Bernard-Soulier Syndrome
Physiology
The critical interaction of glycoprotein (GP) IBbeta with GPIX-a genetic cause of Bernard-Soulier syndrome. (1/69)
Bernard-Soulier syndrome is an uncommon bleeding disorder caused by a quantitative or qualitative defect in the platelet glycoprotein (GP)Ib/IX complex. The complex is composed of four subunits, GPIbalpha, GPIbbeta, GPIX, and GPV. Here we describe the molecular basis of a novel Bernard-Soulier syndrome variant in a patient in whom GPIbalpha and GPIX were undetectable on the platelet surface. DNA sequence analysis showed normal sequence for GPIbalpha, GPIX, and GPV. The GPIbbeta gene has been mapped to the 22q11.2 region of chromosome 22 which was deleted from one chromosome of this patient. There was a single nucleotide deletion within the codon for Ala 80 in GPIbbeta within the other allele. This mutation causes a translational frame shift that encodes for 86 altered amino acids and predicts a premature stop 15 amino acids short of the length of the wild-type protein. Transient coexpression of the mutant GPIbbeta in 293T cells with wild-type GPIbalpha and GPIX resulted in the surface expression of GPIbalpha, but the absence of GPIX. Moreover, when a plasmid encoding the wild-type GPIbbeta was transiently transfected into Chinese hamster ovary cells stably expressing GPalpha, which retain the capacity to reexpress GPIX, there was a significant increase in the surface expression of GPIX. In contrast, when the mutant GPIbbeta was transiently transfected into these cells, GPIX was not reexpressed on the plasma surface. Thus, a deletion of one copy of GPIbbeta and a single nucleotide deletion in the codon for Ala 80 within the remaining GPIbbeta allele causes the Bernard-Soulier phenotype through an interaction of GPIbbeta with GPIX resulting in the absence of GPIbalpha on the plasma membrane. The interaction of GPIbbeta with GPIX is essential for the functional expression of GPIbalpha. (+info)Glycoprotein V-deficient platelets have undiminished thrombin responsiveness and Do not exhibit a Bernard-Soulier phenotype. (2/69)
Adhesion of platelets to extracellular matrix via von Willebrand factor (vWF) and activation of platelets by thrombin are critical steps in hemostasis. Glycoprotein (GP) V is a component of the GPIb-V-IX complex, the platelet receptor for vWF. GPV is also cleaved by thrombin. Deficiency of GPIb or GPIX results in Bernard-Soulier syndrome (BSS), a bleeding disorder in which platelets are giant and have multiple functional defects. Whether GPV-deficiency might also cause BSS is unknown as are the roles of GPV in platelet-vWF interaction and thrombin signaling. We report that GPV-deficient mice developed normally, had no evidence of spontaneous bleeding, and had tail bleeding times that were not prolonged compared with wild-type mice. GPV-deficient platelets were normal in size and structure as assessed by flow cytometry and electron microscopy. GPV-deficient and wild-type platelets were indistinguishable in botrocetin-mediated platelet agglutination and in their ability to adhere to mouse vWF A1 domain. Platelet aggregation and ATP secretion in response to low and high concentrations of thrombin were not decreased in GPV-deficient platelets compared with wild-type. Our results show that (1) GPV is not necessary for GPIb expression and function in platelets and that GPV deficiency is not likely to be a cause of human BSS and (2) GPV is not necessary for robust thrombin signaling. Whether redundancy accounts for the lack of phenotype of GPV-deficiency or whether GPV serves subtle or as yet unprobed functions in platelets or other cells remains to be determined. (+info)Inherited giant platelet disorders. Classification and literature review. (3/69)
Inherited giant platelet disorders are extremely rare. The aim of this article is to review the clinical and laboratory features of this heterogeneous group and to arrive at a working classification. We conducted our literature search using the National Library of Medicine database. A total of 12 clinical entities were described. We classified them into 4 groups depending on the clinical and structural abnormalities. The pathophysiology of these disorders is largely unknown, and more research is needed, particularly in the light of recent advances in laboratory medicine. This review may provide a valuable reference for clinicians and may form a basis for future classification and research. (+info)Generation and rescue of a murine model of platelet dysfunction: the Bernard-Soulier syndrome. (4/69)
The human Bernard-Soulier syndrome is an autosomal recessive disorder of platelet dysfunction presenting with mild thrombocytopenia, circulating "giant" platelets and a bleeding phenotype. The bleeding in patients with the Bernard-Soulier syndrome is disproportionately more severe than suggested by the reduced platelet count and is explained by a defect in primary hemostasis owing to the absence of the platelet glycoprotein (GP) Ib-IX-V membrane receptor. However, the molecular basis for the giant platelet phenotype and thrombocytopenia have remained unresolved but assumed to be linked to an absent receptor complex. We have disrupted the gene encoding the alpha-subunit of mouse GP Ib-IX-V (GP Ibalpha) and describe a murine model recapitulating the hallmark characteristics of the human Bernard-Soulier syndrome. The results demonstrate a direct link between expression of a GP Ib-IX-V complex and normal megakaryocytopoiesis and platelet morphogenesis. Moreover, using transgenic technology the murine Bernard-Soulier phenotype was rescued by expression of a human GP Ibalpha subunit on the surface of circulating mouse platelets. Thus, an in vivo model is defined for analysis of the human GP Ib-IX-V receptor and its role in the processes performed exclusively by megakaryocytes and platelets. (+info)Surface expression of glycoprotein ib alpha is dependent on glycoprotein ib beta: evidence from a novel mutation causing Bernard-Soulier syndrome. (5/69)
Bernard-Soulier syndrome is a rare bleeding disorder caused by a quantitative or qualitative defect in the platelet glycoprotein (GP) Ib-IX-V complex. The complex, which serves as a platelet receptor for von Willebrand factor, is composed of 4 subunits: GPIb alpha, GPIb beta, GPIX, and GPV. We here describe the molecular basis of a novel form of Bernard-Soulier syndrome in a patient in whom the components of the GPIb-IX-V complex were undetectable on the platelet surface. Although confocal imaging confirmed that GPIb alpha was not present on the platelet surface, GPIb alpha was readily detectable in the patient's platelets. Moreover, immunoprecipitation of plasma with specific monoclonal antibodies identified circulating, soluble GPIb alpha. DNA-sequence analysis revealed normal sequences for GPIb alpha and GPIX. There was a G to A substitution at position 159 of the gene encoding GPIb beta, resulting in a premature termination of translation at amino acid 21. Studies of transient coexpression of this mutant, W21stop-GPIb beta, together with wild-type GPIbalpha and GPIX, demonstrated a failure of GPIX expression on the surface of HEK 293T cells. Similar results were obtained with Chinese hamster ovary alpha IX cells, a stable cell line expressing GPIbalpha that retains the capacity to re-express GPIX. Thus, we found that GPIbbeta affects the surface expression of the GPIb-IX complex by failing to support the insertion of GPIb alpha and GPIX into the platelet membrane. (Blood. 2000;96:532-539) (+info)Autosomal-dominant giant platelet syndromes: a hint of the same genetic defect as in Fechtner syndrome owing to a similar genetic linkage to chromosome 22q11-13. (6/69)
Families with 3 different syndromes characterized by autosomal dominant inheritance of low platelet count and giant platelets were studied. Fechtner syndrome is an autosomal-dominant variant of Alport syndrome manifested by nephritis, sensorineural hearing loss, and cataract formation in addition to macrothrombocytopenia and polymorphonuclear inclusion bodies. Sebastian platelet syndrome is an autosomal-dominant macrothrombocytopenia combined with neutrophil inclusions that differ from those found in May-Hegglin syndrome or Chediak-Higashi syndrome or the Dohle bodies described in patients with sepsis. These inclusions are, however, similar to those described in Fechtner syndrome. Other features of Alport syndrome, though, including deafness, cataracts, and nephritis, are absent in Sebastian platelet syndrome. Epstein syndrome is characterized by macrothrombocytopenia without neutrophil inclusions, in addition to the classical Alport manifestations-deafness, cataracts, and nephritis-and it is also inherited in an autosomal-dominant mode. We mapped the disease-causing gene to the long arm of chromosome 22 in an Italian family with Fechtner syndrome, 2 German families with the Sebastian platelet syndrome, and an American family with the Epstein syndrome. Four markers on chromosome 22q yielded an LOD score greater than 2.76. A maximal 2-point LOD score of 3.41 was obtained with the marker D22S683 at a recombination fraction of 0.00. Recombination analysis placed the disease-causing gene in a 3.37-Mb interval between the markers D22S284 and D22S693. The disease-causing gene interval in these 3 syndromes is similar to the interval described recently in an Israeli family with a slightly different Fechtner syndrome than the one described here. Recombination analysis of these 3 syndromes refines the interval containing the disease-causing gene from 5.5 Mb to 3.37 Mb. The clinical likeness and the similar interval containing the disease-causing gene suggest that the 3 different syndromes may arise from a similar genetic defect. (+info)Autosomal dominant macrothrombocytopenia in Italy is most frequently a type of heterozygous Bernard-Soulier syndrome. (7/69)
A form of autosomal dominant macrothrombocytopenia is characterized by mild or no clinical symptoms, normal platelet function, and normal megakaryocyte count. Because this condition has so far received little attention, patients are subject to misdiagnosis and inappropriate therapy. To identify the molecular basis of this disease, 12 Italian families were studied by linkage analysis and mutation screening. Flow cytometry evaluations of platelet membrane glycoproteins (GPs) were also performed. Linkage analysis in 2 large families localized the gene to chromosome 17p, in an interval containing an excellent candidate, the GPIbalpha gene. GPIbalpha, together with other proteins, constitutes the plasma von Willebrand factor (vWF) receptor, which is altered in Bernard-Soulier syndrome (BSS). In 6 of 12 families, a heterozygous Ala156Val missense substitution was identified. Platelet membrane GP studies were performed in 10 patients. Eight were distinguished by a reduction of GPs comparable to that found in a BSS heterozygous condition, whereas the other 2, without the Ala156Val mutation, had a normal content of platelet GPs. In conclusion, the current study provides evidence that most (10 of 12) patients with an original diagnosis of autosomal dominant macrothrombocytopenia shared clinical and molecular features with the heterozygous BSS phenotype. The remaining 2 affected subjects represented patients with "true" autosomal dominant macrothrombocytopenia; the GPIb/IX/V complex was normally distributed on the surface of their platelets. Thus, the diagnosis of heterozygous BSS must always be suspected in patients with inherited thrombocytopenia and platelet macrocytosis. (+info)Increased thrombogenesis and embolus formation in mice lacking glycoprotein V. (8/69)
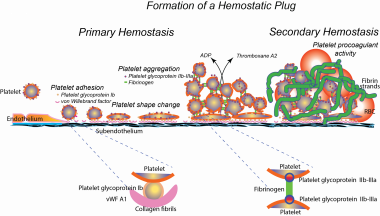
The glycoprotein (GP) Ib-V-IX complex plays a critical role in initiating platelet adhesion to von Willebrand factor (vWF) at the site of vascular injury. The complex also forms a high-affinity binding site for thrombin. Using an intravital microscopy mouse model, it was previously established that vWF plays a critical role in mediating platelet adhesion and thrombus formation following mesenteric arteriolar injury induced by ferric chloride. Further characterization of this model showed that these thrombotic events were also thrombin dependent. Using this vWF- and thrombin-dependent model, this study shows that GP V gene deficiency significantly accelerates both platelet adhesion and thrombus formation in mice following arteriolar injury. The time required for vessel occlusion in GP V-deficient (GP V(-/-)) mice was significantly shorter than that in wild-type mice. Interestingly, large emboli were also produced in GP V(-/-) mice, but not in wild-type mice, causing frequent downstream occlusion. However, when the 2 genotypes were compared in the in vitro perfusion chamber where thrombin was inhibited by heparin, no significant differences were found in either initial single-platelet adhesion or thrombus volume. These results demonstrate that GP V(-/-) mice have accelerated thrombus growth in response to vascular injury and suggest that this is caused by enhanced thrombin-induced platelet activation rather than enhanced binding of GPIb-V-IX to vWF. Absence of GP V also compromises thrombus stability. (+info)Bernard-Soulier Syndrome is a rare autosomal recessive bleeding disorder characterized by a deficiency or dysfunction of the glycoprotein Ib-IX-V complex, which is a crucial component of platelet function. This complex plays a role in the initial adhesion of platelets to the damaged endothelium at the site of blood vessel injury.
The deficiency or dysfunction of this complex leads to abnormalities in platelet aggregation and results in prolonged bleeding times, increased bruising, and excessive blood loss during menstruation, surgery, or trauma. Additionally, individuals with Bernard-Soulier Syndrome often have giant platelets and a decreased platelet count (thrombocytopenia).
The syndrome is named after Jean J. Bernard and Jean-Pierre Soulier, who first described the disorder in 1948. It has an estimated prevalence of about 1 in one million individuals worldwide.
"History, 19th Century" is not a medical term or concept. It refers to the historical events, developments, and figures related to the 1800s in various fields, including politics, culture, science, and technology. However, if you are looking for medical advancements during the 19th century, here's a brief overview:
The 19th century was a period of significant progress in medicine, with numerous discoveries and innovations that shaped modern medical practices. Some notable developments include:
1. Edward Jenner's smallpox vaccine (1796): Although not strictly within the 19th century, Jenner's discovery laid the foundation for vaccination as a preventive measure against infectious diseases.
2. Germ theory of disease: The work of Louis Pasteur, Robert Koch, and others established that many diseases were caused by microorganisms, leading to the development of antiseptic practices and vaccines.
3. Anesthesia: In 1842, Crawford Long first used ether as an anesthetic during surgery, followed by the introduction of chloroform in 1847 by James Simpson.
4. Antisepsis and asepsis: Joseph Lister introduced antiseptic practices in surgery, significantly reducing postoperative infections. Later, the concept of asepsis (sterilization) was developed to prevent contamination during surgical procedures.
5. Microbiology: The development of techniques for culturing and staining bacteria allowed for better understanding and identification of pathogens.
6. Physiology: Claude Bernard's work on the regulation of internal body functions, or homeostasis, contributed significantly to our understanding of human physiology.
7. Neurology: Jean-Martin Charcot made significant contributions to the study of neurological disorders, including multiple sclerosis and Parkinson's disease.
8. Psychiatry: Sigmund Freud developed psychoanalysis, a new approach to understanding mental illnesses.
9. Public health: The 19th century saw the establishment of public health organizations and initiatives aimed at improving sanitation, water quality, and vaccination programs.
10. Medical education reforms: The Flexner Report in 1910 led to significant improvements in medical education standards and practices.
Physiology is the scientific study of the normal functions and mechanisms of living organisms, including all of their biological systems, organs, cells, and biomolecules. It focuses on how various bodily functions are regulated, coordinated, and integrated to maintain a healthy state in an organism. This field encompasses a wide range of areas such as cellular physiology, neurophysiology, cardiovascular physiology, respiratory physiology, renal physiology, endocrine physiology, reproductive physiology, and exercise physiology, among others. Physiologists use a combination of experimental and theoretical approaches to understand the principles underlying normal biological function and to investigate how these functions are altered in various disease states.
I'm sorry for any confusion, but "History, 20th Century" is a broad and complex topic that refers to the events, developments, and transformations that occurred throughout the world during the 1900s. It is not a medical term or concept. If you're interested in learning more about this historical period, I would recommend consulting a history textbook, reputable online resources, or speaking with a historian. They can provide detailed information about the political, social, economic, and cultural changes that took place during the 20th century.
A syndrome, in medical terms, is a set of symptoms that collectively indicate or characterize a disease, disorder, or underlying pathological process. It's essentially a collection of signs and/or symptoms that frequently occur together and can suggest a particular cause or condition, even though the exact physiological mechanisms might not be fully understood.
For example, Down syndrome is characterized by specific physical features, cognitive delays, and other developmental issues resulting from an extra copy of chromosome 21. Similarly, metabolic syndromes like diabetes mellitus type 2 involve a group of risk factors such as obesity, high blood pressure, high blood sugar, and abnormal cholesterol or triglyceride levels that collectively increase the risk of heart disease, stroke, and diabetes.
It's important to note that a syndrome is not a specific diagnosis; rather, it's a pattern of symptoms that can help guide further diagnostic evaluation and management.
 Bernard-Soulier syndrome
Bernard-Soulier syndrome Bernard-Soulier syndrome: MedlinePlus Genetics
Bernard-Soulier syndrome: MedlinePlus Genetics Bernard-Soulier Syndrome Treatment & Management: Approach Considerations
Bernard-Soulier Syndrome Treatment & Management: Approach Considerations PMM.24 The management of Bernard Soulier Syndrome in Pregnancy | ADC Fetal & Neonatal Edition
PMM.24 The management of Bernard Soulier Syndrome in Pregnancy | ADC Fetal & Neonatal Edition Treatment for Bernard-Soulier Syndrome | Bernard-Soulier Syndrome | The Basics | HoG Handbook | Hemophilia of Georgia
Treatment for Bernard-Soulier Syndrome | Bernard-Soulier Syndrome | The Basics | HoG Handbook | Hemophilia of Georgia Bernard - Soulier syndrome is characterized by all except: - Paramedics World QnA
Bernard - Soulier syndrome is characterized by all except: - Paramedics World QnA Bernard-Soulier syndrome, type A2, autosomal dominant (Concept Id: C3277076)
- MedGen - NCBI
Bernard-Soulier syndrome, type A2, autosomal dominant (Concept Id: C3277076)
- MedGen - NCBI Bernard-soulier Syndrome
Bernard-soulier Syndrome Dental management of patients with bleeding disorders - a literature review and case report: Bernard Soulier syndrome
Dental management of patients with bleeding disorders - a literature review and case report: Bernard Soulier syndrome Genetics of familial forms of thrombocytopenia
Genetics of familial forms of thrombocytopenia KAKEN - Research Projects | Study on a new release mechanism abnormality (unresponsiveness to thromboxane A 2) of platelet ...
KAKEN - Research Projects | Study on a new release mechanism abnormality (unresponsiveness to thromboxane A 2) of platelet ... Bio/Data Ristocetin Reagent 1.0 - 1.5mg/mL:Blood, Hematology and Coagulation
| Fisher Scientific
Bio/Data Ristocetin Reagent 1.0 - 1.5mg/mL:Blood, Hematology and Coagulation
| Fisher Scientific Thrombocytopenia Causes
Thrombocytopenia Causes![Recombinant Anti-CD42b antibody [SP202] (ab227669) | Abcam](data:image/png;base64,iVBORw0KGgoAAAANSUhEUgAAABAAAAAQCAYAAAAf8/9hAAABm0lEQVQ4jaWTv0tbURTHP/cl75lqTIiNRFyEJIiUxNB2qf+D6NIuDg7WwcXFxU2yOznYte2klFIqpXVqoXQqgTYZKhURUURTlajJy++Xdx1eeJrmTc/vcuHc8/3cc869V8ileBZkClcSOcW9GUCmFPdmS16n4PfjKp/2KxQbJsmwxnwywOs/RUoNCcB0rI+xAbUb0DQls9tnbO7qHcC3OyWOigbn1RYA0aDXGbD8o8Dmro6qCOYS/Tx6qPHztMbGXx3Zzll5FuJppKe7hULNZD17jQA+TA0xGe21Nh4HmRj2sfjtAoDno36iQdUG2EPM5Gs0WpJ4SL01t7UwHqBPdZ63HTVMaxWOaWBK6Ri3AU8iPSgC9i6bfDmodCS9yhWpGs4AT3piIA3Qrykc6y1+ndV5v1cmX25xWGqy9vua1cyVbRh84GEk4CXk81gVy6WYja4YkpnP/9jaL3eckghrnOgGhZrV57vJCC9G/cB/19jrFXycHuLrUZXtgwpXdZPUoMbLZIA3dx5SMnx7jR0VuNG9/4ICIufaLWT2BlLHjkWr+SchAAAAAElFTkSuQmCC) Recombinant Anti-CD42b antibody [SP202] (ab227669) | Abcam
Recombinant Anti-CD42b antibody [SP202] (ab227669) | Abcam Bleeding Facts | The Haemophilia Society
Bleeding Facts | The Haemophilia Society Functional Platelet Disorders | Choose the Right Test
Functional Platelet Disorders | Choose the Right Test Analýza Českých Genomů pro Teranostiku | Lékařská fakulta Masarykovy univerzity | MED MUNI
Analýza Českých Genomů pro Teranostiku | Lékařská fakulta Masarykovy univerzity | MED MUNI urofacial syndrome - Ontology Browser - Rat Genome Database
urofacial syndrome - Ontology Browser - Rat Genome Database Clinical, Pathological, and Genetic Analysis of Ten Patients with MYH9-Related Disease | Acta Haematologica | Karger Publishers
Clinical, Pathological, and Genetic Analysis of Ten Patients with MYH9-Related Disease | Acta Haematologica | Karger Publishers Expanded Carrier Screening | Thermo Fisher Scientific - US
Expanded Carrier Screening | Thermo Fisher Scientific - US SMART: LRR TYP domain annotation
SMART: LRR TYP domain annotation Platelet Glycoprotein GPIb-IX Complex - Medical Dictionary online-medical-dictionary.org
Platelet Glycoprotein GPIb-IX Complex - Medical Dictionary online-medical-dictionary.org