Family study of inherited syndrome with multiple congenital deformities: symphalangism, carpal and tarsal fusion, brachydactyly, craniosynostosis, strabismus, hip osteochondritis. (1/270)
A syndrome of brachydactyly (absence of some middle or distal phalanges), aplastic or hypoplastic nails, symphalangism (ankylois of proximal interphalangeal joints), synostosis of some carpal and tarsal bones, craniosynostosis, and dysplastic hip joints is reported in five members of an Italian family. It may represent a previously undescribed autosomal dominant trait. (+info)A novel skeletal dysplasia with developmental delay and acanthosis nigricans is caused by a Lys650Met mutation in the fibroblast growth factor receptor 3 gene. (2/270)
We have identified a novel fibroblast growth factor receptor 3 (FGFR3) missense mutation in four unrelated individuals with skeletal dysplasia that approaches the severity observed in thanatophoric dysplasia type I (TD1). However, three of the four individuals developed extensive areas of acanthosis nigricans beginning in early childhood, suffer from severe neurological impairments, and have survived past infancy without prolonged life-support measures. The FGFR3 mutation (A1949T: Lys650Met) occurs at the nucleotide adjacent to the TD type II (TD2) mutation (A1948G: Lys650Glu) and results in a different amino acid substitution at a highly conserved codon in the kinase domain activation loop. Transient transfection studies with FGFR3 mutant constructs show that the Lys650Met mutation causes a dramatic increase in constitutive receptor kinase activity, approximately three times greater than that observed with the Lys650Glu mutation. We refer to the phenotype caused by the Lys650Met mutation as "severe achondroplasia with developmental delay and acanthosis nigricans" (SADDAN) because it differs significantly from the phenotypes of other known FGFR3 mutations. (+info)Non-invasive aortic blood flow measurement in infants during repair of craniosynostosis. (3/270)
We have assessed the potential clinical benefit of a new echo-Doppler device (Dynemo 3000) which provides a continuous measure of aortic blood flow (ABF) using an aortic flowmeter and a paediatric oesophageal probe, during repair of craniosynostosis in infants under general anaesthesia. The data recorded included: ABFi (i = indexed to body surface area), stroke volume (SVi), systemic vascular resistance (TSVRi), pre-ejection period (PEP), left ventricular ejection time (LVET), mean arterial pressure (MAP), heart rate (HR) and central venous pressure (CVP). Data were collected: before (T1) and 3 min after skin incision (T2), at the time of maximal haemorrhage (T3) and at the end of the procedure (T4). Twelve infants (aged 7.0 (range 6-12) months) were included. ABFi, MAP and CVP were significantly lower at T3 compared with T1 (2.0 (0.8) vs 3.0 (0.8) litre min-1 m-2, 46.1 (5.8) vs 65.2 (8.9) mm Hg and 2.8 (1.6) vs 5.2 (2.1) mm Hg; P < 0.05). PEP/LVET ratio was significantly lower at T2 compared with T1 (0.25 (0.05) vs 0.30 (0.06)) and increased at T4 (0.36 (0.04); P < 0.05). These preliminary results suggest that this non-invasive ABF echo-Doppler device may be useful for continuous haemodynamic monitoring during a surgical procedure associated with haemorrhage in infants. (+info)Decreased proliferation and altered differentiation in osteoblasts from genetically and clinically distinct craniosynostotic disorders. (4/270)
Craniosynostoses are a heterogeneous group of disorders characterized by premature fusion of cranial sutures. Mutations in fibroblast growth factor receptors (FGFRs) have been associated with a number of such conditions. Nevertheless, the cellular mechanism(s) involved remain unknown. We analyzed cell proliferation and differentiation in osteoblasts obtained from patients with three genetically and clinically distinct craniosynostoses: Pfeiffer syndrome carrying the FGFR2 C342R substitution, Apert syndrome with FGFR2 P253R change, and a nonsyndromic craniosynostosis without FGFR canonic mutations, as compared with control osteoblasts. Osteoblasts from craniosynostotic patients exhibited a lower proliferation rate than control osteoblasts. P253R and nonsyndromic craniosynostosis osteoblasts showed a marked differentiated phenotype, characterized by high alkaline phosphatase activity, increased mineralization and expression of noncollagenous matrix proteins, associated with high expression and activation of protein kinase Calpha and protein kinase Cepsilon isoenzymes. By contrast, the low proliferation rate of C342R osteoblasts was not associated with a differentiated phenotype. Although they showed higher alkaline phosphatase activity than control, C342R osteoblasts failed to mineralize and expressed low levels of osteopontin and osteonectin and high protein kinase Czeta levels. Stimulation of proliferation and inhibition of differentiation were observed in all cultures on FGF2 treatment. Our results suggest that an anticipated proliferative/differentiative switch, associated with alterations of the FGFR transduction pathways, could be the causative common feature in craniosynostosis and that mutations in distinct FGFR2 domains are associated with an in vitro heterogeneous differentiative phenotype. (+info)Fetal craniofacial structure and intracranial morphology in a case of Apert syndrome. (5/270)
Apert syndrome is characterized by craniosynostosis, midfacial hypoplasia and bilateral syndactyly. We document in detail the intrauterine natural history of Apert syndrome by serial sonographic examination. Ultrasound examination of a 19-week fetus revealed an abnormal appearance of the skull. The subsequent examination including transvaginal brain scanning demonstrated a deformed occipital part of the cerebrum and lateral ventricles, frontal bossing, a low nasal bridge and an abnormal appearance of the fetal hands and feet. The distortion of the fetal profile became progressively worse with advancing gestation. Towards the end of pregnancy, anterior prominence of the cerebrum, ventricles and corpus callosum was demonstrated and mild non-progressive ventriculomegaly was seen. The female 3152-g newborn with the typical facial appearance of Apert syndrome, bilateral syndactyly of the fingers and toes and isolated cleft palate was delivered at 37 weeks. Postnatal three-dimensional computed tomography scan demonstrated the fusion of the coronal suture and a wide mid-line calvarial defect, and cranial magnetic resonance imaging confirmed the prenatal sonographic findings. Although the karyotype was normal, genomic DNA analysis of the fibroblast growth factor receptor 2 revealed Ser252Trp, which is specified in the mutational basis of Apert syndrome. The time course of the prenatal findings in this case may help increase understanding of the intrauterine natural history of Apert syndrome. (+info)Reduction of operating time and blood transfusion for craniosynostosis by simulated surgery using three-dimensional solid models. (6/270)
Preoperative planning of craniofacial synostosis can be achieved through the use of two- or three-dimensional (3D) computed tomography (CT) images and by 3D solid models. The advantage of using 3D models was evaluated by calculating the amount of blood transfused and the operating time for 36 craniosynostosis procedures, 21 planned with 3D models and 15 with CT images performed in the past 7 years. The use of 3D models reduced both blood loss and operating time for fronto-orbital advancement with reshaping, LeFort III advancement, and LeFort IV minus Glabellar advancement; blood loss for fronto-orbital advancement without reshaping; and operating time for total cranial reshaping. (+info)Three-dimensional morphological analysis of isolated metopic synostosis. (7/270)
Morphological differences were quantified in three-dimensions among individuals with untreated isolated metopic synostosis and between those individuals and similar aged-matched normal dry skulls to test two hypotheses: first, that the dysmorphology is a self-correcting condition; and second, that a lack of vertical growth of the skull produces this dysmorphology. Three-dimensional (3D) coordinates were recorded for 22 craniofacial landmarks from CT scans of 15 metopic patients, ranging from 5- to 32-months-old, and of four normal dry skulls, ranging in age from 6- to 36-months-old. The patient population was diagnosed with isolated metopic synostosis at The Johns Hopkins Medical Institutions in Baltimore, Maryland or Children's Hospital in St. Louis, Missouri. Comparisons between the metopic age groups indicate that the trigonocephalic phenotype worsens through time. Between 5 and 14 months, the neurocranium displays an increase in vertical growth. This was followed by a lack of vertical growth between 14 and 32 months. The face displays a lack of vertical growth from 5 to 14 months and an increase in vertical growth after 14 months. Comparisons between the metopic age groups and the normal skulls indicate that the trigonocephalic head is taller superoinferiorly and longer anteroposteriorly. Relative to the normal phenotype, the inferior temporal region in the metopic phenotype is narrow. These findings enabled the rejection of both hypotheses and localized form differences between normal and metopic phenotypes. Based on these results, we suggest that the trigonocephalic phenotype worsens with age and the amount of vertical growth that produces the trigonocephalic phenotype varies throughout growth with respect to location within the skull and age. (+info)Evidence for digenic inheritance in some cases of Antley-Bixler syndrome? (8/270)
The Antley-Bixler syndrome has been thought to be caused by an autosomal recessive gene. However, patients with this phenotype have been reported with a new dominant mutation at the FGFR2 locus as well as in the offspring of mothers taking the antifungal agent fluconazole during early pregnancy. In addition to the craniosynostosis and joint ankylosis which are the clinical hallmarks of the condition, many patients, especially females, have genital abnormalities. We now report abnormalities of steroid biogenesis in seven of 16 patients with an Antley-Bixler phenotype. Additionally, we identify FGFR2 mutations in seven of these 16 patients, including one patient with abnormal steroidogenesis. These findings, suggesting that some cases of Antley-Bixler syndrome are the outcome of two distinct genetic events, allow a hypothesis to be formulated under which we may explain all the differing and seemingly contradictory circumstances in which the Antley-Bixler phenotype has been recognised. (+info)Craniosynostosis is a medical condition that affects the skull of a developing fetus or infant. It is characterized by the premature closure of one or more of the fibrous sutures between the bones of the skull (cranial sutures). These sutures typically remain open during infancy to allow for the growth and development of the brain.
When a suture closes too early, it can restrict the growth of the surrounding bones and cause an abnormal shape of the head. The severity of craniosynostosis can vary depending on the number of sutures involved and the extent of the premature closure. In some cases, craniosynostosis can also lead to increased pressure on the brain, which can cause a range of neurological symptoms.
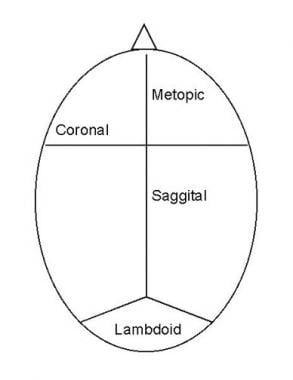
There are several types of craniosynostoses, including:
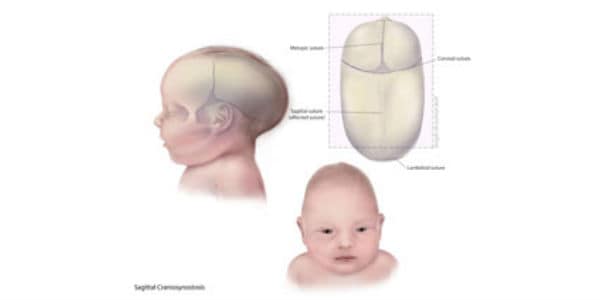
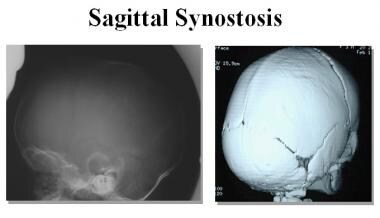
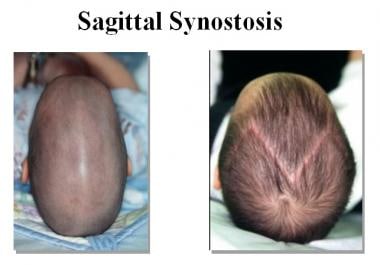
1. Sagittal synostosis: This is the most common type and involves the premature closure of the sagittal suture, which runs from front to back along the top of the head. This can cause the skull to grow long and narrow, a condition known as scaphocephaly.
2. Coronal synostosis: This type involves the premature closure of one or both of the coronal sutures, which run from the temples to the front of the head. When one suture is affected, it can cause the forehead to bulge and the eye socket on that side to sink in (anterior plagiocephaly). When both sutures are affected, it can cause a flattened appearance of the forehead and a prominent back of the head (brachycephaly).
3. Metopic synostosis: This type involves the premature closure of the metopic suture, which runs from the top of the forehead to the bridge of the nose. It can cause a triangular shape of the forehead and a prominent ridge along the midline of the skull (trigonocephaly).
4. Lambdoid synostosis: This is the least common type and involves the premature closure of the lambdoid suture, which runs along the back of the head. It can cause an asymmetrical appearance of the head and face, as well as possible neurological symptoms.
In some cases, multiple sutures may be affected, leading to more complex craniofacial abnormalities. Treatment for craniosynostosis typically involves surgery to release the fused suture(s) and reshape the skull. The timing of the surgery depends on the type and severity of the condition but is usually performed within the first year of life. Early intervention can help prevent further complications, such as increased intracranial pressure and developmental delays.
 Craniosynostosis - Wikipedia
Craniosynostosis - Wikipedia
 Craniosynostosis: MedlinePlus Medical Encyclopedia
Craniosynostosis: MedlinePlus Medical Encyclopedia
 Craniosynostosis
Craniosynostosis
 Metopic craniosynostosis: Types, treatment, and more
Metopic craniosynostosis: Types, treatment, and more
 Craniosynostosis: A Reversible Pathology?
Craniosynostosis: A Reversible Pathology?
 Craniosynostosis Management: Overview, History, Pathogenesis
Craniosynostosis Management: Overview, History, Pathogenesis
 Craniosynostosis - Seattle Children's
Craniosynostosis - Seattle Children's
 Craniosynostosis before and after photos
Craniosynostosis before and after photos
 Surgery Reshapes and Expands Aria's Skull | Craniosynostosis Patient Story
Surgery Reshapes and Expands Aria's Skull | Craniosynostosis Patient Story
 Alliance Maladies Rares - Craniosynostoses isolées
Alliance Maladies Rares - Craniosynostoses isolées
 Syndromic craniosynostosis | Headlines Charity
Syndromic craniosynostosis | Headlines Charity
 Craniosynostosis Facts from the NE Fetal Treatment Program
Craniosynostosis Facts from the NE Fetal Treatment Program
 Navigating your child's diagnosis of Craniosynostosis
Navigating your child's diagnosis of Craniosynostosis
 Craniosynostosis | Conditions | UCSF Benioff Children's Hospitals
Craniosynostosis | Conditions | UCSF Benioff Children's Hospitals
Navigating your child's diagnosis of Craniosynostosis
 Craniosynostosis Fontaine type - About the Disease - Genetic and Rare Diseases Information Center
Craniosynostosis Fontaine type - About the Disease - Genetic and Rare Diseases Information Center
 Clinical dividends from the molecular genetic diagnosis of craniosynostosis. - Radcliffe Department of Medicine
Clinical dividends from the molecular genetic diagnosis of craniosynostosis. - Radcliffe Department of Medicine
 Craniosynostosis Care at Tufts Medical Center
Craniosynostosis Care at Tufts Medical Center
 ASSESSMENT OF INTRACRANIAL VOLUME PRIOR AND AFTER SURGICAL CORRECTION IN PATIENTS WITH CRANIOSYNOSTOSIS | Journal of Population...
ASSESSMENT OF INTRACRANIAL VOLUME PRIOR AND AFTER SURGICAL CORRECTION IN PATIENTS WITH CRANIOSYNOSTOSIS | Journal of Population...
 Clinical Trials : Craniosynostoses
Clinical Trials : Craniosynostoses
Temat: 'CRANIOSYNOSTOSES' - OpacWWW - Prolib Integro
 craniosynostosis | omi-kiddos
craniosynostosis | omi-kiddos
 Craniosynostosis Workshop | Aster
Craniosynostosis Workshop | Aster
 craniosynostosis | Hereditary Ocular Diseases
craniosynostosis | Hereditary Ocular Diseases
 Craniosynostosis Reunion - Pediatric Neurosurgery
Craniosynostosis Reunion - Pediatric Neurosurgery
Pediatric Craniosynostosis: Background, Pathophysiology, Epidemiology
Safety of antifibrinolytics in 6583 pediatric patients having craniosynostosis surgery: A decade of data reported from the...
 Cranial Bones: Function and Anatomy, Diagram, Conditions, Health Tips
Cranial Bones: Function and Anatomy, Diagram, Conditions, Health Tips
Children with craniosynostosis7
- However, most children with craniosynostosis are otherwise healthy and have normal intelligence. (medlineplus.gov)
- Most children with craniosynostosis have only 1 fused suture (single-suture craniosynostosis). (seattlechildrens.org)
- Our team has more experience caring for children with craniosynostosis than any other center in the United States. (seattlechildrens.org)
- Each year we care for more than 400 children with craniosynostosis. (seattlechildrens.org)
- Intracranial volume post cranial expansion surgery using three-dimensional computed tomography scan imaging in children with craniosynostosis. (jptcp.com)
- Services for families are continually improved by direct feedback and design from the Family Advisory Board, made up of parents of children with craniosynostosis. (pediatricneurosurgery.net)
- When children with craniosynostosis, usually complex, also display other body deformities, this is termed syndromic craniosynostosis. (medscape.com)
Single-suture craniosynostosis6
- Single-suture craniosynostosis: a review of neurobehavioral research and theory. (jptcp.com)
- American society of maxillofacial surgeon's outcome study: preoperative and postoperative neurodevelopmental findings in single-suture craniosynostosis. (jptcp.com)
- We conducted a mixed-methods exploratory study between August 2019 and March 2020 to develop a decision aid about surgical treatment for single suture craniosynostosis. (figshare.com)
- Distinct booklets were created to enable focused discussion of treatment options for the 3 major types of single suture craniosynostosis (sagittal, metopic, unicoronal). (figshare.com)
- Three decision aids representing the 3 most common forms of single suture craniosynostosis were developed. (figshare.com)
- We developed a customizable decision aid for single suture craniosynostosis treatment options. (figshare.com)
Type of craniosynostosis6
- Symptoms depend on the type of craniosynostosis. (medlineplus.gov)
- This type of craniosynostosis is caused by mutations in the fibroblast growth factor receptor-2 gene, FGFR2 , located at 10q26. (arizona.edu)
- A child with premature fusion of the sagittal suture, the most common type of craniosynostosis, presents with an elongated head shape, as the skull continues to grow along the lambdoid and coronal sutures (Fig. 2). (cincinnatichildrens.org)
- Each type of craniosynostosis has unique symptoms depending on the skull sutures (joints) and bones that grow together too soon. (universityhealth.com)
- There's a low risk of abnormal brain growth and development with this type of craniosynostosis. (universityhealth.com)
- Lambdoidal synostosis is the least common type of craniosynostosis. (universityhealth.com)
Types of craniosynostosis2
- In this video, Dr. Carrie Heike explains how the different types of craniosynostosis affect a baby's skull. (seattlechildrens.org)
- Although the genetic basis has been established for many types of craniosynostosis , the etiopathogenesis of isolated lambdoid synostosis has not yet been established. (bvsalud.org)
Neurosurgery4
- If your baby is found to have craniosynostosis, he or she will receive care from specialists in many areas including: plastic surgery, neurosurgery, genetics and ophthalmology. (lifespan.org)
- Department of Neurosurgery at Aster Hospitals Bangalore & Aster Medcity, Cochin, is conducting a workshop on surgical management of Craniosynostosis to be held on the 23rd and 24th of January 2022. (asterhospitals.in)
- At TCH, the comprehensive multidisciplinary craniosynostosis surgery program has care organized around the patients' needs, so pediatric neurosurgery and plastic surgery specialists see the patient together in addition to genetics, ophthalmology, social work, anesthesiology, and otolaryngology services as part of the coordinated care. (pediatricneurosurgery.net)
- Also see the Medscape Reference Neurosurgery article Surgery for Craniosynostosis. (medscape.com)
Birth defect7
- Craniosynostosis is a birth defect in which one or more sutures on a baby's head closes earlier than usual. (medlineplus.gov)
- A birth defect which results in a misshapen head, craniosynostosis is more than a cosmetic problem. (medic8.com)
- Craniosynostosis is a birth defect that impacts the formation of a baby's skull. (classlawgroup.com)
- If your child suffers from craniosynostosis after you or a loved one used Prozac while pregnant, and you would like to receive a free consultation with our Prozac birth defect lawyers , please fill out the form on the right or call toll-free (866) 981-4800. (classlawgroup.com)
- Craniosynostosis is a type of birth defect where one or more of the sutures in between a child's skull closes before the brain is fully developed. (drtotonchi.com)
- Craniosynostosis is a birth defect in which the bones in a baby's skull join together too early. (raisingboyswithlove.com)
- Craniosynostosis is a birth defect affecting one or more of the joints or sutures of a baby's skull, causing them to close prematurely. (stanford.edu)
Cleft lip an2
- His clinical areas of focus in craniofacial surgery include cleft lip and palate, craniosynostosis, frontonasal encephalocele and complex Tessier clefts. (cappskids.org)
- As a member of the Cleft and Craniofacial Center at Boston Children's Hospital, Dr. Meara has a special interest in cleft lip and palate and craniofacial diagnoses including syndromic and non-syndromic craniosynostosis, frontonasal encephalocele and complex Tessier facial cleft. (cappskids.org)
Neurodevelopmental2
- Some evidence links metopic craniosynostosis with neurodevelopmental delays, meaning that some children with the condition reach their milestones later than children of a similar age who do not have this condition. (medicalnewstoday.com)
- [ 1 ] Plain radiography, ultrasonography, computed tomography (CT) scanning, and magnetic resonance imaging (MRI) can be used in the evaluation of craniosynostosis, as can neurodevelopmental testing. (medscape.com)
Nonsyndromic4
- Radiomorphologic profiles of nonsyndromic sagittal craniosynostosis. (wroc.pl)
- Mutations in several genes have been identified in patients with isolated nonsyndromic coronal craniosynostosis. (msdmanuals.com)
- BACKGROUND: Surgical intervention during infancy for both syndromic and nonsyndromic patients with craniosynostosis remains the criterion standard of treatment with the 2 main options being open vault remodeling versus minimally invasive surgery. (cornell.edu)
- Although many forms of syndromic and nonsyndromic craniosynostosis demonstrate an inherited pattern, few articles have reported lambdoid craniosynostosis in the same family . (bvsalud.org)
Apert3
- If there is any documented family history of either craniosynostosis or genetic variations such as Crouton or Apert Syndrome. (medic8.com)
- Coronal craniosynostosis is commonly associated with facial and extracranial anomalies within the context of Crouzon, Muenke, Pfeiffer, Saethre-Chotzen, Carpenter, or Apert syndromes. (msdmanuals.com)
- Apert syndrome is a rare type I acrocephalosyndactyly syndrome characterized by craniosynostosis, severe syndactyly of the hands and feet, and dysmorphic facial features. (bvsalud.org)
Symptoms6
- Learning difficulties, headaches, visual defects and other symptoms may all appear in childhood as a manifestation of craniosynostosis. (medic8.com)
- The majority of children presenting with these symptoms will not have craniosynostosis but it's important to get the diagnosis ruled out. (medic8.com)
- When Do Symptoms of Craniosynostosis Fontaine type Begin? (nih.gov)
- The following symptoms are commonly associated with craniosynostosis. (classlawgroup.com)
- What are the symptoms for a child with craniosynostosis? (drtotonchi.com)
- Because of the variations in presentation of the disease and symptoms associated with craniosynostosis patient's need to be evaluated jointly by a pediatric neurosurgeon and plastic surgery teams. (drtotonchi.com)
Cranial10
- The two continue to work together, but now treat patients like Aria in the newly formed Craniosynostosis and Cranial Reconstruction Center. (cincinnatichildrens.org)
- A dozen years have passed since the first genetic lesion was identified in a family with craniosynostosis, the premature fusion of the cranial sutures. (ox.ac.uk)
- Aim and Objectives: To assess the intracranial volume (ICV) in craniosynostosis patients after the cranial reshaping surgeries and comparing the preoperative values with the postoperative and normal values. (jptcp.com)
- Craniosynostosis consists of premature fusion of 1 or more cranial sutures, often resulting in an abnormal head shape. (medscape.com)
- Traditionally, craniosynostosis is treated via an incision on the infant's scalp to access the cranial bones. (drtotonchi.com)
- Craniosynostosis refers to the premature fusion or ossification of the cranial sutures and can occur from genetic etiologies, as well as from some metabolic disorders and mechanical changes, such as in a child with shunted hydrocephalus . (cincinnatichildrens.org)
- Abnormal early fusion of one or more of the cranial sutures, craniosynostosis, is diagnosed in roughly 1 out of every 2000 to 2500 babies around the world. (craniosacraltherapyny.com)
- Craniosynostosis is a primary abnormality of skull growth involving premature fusion of the cranial sutures such that the growth velocity of the skull often cannot match that of the developing brain. (nih.gov)
- METHODS: A retrospective review of all patients who underwent primary open cranial vault repair for craniosynostosis by a single surgeon (J.A.A.) at New York-Presbyterian Hospital from 1995 to 2015 was performed. (cornell.edu)
- Our data suggest that the ERF level is an important regulator of cranial bone development and that pharmacological modulation of its activity may represent a valid intervention approach both in CRS4 and in other syndromic forms of craniosynostosis mediated by the FGFR-RAS-ERK-ERF pathway. (ox.ac.uk)
Followed in coronal1
- The workshop shall comprise didactic lectures on preoperative assessment, ophthalmological evaluation, intra and perioperative management and surgical steps to be followed in coronal and sagittal craniosynostosis. (asterhospitals.in)
Coronal and sagittal1
- Superior view of 3-dimensional CT scan of a child with metopic craniosynostosis and patent coronal and sagittal sutures. (medscape.com)
Diagnosis5
- This article should familiarize the reader with the pathology, manifestations, diagnosis, and surgical treatment of craniosynostosis. (medscape.com)
- Clinical dividends from the molecular genetic diagnosis of craniosynostosis. (ox.ac.uk)
- Craniofacial surgery for craniosynostosis: challenges in diagnosis, management and long-term outcome. (jptcp.com)
- In this case, a mother brought her 3 week old baby girl to a chiropractor following a medical diagnosis of craniosynostosis by her obstetrician and pediatrician. (craniosacraltherapyny.com)
- The review of his father 's surgical record from 33 years ago and of his computed tomographic scan ordered by our team confirmed the diagnosis of previous lambdoid craniosynostosis . (bvsalud.org)
Form of craniosynostosis2
- This is the most common form of craniosynostosis. (wikipedia.org)
- the extent of the deformity depends on the form of craniosynostosis. (medic8.com)
Cases of craniosynostosis2
- in cases of craniosynostosis where just one suture is affected, just 15% of infants suffer raised ICP. (medic8.com)
- Measuring intracranial volume in early-presenting cases of craniosynostosis would be the cornerstone in determining the optimal time for surgery on clinical terms. (jptcp.com)
Features of craniosynostosis1
- The features of craniosynostosis' particular phenotype are determined by which suture is closed. (wikipedia.org)
Evaluation of craniosynostosis1
- Three-dimensional stereophotogrammetry in the evaluation of craniosynostosis: current and potential use cases. (jptcp.com)
Sagittal suture1
- Craniosynostosis of the sagittal suture is the most common type. (seattlechildrens.org)
Open craniosynostosis3
- Metopic Strip can oftentimes be treated non-surgically and is a condition that our highly-trained physicians diagnose and distinguish from metopic open craniosynostosis. (childrens.com)
- Twenty-Year Outcome Experience With Open Craniosynostosis Repairs: An Analysis of Reoperation and Complication Rates. (cornell.edu)
- Despite this, there is a paucity of literature on the long-term outcomes of contemporary open craniosynostosis repair. (cornell.edu)
Syndromic craniosynostosis1
- Syndromic craniosynostosis is sometimes referred to as 'complex' or 'multi suture' craniosynostosis and is where there are physical features or problems affecting other parts of the body, which follow recognisable patterns. (headlines.org.uk)
Syndrome4
- Craniosynostosis is part of a syndrome in 15% to 40% of affected patients, but it usually occurs as an isolated condition. (wikipedia.org)
- In some cases, craniosynostosis is associated with an underlying syndrome. (lifespan.org)
- The long-term outcome depends on the type and severity of the craniosynostosis, and whether it is part of a larger syndrome. (lifespan.org)
- Il s'agit de la première série de cas du syndrome de Sanjad-Sakati confirmés génétiquement en Jordanie. (who.int)
Surgery16
- Craniosynostosis, and the consequent skull shape deformities, is treated with surgery including osteotomies of the fused sutures. (nih.gov)
- Additionally, in 7 patients (8%) a new suture appeared in a part of the suture that had a discernible suture prior to surgery.In conclusion, in this consecutive and well-defined patient cohort operated for craniosynostosis, the formation of a neosuture is not a rare, and speculatively not a random, event. (nih.gov)
- Craniosynostosis is most often treated with surgery between five months and one year of age, depending on the type of synostosis. (lifespan.org)
- There are two broad categories of surgery for craniosynostosis. (ucsfbenioffchildrens.org)
- Children who have had craniosynostosis surgery played alongside siblings and friends. (pediatricneurosurgery.net)
- Among the children dressed in comic book superhero t-shirts and assorted capes, a number had healed scars from craniosynostosis surgery barely visible in their hair. (pediatricneurosurgery.net)
- TCH is one of the largest craniosynostosis programs in the region and the country, offering a spectrum of treatment strategies including minimally invasive surgery and complex reconstructions. (pediatricneurosurgery.net)
- We just think it's wonderful to have events like this for kids and families," said one of the mothers, whose son recently reached one-year milestone after his surgery for craniosynostosis. (pediatricneurosurgery.net)
- The primary outcome is the rate of postoperative complications possibly attributable to antifibrinolytic use (seizures, seizure-like activity, and thromboembolic events) in infants and children undergoing craniosynostosis surgery who did or did not receive antifibrinolytics. (northwestern.edu)
- Destined to be the definitive reference in this complex surgical area, Endoscopic Craniosynostosis Surgery is the first single resource to offer complete coverage of techniques, outcomes, complications, and results when treating patients with craniosynostosis endoscopically. (americanhhm.com)
- Patients who have craniosynostosis will require surgery to separate the bone and reshape the skull. (drtotonchi.com)
- If the condition hasn't caused any brain abnormalities, then the brain will have sufficient space to grow and develop after craniosynostosis surgery is performed. (drtotonchi.com)
- Most head shape deformities are not due to craniosynostosis, and proper evaluation is required to differentiate between craniosynostosis and other causes of head shape deformities not requiring surgery but still needing treatment. (drtotonchi.com)
- In this case study, a 3 week old girl who was medically diagnosed with craniosynostosis was brought to a chiropractor in an attempt to help avoid skull surgery. (craniosacraltherapyny.com)
- According to the patient's mother, she was advised that the medical care plan was to monitor her child for 3 months and if craniosynostosis progressed further, surgery would have to be performed on her baby's skull. (craniosacraltherapyny.com)
- At presentation, the father reported skull surgery during his infancy for unilateral lambdoid craniosynostosis . (bvsalud.org)
Syndromes1
- Children with these syndromes have other medical conditions besides craniosynostosis. (seattlechildrens.org)
Deformity6
- Using this law, the pattern of skull deformity in craniosynostosis often may be predicted. (wikipedia.org)
- Craniosynostosis results in head deformity that can be severe and permanent if it is not corrected. (medlineplus.gov)
- Craniosynostosis can be subdivided into a number of separate categories, differentiated by the sutures which are involved in the deformity. (medic8.com)
- In craniosynostosis , however, two or more bones of the skull grow together, which causes problems with head growth and could lead to serious complications if left untreated, including dangerous pressure on the brain and head deformity. (cincinnatichildrens.org)
- Treatment for craniosynostosis is required to prevent the psychosocial implications of having a major deformity and in many cases to prevent elevated brain pressure. (ucsfbenioffchildrens.org)
- Coronal craniosynostosis is the second most common type and can be bilateral, causing a short and broad skull (brachycephaly), or unilateral, causing a diagonal skull deformity (plagiocephaly). (msdmanuals.com)
Complications2
- Craniosynostosis: an analysis of the timing, treatment, and complications in 164 consecutive patients. (jptcp.com)
- Craniosynostosis requires surgical treatment to avoid later complications. (healthline.com)
Genetic2
- 2013). For discussion of genetic heterogeneity of craniosynostosis, see CRS1 (123100). (nih.gov)
- The genetic pathogenesis of lambdoid craniosynostosis will be discussed. (bvsalud.org)
Multiple sutures3
- Complex or compound craniosynostosis is used to describe premature fusion of multiple sutures. (medscape.com)
- It can occur in primary craniosynostosis when multiple sutures fuse. (medscape.com)
- Craniosynostosis can affect multiple sutures in the baby's skull and in some cases is associated with a brain abnormality that can prevent the brain from growing normally. (stanford.edu)
Abnormal5
- Craniosynostosis is the premature and abnormal fusion of one of the six suture lines that form the living skull (see the images below), resulting in an abnormal head shape from aberrant bone growth patterns. (medscape.com)
- Craniosynostosis is the premature closure of one or more of these open areas, resulting in the abnormal shaping of the head and face. (wroc.pl)
- Primary craniosynostosis: Although the major morbidity is due to the abnormal shape of the skull, intracranial pressure can be elevated. (medscape.com)
- Craniosynostosis is a condition where the skull prematurely ossifies (turns to bone) resulting in abnormal development of the shape of the head and face and in some cases increased pressure on the brain that can result in visual impairment or developmental delays. (earlyyearsshop.ie)
- Should the growth process happen too early, or in an abnormal manner, the resulting condition is called craniosynostosis. (craniosacraltherapyny.com)
Trigonocephaly1
- Metopic craniosynostosis, or trigonocephaly, occurs when the metopic suture fuses too early. (medicalnewstoday.com)
Premature closure2
- Craniosynostosis is a congenital anomaly that results from the premature closure of one or more sutures. (medicalnewstoday.com)
- The appearance of a new suture long after the normal time period for suture formation in utero indicates that the craniosynostosis may just as well be caused by disturbed formation of the suture as actual premature closure. (nih.gov)
Occurs6
- Craniosynostosis occurs in one in 2000 births. (wikipedia.org)
- Metopic craniosynostosis is a rare type that occurs in about 15% of craniosynostosis cases. (medicalnewstoday.com)
- In this article, we examine metopic craniosynostosis and how often it occurs. (medicalnewstoday.com)
- Metopic craniosynostosis occurs in around 1 in 5,000-15,000 live births. (medicalnewstoday.com)
- Craniosynostosis occurs when one or more sutures close (or "fuse") prematurely. (lifespan.org)
- Craniosynostosis is a craniofacial abnormality that occurs in a variety of forms. (wroc.pl)
Genes4
- Children born with craniosynostosis have a distinct phenotype, i.e., appearance-observable traits caused by the expression of a condition's genes. (wikipedia.org)
- Subsequently, mutations in the FGFR2, FGFR3, TWIST1, and EFNB1 genes have been shown to account for approximately 25% of craniosynostosis, whilst several additional genes make minor contributions. (ox.ac.uk)
- Plea for systematic prenatal genes panel testing when facing isolated craniosynostosis on fetal imaging. (wroc.pl)
- Several genes have been implicated in sagittal craniosynostosis, but chromosomal microarray analysis is not typically necessary unless developmental delays or other congenital anomalies are present. (msdmanuals.com)
Baby's skull1
- Craniosynostosis is when 1 or more of the soft fibrous seams (sutures) in a baby's skull close earlier than normal. (seattlechildrens.org)
Syndactyly1
- Type 1 has the more typical features with midface hypoplasia, broad thumbs and toes, craniosynostosis, and often some degree of syndactyly. (arizona.edu)
Abnormality1
- He presented at 3 years of age with right unilateral lambdoid craniosynostosis with facial asymmetry and lateral deviation of his jaw , with occlusal abnormality. (bvsalud.org)
Metopic suture2
- Closure of the metopic suture before an infant is 3 months old will cause metopic craniosynostosis. (medicalnewstoday.com)
- A child with metopic craniosynostosis with ridging at the site of the metopic suture and hypoplasia of the orbits. (medscape.com)
Lambdoid4
- Familial lambdoid craniosynostosis between father and son. (bvsalud.org)
- Lambdoid craniosynostosis is an uncommon condition, with an incidence of 1 per 33,000 live births . (bvsalud.org)
- Only 2 previous cases of familial isolated lambdoid craniosynostosis have been previously described in literature . (bvsalud.org)
- We report the third case of inherited unilateral lambdoid craniosynostosis . (bvsalud.org)
Phenotype1
- Here, we analyze the onset and development of the craniosynostosis phenotype in an Erf-insufficient mouse model and evaluate the potential of the residual Erf activity augmented by pharmacological compounds to ameliorate the disease. (ox.ac.uk)
Unicoronal1
- Preoperative photograph of a child with unicoronal craniosynostosis. (medscape.com)
Mutations2
- About 25% of coronal craniosynostosis cases are syndromic and due to single-gene mutations or chromosomal defects. (msdmanuals.com)
- Mutations in TCF12 , encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. (msdmanuals.com)
Commonly1
- It may result from a primary defect of ossification (primary craniosynostosis) or, more commonly, from a failure of brain growth (secondary craniosynostosis). (medscape.com)
Patients5
- When they first saw Aria, both Pan and Stevenson were working together in a joint clinic treating patients with craniosynostosis and other congenital and acquired skull abnormalities. (cincinnatichildrens.org)
- This study included a list of all 130 patients who underwent craniosynostosis corrective surgeries and identified retrospectively and prospectively at Abu El-Rish Hospital during a 4-year window from 2017 to 2021. (jptcp.com)
- Guideline for care of patients with the diagnoses of craniosynostosis: working group on craniosynostosis. (jptcp.com)
- Intracranial volume in patients with nonsyndromal craniosynostosis. (jptcp.com)
- Dr. Ali Totonchi offers his expertise for treatment of his craniosynostosis patients in Cleveland, Westlake & Lyndhurst. (drtotonchi.com)
Bone2
- Craniosynostosis is a condition in which one or more of the fibrous sutures in a young infant's skull prematurely fuses by turning into bone (ossification), thereby changing the growth pattern of the skull. (wikipedia.org)
- Other secondary causes of craniosynostosis include systemic disorders that affect bone metabolism such as rickets and hypercalcemia (see Causes). (medscape.com)