appropriate management of this complex condition.
Glucan 1,4-alpha-glucosidase, also known as amyloglucosidase or glucoamylase, is an enzyme that catalyzes the hydrolysis of 1,4-glycosidic bonds in starch and other oligo- and polysaccharides, breaking them down into individual glucose molecules. This enzyme specifically acts on the alpha (1->4) linkages found in amylose and amylopectin, two major components of starch. It is widely used in various industrial applications, including the production of high fructose corn syrup, alcoholic beverages, and as a digestive aid in some medical supplements.
Antiporters, also known as exchange transporters, are a type of membrane transport protein that facilitate the exchange of two or more ions or molecules across a biological membrane in opposite directions. They allow for the movement of one type of ion or molecule into a cell while simultaneously moving another type out of the cell. This process is driven by the concentration gradient of one or both of the substances being transported. Antiporters play important roles in various physiological processes, including maintaining electrochemical balance and regulating pH levels within cells.
Glucose-6-phosphate (G6P) is a vital intermediate compound in the metabolism of glucose, which is a simple sugar that serves as a primary source of energy for living organisms. G6P plays a critical role in both glycolysis and gluconeogenesis pathways, contributing to the regulation of blood glucose levels and energy production within cells.
In biochemistry, glucose-6-phosphate is defined as:
A hexose sugar phosphate ester formed by the phosphorylation of glucose at the 6th carbon atom by ATP in a reaction catalyzed by the enzyme hexokinase or glucokinase. This reaction is the first step in both glycolysis and glucose storage (glycogen synthesis) processes, ensuring that glucose can be effectively utilized for energy production or stored for later use.
G6P serves as a crucial metabolic branch point, leading to various pathways such as:
1. Glycolysis: In the presence of sufficient ATP and NAD+ levels, G6P is further metabolized through glycolysis to generate pyruvate, which enters the citric acid cycle for additional energy production in the form of ATP, NADH, and FADH2.
2. Gluconeogenesis: During periods of low blood glucose levels, G6P can be synthesized back into glucose through the gluconeogenesis pathway, primarily occurring in the liver and kidneys. This process helps maintain stable blood glucose concentrations and provides energy to cells when dietary intake is insufficient.
3. Pentose phosphate pathway (PPP): A portion of G6P can be shunted into the PPP, an alternative metabolic route that generates NADPH, ribose-5-phosphate for nucleotide synthesis, and erythrose-4-phosphate for aromatic amino acid production. The PPP is essential in maintaining redox balance within cells and supporting biosynthetic processes.
Overall, glucose-6-phosphate plays a critical role as a central metabolic intermediate, connecting various pathways to regulate energy homeostasis, redox balance, and biosynthesis in response to cellular demands and environmental cues.
A liver cell adenoma is a benign tumor that develops in the liver and is composed of cells similar to those normally found in the liver (hepatocytes). These tumors are usually solitary, but multiple adenomas can occur, especially in women who have taken oral contraceptives for many years. Liver cell adenomas are typically asymptomatic and are often discovered incidentally during imaging studies performed for other reasons. In rare cases, they may cause symptoms such as abdominal pain or discomfort, or complications such as bleeding or rupture. Treatment options include monitoring with periodic imaging studies or surgical removal of the tumor.
Collagen type VII is a type of collagen that is a major component of the anchoring fibrils, which are structures that help to attach the epidermis (the outermost layer of the skin) to the dermis (the layer of skin directly below the epidermis). Collagen type VII is composed of three identical chains that are encoded by the COL7A1 gene. Mutations in this gene can lead to a group of inherited blistering disorders known as autosomal recessive dystrophic epidermolysis bullosa, which is characterized by fragile skin and mucous membranes that blister and tear easily, often from minor trauma or friction.
1,4-Alpha-Glucan Branching Enzyme (GBE) is an enzyme that plays a crucial role in the synthesis of glycogen, a complex carbohydrate that serves as the primary form of energy storage in animals and fungi. GBE catalyzes the transfer of a segment of a linear glucose chain (alpha-1,4 linkage) to an alpha-1,6 position on another chain, creating branches in the glucan molecule. This branching process enhances the solubility and compactness of glycogen, allowing it to be stored more efficiently within cells.
Defects in GBE are associated with a group of genetic disorders known as glycogen storage diseases type IV (GSD IV), also called Andersen's disease. This autosomal recessive disorder is characterized by the accumulation of abnormally structured glycogen in various tissues, particularly in the liver and muscles, leading to progressive liver failure, muscle weakness, cardiac complications, and sometimes neurological symptoms.
Glycogen Storage Disease Type IIb, also known as Pompe Disease, is a genetic disorder caused by a deficiency of the enzyme acid alpha-glucosidase (GAA). This enzyme is responsible for breaking down glycogen, a complex carbohydrate, into glucose within lysosomes. When GAA activity is lacking, glycogen accumulates in various tissues, including muscle and nerve cells, leading to cellular dysfunction and damage.
Type IIb Pompe Disease is characterized by progressive muscle weakness and hypertrophy (enlargement) of the heart muscle (cardiomyopathy). This form of the disease typically presents in infancy or early childhood and can progress rapidly, often resulting in severe cardiac complications and respiratory failure if left untreated.
Early diagnosis and treatment with enzyme replacement therapy (ERT) can significantly improve outcomes for individuals with Type IIb Pompe Disease. ERT involves administering recombinant human GAA to replace the deficient enzyme, helping to reduce glycogen accumulation in tissues and alleviate symptoms.
Fructose-1,6-diphosphatase deficiency is a rare inherited metabolic disorder that affects the body's ability to metabolize carbohydrates, particularly fructose and glucose. This enzyme deficiency results in an accumulation of certain metabolic intermediates, which can cause a variety of symptoms, including hypoglycemia (low blood sugar), lactic acidosis, hyperventilation, and seizures. The condition is typically diagnosed in infancy or early childhood and is treated with a diet low in fructose and other sugars that can't be metabolized properly due to the enzyme deficiency. If left untreated, the disorder can lead to serious complications, such as brain damage and death.
 Phosphofructokinase deficiency
Phosphofructokinase deficiency
PFKM
Glycogen storage disease
List of MeSH codes (C05)
Glycogen storage disease type III
Glycogen debranching enzyme
Glycogen phosphorylase
Pseudoathletic appearance
Glycogen storage disease type 0
Phosphoglycerate mutase
Glucose-6-phosphate exchanger SLC37A4
Metabolic myopathy
PYGL
G6PC2
PHKG2
Glycogen branching enzyme
Corn starch
G6PC
Glycogen storage disease type V
Phosphorylase kinase, alpha 1
Glycogen storage disease type IV
Joannes Cassianus Pompe
List of MeSH codes (C16)
Peter J. Taub
Hepatocellular adenoma
Medical genetics of Jews
Lactate dehydrogenase
Norwegian Forest cat
Lafora disease
Neutropenia
 Glycogen storage disease type VII: MedlinePlus Genetics
Glycogen storage disease type VII: MedlinePlus Genetics
 Type VII Glycogen Storage Disease: Practice Essentials, Background, Pathophysiology
Type VII Glycogen Storage Disease: Practice Essentials, Background, Pathophysiology
Glycogen Storage Diseases Types I-VII: Background, Pathophysiology, Etiology
Type VII Glycogen Storage Disease Differential Diagnoses
Type VII Glycogen Storage Disease: Background, Pathophysiology, Epidemiology
Phosphofructokinase deficiency - Wikipedia
 Glycogen Storage Disease Type VII (GSD VII) | Syndromes: Rapid Recognition and Perioperative Implications |...
Glycogen Storage Disease Type VII (GSD VII) | Syndromes: Rapid Recognition and Perioperative Implications |...
 Shailendra B. Patel
Shailendra B. Patel
 PFKM Enzyme Human Recombinant | Phosphohexokinase | ProSpec
PFKM Enzyme Human Recombinant | Phosphohexokinase | ProSpec
Genetics of Glycogen-Storage Disease Type II (Pompe Disease) Medication: Enzyme replacement, Pharmacologic Chaperones
Embark Dog DNA Test - Breed
Embark Dog DNA Test - Breed
 Glycogen debranching enzyme - wikidoc
Glycogen debranching enzyme - wikidoc
Specific PHGKB|Rare Diseases PHGKB|PHGKB
 Rabies Certificate - Veripet EZ-Health
Rabies Certificate - Veripet EZ-Health
 World Endocrinology Congress 2018 | Endocrinology Conferences 2018 | Diabetes Conferences 2018 | Diabetes Symposium | Endocrine...
World Endocrinology Congress 2018 | Endocrinology Conferences 2018 | Diabetes Conferences 2018 | Diabetes Symposium | Endocrine...
 Embark Dog DNA Test Kit: Dog Breed & Health Test
- Embark Vet
Embark Dog DNA Test Kit: Dog Breed & Health Test
- Embark Vet
 Association of the congenital neuromuscular form of glycogen storage disease type IV with a large deletion and recurrent...
Association of the congenital neuromuscular form of glycogen storage disease type IV with a large deletion and recurrent...
 Metabolic Disease - body, last, causes
Metabolic Disease - body, last, causes
Muscular Dystrophy, Emery-Dreifuss | Profiles RNS
MH DELETED MN ADDED MN
 MH DELETED MN ADDED MN
MH DELETED MN ADDED MN
MH DELETED MN ADDED MN
MH DELETED MN ADDED MN
MH DELETED MN ADDED MN
 Expanded Carrier Screening | Thermo Fisher Scientific - US
Expanded Carrier Screening | Thermo Fisher Scientific - US
 Glycogen storage disease type II (NORD): Video | Osmosis
Glycogen storage disease type II (NORD): Video | Osmosis
 Glycogen Storage Diseases - Children's Health Issues - MSD Manual Consumer Version
Glycogen Storage Diseases - Children's Health Issues - MSD Manual Consumer Version
Mucopolysaccharidoses Types I-VII Differential Diagnoses
Enzyme28
- This gene provides instructions for making one piece (the PFKM subunit) of an enzyme called phosphofructokinase, which plays a role in the breakdown of glycogen . (medlineplus.gov)
- Glycogen storage disease (GSD) VII (Tarui disease) is an autosomal recessive disorder caused by a deficiency of phosphofructokinase (PFK), the enzyme that catalyzes the rate-limiting step in glycolysis. (medscape.com)
- Glycogen storage disease (GSD) is the result of an enzyme defect. (medscape.com)
- Enzyme deficiency results in glycogen accumulation in tissues, especially liver and skeletal muscle cells. (medscape.com)
- Although at least 14 unique GSDs are discussed in the literature, the 4 that cause clinically significant muscle weakness are Pompe disease ( GSD type II , acid maltase deficiency ), Cori disease ( GSD type III , debranching enzyme deficiency), McArdle disease ( GSD type V , myophosphorylase deficiency), and Tarui disease (GSD type VII, phosphofructokinase deficiency). (medscape.com)
- These inherited enzyme defects usually present in childhood, although some, such as McArdle disease and Pompe disease, have separate adult-onset forms. (medscape.com)
- For practical purposes, depending on the enzyme activity and the presence of mutations in the G6Pase and T genes, respectively, GSD type I may be subdivided into 2 major forms. (medscape.com)
- Its clinical presentation clearly differs from other forms of GSD, because it is caused by the deficiency of the lysosomal enzyme, alpha-1,4-glucosidase, leading to the pathologic accumulation of normally structured glycogen within the lysosomes of most tissues, differs Three forms of the disease exist: infantile-onset, late-onset juvenile and adult onset. (medscape.com)
- It is an autosomal recessive disorder in which there is an AGL gene mutations which causes deficiency in glycogen debranchinging enzyme and limited storage of dextrin. (medscape.com)
- GSD type IV, also known as amylopectinosis, Glycogen Branching enzyme deficiency (GBE) or Andersen disease, is a rare disease that leads to early death. (medscape.com)
- Enzyme deficiency results in glycogen accumulation in tissues. (medscape.com)
- With an enzyme defect, carbohydrate metabolic pathways are blocked and excess glycogen accumulates in affected tissues. (medscape.com)
- citation needed] Phosphofructokinase is a tetrameric enzyme that consists of three types of subunits: PFKL (liver), PFKM (muscle), and PFKP (platelet). (wikipedia.org)
- Enzyme replacement therapies are available for all age groups (ie, infantile [early onset] or late onset [juvenile/adult]) affected by Pompe disease. (medscape.com)
- Indicated in combination with miglustat (Opfolda) for adults with late-onset Pompe disease (lysosomal acid alpha-glucosidase [GAA] deficiency) who weigh ≥40 kg and are not improving on their current enzyme replacement therapy (ERT). (medscape.com)
- A debranching enzyme is a molecule that helps facilitate the breakdown of glycogen , which serves as a store of glucose in the body, through glucosyltransferase and glucosidase activity. (wikidoc.org)
- [1] When glycogen breakdown is compromised by mutations in the glycogen debranching enzyme, metabolic diseases such as Glycogen storage disease type III can result. (wikidoc.org)
- When glucosyltransferase and glucosidase are catalyzed by distinct enzymes, "glycogen debranching enzyme" usually refers to the glucosidase enzyme . (wikidoc.org)
- Glycogen debranching enzymes assist phosphorylase, the primary enzyme involved in glycogen breakdown , mobilize glycogen stores. (wikidoc.org)
- Anderson disease, also known as glycogen storage disease type IV (MIM 232500), is a rare autosomal recessive disorder caused by a deficiency of glycogen branching enzyme. (nih.gov)
- Pompe disease , also called glycogen storage disease type II, is a genetically inherited condition caused by insufficient functioning of an enzyme called lysosomal acid alpha-1,4-glucosidase, or just acid alpha-glucosidase, and it's caused by a mutation of the GAA gene. (osmosis.org)
- Now for some reason, and it's not really understood why, but small amounts of glycogen end up in the lysosomes, where it's broken down by an enzyme called acid alpha-glucosidase, to release glucose from the glycogen chain. (osmosis.org)
- Glycogen storage diseases are caused by the lack of an enzyme needed to change glucose into glycogen and break down glycogen into glucose. (msdmanuals.com)
- In the GSD type1b, the enzyme glucose-6-phosphatase (G6P) cannot be transported across the microsomal membrane in the liver, and the glycogen cannot be metabolized [ 3 ]. (opendentistryjournal.com)
- Oxidative stress and inflammation in mucopolysaccharidosis type IVA patients treated with enzyme replacement therapy. (medscape.com)
- The lysosomal storage disorders are hereditary metabolic disorders characterized by autosomal recessive inheritance, mainly caused by deficiency of an enzyme responsible for the intra-lysosomal breakdown of various substrates and products of cellular metabolism. (bvsalud.org)
- Molecular genetic testing of LSDs is required for diagnostic confirmation when lysosomal enzyme assays are not available or not feasible to perform, and for the identification of the disease causing genetic variants. (bvsalud.org)
- Glycogen storage diseases (GSDs) are a group of inborn errors of metabolism, typically caused by enzyme defects, resulting in a buildup of glycogen in the liver, muscles, and other organs. (arupconsult.com)
Glucose22
- Glycogen can be broken down rapidly into the simple sugar glucose when energy is needed, for instance to maintain normal blood glucose levels between meals or for energy during exercise. (medlineplus.gov)
- These enzymes normally catalyze reactions that ultimately convert glycogen compounds to glucose. (medscape.com)
- One form, von Gierke disease ( GSD type Ia , glucose-6-phosphatase deficiency), causes clinically significant end-organ disease with significant morbidity. (medscape.com)
- Glycogen storage disease (GSD) type I, also known as von Gierke disease, is a group of inherited autosomal recessive metabolic disorders of the glucose-6- phosphatase system which helps maintain glucose homeostasis. (medscape.com)
- [ 1 ] In 1952, Cori and Cori demonstrated that glucose-6-phosphatase (G6Pase) deficiency was a cause of GSD type I. (medscape.com)
- [ 2 ] In 1978, Narisawa et al proposed that a transport defect of glucose-6-phosphate (G6P) into the microsomal compartment may be present in some patients with GSD type I. (medscape.com)
- GSD type Id is deficiency in a transporter that translocates free glucose molecules from microsomes into the cytosol. (medscape.com)
- glucose cannot be used as a source of energy and glycogen accumulates because of impaired degradation and/or excess synthesis. (mhmedical.com)
- Together with phosphorylases , debranching enzymes mobilize glucose reserves from glycogen deposits in the muscles and liver. (wikidoc.org)
- Glycogen breakdown is highly regulated in the body, especially in the liver , by various hormones including insulin and glucagon , to maintain a homeostatic balance of blood-glucose levels. (wikidoc.org)
- Together with phosphorylase , glycogen debranching enzymes function in glycogen breakdown and glucose mobilization. (wikidoc.org)
- When phosphorylase has digested a glycogen branch down to four glucose residues, it will not remove further residues. (wikidoc.org)
- Phosphorylase can only cleave α-1,4- glycosidic bond between adjacent glucose molecules in glycogen but branches exist as α-1,6 linkages. (wikidoc.org)
- 4-α-D-glucanotransferase ( EC 2.4.1.25 ), or glucosyltransferase , transfers three glucose residues from the four-residue glycogen branch to a nearby branch. (wikidoc.org)
- Amylo-α-1,6-glucosidase ( EC 3.2.1.33 ), or glucosidase , cleaves the remaining alpha-1,6 linkage, producing glucose and a linear chain of glycogen. (wikidoc.org)
- It is thought to proceed through a two step acid base assistance type mechanism, with an oxocarbenium ion intermediate, and retention of configuration in glucose. (wikidoc.org)
- When the body needs energy, it breaks down the glycogen into glucose, a form of sugar. (humanillnesses.com)
- Glucose is used for energy by most cells of the body , and it's stored inside the cells as a compact, branch-shaped molecule called glycogen . (osmosis.org)
- Any glucose that is not used immediately for energy is held in reserve in the liver, muscles, and kidneys in the form of glycogen and is released when needed by the body. (msdmanuals.com)
- Specific enzymes breakdown molecules of glycogen into glucose, which is used as a source of energy by the organism. (opendentistryjournal.com)
- Normally your enzymes break carbohydrates down into glucose (a type of sugar). (medlineplus.gov)
- One of the patients showed distinctively lower glucose levels and higher lactate and ketone body contents, suggesting poorer metabolic control of the disease compared with other patients. (ua.pt)
Liver14
- Glycogen is a complex multibranched polysaccharide that acts as energy storage in the human body and is formed mainly in the liver and skeletal muscles. (medscape.com)
- GSD type Ia demonstrates deficient G6Pase activity in the fresh and frozen liver tissue. (medscape.com)
- The disease presents with variable cardiac muscle, skeletal muscle and liver involvement and has different subtypes. (medscape.com)
- For instance, the body's tissues store a carbohydrate called glycogen (GLY-ko-jen) in the liver * and the muscles. (humanillnesses.com)
- Now, normally, glycogen is found in the largest amounts in the cytoplasm of liver cells and all three types of muscle cell . (osmosis.org)
- that occur when there is a defect in the enzymes that are involved in the metabolism of glycogen, often resulting in growth abnormalities, weakness, a large liver, low blood sugar, and confusion. (msdmanuals.com)
- For types I, III, and VI, symptoms are low levels of sugar in the blood ( hypoglycemia ) and protrusion of the abdomen (because excess or abnormal glycogen may enlarge the liver). (msdmanuals.com)
- The glycogen accumulates in several organs, such as liver and kidneys [ 4 , 5 ], and the disease leads to systemic and intraoral manifestations. (opendentistryjournal.com)
- Glycogen storage disease type VI is a type of glycogen storage disease caused by a deficiency in liver glycogen phosphorylase . (chemeurope.com)
- Other individuals have a multitude of the most severe symptoms of end-stage liver disease and a limited chance for survival. (medscape.com)
- Specific medical therapies may be applied to many liver diseases in an effort to diminish symptoms and to prevent or forestall the development of cirrhosis. (medscape.com)
- Numerous treatment strategies for acute liver failure simply prevent complications and decelerate disease progression. (springer.com)
- Although liver failure can be treated via hepatocyte transplantation, it also faces multiple problems comprising the shortage of high-quality hepatocytes sources, rejection of allogeneic transplants, difficulty to expand, and losing hepatic characteristics in vitro [ 7 , 8 ]. (springer.com)
- You also have about 500 grams of carbs stored as glycogen in muscle and liver tissue. (carbmanager.com)
Inherited disorder1
- Glycogen storage disease type VII (GSDVII) is an inherited disorder caused by an inability to break down a complex sugar called glycogen in muscle cells. (medlineplus.gov)
Accumulates1
- In individuals with Pompe, glycogen mostly accumulates in the lysosomes of those cells. (osmosis.org)
Mutations4
- Raben N, Sherman JB, Adams E. Various classes of mutations in patients with phosphofructokinase deficiency (Tarui''s disease). (medscape.com)
- Mutations in PFKM gene have been related with glycogen storage disease type VII, also identified as Tarui disease. (prospecbio.com)
- This case report also highlights the need for a more comprehensive search for large deletion mutations associated with glycogen storage disease type IV, especially if routine GBE1 gene sequencing results are equivocal. (nih.gov)
- Because a breed group is genetically related, mutations and diseases in ancestors or within a breed put all breeds within that group at risk for the same inherited disorders. (pawprintgenetics.com)
Form of glycogen1
- About 1 in 25,000 infants has some form of glycogen storage disease. (msdmanuals.com)
Phosphofructokinase deficiency6
- Clinical features and new molecular findings in muscle phosphofructokinase deficiency (GSD type VII). (medscape.com)
- Muscle phosphofructokinase deficiency (Tarui''s disease): report of a case. (medscape.com)
- Garcia et al investigated the effects of phosphofructokinase deficiency in tissue other than skeletal muscle on the pathogenesis of GSD type VII. (medscape.com)
- Classic phosphofructokinase deficiency is the most common type of this disorder. (wikipedia.org)
- Symptoms of phosphofructokinase deficiency can closely resemble those of other metabolic diseases, include deficiencies of phosphoglycerate kinase, phosphoglycerate mutase, lactate dehydrogenase, beta-enolase and aldolase A. Thus, proper diagnosis is important to determine a treatment plan. (wikipedia.org)
- One of the four glycogen storage diseases characterized by phosphofructokinase deficiency in the muscles and associated with abnormal deposition of glycogen in muscle tissues, exercise intolerance, and anemia. (mhmedical.com)
Metabolism3
- Dr. Garrod called these diseases 'inborn errors of metabolism,' a name that persists to this day. (humanillnesses.com)
- The 7 patients in the first group had an IMD involving energy metabolism. (pediatriconcall.com)
- The metabolic myopathies (MM) are a group of muscle disorders resulting from failed energy production related to defects in glycogen, lipid or mitochondrial metabolism. (pediatriconcall.com)
Patients17
- These subtypes are clinically indistinguishable from one another, except for the fact that patients with GSD type Ib have altered neutrophil functions predisposing them to gram-positive bacterial infections. (medscape.com)
- GSD IIIa is the most common subtype, affecting about 85% of patients with this disease. (medscape.com)
- GSD IIIb is less severe and less common, affecting 15% of patients with the disease. (medscape.com)
- Differentiating patients with GSD type III from those with GSD type I solely on the basis of physical findings is not easy. (medscape.com)
- The long-term outcome of patients with glycogen storage diseases. (medscape.com)
- Myozyme has been shown to improve ventilator-free survival in patients with infantile-onset Pompe disease compared with untreated historical controls. (medscape.com)
- Indicated for treatment of patients aged 1 year and older with late-onset Pompe disease. (medscape.com)
- Natural history of extensive Mongolian spots in mucopolysaccharidosis type II (Hunter syndrome): a survey among 52 Japanese patients. (medscape.com)
- This classification is fundamental to the type of marketing authorization (MA), and therefore to the controls to be performed, from preclinical stages through clinical trials to pharmacovigilance, to meet the safety requirements for patients. (frontiersin.org)
- AIM: In many countries, adult clinics specifically dedicated to adult patients with lysosomal storage diseases (LSDs) do not exist. (bvsalud.org)
- RESULTS: A total of 23 LSD patients and parents of a patient with mucopolysaccharidosis type-3b with intellectual deficit were interviewed, with 84.6% of patients diagnosed after the age of 18 years and 18% of patients diagnosed before the age of 18 years desiring management by adult physicians. (bvsalud.org)
- When your patients are looking to understand if they are a carrier for specific genetic conditions like cystic fibrosis, spinal muscular atrophy, fragile X syndrome, or Tay-Sachs disease, appropriate genetic screening and actionable results are essential . (questwomenshealth.com)
- For example, in familial hypercholesterolemia, enzymes do not receive the signals that typically inhibit cholesterol synthesis, so that excessive production of cholesterol occurs, leading to early coronary vascular disease and strokes in patients. (newworldencyclopedia.org)
- A total of 138 patients were included, 7 with an IMD and 131 without. (pediatriconcall.com)
- 6,7 Patients with metabolic myopathies may present with indolent myopathic features, exercise intolerance or recurrent rhabdomyolysis. (pediatriconcall.com)
- CV004 trade name] may also be used in the treatment of coronavirus disease 2019 (COVID-19) in adult and adolescent patients (aged 12 years and older with body weight of at least 40 kg) who require supplemental oxygen therapy. (who.int)
- In 1999, after a 1998 enterovirus 71 epidemic, the infection that required intensive care, and 7 of those Taiwan Centers for Disease Control established a nation- patients died. (cdc.gov)
Glycogenosis3
- Finsterer J, Stollberger C, Kopsa W. Neurologic and cardiac progression of glycogenosis type VII over an eight-year period. (medscape.com)
- Finsterer J, Stollberger C. Progressive mitral valve thickening and progressive muscle cramps as manifestations of glycogenosis VII (Tarui's Disease). (medscape.com)
- Glycogenosis type VII. (mhmedical.com)
Tarui3
- Tarui disease resolves with rest, and, although no specific treatment exists, the condition may not progress to severe disability. (medscape.com)
- Tarui disease is an autosomal recessive condition. (medscape.com)
- Also named Tarui disease after the Japanese physician Seiichiro Tarui (born in 1927) who first described the disease in 1965. (mhmedical.com)
20231
- 30(7): 450-454, 2023 Oct. (bvsalud.org)
Hepatic1
- Nutrition therapy for hepatic glycogen storage diseases. (medscape.com)
Syndrome5
- The mild form of mucopolysaccharidosis type I (Scheie syndrome) is associated with increased ascending aortic stiffness. (medscape.com)
- High prevalence of carpal tunnel syndrome in children with mucopolysaccharidosis type II (Hunter syndrome). (medscape.com)
- Dodsworth Ch, Burton B K,. Increased incidence of neonatal respiratory distress in infants withmucopolysaccharidosis type II (MPS II, Hunter syndrome). (medscape.com)
- The Ashkenazi Jewish Panel includes the following diseases: Bloom syndrome, Canavan disease, Fanconi anemia type C, familial dysautonomia, Gaucher disease, glycogen storage disease type 1a, Mucolipidosis IV, Neimann-Pick disease, and Tay-Sachs disease. (cdc.gov)
- AIDS-like syndrome: AIDS-like disease (illness) (syndrome) ARC AIDS-related complex Pre-AIDS AIDS-related conditions Prodromal-AIDS 3. (cdc.gov)
Accumulation1
- At the opportunity, the pathologist visualized glycogen accumulation in vesicles inside the cardiac fibers1. (bvsalud.org)
Tissues1
- A metabolic (met-a-BOLL-ik) disease is a condition that interferes with the bodys chemical processes involved in growth, maintenance of healthy tissues, disposal of waste products, and production of energy to fuel body functions. (humanillnesses.com)
Lysosomes3
- It binds to mannose-6-phosphate receptors and then is transported into lysosomes, then undergoes proteolytic cleavage that results in increased enzymatic activity and ability to cleave glycogen. (medscape.com)
- In Pompe disease , a mutation of the GAA gene prevents the production of enough functional acid alpha-glucosidase, and as a result, lysosomes can't break down glycogen . (osmosis.org)
- This leads to a buildup of glycogen within the cytoplasm and lysosomes, and that leads to cellular damage and destruction. (osmosis.org)
Lipid1
- Some severe diseases, such as many of the lipid storage diseases, currently have no effective therapy. (newworldencyclopedia.org)
Biochemical1
- A metabolic disorder is any disease or disorder that negatively affects the biochemical reactions through which individual animal cells process nutrient molecules (such as the components of carbohydrates , proteins , and fats ) to yield energy or perform the functions necessary to sustain life (such as building complex molecules and creating cellular structure). (newworldencyclopedia.org)
Disorders4
- The glycogen storage disease (GSD) is a group of inherited disorders that involve deficiencies in the enzymes that metabolize glycogen. (opendentistryjournal.com)
- Background & objectives: Lysosomal storage disorders (LSDs) are genetic metabolic disorders which result from deficiency of lysosomal enzymes or defects in other lysosomal components. (bvsalud.org)
- BACKGROUND: GD and ASMD are lysosomal storage disorders that enter into differential diagnosis due to the possible overlap in their clinical manifestations. (bvsalud.org)
- there are many types and subtypes, and other disorders may have overlapping phenotypes. (arupconsult.com)
Abnormal1
- Glycogen deposited in these organs has an abnormal structure. (medscape.com)
Lysosomal1
- GSD type II, also known as alpha glucosidase deficiency (GAA, acid maltase deficiency) or Pompe disease, is a prototypic lysosomal disease. (medscape.com)
Excess1
- When you eat a high-carb meal, your blood sugar spikes, and insulin arrives to store that excess sugar as glycogen or fat. (carbmanager.com)
Skeletal muscles1
- As a result, no functional phosphofructokinase is formed in skeletal muscles, and glycogen cannot be completely broken down. (medlineplus.gov)
McArdle1
- Pharmacological and Nutritional Treatment for McArdle Disease (Glycogen Storage Disease Type V)." Evidence-Based Medicine Guidelines , Duodecim Medical Publications Limited, 2019. (unboundmedicine.com)
Hereditary1
- In lectures delivered in 1908, Garrod described several hereditary diseases that are caused by too little or complete lack of certain enzymes (EN-zimes). (humanillnesses.com)
Pompe11
- Pompe initially described the disease in 1932. (medscape.com)
- Pompe disease). (medscape.com)
- Replaces rhGAA, which is deficient or lacking in persons with Pompe disease. (medscape.com)
- It has not been adequately studied for treatment of other forms of Pompe disease. (medscape.com)
- Lumizyme is indicated for infantile-onset Pompe disease and also for late (non-infantile) Pompe disease. (medscape.com)
- Pompe disease: early diagnosis and early treatment make a difference. (medscape.com)
- Clinical features and predictors for disease natural progression in adults with Pompe disease: a nationwide prospective observational study. (medscape.com)
- Pompe disease is an autosomal recessive condition - so in other words, both parents must be carriers. (osmosis.org)
- to investigate nursing team knowledge and practices regarding care for children with Pompe disease in intensive care. (bvsalud.org)
- Pompe en cuidados intensivos. (bvsalud.org)
- Pompe Disease (PD) was discovered in 1932 by pathologist Joannes Cassianus Pompe, during the autopsy of a seven-month-old child who died from idiopathic myocardial hypertrophy. (bvsalud.org)
Muscles1
- Muscles that do not have access to glycogen as an energy source become weakened and cramped following moderate strain, such as exercise, and in some cases, begin to break down. (medlineplus.gov)
Enzymes3
- Proteins that catalyze both functions are referred to as glycogen debranching enzymes (GDEs). (wikidoc.org)
- Thus the debranching enzymes, transferase and α-1,6- glucosidase converts the branched glycogen structure into a linear one, paving the way for further cleavage by phosphorylase. (wikidoc.org)
- The GSD is a rare autosomal recessive disease, and it exists in a variety of forms in accordance to specific enzymes involved [ 2 ]. (opendentistryjournal.com)
Carbohydrates2
- Treatment depends on the type of glycogen storage disease and usually involves regulating the intake of carbohydrates. (msdmanuals.com)
- Carbohydrates Carbohydrates, proteins, and fats are the main types of macronutrients in food (nutrients that are required daily in large quantities). (msdmanuals.com)
Cleave1
- [7] This is a common method through which to cleave bonds, with an acid below the site of hydrolysis to lend a proton and a base above to deprotinate a water which can then act as a nucleophile . (wikidoc.org)
Phenotypic1
- The table below shows human diseases predicted to be associated to Mcm3 by phenotypic similarity . (mousephenotype.org)
20161
- BMW boasts that the all-new 2016 7 Series features 24 new innovations, and that half of those are segment exclusives. (charitycar.us)
Clinical5
- Clinical features similar to GSD type V. Temporary weakness and painful muscle cramps occur after exercise. (mhmedical.com)
- Three clinical forms have been described: classic, infantile onset, and late onset type. (mhmedical.com)
- Glycogen storage disease type IV has a broad clinical spectrum ranging from a perinatal lethal form to a nonprogressive later-onset disease in adults. (nih.gov)
- The patient should be observed closely for signs that the dose may need to be altered, such as changes in clinical status resulting from disease remissions or exacerbations. (who.int)
- Clinical information was collected for 202 settings ( 5-7 ). (cdc.gov)
Breakdown1
- A lack of glycogen breakdown interferes with the function of muscle cells. (medlineplus.gov)
Infants1
- PFK deficient infants also often have some type of respiratory issue. (wikipedia.org)
Gierke1
- Von Gierke described the first patient with GSD type I in 1929 under the name hepatonephromegalia glycogenica. (medscape.com)
20211
- J Mol Diag 2021 23:1501-1506 external icon ) ClinGen Variant Curation Expert Panels nominated 546 pathogenic and difficult to detect variants ( link to table of variants excel icon ) in 84 disease-associated genes ( link to table of genes word icon ). (cdc.gov)
Cori1
- GSD type III is also known as Forbes-Cori disease or limit dextrinosis. (medscape.com)
Specimens1
- GSD type Ib demonstrates normal G6Pase activity in the frozen tissue samples and lowered activity in the fresh specimens. (medscape.com)
Severe2
- Many of the more severe symptoms found in the classic type of this disease are absent in the late-onset form. (wikipedia.org)
- Doses of less than 0.4 mg may be sufficient in less severe conditions while severe and life-threatening diseases may require up to 20 mg or more a day. (who.int)
Acid3
- This type presents with exercise-induced muscle cramps and weakness (sometimes rhabdomyolysis), myoglobinuria, as well as with haemolytic anaemia causing dark urine a few hours later.Hyperuricemia is common, due to the kidneys' inability to process uric acid following damage resulting from processing myoglobin. (wikipedia.org)
- Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. (medscape.com)
- Genes Genes are segments of deoxyribonucleic acid (DNA) that contain the code for a specific protein that functions in one or more types of cells in the body or the code for functional ribonucleic. (msdmanuals.com)
NORD2
- Please note that NORD provides this information for the benefit of the rare disease community. (rarediseases.org)
- NORD is not a medical provider or health care facility and thus can neither diagnose any disease or disorder nor endorse or recommend any specific medical treatments. (rarediseases.org)
Pharmacological1
- Miglustat alone has no pharmacological activity in cleaving glycogen. (medscape.com)