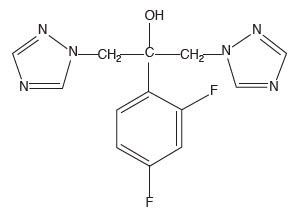
Astemizole
Terfenadine
Histamine H1 Antagonists
Triprolidine
Ether-A-Go-Go Potassium Channels
Anti-Allergic Agents
Chlorpheniramine
Long QT Syndrome
Electrocardiography
Congenital Disorders of Glycosylation
Syncope
Risk of ventricular arrhythmias associated with nonsedating antihistamine drugs. (1/48)
AIMS: To quantify and compare the incidence of ventricular arrhythniias associated with the use of five nonsedating antihistamines: acrivastine, astemizole, cetirizine, loratadine and terfenadine. The effects of age, sex, dose, duration of treatment, and the interaction with P450 inhibitor drugs were also examined. METHODS: We carried out a cohort study with a nested case-control analysis using the UK-based General Practice Research database (GPRD). The study cohort included persons aged less than 80 years old who received their first prescription for any of the five study drugs between January 1, 1992 and September 30, 1996. We estimated relative risks and 95% confidence intervals of idiopathic ventricular arrhythmias with current use of antihistamines as compared with non use. RESULTS: The study cohort included 197425 persons who received 513012 prescriptions. Over the study period 18 valid cases of idiopathic ventricular arrhythmias were detected. Nine occurred during the current use of any antihistamine, resulting in a crude incidence of 1.9 per 10000 person-years (95%CI: 1.0-3.6) and a relative risk of 4.2 (95%CI: 1.5-11.8) as compared with non use. Astemizole presented the highest relative risk (RR= 19.0; 95%CI: 4.8-76.0) of all study drugs, while terfenadine (RR=2.1; 95%CI:0.5-8.5) was in the range of other nonsedating antihistamines. Older age was associated with a greater risk of ventricular arrhythmias (RR=7.4; 95%CI: 2.6-21.4) and seemed to increase the effect of antihistamines (RR=6.4; 95%CI: 1.7-24.8). The proportions of high dose terfenadine and the concomitant use with P450 inhibitors among current users of terfenadine were 2.7% and 3.4%, respectively over the study period with no single case of ventricular arrhythmias occurring in the presence of these two risk factors. CONCLUSIONS: The use of nonsedating antihistamines increases the risk of ventricular arrhythmias by a factor of four in the general population. Yet, the absolute effect is quite low requiring 57000 prescriptions, or 5300 person-years of use for one case to occur. The risk associated with terfenadine was no different from that with other nonsedating antihistamines. (+info)Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. (2/48)
The chromosome 7-linked form of congenital long QT syndrome (LQT2) is caused by mutations in the human ether-a-go-go-related gene (HERG) that encodes the rapidly activating delayed rectifier potassium channel. One mechanism for the loss of normal channel function in LQT2 is defective protein trafficking, which results in the failure of the channel protein to reach the plasma membrane. Here we show that the N470D LQT2 mutant protein is trafficking-deficient when expressed at 37 degrees C in HEK293 cells, whereas at 27 degrees C its trafficking to the plasma membrane and channel function are markedly improved. We further show that the antiarrhythmic drug E-4031, which selectively blocks HERG channels, also corrects defective protein trafficking of the N470D mutant and can restore the generation of HERG current. Similar findings were obtained with the drugs astemizole and cisapride, as well as with high concentrations of glycerol. The effect of E-4031 on HERG protein trafficking was concentration-dependent and required low drug concentrations (saturation present at 5 microM), developed rapidly with drug exposure, and occurred post-translationally. These findings suggest that protein misfolding leading to defective trafficking of some HERG LQT mutations may be corrected by specific pharmacological strategies. (+info)Redox state dependency of HERGS631C channel pharmacology: relation to C-type inactivation. (3/48)
The S631C mutation in human ether-a-go-go-related gene (HERG) channels has previously been reported to disrupt C-type inactivation and ion-selectivity when Cys-631 is in the oxidized state. In this study, we report the relation between pharmacology and C-type inactivation for HERGS631C channels. We demonstrate that HERGS631C in its reduced state is fully blocked by 1 microM astemizole, terfenadine and dofetilide, similar to wild-type HERG channels. In contrast, oxidized HERGS631C is insensitive for these blockers. Our results suggest that an interaction with HERG channels in the inactivated state might be a common mechanism to a variety of drugs known to block HERG channels with high affinity. (+info)Pharmacological blockade of ERG K(+) channels and Ca(2+) influx through store-operated channels exerts opposite effects on intracellular Ca(2+) oscillations in pituitary GH(3) cells. (4/48)
In the present study, the effects on intracellular calcium concentration ([Ca(2+)](i)) oscillations of the blockade of ether-a-go-go-related gene (ERG) K(+) channels and of Ca(2+) influx through store-operated channels (SOC) activated by [Ca(2+)](i) store depletion have been studied in GH(3) cells by means of a combination of single-cell fura-2 microfluorimetry and whole-cell mode of the patch-clamp technique. Nanomolar concentrations (1-30 nM) of the piperidinic second-generation antihistamines terfenadine and astemizole and of the class III antiarrhythmic methanesulfonanilide dofetilide, by blocking ERG K(+) channels, increased the frequency and the amplitude of [Ca(2+)](i) oscillations in resting oscillating GH(3) cells. These compounds also induced the appearance of an oscillatory pattern of [Ca(2+)](i) in a subpopulation of nonoscillating GH(3) cells. The effects of ERG K(+) channel blockade on [Ca(2+)](i) oscillations appeared to be due to the activation of L-type Ca(2+) channels, because they were prevented by 300 nM nimodipine. By contrast, the piperazinic second-generation antihistamine cetirizine (0.01-30 microM), which served as a negative control, failed to affect ERG K(+) channels and did not interfere with [Ca(2+)](i) oscillations in GH(3) cells. Interestingly, micromolar concentrations of terfenadine and astemizole (0.3-30 microM), but not of dofetilide (10-100 microM), produced an inhibition of the spontaneous oscillatory pattern of [Ca(2+)](i) changes. This effect was possibly related to an inhibition of SOC, because these compounds inhibited the increase of [Ca(2+)](i) achieved by extracellular calcium reintroduction after intracellular calcium store depletion with the sarcoplasmic or endoplasmic reticulum calcium ATPase pump inhibitor thapsigargin (10 microM) in an extracellular calcium-free medium. The same inhibitory effect on [Ca(2+)](i) oscillations and SOC was observed with the first-generation antihistamine hydroxyzine (1-30 microM), the more hydrophobic metabolic precursor of cetirizine. Collectively, the results of the present study obtained with compounds that interfere in a different concentration range with ERG K(+) channels or SOC suggest that 1) ERG K(+) channels play a relevant role in controlling the oscillatory pattern of [Ca(2+)](i) in resting GH(3) cells and 2) the inhibition of SOC might induce an opposite effect, i.e., an inhibition of [Ca(2+)](i) oscillations. (+info)Inhibition of HERG1 K(+) channels by the novel second-generation antihistamine mizolastine. (5/48)
1. Ventricular arrhythmias are rare but life-threatening side effects of therapy with the second-generation H(1) receptor antagonists terfenadine and astemizole. Blockade of the K(+) channels encoded by the Human Ether-a-go-go-Related Gene 1 (HERG1) K(+) channels, which is the molecular basis of the cardiac repolarizing current I(Kr), by prolonging cardiac repolarization, has been recognized as the mechanism underlying the cardiac toxicity of these compounds. 2. In the present study, the potential blocking ability of the novel second-generation H(1) receptor antagonist mizolastine of the HERG1 K(+) channels heterologously expressed in Xenopus oocytes and in HEK 293 cells or constitutively present in SH-SY5Y human neuroblastoma cells has been examined and compared to that of astemizole. 3. Mizolastine blocked HERG1 K(+) channels expressed in Xenopus oocytes with an estimated IC(50) of 3.4 microM. Mizolastine blockade was characterized by a fast dissociation rate when compared to that of astemizole; when fitted to a monoexponential function, the time constants for drug dissociation from the K(+) channel were 72.4+/-11.9 s for 3 microM mizolastine, and 1361+/-306 s for 1 microM astemizole. 4. In human embryonic kidney 293 cells (HEK 293 cells) stably transfected with HERG1 cDNA, extracellular application of mizolastine exerted a dose-related inhibitory action on I(HERG1), with an IC(50) of 350+/-76 nM. Furthermore, mizolastine dose-dependently inhibited HERG1 K(+) channels constitutively expressed in SH-SY5Y human neuroblastoma clonal cells. 5. The results of the present study suggest that the novel second-generation H(1) receptor antagonist mizolastine, in concentrations higher than those achieved in vivo during standard therapy, is able to block in some degree both constitutively and heterologously expressed HERG1 K(+) channels, and confirm the heterogeneity of molecules belonging to this therapeutical class with respect to their HERG1-inhibitory action. (+info)Acute canine model for drug-induced Torsades de Pointes in drug safety evaluation-influences of anesthesia and validation with quinidine and astemizole. (6/48)
An acute in vivo model for drug-induced torsades de pointes (TdP) for use in safety evaluation of drugs was developed using dogs with acute complete atrioventricular (AV) block. In order to study the effects of anesthetic agents on the inducibility of TdP, arrhythmias were induced by programmed electrical stimulation (PES) before and after cumulative intravenous administration of quinidine under anesthesia with sodium pentobarbital, halothane, or isoflurane. Both prolongation of the QTc and the incidence of TdP were greatest in dogs anesthetized with halothane and were smallest in those given pentobarbital, suggesting that halothane is the most suitable anesthetic for this TdP model. To further validate this model, astemizole was administered intravenously to other dogs under halothane anesthesia. Astemizole at 0.3 mg/kg caused slight prolongation of the QT interval but did not induce any arrhythmias. At 1 mg/kg, however, TdP were induced in 5 of 10 animals and in an additional 2 animals at 3 mg/kg. Single and multiple ectopic beats preceded the induction of TdP, and the ectopic beats were observed in a dose-dependent manner. The plasma concentrations of quinidine in dogs with TdP were equivalent to or less than quinidine levels in humans with TdP, while those of astemizole were higher in dogs. In conclusion, this acute canine model of TdP with halothane anesthesia, complete AV block, PES, and simultaneous measurements of plasma drug concentration would be valuable for assessing the risk of drugs, especially I(Kr) blockers, to induce TdP in humans. (+info)Involvement of multiple human cytochromes P450 in the liver microsomal metabolism of astemizole and a comparison with terfenadine. (7/48)
AIMS: The aims of the present study were to investigate the metabolism of astemizole in human liver microsomes, to assess possible pharmacokinetic drug-interactions with astemizole and to compare its metabolism with terfenadine, a typical H1 receptor antagonist known to be metabolized predominantly by CYP3A4. METHODS: Astemizole or terfenadine were incubated with human liver microsomes or recombinant cytochromes P450 in the absence or presence of chemical inhibitors and antibodies. RESULTS: Troleandomycin, a CYP3A4 inhibitor, markedly reduced the oxidation of terfenadine (26% of controls) in human liver microsomes, but showed only a marginal inhibition on the oxidation of astemizole (81% of controls). Three metabolites of astemizole were detected in a liver microsomal system, i.e. desmethylastemizole (DES-AST), 6-hydroxyastemizole (6OH-AST) and norastemizole (NOR-AST) at the ratio of 7.4 : 2.8 : 1. Experiments with recombinant P450s and antibodies indicate a negligible role for CYP3A4 on the main metabolic route of astemizole, i.e. formation of DES-AST, although CYP3A4 may mediate the relatively minor metabolic routes to 6OH-AST and NOR-AST. Recombinant CYP2D6 catalysed the formation of 6OH-AST and DES-AST. Studies with human liver microsomes, however, suggest a major role for a mono P450 in DES-AST formation. CONCLUSIONS: In contrast to terfenadine, a minor role for CYP3A4 and involvement of multiple P450 isozymes are suggested in the metabolism of astemizole. These differences in P450 isozymes involved in the metabolism of astemizole and terfenadine may associate with distinct pharmacokinetic influences observed with coadministration of drugs metabolized by CYP3A4. (+info)The binding site for channel blockers that rescue misprocessed human long QT syndrome type 2 ether-a-gogo-related gene (HERG) mutations. (8/48)
Mutations in the human ether-a-gogo-related gene (HERG) K(+) channel gene cause chromosome 7-linked long QT syndrome type 2 (LQT2), which is characterized by a prolonged QT interval in the electrocardiogram and an increased susceptibility to life-threatening cardiac arrhythmias. LQT2 mutations produce loss-of-function phenotypes and reduce I(Kr) currents either by the heteromeric assembly of non- or malfunctioning channel subunits with wild type subunits at the cell surface or by retention of misprocessed mutant HERG channels in the endoplasmic reticulum. Misprocessed mutations often encode for channel proteins that are functional upon incorporation into the plasma membrane. As a result the pharmacological correction of folding defects and restoration of protein function are of considerable interest. Here we report that the trafficking-deficient pore mutation HERG G601S was rescued by a series of HERG channel blockers that increased cell surface expression. Rescue by these pharmacological chaperones varied directly with their blocking potency. We used structure-activity relationships and site-directed mutagenesis to define the binding site of the pharmacological chaperones. We found that binding occurred in the inner cavity and correlated with hydrophobicity and cationic charge. Rescue was domain-restricted because the trafficking of two misprocessed mutations in the C terminus, HERG F805C and HERG R823W, was not restored by channel blockers. Our findings represent a first step toward the design of pharmacological chaperones that will rescue HERG K(+) channels without block. (+info)Astemizole is a second-generation antihistamine that was previously used to treat symptoms associated with allergies, such as hay fever, hives, and other allergic skin reactions. It works by blocking the action of histamine, a substance in the body that causes allergic symptoms. However, astemizole has been withdrawn from the market in many countries due to rare but serious side effects on the heart.
Terfenadine is an antihistamine medication that has been used to treat symptoms of allergies such as hay fever, hives, and other allergic reactions. It works by blocking the action of histamine, a substance in the body that causes allergic symptoms. Terfenadine was first approved for use in the United States in 1985, but it is no longer available in many countries due to concerns about rare but serious side effects related to heart rhythm disturbances. It has been replaced by other antihistamines that are considered safer and more effective.
Histamine H1 antagonists, also known as H1 blockers or antihistamines, are a class of medications that work by blocking the action of histamine at the H1 receptor. Histamine is a chemical mediator released by mast cells and basophils in response to an allergic reaction or injury. It causes various symptoms such as itching, sneezing, runny nose, and wheal and flare reactions (hives).
H1 antagonists prevent the binding of histamine to its receptor, thereby alleviating these symptoms. They are commonly used to treat allergic conditions such as hay fever, hives, and eczema, as well as motion sickness and insomnia. Examples of H1 antagonists include diphenhydramine (Benadryl), loratadine (Claritin), cetirizine (Zyrtec), and doxylamine (Unisom).
Butyrophenones are a group of synthetic antipsychotic drugs that are primarily used to treat symptoms of schizophrenia and other psychotic disorders. They act as dopamine receptor antagonists, which means they block the action of dopamine, a neurotransmitter in the brain associated with mood, motivation, and pleasure.
Some examples of butyrophenones include haloperidol, droperidol, and benperidol. These drugs are known for their potent antipsychotic effects and can also be used to manage agitation, aggression, and other behavioral disturbances in patients with various psychiatric and neurological disorders.
In addition to their antipsychotic properties, butyrophenones have been used off-label for their sedative and analgesic effects. However, they are associated with a range of side effects, including extrapyramidal symptoms (EPS), such as involuntary muscle spasms and tremors, as well as other neurological and cardiovascular adverse reactions. Therefore, their use is typically reserved for cases where other treatments have been ineffective or contraindicated.
Triprolidine is an antihistamine medication that is used to relieve symptoms caused by allergies, such as runny nose, sneezing, and itchy or watery eyes. It works by blocking the action of histamine, a substance in the body that causes allergic symptoms. Triprolidine may also be used to help relieve symptoms of motion sickness.
It is important to note that this definition is for informational purposes only and should not be taken as medical advice. If you have any questions about triprolidine or its use, it is best to consult with a healthcare professional.
Ether-à-go-go (EAG) potassium channels are a type of voltage-gated potassium channel that are widely expressed in the heart, brain, and other tissues. They are named after the ethereal dance movements observed in fruit flies with mutations in these channels.
EAG potassium channels play important roles in regulating electrical excitability and signaling in excitable cells. In the heart, they help to control the duration of the action potential and the refractory period, which is critical for maintaining normal heart rhythm. In the brain, they are involved in regulating neuronal excitability and neurotransmitter release.
Mutations in EAG potassium channels have been associated with various human diseases, including cardiac arrhythmias, epilepsy, and bipolar disorder. The medical definition of "Ether-A-Go-Go Potassium Channels" refers to the genetic components that make up these channels and their role in physiological processes and disease states.
Anti-allergic agents, also known as antihistamines, are a class of medications used to treat allergies. They work by blocking the action of histamine, a substance in the body that is released during an allergic reaction and causes symptoms such as itching, sneezing, runny nose, and watery eyes.
There are two main types of antihistamines: first-generation and second-generation. First-generation antihistamines, such as diphenhydramine (Benadryl) and chlorpheniramine (Chlor-Trimeton), can cause drowsiness and other side effects, such as dry mouth and blurred vision. They are typically used for the treatment of short-term symptoms, such as those caused by seasonal allergies or a mild reaction to an insect bite.
Second-generation antihistamines, such as loratadine (Claritin) and cetirizine (Zyrtec), are less likely to cause drowsiness and other side effects. They are often used for the long-term treatment of chronic allergies, such as those caused by dust mites or pet dander.
In addition to their use in treating allergies, antihistamines may also be used to treat symptoms of motion sickness, insomnia, and anxiety. It is important to follow the instructions on the label when taking antihistamines and to talk to a healthcare provider if you have any questions or concerns about using these medications.
Chlorpheniramine is an antihistamine medication that is used to relieve allergic symptoms caused by hay fever, hives, and other allergies. It works by blocking the action of histamine, a substance in the body that causes allergic symptoms. Chlorpheniramine is available in various forms, including tablets, capsules, syrup, and injection.
Common side effects of chlorpheniramine include drowsiness, dry mouth, blurred vision, and dizziness. It may also cause more serious side effects such as rapid heartbeat, difficulty breathing, and confusion, especially in elderly people or those with underlying medical conditions. Chlorpheniramine should be used with caution and under the supervision of a healthcare provider, particularly in children, pregnant women, and people with medical conditions such as glaucoma, enlarged prostate, and respiratory disorders.
It is important to follow the dosage instructions carefully when taking chlorpheniramine, as taking too much can lead to overdose and serious complications. If you experience any unusual symptoms or have concerns about your medication, it is best to consult with a healthcare provider.
Long QT syndrome (LQTS) is a cardiac electrical disorder characterized by a prolonged QT interval on the electrocardiogram (ECG), which can potentially trigger rapid, chaotic heartbeats known as ventricular tachyarrhythmias, such as torsades de pointes. These arrhythmias can be life-threatening and lead to syncope (fainting) or sudden cardiac death. LQTS is often congenital but may also be acquired due to certain medications, medical conditions, or electrolyte imbalances. It's essential to identify and manage LQTS promptly to reduce the risk of severe complications.
Electrocardiography (ECG or EKG) is a medical procedure that records the electrical activity of the heart. It provides a graphic representation of the electrical changes that occur during each heartbeat. The resulting tracing, called an electrocardiogram, can reveal information about the heart's rate and rhythm, as well as any damage to its cells or abnormalities in its conduction system.
During an ECG, small electrodes are placed on the skin of the chest, arms, and legs. These electrodes detect the electrical signals produced by the heart and transmit them to a machine that amplifies and records them. The procedure is non-invasive, painless, and quick, usually taking only a few minutes.
ECGs are commonly used to diagnose and monitor various heart conditions, including arrhythmias, coronary artery disease, heart attacks, and electrolyte imbalances. They can also be used to evaluate the effectiveness of certain medications or treatments.
The KCNQ1 potassium channel, also known as the Kv7.1 channel, is a voltage-gated potassium ion channel that plays a crucial role in the regulation of electrical excitability in cardiac myocytes and inner ear epithelial cells. In the heart, it helps to control the duration and frequency of action potentials, thereby contributing to the maintenance of normal cardiac rhythm. Mutations in the KCNQ1 gene can lead to various cardiac disorders, such as long QT syndrome type 1 and familial atrial fibrillation. In the inner ear, it helps regulate potassium homeostasis and is essential for hearing and balance functions. Dysfunction of this channel has been linked to deafness and balance disorders.
Congenital Disorders of Glycosylation (CDG) are a group of genetic disorders that affect the body's ability to add sugar molecules (glycans) to proteins and lipids. This process, known as glycosylation, is essential for the proper functioning of many cellular processes, including protein folding, trafficking, and signaling.
CDG can be caused by mutations in genes that are involved in the synthesis or transport of glycans. These genetic defects can lead to abnormal glycosylation patterns, which can result in a wide range of clinical manifestations, including developmental delay, intellectual disability, seizures, movement disorders, hypotonia, coagulation abnormalities, and multi-organ involvement.
CDG are typically classified into two main types: type I CDG, which involves defects in the synthesis of the lipid-linked oligosaccharide precursor used for N-glycosylation, and type II CDG, which involves defects in the processing and transfer of glycans to proteins.
The diagnosis of CDG is often based on clinical features, laboratory tests, and genetic analysis. Treatment is typically supportive and multidisciplinary, focusing on addressing specific symptoms and improving quality of life. In some cases, dietary modifications or supplementation with mannose or other sugars may be beneficial.
Syncope is a medical term defined as a transient, temporary loss of consciousness and postural tone due to reduced blood flow to the brain. It's often caused by a drop in blood pressure, which can be brought on by various factors such as dehydration, emotional stress, prolonged standing, or certain medical conditions like heart diseases, arrhythmias, or neurological disorders.
During a syncope episode, an individual may experience warning signs such as lightheadedness, dizziness, blurred vision, or nausea before losing consciousness. These episodes usually last only a few minutes and are followed by a rapid, full recovery. However, if left untreated or undiagnosed, recurrent syncope can lead to severe injuries from falls or even life-threatening conditions related to the underlying cause.
Cardiac arrhythmias are abnormal heart rhythms that result from disturbances in the electrical conduction system of the heart. The heart's normal rhythm is controlled by an electrical signal that originates in the sinoatrial (SA) node, located in the right atrium. This signal travels through the atrioventricular (AV) node and into the ventricles, causing them to contract and pump blood throughout the body.
An arrhythmia occurs when there is a disruption in this electrical pathway or when the heart's natural pacemaker produces an abnormal rhythm. This can cause the heart to beat too fast (tachycardia), too slow (bradycardia), or irregularly.
There are several types of cardiac arrhythmias, including:
1. Atrial fibrillation: A rapid and irregular heartbeat that starts in the atria (the upper chambers of the heart).
2. Atrial flutter: A rapid but regular heartbeat that starts in the atria.
3. Supraventricular tachycardia (SVT): A rapid heartbeat that starts above the ventricles, usually in the atria or AV node.
4. Ventricular tachycardia: A rapid and potentially life-threatening heart rhythm that originates in the ventricles.
5. Ventricular fibrillation: A chaotic and disorganized electrical activity in the ventricles, which can be fatal if not treated immediately.
6. Heart block: A delay or interruption in the conduction of electrical signals from the atria to the ventricles.
Cardiac arrhythmias can cause various symptoms, such as palpitations, dizziness, shortness of breath, chest pain, and fatigue. In some cases, they may not cause any symptoms and go unnoticed. However, if left untreated, certain types of arrhythmias can lead to serious complications, including stroke, heart failure, or even sudden cardiac death.
Treatment for cardiac arrhythmias depends on the type, severity, and underlying causes. Options may include lifestyle changes, medications, cardioversion (electrical shock therapy), catheter ablation, implantable devices such as pacemakers or defibrillators, and surgery. It is essential to consult a healthcare professional for proper evaluation and management of cardiac arrhythmias.
 Astemizole
Astemizole
Pulmonary hypertension
Aquagenic urticaria
Benzimidazole
Nefazodone
Azatadine
Lichen nitidus
CYP2J2
Creutzfeldt-Jakob disease
Chlorphenamine
Diphenhydramine
CYP3A4
Hydrogen potassium ATPase
Erythromycin
Antihistamine
HSV epigenetics
Tricyclic antidepressant
Bilastine
Clarithromycin
Antiviral drug
List of MeSH codes (D03)
QT interval
Indapamide
FIASMA
Lansoprazole
List of drugs: As-Az
ATC code R06
H1 antagonist
Astemizole - Wikipedia
 With allergy drugs such as astemizole, how about claritin (loratadine)? | HealthTap Online Doctor
With allergy drugs such as astemizole, how about claritin (loratadine)? | HealthTap Online Doctor
Astemizole - wikidoc
 Anticancer effects of ikarugamycin and astemizole identified in a screen for stimulators of cellular immune responses | Journal...
Anticancer effects of ikarugamycin and astemizole identified in a screen for stimulators of cellular immune responses | Journal...
Interactions between Astemizole Oral and qt-prolonging-agents-cilostazol
 Selpercatinib (Oral Route) Description and Brand Names - Mayo Clinic
Selpercatinib (Oral Route) Description and Brand Names - Mayo Clinic
 Duo-Vil 2-25 Advanced Patient Information - Drugs.com
Duo-Vil 2-25 Advanced Patient Information - Drugs.com
Toremifene Advanced Patient Information - Drugs.com
Ciprofloxacin (Otic Route) Description and Brand Names - Mayo Clinic
 Appendix -- Characteristics of Available Antiretroviral Drugs
Appendix -- Characteristics of Available Antiretroviral Drugs
 Medications and other Agents that Increase Sensitivity to Light | Wisconsin Department of Health Services
Medications and other Agents that Increase Sensitivity to Light | Wisconsin Department of Health Services
 Drug Price List: Generics and Matching Brands
Drug Price List: Generics and Matching Brands
 Propulsid (Cisapride (Removed from US Market)): Uses, Dosage, Side Effects, Interactions, Warning
Propulsid (Cisapride (Removed from US Market)): Uses, Dosage, Side Effects, Interactions, Warning
 Torsade de Pointes: Overview, Pathophysiology, Etiology of Torsade
Torsade de Pointes: Overview, Pathophysiology, Etiology of Torsade
 Clarithromycin by Pro Doc Limitée - Uses, Side Effects, Interactions - MedBroadcast.com
Clarithromycin by Pro Doc Limitée - Uses, Side Effects, Interactions - MedBroadcast.com
Cough, Cold, and Allergy Preparation Toxicity: Practice Essentials, Background, Pathophysiology
Antihistamines
Long QT Syndrome: Practice Essentials, Background, Etiopathophysiology
 A low-cost smartphone fluorescence microscope for research, life science education, and STEM outreach | Scientific Reports
A low-cost smartphone fluorescence microscope for research, life science education, and STEM outreach | Scientific Reports
 Appendix -- Characteristics of Available Antiretroviral Drugs
Appendix -- Characteristics of Available Antiretroviral Drugs
 ITRACAP - MyDr.com.au
ITRACAP - MyDr.com.au
 Human Metabolome Database: Showing Protein Nuclear factor NF-kappa-B p105 subunit (HMDBP02145)
Human Metabolome Database: Showing Protein Nuclear factor NF-kappa-B p105 subunit (HMDBP02145)
 Nursing Pharmacology: Antifungal
Nursing Pharmacology: Antifungal
 Sandoz Voriconazole - Pharmasave - Pharmasave
Sandoz Voriconazole - Pharmasave - Pharmasave
Nursing Pharmacology: Antifungal Drugs
 DailyMed - ERY-TAB- erythromycin tablet, delayed release
DailyMed - ERY-TAB- erythromycin tablet, delayed release
 Metryl - Side Effects, Uses, Dosage, Overdose, Pregnancy, Alcohol | RxWiki
Metryl - Side Effects, Uses, Dosage, Overdose, Pregnancy, Alcohol | RxWikiTerfenadine3
- certain antihistamines such as astemizole (Hismanal)(no longer available in the U.S.) and terfenadine (Seldane)(no longer available in the U.S. (medlineplus.gov)
- In a comparative study the new H 1 -receptor antagonists, astemizole, terfenadine and cetirizine were more effective than older drugs, with a lesser adverse effect profile. (soton.ac.uk)
- Case reports of this complication are particularly common for astemizole and terfenadine. (wikitox.org)
Hismanal2
- Astemizole (marketed under the brand name Hismanal, developmental code R43512) was a second-generation antihistamine drug that has a long duration of action. (wikipedia.org)
- Astemizole ( hismanal ) was a popular, non sedating, once a day antihistamine for allergy symptoms like runny, itchy nose, itchy eyes , sneezing etc. (healthtap.com)
Histamine H1-receptor antagon1
- Astemizole is a histamine H1-receptor antagonist. (wikipedia.org)
Antihistamine1
- However, astemizole is an antihistamine that has potential for use as an anti-prion drug. (infectioncontroltoday.com)
Tacrolimus2
- The two compounds are already marketed as the drugs tacrolimus and astemizole. (infectioncontroltoday.com)
- The first author of the study, Unique Drug Screening Approach for Prion Diseases Identifies Tacrolimus and Astemizole as Antiprion Agents, is Yervand Eduard Karapetyan of The Scripps Research Institute. (infectioncontroltoday.com)
Antagonist1
- In contrast, astemizole acts as a histamine H1 receptor (H1R1) antagonist to activate T cells in a non-specific, DC-independent fashion. (bmj.com)
Blood-brain b3
- Despite some earlier reports that astemizole does not cross the blood-brain barrier, several studies have shown high permeability and high binding to protein folds associated with Alzheimer's. (wikipedia.org)
- Astemizole does not cross the blood-brain barrier , and H1 receptor binding is mostly in the peripheral rather than central nervous system (CNS depression is thus minimal). (wikidoc.org)
- Astemizole not only crosses the blood-brain barrier, but works effectively at a relatively low concentration. (infectioncontroltoday.com)
Drugs3
- With allergy drugs such as astemizole, how about claritin (loratadine)? (healthtap.com)
- With allergy drugs such as astemizole can I take claritin (loratadine)? (healthtap.com)
- Results The screening of three drug libraries relevant to known signaling pathways, FDA (Food and Drug Administration)-approved drugs and neuroendocrine factors yielded two major hits, astemizole and ikarugamycin. (bmj.com)
Oral2
- Astemizole has an oral LD50 of approximately 2052 mg/kg (in mice). (wikipedia.org)
- Astemizole has an oral LD 50 of approximately 2052mg/kg (in mice). (wikidoc.org)
Immune2
- Of note, astemizole enhanced the CD8 + /Foxp3 + ratio in the tumor immune infiltrate as well as IFN-γ production by local CD8 + T lymphocytes. (bmj.com)
- The combination of astemizole and oxaliplatin was able to cure the majority of mice bearing orthotopic non-small cell lung cancers (NSCLC), then inducing a state of protective long-term immune memory. (bmj.com)
Market1
- Astemizole is off the market as it caused arrythmias (irregular heart beats). (healthtap.com)
Effects2
- Astemizole may also act on histamine H3 receptors, thereby producing adverse effects. (wikipedia.org)
- Perinatal astemizole exposure in the rat throughout gestation: long-term behavioral and anatomic effects associated with reproduction. (bvsalud.org)
Drug1
- 8. Migliani R, Clouzeau J, Decousser JW, and astemizole before they were with- Ravelomanana N, Rasamoelisoa J, goal in drug therapy decision-making. (cdc.gov)
Action1
- So future studies on the mode of action of astemizole may uncover potentially new therapeutic targets for prion diseases and similar disorders. (infectioncontroltoday.com)
Studies1
- [2] Recent studies have also suggested anti-malarial properties of astemizole. (wikidoc.org)
Inhibitors1
- When astemizole was co-administered with CYP3A4 inhibitors, the plasma of astermizole concentration was higher and may be more likely to cause ADRs. (medscape.com)
Antihistamine drug1
- We recently identified astemizole (AST), an antihistamine drug, as a cholesterol trafficking inhibitor from a phenotypic screen. (johnshopkins.edu)
Chlorpheniramine1
- 9. A comparison of astemizole and chlorpheniramine in dermographic urticaria. (nih.gov)
Histamine-induced1
- 4. The effects of astemizole on histamine-induced weal and flare. (nih.gov)
Cardiotoxicity1
- Astemizole: Increased risk of cardiotoxicity and arrhythmias. (pediatriconcall.com)
Prolongation1
- showed in the animal models that astemizole at 0.3 mg/kg caused slight prolongation of the QT interval but did not induce any arrhythmias. (medscape.com)
Vitro1
- In vitro experiments indicated that both astemizole and demethylated astemizole were strong potassium channel blockers that reduced the conductivity of the delayed rectifier potassium channel (Ikr channel, encoded by hERG ), thereby triggering LQTS. (medscape.com)
CYP3A41
- [ 18 ] Orally taken astemizole was O -demethylated by CYP2J2 and CYP3A4 to form demethylated astemizole. (medscape.com)
Potassium1
- Astemizole was a strong potassium channel blocker that can trigger long QT syndrome (LQTS). (medscape.com)
19831
- Astemizole was first marketed in the UK in 1983 and approved by the FDA in the USA in 1988. (medscape.com)
19991
- However, Johnson & Johnson voluntarily withdrew astemizole from the global market in 1999. (medscape.com)
Clinical1
- 6. Clinical profile of astemizole. (nih.gov)
Adverse1
- Astemizole may also act on histamine H3 receptors, thereby producing adverse effects. (wikipedia.org)
Treatment4
- 2. [Treatment of urticaria with the new H1 antihistaminic astemizole--with special reference to the time of onset of effect and maximum effect]. (nih.gov)
- 8. [Treatment of chronic urticaria with a new synthetic antihistamine: astemizole]. (nih.gov)
- 13. Prolonged benefit in the treatment of chronic idiopathic urticaria with astemizole. (nih.gov)
- 18. Treatment of chronic idiopathic urticaria with astemizole. (nih.gov)