Apoproteins
Apolipoproteins
Lipoproteins, HDL
Apoenzymes
Lipoproteins, VLDL
Apolipoproteins B
Lipoproteins
Lipoproteins, LDL
Apolipoproteins A
Chylomicrons
Apolipoprotein A-I
Enediynes
Electrophoresis, Polyacrylamide Gel
Apolipoproteins E
Heme
Tetrapyrroles
Cholesterol
Phycobilins
Phycocyanin
Flavodoxin
Triglycerides
Retinaldehyde
Lipoproteins, HDL3
Cholesterol Esters
Apolipoproteins C
Spectrophotometry
Liver
Lipoproteins, HDL2
Pulmonary Surfactants
Urease
Ultracentrifugation
Abetalipoproteinemia
Amino Acid Sequence
Apolipoprotein A-II
Phospholipids
Light-Harvesting Protein Complexes
Chlorophyll
Pulmonary Surfactant-Associated Proteins
Helicobacter mustelae
Flavin-Adenine Dinucleotide
Immunoelectrophoresis
Molecular Sequence Data
Protein Conformation
Phosphatidylcholine-Sterol O-Acyltransferase
Amino Acids
Immunodiffusion
Cytochrome P-450 CYP2B1
Flavin Mononucleotide
Circular Dichroism
Lipids
Apolipoprotein B-100
Protein Binding
Apolipoprotein C-III
Chemistry
Chemical Phenomena
Hypolipoproteinemias
Hyperlipoproteinemia Type IV
Apolipoprotein B-48
Enterobacter aerogenes
Lipoprotein(a)
Flavoproteins
Cytochrome c Group
Metalloproteins
Hyperlipoproteinemias
Iron-Sulfur Proteins
Chromatography, Gel
Urobilin
Binding Sites
Spectrum Analysis
Phosphatidylcholines
Photosynthetic Reaction Center Complex Proteins
Blood Protein Disorders
Oxidoreductases, N-Demethylating
Chyle
Oxidation-Reduction
Bromotrichloromethane
Fats, Unsaturated
Dietary Fats
Escherichia coli
Biliverdine
Cytochrome P-450 Enzyme System
Proteolipids
Bile Pigments
Spectrophotometry, Ultraviolet
Carrier Proteins
Centrifugation, Density Gradient
Flavins
Iron
Leghemoglobin
Electron Spin Resonance Spectroscopy
Copper
Nickel
Microscopy, Electron
Receptors, LDL
Myoglobin
Phycoerythrin
Cytochromes
D-Aspartate Oxidase
Egg Yolk
Receptors, Lipoprotein
Lipoprotein Lipase
D-Amino-Acid Oxidase
Allylisopropylacetamide
Immunoelectrophoresis, Two-Dimensional
Rats, Inbred Strains
Base Sequence
Oleic Acids
Diatomaceous Earth
Apolipoproteins D
Rod Opsins
Surface Tension
Trypsin
Bacteriorhodopsins
Plant Proteins
Models, Molecular
Chloroplasts
Electrophoresis, Agar Gel
Rhodopsin
Radioimmunoassay
Ethinyl Estradiol
Hemeproteins
Cloning, Molecular
Halobacterium
Rhodospirillales
Chromatography, Agarose
Ferredoxins
Rabbits
Magnetic Resonance Spectroscopy
Cattle
Mesentery
Protein Denaturation
Holoenzymes
Lyases
Hemin
Carotenoids
Zinc
Plastocyanin
Phytochrome A
Structure-Activity Relationship
RNA, Messenger
5-Aminolevulinate Synthetase
Cholesterol, HDL
Chromatography, Affinity
Chromatography, High Pressure Liquid
Apolipoprotein C-II
Hyperlipoproteinemia Type III
Isoelectric Focusing
Lecithin Acyltransferase Deficiency
Coenzymes
Photosystem II Protein Complex
Cobalt
Aryl Hydrocarbon Hydroxylases
Cholesterol, LDL
Schiff Bases
Macromolecular Substances
Oleic Acid
Mutation
Hydrogen-Ion Concentration
Molecular Structure
Temperature
Plants
Isotope Labeling
Biological Transport
Thioctic Acid
Arteriosclerosis
Hydroxylamine
Dithionitrobenzoic Acid
Pulmonary Alveolar Proteinosis
Photosystem I Protein Complex
Transducin
Cytochrome b Group
Thromboplastin
Antibiotics, Antineoplastic
Chickens
Cells, Cultured
Phenobarbital
Iodine Radioisotopes
Cycloheximide
Apoferritins
Peptides
Apoproteins are the protein components of lipoprotein complexes, which are responsible for transporting fat molecules, such as cholesterol and triglycerides, throughout the body. Apoproteins play a crucial role in the metabolism of lipids by acting as recognition signals that allow lipoproteins to interact with specific receptors on cell surfaces.
There are several different types of apoproteins, each with distinct functions. For example, apolipoprotein A-1 (apoA-1) is the major protein component of high-density lipoproteins (HDL), which are responsible for transporting excess cholesterol from tissues to the liver for excretion. Apolipoprotein B (apoB) is a large apoprotein found in low-density lipoproteins (LDL), very low-density lipoproteins (VLDL), and lipoprotein(a). ApoB plays a critical role in the assembly and secretion of VLDL from the liver, and it also mediates the uptake of LDL by cells.
Abnormalities in apoprotein levels or function can contribute to the development of various diseases, including cardiovascular disease, diabetes, and Alzheimer's disease. Therefore, measuring apoprotein levels in the blood can provide valuable information for diagnosing and monitoring these conditions.
Apolipoproteins are a group of proteins that are associated with lipids (fats) in the body and play a crucial role in the metabolism, transportation, and regulation of lipids. They are structural components of lipoprotein particles, which are complexes of lipids and proteins that transport lipids in the bloodstream.
There are several types of apolipoproteins, including ApoA, ApoB, ApoC, ApoD, ApoE, and others. Each type has a specific function in lipid metabolism. For example, ApoA is a major component of high-density lipoprotein (HDL), often referred to as "good cholesterol," and helps remove excess cholesterol from cells and tissues and transport it to the liver for excretion. ApoB, on the other hand, is a major component of low-density lipoprotein (LDL), or "bad cholesterol," and plays a role in the delivery of cholesterol to cells and tissues.
Abnormal levels of apolipoproteins or dysfunctional forms of these proteins have been linked to various diseases, including cardiovascular disease, Alzheimer's disease, and metabolic disorders such as diabetes. Therefore, measuring apolipoprotein levels in the blood can provide valuable information for diagnosing and monitoring these conditions.
High-Density Lipoproteins (HDL) are a type of lipoprotein that play a crucial role in the transportation and metabolism of cholesterol in the body. They are often referred to as "good" cholesterol because they help remove excess cholesterol from cells and carry it back to the liver, where it can be broken down and removed from the body. This process is known as reverse cholesterol transport.
HDLs are composed of a lipid core containing cholesteryl esters and triglycerides, surrounded by a shell of phospholipids, free cholesterol, and apolipoproteins, primarily apoA-I. The size and composition of HDL particles can vary, leading to the classification of different subclasses of HDL with varying functions and metabolic fates.
Elevated levels of HDL have been associated with a lower risk of developing cardiovascular diseases, while low HDL levels increase the risk. However, it is essential to consider that HDL function and quality may be more important than just the quantity in determining cardiovascular risk.
An apoenzyme is the protein component of an enzyme that is responsible for its catalytic activity. It combines with a cofactor, which can be either an organic or inorganic non-protein molecule, to form the active enzyme. The cofactor can be a metal ion or a small organic molecule called a coenzyme.
The term "apoenzyme" is used to describe the protein portion of an enzyme after it has lost its cofactor. When the apoenzyme combines with the cofactor, the active holoenzyme is formed, which is capable of carrying out the specific biochemical reaction for which the enzyme is responsible.
In some cases, the loss of a cofactor can result in the complete loss of enzymatic activity, while in other cases, the apoenzyme may retain some residual activity. The relationship between an apoenzyme and its cofactor is specific, meaning that each cofactor typically only binds to and activates one particular type of apoenzyme.
VLDL (Very Low-Density Lipoproteins) are a type of lipoprotein that play a crucial role in the transport and metabolism of fat molecules, known as triglycerides, in the body. They are produced by the liver and consist of a core of triglycerides surrounded by a shell of proteins called apolipoproteins, phospholipids, and cholesterol.
VLDL particles are responsible for delivering fat molecules from the liver to peripheral tissues throughout the body, where they can be used as an energy source or stored for later use. During this process, VLDL particles lose triglycerides and acquire more cholesterol, transforming into intermediate-density lipoproteins (IDL) and eventually low-density lipoproteins (LDL), which are also known as "bad" cholesterol.
Elevated levels of VLDL in the blood can contribute to the development of cardiovascular disease due to their association with increased levels of triglycerides and LDL cholesterol, as well as decreased levels of high-density lipoproteins (HDL), which are considered "good" cholesterol.
Apolipoprotein B (ApoB) is a type of protein that plays a crucial role in the metabolism of lipids, particularly low-density lipoprotein (LDL) or "bad" cholesterol. ApoB is a component of LDL particles and serves as a ligand for the LDL receptor, which is responsible for the clearance of LDL from the bloodstream.
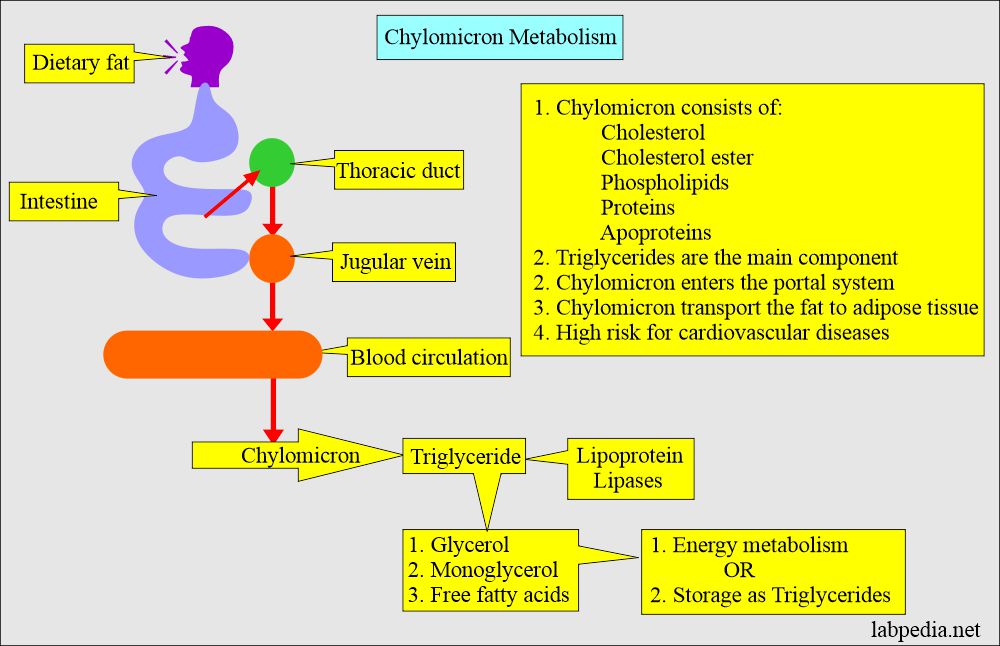
There are two main forms of ApoB: ApoB-100 and ApoB-48. ApoB-100 is found in LDL particles, very low-density lipoprotein (VLDL) particles, and chylomicrons, while ApoB-48 is only found in chylomicrons, which are produced in the intestines and responsible for transporting dietary lipids.
Elevated levels of ApoB are associated with an increased risk of cardiovascular disease (CVD), as they indicate a higher concentration of LDL particles in the bloodstream. Therefore, measuring ApoB levels can provide additional information about CVD risk beyond traditional lipid profile tests that only measure total cholesterol, LDL cholesterol, HDL cholesterol, and triglycerides.
Lipoproteins are complex particles composed of multiple proteins and lipids (fats) that play a crucial role in the transport and metabolism of fat molecules in the body. They consist of an outer shell of phospholipids, free cholesterols, and apolipoproteins, enclosing a core of triglycerides and cholesteryl esters.
There are several types of lipoproteins, including:
1. Chylomicrons: These are the largest lipoproteins and are responsible for transporting dietary lipids from the intestines to other parts of the body.
2. Very-low-density lipoproteins (VLDL): Produced by the liver, VLDL particles carry triglycerides to peripheral tissues for energy storage or use.
3. Low-density lipoproteins (LDL): Often referred to as "bad cholesterol," LDL particles transport cholesterol from the liver to cells throughout the body. High levels of LDL in the blood can lead to plaque buildup in artery walls and increase the risk of heart disease.
4. High-density lipoproteins (HDL): Known as "good cholesterol," HDL particles help remove excess cholesterol from cells and transport it back to the liver for excretion or recycling. Higher levels of HDL are associated with a lower risk of heart disease.
Understanding lipoproteins and their roles in the body is essential for assessing cardiovascular health and managing risks related to heart disease and stroke.
Low-density lipoproteins (LDL), also known as "bad cholesterol," are a type of lipoprotein that carry cholesterol and other fats from the liver to cells throughout the body. High levels of LDL in the blood can lead to the buildup of cholesterol in the walls of the arteries, which can increase the risk of heart disease and stroke.
Lipoproteins are complex particles composed of proteins (apolipoproteins) and lipids (cholesterol, triglycerides, and phospholipids) that are responsible for transporting fat molecules around the body in the bloodstream. LDL is one type of lipoprotein, along with high-density lipoproteins (HDL), very low-density lipoproteins (VLDL), and chylomicrons.
LDL particles are smaller than HDL particles and can easily penetrate the artery walls, leading to the formation of plaques that can narrow or block the arteries. Therefore, maintaining healthy levels of LDL in the blood is essential for preventing cardiovascular disease.
Apolipoprotein A (apoA) is a type of apolipoprotein that is primarily associated with high-density lipoproteins (HDL), often referred to as "good cholesterol." There are several subtypes of apoA, including apoA-I, apoA-II, and apoA-IV.
ApoA-I is the major protein component of HDL particles and plays a crucial role in reverse cholesterol transport, which is the process by which excess cholesterol is removed from tissues and delivered to the liver for excretion. Low levels of apoA-I have been linked to an increased risk of cardiovascular disease.
ApoA-II is another protein component of HDL particles, although its function is less well understood than that of apoA-I. Some studies suggest that apoA-II may play a role in regulating the metabolism of HDL particles.
ApoA-IV is found in both HDL and chylomicrons, which are lipoprotein particles that transport dietary lipids from the intestine to the liver. The function of apoA-IV is not well understood, but it may play a role in regulating appetite and energy metabolism.
Overall, apolipoproteins A are important components of HDL particles and play a critical role in maintaining healthy lipid metabolism and reducing the risk of cardiovascular disease.
Chylomicrons are a type of lipoprotein that are responsible for carrying dietary lipids, such as triglycerides and cholesterol, from the intestines to other parts of the body through the lymphatic system and bloodstream. They are the largest lipoproteins and are composed of an outer layer of phospholipids, free cholesterol, and apolipoproteins, which surrounds a core of triglycerides and cholesteryl esters. Chylomicrons are produced in the intestinal mucosa after a meal containing fat, and their production is stimulated by the hormone cholecystokinin. Once in the bloodstream, chylomicrons interact with other lipoproteins and enzymes to deliver their lipid cargo to various tissues, including muscle and adipose tissue, where they are used for energy or stored for later use.
Apolipoprotein A-I (ApoA-I) is a major protein component of high-density lipoproteins (HDL) in human plasma. It plays a crucial role in the metabolism and transport of lipids, particularly cholesterol, within the body. ApoA-I facilitates the formation of HDL particles, which are involved in the reverse transport of cholesterol from peripheral tissues to the liver for excretion. This process is known as reverse cholesterol transport and helps maintain appropriate cholesterol levels in the body. Low levels of ApoA-I or dysfunctional ApoA-I have been associated with an increased risk of developing cardiovascular diseases.
Enediynes are a class of organic compounds that contain an unsaturated hydrocarbon structure consisting of two double bonds separated by a single bond, forming a core structural unit of R-C=C=C=C-R'. This unique arrangement gives enediynes significant chemical reactivity and has been the basis for their development as antitumor agents.
Enediynes can undergo a cyclization reaction known as the Bergman cyclization, which generates a highly reactive 1,4-diradical species capable of causing significant damage to DNA and other cellular components. This property has been exploited in the design of enediyne-based anticancer drugs, such as neocarzinostatin and calicheamicin, that can selectively target and destroy cancer cells while minimizing harm to normal tissues.
It is important to note that this definition is a general description of the chemical structure and properties of enediynes, and it does not provide specific medical advice or recommendations for treatment. If you have any questions about enediynes or their potential use in medicine, please consult with a qualified healthcare professional.
Lymph is a colorless, transparent fluid that circulates throughout the lymphatic system, which is a part of the immune and circulatory systems. It consists of white blood cells called lymphocytes, proteins, lipids, glucose, electrolytes, hormones, and waste products. Lymph plays an essential role in maintaining fluid balance, absorbing fats from the digestive tract, and defending the body against infection by transporting immune cells to various tissues and organs. It is collected from tissues through lymph capillaries and flows through increasingly larger lymphatic vessels, ultimately returning to the bloodstream via the subclavian veins in the chest region.
Electrophoresis, polyacrylamide gel (EPG) is a laboratory technique used to separate and analyze complex mixtures of proteins or nucleic acids (DNA or RNA) based on their size and electrical charge. This technique utilizes a matrix made of cross-linked polyacrylamide, a type of gel, which provides a stable and uniform environment for the separation of molecules.
In this process:
1. The polyacrylamide gel is prepared by mixing acrylamide monomers with a cross-linking agent (bis-acrylamide) and a catalyst (ammonium persulfate) in the presence of a buffer solution.
2. The gel is then poured into a mold and allowed to polymerize, forming a solid matrix with uniform pore sizes that depend on the concentration of acrylamide used. Higher concentrations result in smaller pores, providing better resolution for separating smaller molecules.
3. Once the gel has set, it is placed in an electrophoresis apparatus containing a buffer solution. Samples containing the mixture of proteins or nucleic acids are loaded into wells on the top of the gel.
4. An electric field is applied across the gel, causing the negatively charged molecules to migrate towards the positive electrode (anode) while positively charged molecules move toward the negative electrode (cathode). The rate of migration depends on the size, charge, and shape of the molecules.
5. Smaller molecules move faster through the gel matrix and will migrate farther from the origin compared to larger molecules, resulting in separation based on size. Proteins and nucleic acids can be selectively stained after electrophoresis to visualize the separated bands.
EPG is widely used in various research fields, including molecular biology, genetics, proteomics, and forensic science, for applications such as protein characterization, DNA fragment analysis, cloning, mutation detection, and quality control of nucleic acid or protein samples.
Apolipoprotein E (ApoE) is a protein involved in the metabolism of lipids, particularly cholesterol. It is produced primarily by the liver and is a component of several types of lipoproteins, including very low-density lipoproteins (VLDL) and high-density lipoproteins (HDL).
ApoE plays a crucial role in the transport and uptake of lipids in the body. It binds to specific receptors on cell surfaces, facilitating the delivery of lipids to cells for energy metabolism or storage. ApoE also helps to clear cholesterol from the bloodstream and is involved in the repair and maintenance of tissues.
There are three major isoforms of ApoE, designated ApoE2, ApoE3, and ApoE4, which differ from each other by only a few amino acids. These genetic variations can have significant effects on an individual's risk for developing certain diseases, particularly cardiovascular disease and Alzheimer's disease. For example, individuals who inherit the ApoE4 allele have an increased risk of developing Alzheimer's disease, while those with the ApoE2 allele may have a reduced risk.
In summary, Apolipoprotein E is a protein involved in lipid metabolism and transport, and genetic variations in this protein can influence an individual's risk for certain diseases.
Heme is not a medical term per se, but it is a term used in the field of medicine and biology. Heme is a prosthetic group found in hemoproteins, which are proteins that contain a heme iron complex. This complex plays a crucial role in various biological processes, including oxygen transport (in hemoglobin), electron transfer (in cytochromes), and chemical catalysis (in peroxidases and catalases).
The heme group consists of an organic component called a porphyrin ring, which binds to a central iron atom. The iron atom can bind or release electrons, making it essential for redox reactions in the body. Heme is also vital for the formation of hemoglobin and myoglobin, proteins responsible for oxygen transport and storage in the blood and muscles, respectively.
In summary, heme is a complex organic-inorganic structure that plays a critical role in several biological processes, particularly in electron transfer and oxygen transport.
Tetrapyrroles are a class of organic compounds that contain four pyrrole rings joined together in a macrocyclic structure. They are important in biology because they form the core structure of many essential cofactors and prosthetic groups in proteins, including heme, chlorophyll, and cobalamin (vitamin B12).
Heme is a tetrapyrrole that contains iron and is a crucial component of hemoglobin, the protein responsible for oxygen transport in red blood cells. Chlorophyll is another tetrapyrrole that contains magnesium and plays a vital role in photosynthesis, the process by which plants convert light energy into chemical energy. Cobalamin contains cobalt and is essential for DNA synthesis, fatty acid metabolism, and neurotransmitter synthesis.
Abnormalities in tetrapyrrole biosynthesis can lead to various diseases, such as porphyrias, which are characterized by the accumulation of toxic intermediates in the heme biosynthetic pathway.
Cholesterol is a type of lipid (fat) molecule that is an essential component of cell membranes and is also used to make certain hormones and vitamins in the body. It is produced by the liver and is also obtained from animal-derived foods such as meat, dairy products, and eggs.
Cholesterol does not mix with blood, so it is transported through the bloodstream by lipoproteins, which are particles made up of both lipids and proteins. There are two main types of lipoproteins that carry cholesterol: low-density lipoproteins (LDL), also known as "bad" cholesterol, and high-density lipoproteins (HDL), also known as "good" cholesterol.
High levels of LDL cholesterol in the blood can lead to a buildup of cholesterol in the walls of the arteries, increasing the risk of heart disease and stroke. On the other hand, high levels of HDL cholesterol are associated with a lower risk of these conditions because HDL helps remove LDL cholesterol from the bloodstream and transport it back to the liver for disposal.
It is important to maintain healthy levels of cholesterol through a balanced diet, regular exercise, and sometimes medication if necessary. Regular screening is also recommended to monitor cholesterol levels and prevent health complications.
In the context of medicine and pharmacology, "kinetics" refers to the study of how a drug moves throughout the body, including its absorption, distribution, metabolism, and excretion (often abbreviated as ADME). This field is called "pharmacokinetics."
1. Absorption: This is the process of a drug moving from its site of administration into the bloodstream. Factors such as the route of administration (e.g., oral, intravenous, etc.), formulation, and individual physiological differences can affect absorption.
2. Distribution: Once a drug is in the bloodstream, it gets distributed throughout the body to various tissues and organs. This process is influenced by factors like blood flow, protein binding, and lipid solubility of the drug.
3. Metabolism: Drugs are often chemically modified in the body, typically in the liver, through processes known as metabolism. These changes can lead to the formation of active or inactive metabolites, which may then be further distributed, excreted, or undergo additional metabolic transformations.
4. Excretion: This is the process by which drugs and their metabolites are eliminated from the body, primarily through the kidneys (urine) and the liver (bile).
Understanding the kinetics of a drug is crucial for determining its optimal dosing regimen, potential interactions with other medications or foods, and any necessary adjustments for special populations like pediatric or geriatric patients, or those with impaired renal or hepatic function.
Phycobilins are linear tetrapyrrole chromophores found in cyanobacteria, red algae, and glaucophytes. They are the light-harvesting pigments associated with phycobiliproteins in the phycobilisome complex, which is a type of antenna system used to capture light for photosynthesis. The main types of phycobilins are phycocyanobilin, phycoerythrobilin, and allophycocyanobilin. These pigments absorb light in the blue-green to red region of the electromagnetic spectrum and transfer the energy to chlorophyll a for use in photosynthesis. Phycobilins are also used as fluorescent labels in various biochemical and medical research applications.
Phycocyanin is a pigment-protein complex found in cyanobacteria and some types of algae, such as Spirulina. It belongs to the family of phycobiliproteins and plays a crucial role in the light-harvesting process during photosynthesis. Phycocyanin absorbs light in the orange and red regions of the visible spectrum and transfers the energy to chlorophyll for use in photosynthesis. It has been studied for its potential health benefits, including antioxidant, anti-inflammatory, and neuroprotective properties. However, more research is needed to fully understand its effects and potential therapeutic uses.
Flavodoxin is not strictly a medical term, but it is a term used in biochemistry and molecular biology. Flavodoxins are small electron transfer proteins that contain a non-heme iron atom bound to a organic molecule called flavin mononucleotide (FMN). They play a role in various biological processes such as photosynthesis, nitrogen fixation and respiration where they function as electron carriers. Flavodoxins can undergo reversible oxidation and reduction, and this property allows them to transfer electrons between different enzymes during metabolic reactions. They are not specific to human physiology, but can be found in various organisms including bacteria, algae, and plants.
Triglycerides are the most common type of fat in the body, and they're found in the food we eat. They're carried in the bloodstream to provide energy to the cells in our body. High levels of triglycerides in the blood can increase the risk of heart disease, especially in combination with other risk factors such as high LDL (bad) cholesterol, low HDL (good) cholesterol, and high blood pressure.
It's important to note that while triglycerides are a type of fat, they should not be confused with cholesterol, which is a waxy substance found in the cells of our body. Both triglycerides and cholesterol are important for maintaining good health, but high levels of either can increase the risk of heart disease.
Triglyceride levels are measured through a blood test called a lipid panel or lipid profile. A normal triglyceride level is less than 150 mg/dL. Borderline-high levels range from 150 to 199 mg/dL, high levels range from 200 to 499 mg/dL, and very high levels are 500 mg/dL or higher.
Elevated triglycerides can be caused by various factors such as obesity, physical inactivity, excessive alcohol consumption, smoking, and certain medical conditions like diabetes, hypothyroidism, and kidney disease. Medications such as beta-blockers, steroids, and diuretics can also raise triglyceride levels.
Lifestyle changes such as losing weight, exercising regularly, eating a healthy diet low in saturated and trans fats, avoiding excessive alcohol consumption, and quitting smoking can help lower triglyceride levels. In some cases, medication may be necessary to reduce triglycerides to recommended levels.
Retinaldehyde, also known as retinal, is a form of vitamin A that is essential for vision. It is the aldehyde form of retinol (vitamin A alcohol) and is involved in the visual cycle, where it plays a crucial role in the process of converting light into electrical signals that are sent to the brain.
When light hits the retina, it activates a protein called rhodopsin, which contains retinaldehyde as one of its components. This activation causes a chemical change in retinaldehyde, leading to the generation of an electrical signal that is transmitted to the brain via the optic nerve.
Retinaldehyde is also involved in other physiological processes, including the regulation of gene expression and cell growth and differentiation. It can be synthesized in the body from beta-carotene, a pigment found in fruits and vegetables, or obtained directly from animal sources such as liver, fish liver oil, and dairy products.
HDL3 (High-Density Lipoprotein 3) is a type of lipoprotein that plays a role in the transport and metabolism of cholesterol in the body. HDLs are commonly known as "good cholesterol" because they help remove excess cholesterol from cells and carry it back to the liver, where it can be broken down and removed from the body.
HDL3 is one of the subclasses of HDL based on its density and size. It is denser than HDL2 but less dense than HDL1. HDL3 is smaller in size and contains a higher proportion of protein to lipid compared to other HDL subclasses. It is also more efficient in reverse cholesterol transport, which is the process of removing cholesterol from tissues and delivering it to the liver for excretion.
It's worth noting that while high levels of HDL are generally associated with a lower risk of heart disease, recent research suggests that the relationship between HDL and cardiovascular health may be more complex than previously thought.
Cholesteryl esters are formed when cholesterol, a type of lipid (fat) that is important for the normal functioning of the body, becomes combined with fatty acids through a process called esterification. This results in a compound that is more hydrophobic (water-repelling) than cholesterol itself, which allows it to be stored more efficiently in the body.
Cholesteryl esters are found naturally in foods such as animal fats and oils, and they are also produced by the liver and other cells in the body. They play an important role in the structure and function of cell membranes, and they are also precursors to the synthesis of steroid hormones, bile acids, and vitamin D.
However, high levels of cholesteryl esters in the blood can contribute to the development of atherosclerosis, a condition characterized by the buildup of plaque in the arteries, which can increase the risk of heart disease and stroke. Cholesteryl esters are typically measured as part of a lipid profile, along with other markers such as total cholesterol, HDL cholesterol, and triglycerides.
Apolipoprotein C (apoC) is a group of proteins that are associated with lipoproteins, which are complex particles composed of lipids and proteins that play a crucial role in the transport and metabolism of lipids in the body. There are three main types of apoC proteins: apoC-I, apoC-II, and apoC-III.
ApoC-I is involved in the regulation of lipoprotein metabolism and has been shown to inhibit the activity of cholesteryl ester transfer protein (CETP), which is an enzyme that facilitates the transfer of cholesteryl esters from high-density lipoproteins (HDL) to low-density lipoproteins (LDL) and very low-density lipoproteins (VLDL).
ApoC-II is a cofactor for lipoprotein lipase, an enzyme that hydrolyzes triglycerides in chylomicrons and VLDL, leading to the formation of smaller, denser lipoproteins. A deficiency in apoC-II can lead to hypertriglyceridemia, a condition characterized by elevated levels of triglycerides in the blood.
ApoC-III is also involved in the regulation of lipoprotein metabolism and has been shown to inhibit the activity of lipoprotein lipase and CETP. Elevated levels of apoC-III have been associated with an increased risk of cardiovascular disease, possibly due to its effects on lipoprotein metabolism.
In summary, apolipoprotein C is a group of proteins that are involved in the regulation of lipoprotein metabolism and have important roles in the transport and metabolism of lipids in the body.
Spectrophotometry is a technical analytical method used in the field of medicine and science to measure the amount of light absorbed or transmitted by a substance at specific wavelengths. This technique involves the use of a spectrophotometer, an instrument that measures the intensity of light as it passes through a sample.
In medical applications, spectrophotometry is often used in laboratory settings to analyze various biological samples such as blood, urine, and tissues. For example, it can be used to measure the concentration of specific chemicals or compounds in a sample by measuring the amount of light that is absorbed or transmitted at specific wavelengths.
In addition, spectrophotometry can also be used to assess the properties of biological tissues, such as their optical density and thickness. This information can be useful in the diagnosis and treatment of various medical conditions, including skin disorders, eye diseases, and cancer.
Overall, spectrophotometry is a valuable tool for medical professionals and researchers seeking to understand the composition and properties of various biological samples and tissues.
The liver is a large, solid organ located in the upper right portion of the abdomen, beneath the diaphragm and above the stomach. It plays a vital role in several bodily functions, including:
1. Metabolism: The liver helps to metabolize carbohydrates, fats, and proteins from the food we eat into energy and nutrients that our bodies can use.
2. Detoxification: The liver detoxifies harmful substances in the body by breaking them down into less toxic forms or excreting them through bile.
3. Synthesis: The liver synthesizes important proteins, such as albumin and clotting factors, that are necessary for proper bodily function.
4. Storage: The liver stores glucose, vitamins, and minerals that can be released when the body needs them.
5. Bile production: The liver produces bile, a digestive juice that helps to break down fats in the small intestine.
6. Immune function: The liver plays a role in the immune system by filtering out bacteria and other harmful substances from the blood.
Overall, the liver is an essential organ that plays a critical role in maintaining overall health and well-being.
HDL2 (High-Density Lipoprotein 2) is a type of lipoprotein that plays a role in the transportation and metabolism of cholesterol in the body. HDL particles are responsible for picking up excess cholesterol from tissues and cells throughout the body and transporting it back to the liver, where it can be broken down and removed from the body. This process is known as reverse cholesterol transport.
HDL2 is one of the subclasses of HDL particles, which are classified based on their size, density, and composition. HDL2 particles are larger and denser than other HDL subclasses, such as HDL3. They have a higher proportion of cholesteryl esters to phospholipids and apolipoproteins compared to other HDL subclasses.
Elevated levels of HDL2 have been associated with a lower risk of cardiovascular disease, while low levels of HDL2 have been linked to an increased risk of heart disease. However, the exact role of HDL2 in cardiovascular health and disease is still being studied and understood.
Pulmonary surfactants are a complex mixture of lipids and proteins that are produced by the alveolar type II cells in the lungs. They play a crucial role in reducing the surface tension at the air-liquid interface within the alveoli, which helps to prevent collapse of the lungs during expiration. Surfactants also have important immunological functions, such as inhibiting the growth of certain bacteria and modulating the immune response. Deficiency or dysfunction of pulmonary surfactants can lead to respiratory distress syndrome (RDS) in premature infants and other lung diseases.
Urease is an enzyme that catalyzes the hydrolysis of urea into ammonia and carbon dioxide. It is found in various organisms, including bacteria, fungi, and plants. In medicine, urease is often associated with certain bacterial infections, such as those caused by Helicobacter pylori, which can produce large amounts of this enzyme. The presence of urease in these infections can lead to increased ammonia production, contributing to the development of gastritis and peptic ulcers.
Phytochrome is a photoreceptor protein responsible for detecting and mediating responses to different wavelengths of light, primarily red and far-red, in plants and some microorganisms. It plays a crucial role in various physiological processes such as seed germination, stem elongation, leaf expansion, chlorophyll production, and flowering.
The phytochrome protein exists in two interconvertible forms: Pr (the red-light-absorbing form) and Pfr (the far-red-light-absorbing form). The conversion between these forms regulates the downstream signaling pathways that control plant growth and development. Red light (around 660 nm) promotes the formation of the Pfr form, while far-red light (around 730 nm) converts it back to the Pr form. This reversible photoresponse allows plants to adapt their growth patterns based on the quality and duration of light they receive.
Ultracentrifugation is a medical and laboratory technique used for the separation of particles of different sizes, densities, or shapes from a mixture based on their sedimentation rates. This process involves the use of a specialized piece of equipment called an ultracentrifuge, which can generate very high centrifugal forces, much greater than those produced by a regular centrifuge.
In ultracentrifugation, a sample is placed in a special tube and spun at extremely high speeds, causing the particles within the sample to separate based on their size, shape, and density. The larger or denser particles will sediment faster and accumulate at the bottom of the tube, while smaller or less dense particles will remain suspended in the solution or sediment more slowly.
Ultracentrifugation is a valuable tool in various fields, including biochemistry, molecular biology, and virology. It can be used to purify and concentrate viruses, subcellular organelles, membrane fractions, ribosomes, DNA, and other macromolecules from complex mixtures. The technique can also provide information about the size, shape, and density of these particles, making it a crucial method for characterizing and studying their properties.
Abetalipoproteinemia is a rare inherited genetic disorder that affects the way the body absorbs and metabolizes fats and fat-soluble vitamins. It is caused by mutations in the genes responsible for producing proteins involved in the formation and transport of beta-lipoproteins, which are necessary for the absorption of dietary fats and cholesterol from the intestines.
Individuals with abetalipoproteinemia are unable to produce adequate levels of these lipoproteins, leading to a deficiency in fat-soluble vitamins (A, D, E, and K) and an accumulation of fats in the intestines. This results in various symptoms such as steatorrhea (fatty, foul-smelling stools), malabsorption, diarrhea, failure to thrive, and neurological issues due to vitamin E deficiency.
The disorder is typically diagnosed in infancy or early childhood and requires lifelong dietary management, including a low-fat diet and supplementation with fat-soluble vitamins. Early intervention can help prevent the progression of neurological symptoms and improve overall prognosis.
Molecular weight, also known as molecular mass, is the mass of a molecule. It is expressed in units of atomic mass units (amu) or daltons (Da). Molecular weight is calculated by adding up the atomic weights of each atom in a molecule. It is a useful property in chemistry and biology, as it can be used to determine the concentration of a substance in a solution, or to calculate the amount of a substance that will react with another in a chemical reaction.
An amino acid sequence is the specific order of amino acids in a protein or peptide molecule, formed by the linking of the amino group (-NH2) of one amino acid to the carboxyl group (-COOH) of another amino acid through a peptide bond. The sequence is determined by the genetic code and is unique to each type of protein or peptide. It plays a crucial role in determining the three-dimensional structure and function of proteins.
Apolipoprotein A-II (ApoA-II) is a protein component of high-density lipoproteins (HDL), often referred to as "good cholesterol." It is one of the major apolipoproteins in HDL and plays a role in the structure, metabolism, and function of HDL particles. ApoA-II is produced primarily in the liver and intestine and helps facilitate the transport of cholesterol from tissues to the liver for excretion. Additionally, ApoA-II has been shown to have anti-inflammatory properties and may play a role in the regulation of the immune response.
Phospholipids are a major class of lipids that consist of a hydrophilic (water-attracting) head and two hydrophobic (water-repelling) tails. The head is composed of a phosphate group, which is often bound to an organic molecule such as choline, ethanolamine, serine or inositol. The tails are made up of two fatty acid chains.
Phospholipids are a key component of cell membranes and play a crucial role in maintaining the structural integrity and function of the cell. They form a lipid bilayer, with the hydrophilic heads facing outwards and the hydrophobic tails facing inwards, creating a barrier that separates the interior of the cell from the outside environment.
Phospholipids are also involved in various cellular processes such as signal transduction, intracellular trafficking, and protein function regulation. Additionally, they serve as emulsifiers in the digestive system, helping to break down fats in the diet.
Light-harvesting protein complexes are specialized structures in photosynthetic organisms, such as plants, algae, and some bacteria, that capture and transfer light energy to the reaction centers where the initial chemical reactions of photosynthesis occur. These complexes consist of proteins and pigments (primarily chlorophylls and carotenoids) arranged in a way that allows them to absorb light most efficiently. The absorbed light energy is then converted into electrical charges, which are transferred to the reaction centers for further chemical reactions leading to the production of organic compounds and oxygen. The light-harvesting protein complexes play a crucial role in initiating the process of photosynthesis and optimizing its efficiency by capturing and distributing light energy.
Chlorophyll is a green pigment found in the chloroplasts of photosynthetic plants, algae, and some bacteria. It plays an essential role in light-dependent reactions of photosynthesis by absorbing light energy, primarily from the blue and red parts of the electromagnetic spectrum, and converting it into chemical energy to fuel the synthesis of carbohydrates from carbon dioxide and water. The structure of chlorophyll includes a porphyrin ring, which binds a central magnesium ion, and a long phytol tail. There are several types of chlorophyll, including chlorophyll a and chlorophyll b, which have distinct absorption spectra and slightly different structures. Chlorophyll is crucial for the process of photosynthesis, enabling the conversion of sunlight into chemical energy and the release of oxygen as a byproduct.
Pulmonary surfactant-associated proteins are a group of proteins that are found in the pulmonary surfactant, a complex mixture of lipids and proteins that coats the inside surfaces of the alveoli in the lungs. The primary function of pulmonary surfactant is to reduce the surface tension at the air-liquid interface in the alveoli, which facilitates breathing by preventing collapse of the alveoli during expiration.
There are four main pulmonary surfactant-associated proteins, designated as SP-A, SP-B, SP-C, and SP-D. These proteins play important roles in maintaining the stability and function of the pulmonary surfactant film, as well as participating in host defense mechanisms in the lungs.
SP-A and SP-D are members of the collectin family of proteins and have been shown to have immunomodulatory functions, including binding to pathogens and modulating immune cell responses. SP-B and SP-C are hydrophobic proteins that play critical roles in reducing surface tension at the air-liquid interface and maintaining the stability of the surfactant film.
Deficiencies or dysfunction of pulmonary surfactant-associated proteins have been implicated in various lung diseases, including respiratory distress syndrome (RDS) in premature infants, chronic interstitial lung diseases, and pulmonary fibrosis.
Helicobacter mustelae is a gram-negative, spiral-shaped bacterium that colonizes the stomach of ferrets and some other mustelids. It is closely related to Helicobacter pylori, a bacterium known to cause gastritis, peptic ulcers, and gastric cancer in humans.
H. mustelae has been observed to cause similar gastrointestinal diseases in ferrets, making it an important model organism for studying H. pylori infection and related pathologies. Like H. pylori, H. mustelae produces urease, which helps it neutralize the acidic environment of the stomach and facilitates its survival and colonization.
While there is no direct evidence suggesting that H. mustelae can infect humans, researchers use this bacterium to study the pathogenesis and immune responses associated with Helicobacter infections, contributing to a better understanding of gastric diseases caused by these bacteria.
Flavin-Adenine Dinucleotide (FAD) is a coenzyme that plays a crucial role in various metabolic processes, particularly in the electron transport chain where it functions as an electron carrier in oxidation-reduction reactions. FAD is composed of a flavin moiety, riboflavin or vitamin B2, and adenine dinucleotide. It can exist in two forms: an oxidized form (FAD) and a reduced form (FADH2). The reduction of FAD to FADH2 involves the gain of two electrons and two protons, which is accompanied by a significant conformational change that allows FADH2 to donate its electrons to subsequent components in the electron transport chain, ultimately leading to the production of ATP, the main energy currency of the cell.
Immunoelectrophoresis (IEP) is a laboratory technique used in the field of clinical pathology and immunology. It is a method for separating and identifying proteins, particularly immunoglobulins or antibodies, in a sample. This technique combines the principles of electrophoresis, which separates proteins based on their electric charge and size, with immunological reactions, which detect specific proteins using antigen-antibody interactions.
In IEP, a protein sample is first separated by electrophoresis in an agarose or agar gel matrix on a glass slide or in a test tube. After separation, an antibody specific to the protein of interest is layered on top of the gel and allowed to diffuse towards the separated proteins. This creates a reaction between the antigen (protein) and the antibody, forming a visible precipitate at the point where they meet. The precipitate line's position and intensity can then be analyzed to identify and quantify the protein of interest.
Immunoelectrophoresis is particularly useful in diagnosing various medical conditions, such as immunodeficiency disorders, monoclonal gammopathies (like multiple myeloma), and other plasma cell dyscrasias. It can help detect abnormal protein patterns, quantify specific immunoglobulins, and identify the presence of M-proteins or Bence Jones proteins, which are indicative of monoclonal gammopathies.
Molecular sequence data refers to the specific arrangement of molecules, most commonly nucleotides in DNA or RNA, or amino acids in proteins, that make up a biological macromolecule. This data is generated through laboratory techniques such as sequencing, and provides information about the exact order of the constituent molecules. This data is crucial in various fields of biology, including genetics, evolution, and molecular biology, allowing for comparisons between different organisms, identification of genetic variations, and studies of gene function and regulation.
Protein conformation refers to the specific three-dimensional shape that a protein molecule assumes due to the spatial arrangement of its constituent amino acid residues and their associated chemical groups. This complex structure is determined by several factors, including covalent bonds (disulfide bridges), hydrogen bonds, van der Waals forces, and ionic bonds, which help stabilize the protein's unique conformation.
Protein conformations can be broadly classified into two categories: primary, secondary, tertiary, and quaternary structures. The primary structure represents the linear sequence of amino acids in a polypeptide chain. The secondary structure arises from local interactions between adjacent amino acid residues, leading to the formation of recurring motifs such as α-helices and β-sheets. Tertiary structure refers to the overall three-dimensional folding pattern of a single polypeptide chain, while quaternary structure describes the spatial arrangement of multiple folded polypeptide chains (subunits) that interact to form a functional protein complex.
Understanding protein conformation is crucial for elucidating protein function, as the specific three-dimensional shape of a protein directly influences its ability to interact with other molecules, such as ligands, nucleic acids, or other proteins. Any alterations in protein conformation due to genetic mutations, environmental factors, or chemical modifications can lead to loss of function, misfolding, aggregation, and disease states like neurodegenerative disorders and cancer.
Phosphatidylcholine-Sterol O-Acyltransferase (PCOAT, also known as Sterol O-Acyltransferase 1 or SOAT1) is an enzyme that plays a crucial role in the regulation of cholesterol metabolism. It is located in the endoplasmic reticulum and is responsible for the transfer of acyl groups from phosphatidylcholine to cholesterol, forming cholesteryl esters. This enzymatic reaction results in the storage of excess cholesterol in lipid droplets, preventing its accumulation in the cell membrane and potentially contributing to the development of atherosclerosis if not properly regulated.
Defects or mutations in PCOAT can lead to disruptions in cholesterol homeostasis, which may contribute to various diseases such as cardiovascular disorders, metabolic syndrome, and neurodegenerative conditions. Therefore, understanding the function and regulation of this enzyme is essential for developing therapeutic strategies aimed at managing cholesterol-related disorders.
Amino acids are organic compounds that serve as the building blocks of proteins. They consist of a central carbon atom, also known as the alpha carbon, which is bonded to an amino group (-NH2), a carboxyl group (-COOH), a hydrogen atom (H), and a variable side chain (R group). The R group can be composed of various combinations of atoms such as hydrogen, oxygen, sulfur, nitrogen, and carbon, which determine the unique properties of each amino acid.
There are 20 standard amino acids that are encoded by the genetic code and incorporated into proteins during translation. These include:
1. Alanine (Ala)
2. Arginine (Arg)
3. Asparagine (Asn)
4. Aspartic acid (Asp)
5. Cysteine (Cys)
6. Glutamine (Gln)
7. Glutamic acid (Glu)
8. Glycine (Gly)
9. Histidine (His)
10. Isoleucine (Ile)
11. Leucine (Leu)
12. Lysine (Lys)
13. Methionine (Met)
14. Phenylalanine (Phe)
15. Proline (Pro)
16. Serine (Ser)
17. Threonine (Thr)
18. Tryptophan (Trp)
19. Tyrosine (Tyr)
20. Valine (Val)
Additionally, there are several non-standard or modified amino acids that can be incorporated into proteins through post-translational modifications, such as hydroxylation, methylation, and phosphorylation. These modifications expand the functional diversity of proteins and play crucial roles in various cellular processes.
Amino acids are essential for numerous biological functions, including protein synthesis, enzyme catalysis, neurotransmitter production, energy metabolism, and immune response regulation. Some amino acids can be synthesized by the human body (non-essential), while others must be obtained through dietary sources (essential).
Immunodiffusion is a laboratory technique used in immunology to detect and measure the presence of specific antibodies or antigens in a sample. It is based on the principle of diffusion, where molecules move from an area of high concentration to an area of low concentration until they reach equilibrium. In this technique, a sample containing an unknown quantity of antigen or antibody is placed in a gel or agar medium that contains a known quantity of antibody or antigen, respectively.
The two substances then diffuse towards each other and form a visible precipitate at the point where they meet and reach equivalence, which indicates the presence and quantity of the specific antigen or antibody in the sample. There are several types of immunodiffusion techniques, including radial immunodiffusion (RID) and double immunodiffusion (Ouchterlony technique). These techniques are widely used in diagnostic laboratories to identify and measure various antigens and antibodies, such as those found in infectious diseases, autoimmune disorders, and allergic reactions.
Cytochrome P-450 CYP2B1 is a specific isoform of the cytochrome P-450 enzyme system, which is involved in the metabolism of drugs and other xenobiotics in the liver. This particular isoenzyme is primarily found in rats and is responsible for the metabolism of a variety of substrates, including certain drugs, steroids, and environmental toxins.
The cytochrome P-450 system is a group of enzymes located in the endoplasmic reticulum of cells, particularly in the liver. These enzymes play a crucial role in the metabolism of various substances, including drugs, hormones, and toxins. They work by catalyzing oxidation-reduction reactions that convert lipophilic compounds into more hydrophilic ones, which can then be excreted from the body.
CYP2B1 is one of many isoforms of cytochrome P-450, and it has a preference for certain types of substrates. It is involved in the metabolism of drugs such as cyclophosphamide, ifosfamide, and methadone, as well as steroids like progesterone and environmental toxins like pentachlorophenol.
It's important to note that while CYP2B1 is an essential enzyme in rats, its human counterpart, CYP2B6, plays a similar role in drug metabolism in humans. Understanding the function and regulation of these enzymes can help in predicting drug interactions, designing new drugs, and tailoring therapies to individual patients based on their genetic makeup.
Flavin Mononucleotide (FMN) is a coenzyme that plays a crucial role in biological oxidation-reduction reactions. It is derived from the vitamin riboflavin (also known as vitamin B2) and is composed of a flavin molecule bonded to a nucleotide. FMN functions as an electron carrier, accepting and donating electrons in various metabolic pathways, including the citric acid cycle and the electron transport chain, which are essential for energy production in cells. It also participates in the detoxification of harmful substances and contributes to the maintenance of cellular redox homeostasis. FMN can exist in two forms: the oxidized form (FMN) and the reduced form (FMNH2), depending on its involvement in redox reactions.
Circular dichroism (CD) is a technique used in physics and chemistry to study the structure of molecules, particularly large biological molecules such as proteins and nucleic acids. It measures the difference in absorption of left-handed and right-handed circularly polarized light by a sample. This difference in absorption can provide information about the three-dimensional structure of the molecule, including its chirality or "handedness."
In more technical terms, CD is a form of spectroscopy that measures the differential absorption of left and right circularly polarized light as a function of wavelength. The CD signal is measured in units of millidegrees (mdeg) and can be positive or negative, depending on the type of chromophore and its orientation within the molecule.
CD spectra can provide valuable information about the secondary and tertiary structure of proteins, as well as the conformation of nucleic acids. For example, alpha-helical proteins typically exhibit a strong positive band near 190 nm and two negative bands at around 208 nm and 222 nm, while beta-sheet proteins show a strong positive band near 195 nm and two negative bands at around 217 nm and 175 nm.
CD spectroscopy is a powerful tool for studying the structural changes that occur in biological molecules under different conditions, such as temperature, pH, or the presence of ligands or other molecules. It can also be used to monitor the folding and unfolding of proteins, as well as the binding of drugs or other small molecules to their targets.
Lipids are a broad group of organic compounds that are insoluble in water but soluble in nonpolar organic solvents. They include fats, waxes, sterols, fat-soluble vitamins (such as vitamins A, D, E, and K), monoglycerides, diglycerides, triglycerides, and phospholipids. Lipids serve many important functions in the body, including energy storage, acting as structural components of cell membranes, and serving as signaling molecules. High levels of certain lipids, particularly cholesterol and triglycerides, in the blood are associated with an increased risk of cardiovascular disease.
Apolipoprotein B-100 (apoB-100) is a large protein component of low-density lipoprotein (LDL), also known as "bad cholesterol." It plays a crucial role in the metabolism and transport of fats and cholesterol in the body. ApoB-100 is responsible for the binding of LDL to specific receptors on cell surfaces, facilitating the uptake of lipoprotein particles by cells. Elevated levels of apoB-100 in the blood are associated with an increased risk of developing cardiovascular diseases, such as atherosclerosis and coronary artery disease.
Protein binding, in the context of medical and biological sciences, refers to the interaction between a protein and another molecule (known as the ligand) that results in a stable complex. This process is often reversible and can be influenced by various factors such as pH, temperature, and concentration of the involved molecules.
In clinical chemistry, protein binding is particularly important when it comes to drugs, as many of them bind to proteins (especially albumin) in the bloodstream. The degree of protein binding can affect a drug's distribution, metabolism, and excretion, which in turn influence its therapeutic effectiveness and potential side effects.
Protein-bound drugs may be less available for interaction with their target tissues, as only the unbound or "free" fraction of the drug is active. Therefore, understanding protein binding can help optimize dosing regimens and minimize adverse reactions.
Zinostatin is not a widely recognized or commonly used term in medicine. However, it appears to be a brand name for a formulation of the anti-cancer drug Neocarzinostatin (NCS). Neocarzinostatin is a protein produced by the bacterium Streptomyces carzinostaticus and has been studied for its potential to inhibit the growth of various types of cancer cells.
Zinostatin is specifically used in the treatment of hepatocellular carcinoma (HCC), which is a type of liver cancer. It is administered via arterial infusion, where the drug is delivered directly into the hepatic artery that supplies blood to the liver. This method allows for higher concentrations of the drug to reach the tumor site while minimizing systemic exposure and potential side effects.
It's important to note that medical terminology can vary by region and context, so it's possible that "Zinostatin" may not be a term used in all medical communities or for all purposes. Always consult with a healthcare professional or trusted medical source for accurate information.
Apolipoprotein C-III (APOC3) is a protein that is produced in the liver and circulates in the bloodstream. It is a component of certain lipoproteins, including very low-density lipoproteins (VLDL) and chylomicrons, which are responsible for transporting fat molecules, such as triglycerides and cholesterol, throughout the body.
APOC3 plays a role in regulating the metabolism of these lipoproteins. Specifically, it inhibits the activity of an enzyme called lipoprotein lipase, which breaks down triglycerides in VLDL and chylomicrons. As a result, high levels of APOC3 can lead to an increase in triglyceride levels in the blood, which is a risk factor for cardiovascular disease.
Genetic variations in the APOC3 gene have been associated with differences in triglyceride levels and risk of cardiovascular disease. Some studies have suggested that reducing APOC3 levels through genetic editing or other means may be a promising strategy for lowering triglycerides and reducing the risk of heart disease.
In the context of medicine, "chemistry" often refers to the field of study concerned with the properties, composition, and structure of elements and compounds, as well as their reactions with one another. It is a fundamental science that underlies much of modern medicine, including pharmacology (the study of drugs), toxicology (the study of poisons), and biochemistry (the study of the chemical processes that occur within living organisms).
In addition to its role as a basic science, chemistry is also used in medical testing and diagnosis. For example, clinical chemistry involves the analysis of bodily fluids such as blood and urine to detect and measure various substances, such as glucose, cholesterol, and electrolytes, that can provide important information about a person's health status.
Overall, chemistry plays a critical role in understanding the mechanisms of diseases, developing new treatments, and improving diagnostic tests and techniques.
Hyperlipidemias are a group of disorders characterized by an excess of lipids (fats) or lipoproteins in the blood. These include elevated levels of cholesterol, triglycerides, or both. Hyperlipidemias can be inherited (primary) or caused by other medical conditions (secondary). They are a significant risk factor for developing cardiovascular diseases, such as atherosclerosis and coronary artery disease.
There are two main types of lipids that are commonly measured in the blood: low-density lipoprotein (LDL) cholesterol, often referred to as "bad" cholesterol, and high-density lipoprotein (HDL) cholesterol, known as "good" cholesterol. High levels of LDL cholesterol can lead to the formation of plaques in the arteries, which can narrow or block them and increase the risk of heart attack or stroke. On the other hand, high levels of HDL cholesterol are protective because they help remove LDL cholesterol from the bloodstream.
Triglycerides are another type of lipid that can be measured in the blood. Elevated triglyceride levels can also contribute to the development of cardiovascular disease, particularly when combined with high LDL cholesterol and low HDL cholesterol levels.
Hyperlipidemias are typically diagnosed through a blood test that measures the levels of various lipids and lipoproteins in the blood. Treatment may include lifestyle changes, such as following a healthy diet, getting regular exercise, losing weight, and quitting smoking, as well as medication to lower lipid levels if necessary.
Chemical phenomena refer to the changes and interactions that occur at the molecular or atomic level when chemicals are involved. These phenomena can include chemical reactions, in which one or more substances (reactants) are converted into different substances (products), as well as physical properties that change as a result of chemical interactions, such as color, state of matter, and solubility. Chemical phenomena can be studied through various scientific disciplines, including chemistry, biochemistry, and physics.
Hypolipoproteinemias are a group of genetic disorders characterized by low levels of lipoproteins in the blood. Lipoproteins are complex particles composed of proteins and lipids that play a crucial role in the transport and metabolism of fat molecules, such as cholesterol and triglycerides, in the body.
There are several types of hypolipoproteinemias, each associated with deficiencies in specific lipoproteins:
1. Hypobetalipoproteinemia: This disorder is characterized by low levels of beta-lipoproteins, also known as low-density lipoproteins (LDL), or "bad" cholesterol. It can lead to decreased absorption of fat-soluble vitamins and an increased risk of fatty liver disease.
2. Abetalipoproteinemia: This is a rare autosomal recessive disorder characterized by the absence of beta-lipoproteins and apolipoprotein B, which results in very low levels of LDL cholesterol and high-density lipoproteins (HDL), or "good" cholesterol. It can lead to fat malabsorption, neurological symptoms, and retinal degeneration.
3. Tangier disease: This disorder is caused by a deficiency in apolipoprotein A-I and results in low levels of HDL cholesterol. It can cause enlarged orange-colored tonsils, neuropathy, and an increased risk of coronary artery disease.
4. Familial hypoalphalipoproteinemia: This disorder is characterized by low levels of HDL cholesterol due to a deficiency in apolipoprotein A-I or A-II. It can increase the risk of premature coronary artery disease.
It's important to note that while some hypolipoproteinemias are associated with an increased risk of cardiovascular disease, others may actually protect against it due to reduced levels of atherogenic lipoproteins. Treatment for these disorders typically involves dietary modifications and supplementation of fat-soluble vitamins and essential fatty acids. In some cases, medication may be necessary to manage symptoms or prevent complications.
Dietary cholesterol is a type of cholesterol that comes from the foods we eat. It is present in animal-derived products such as meat, poultry, dairy products, and eggs. While dietary cholesterol can contribute to an increase in blood cholesterol levels for some people, it's important to note that saturated and trans fats have a more significant impact on blood cholesterol levels than dietary cholesterol itself.
The American Heart Association recommends limiting dietary cholesterol intake to less than 300 milligrams per day for most people, and less than 200 milligrams per day for those with a history of heart disease or high cholesterol levels. However, individual responses to dietary cholesterol can vary, so it's essential to monitor blood cholesterol levels and adjust dietary habits accordingly.
Hyperlipoproteinemia Type IV is a genetic disorder characterized by an increased level of very low-density lipoproteins (VLDL) in the blood. This leads to elevated levels of triglycerides, which are a type of fat found in the blood. The condition is also sometimes referred to as "Fredrickson Type IV."
People with Hyperlipoproteinemia Type IV have an increased risk of developing pancreatitis, a potentially life-threatening inflammation of the pancreas, due to high levels of triglycerides. They may also have an increased risk of cardiovascular disease due to elevated levels of VLDL and other atherogenic lipoproteins.
The condition is usually inherited in an autosomal dominant manner, meaning that a child has a 50% chance of inheriting the disorder if one parent has it. However, some cases may be caused by mutations in multiple genes or by environmental factors such as obesity, diabetes, and excessive alcohol consumption.
Treatment for Hyperlipoproteinemia Type IV typically involves lifestyle modifications such as weight loss, exercise, and dietary changes to reduce triglyceride levels. In some cases, medication may be necessary to control the condition.
Apolipoprotein B-48 (apoB-48) is a protein component of chylomicrons, which are lipoprotein particles responsible for carrying dietary fat and cholesterol from the intestines to other parts of the body. ApoB-48 is produced in the intestines and is a shorter version of apolipoprotein B-100 (apoB-100), which is a component of low-density lipoproteins (LDL) or "bad cholesterol."
Chylomicrons are assembled and secreted by intestinal cells after a meal, and apoB-48 is essential for the formation and function of these particles. ApoB-48-containing chylomicrons transport dietary lipids to various tissues, including the liver, where they contribute to the maintenance of lipid homeostasis.
Elevated levels of apoB-48 in the blood have been associated with an increased risk of cardiovascular disease, particularly in individuals with familial chylomicronemia syndrome (FCS), a rare genetic disorder characterized by severely elevated triglyceride levels due to impaired clearance of chylomicrons.
"Enterobacter aerogenes" is a species of gram-negative, facultatively anaerobic, rod-shaped bacteria that are commonly found in the environment, including in soil, water, and vegetation. In medical contexts, E. aerogenes is often considered an opportunistic pathogen, meaning it can cause infection in individuals with compromised immune systems or underlying health conditions.
E. aerogenes is a member of the family Enterobacteriaceae and is closely related to other pathogens such as Klebsiella pneumoniae and Escherichia coli. It is known for its ability to produce large amounts of gas, including carbon dioxide and hydrogen sulfide, which can contribute to its virulence and make it difficult to identify using traditional biochemical tests.
E. aerogenes can cause a variety of infections, including urinary tract infections, pneumonia, bacteremia, and wound infections. It is often resistant to multiple antibiotics, which can make treatment challenging. In recent years, there has been an increase in the number of E. aerogenes isolates that are resistant to carbapenems, a class of antibiotics that are often used as a last resort for treating serious bacterial infections.
Flavoproteins are a type of protein molecule that contain noncovalently bound flavin mononucleotide (FMN) or flavin adenine dinucleotide (FAD) as cofactors. These flavin cofactors play a crucial role in redox reactions, acting as electron carriers in various metabolic pathways such as cellular respiration and oxidative phosphorylation. Flavoproteins are involved in several biological processes, including the breakdown of fatty acids, amino acids, and carbohydrates, as well as the synthesis of steroids and other lipids. They can also function as enzymes that catalyze various redox reactions, such as oxidases, dehydrogenases, and reductases. Flavoproteins are widely distributed in nature and found in many organisms, from bacteria to humans.
Cytochrome c is a small protein that is involved in the electron transport chain, a key part of cellular respiration in which cells generate energy in the form of ATP. Cytochrome c contains a heme group, which binds to and transports electrons. The cytochrome c group refers to a class of related cytochromes that have similar structures and functions. These proteins are found in the mitochondria of eukaryotic cells (such as those of plants and animals) and in the inner membranes of bacteria. They play a crucial role in the production of energy within the cell, and are also involved in certain types of programmed cell death (apoptosis).
Metalloproteins are proteins that contain one or more metal ions as a cofactor, which is required for their biological activity. These metal ions play crucial roles in the catalytic function, structural stability, and electron transfer processes of metalloproteins. The types of metals involved can include iron, zinc, copper, magnesium, calcium, or manganese, among others. Examples of metalloproteins are hemoglobin (contains heme-bound iron), cytochrome c (contains heme-bound iron and functions in electron transfer), and carbonic anhydrase (contains zinc and catalyzes the conversion between carbon dioxide and bicarbonate).
Hyperlipoproteinemias are medical conditions characterized by elevated levels of lipoproteins in the blood. Lipoproteins are particles that consist of proteins and lipids, which are responsible for transporting all fat molecules, such as cholesterol and triglycerides, around the body within the water outside cells. These lipids cannot dissolve in the blood, so they must be carried by these lipoprotein particles.
There are several types of hyperlipoproteinemias, classified based on the type of lipoprotein that is elevated and the pattern of inheritance. The most commonly recognized classification system is the Fredrickson classification, which includes five main types:
1. Type I - characterized by an excess of chylomicrons, a type of lipoprotein that carries dietary lipids, leading to extremely high levels of triglycerides in the blood. This rare disorder is usually caused by genetic mutations.
2. Type II - divided into two subtypes:
a. Type IIa - characterized by elevated LDL (low-density lipoprotein), or "bad" cholesterol, levels and often associated with premature cardiovascular disease. This condition can be caused by genetic factors, lifestyle choices, or both.
b. Type IIb - marked by increased levels of both LDL cholesterol and VLDL (very low-density lipoprotein), which leads to elevated triglycerides and cholesterol in the blood. This subtype can also be influenced by genetic factors, lifestyle choices, or both.
3. Type III - known as broad beta disease or remnant removal disease, this condition is characterized by an abnormal accumulation of remnant particles from VLDL and IDL (intermediate-density lipoprotein) metabolism, leading to increased levels of both cholesterol and triglycerides. This disorder can be caused by genetic mutations or secondary factors like diabetes, obesity, or hypothyroidism.
4. Type IV - characterized by elevated VLDL particles and high triglyceride levels in the blood. This condition is often associated with metabolic syndrome, obesity, diabetes, and alcohol consumption.
5. Type V - marked by increased VLDL and chylomicrons (lipoprotein particles that transport dietary lipids) in the blood, leading to extremely high triglyceride levels. This rare condition can be caused by genetic factors or secondary factors like diabetes, obesity, alcohol consumption, or uncontrolled lipid absorption.
It is important to note that these types are not mutually exclusive and can coexist in various combinations. Additionally, lifestyle choices such as diet, exercise, smoking, and alcohol consumption can significantly impact lipoprotein levels and contribute to the development of dyslipidemia (abnormal lipid levels).
Iron-sulfur proteins are a group of metalloproteins that contain iron and sulfur atoms in their active centers. These clusters of iron and sulfur atoms, also known as iron-sulfur clusters, can exist in various forms, including Fe-S, 2Fe-2S, 3Fe-4S, and 4Fe-4S structures. The iron atoms are coordinated to the protein through cysteine residues, while the sulfur atoms can be in the form of sulfide (S2-) or sulfane (-S-).
These proteins play crucial roles in many biological processes, such as electron transfer, redox reactions, and enzyme catalysis. They are found in various organisms, from bacteria to humans, and are involved in a wide range of cellular functions, including energy metabolism, photosynthesis, nitrogen fixation, and DNA repair.
Iron-sulfur proteins can be classified into several categories based on their structure and function, such as ferredoxins, Rieske proteins, high-potential iron-sulfur proteins (HiPIPs), and radical SAM enzymes. Dysregulation or mutations in iron-sulfur protein genes have been linked to various human diseases, including neurodegenerative disorders, cancer, and mitochondrial disorders.
Gel chromatography is a type of liquid chromatography that separates molecules based on their size or molecular weight. It uses a stationary phase that consists of a gel matrix made up of cross-linked polymers, such as dextran, agarose, or polyacrylamide. The gel matrix contains pores of various sizes, which allow smaller molecules to penetrate deeper into the matrix while larger molecules are excluded.
In gel chromatography, a mixture of molecules is loaded onto the top of the gel column and eluted with a solvent that moves down the column by gravity or pressure. As the sample components move down the column, they interact with the gel matrix and get separated based on their size. Smaller molecules can enter the pores of the gel and take longer to elute, while larger molecules are excluded from the pores and elute more quickly.
Gel chromatography is commonly used to separate and purify proteins, nucleic acids, and other biomolecules based on their size and molecular weight. It is also used in the analysis of polymers, colloids, and other materials with a wide range of applications in chemistry, biology, and medicine.
Urobilin is a pigment produced in the liver as a byproduct of the breakdown of bilirubin, which is a waste product resulting from the breakdown of hemoglobin in red blood cells. Some urobilin is excreted through the bile into the intestines, where it can be converted by bacteria into stercobilin, another pigment responsible for the brown color of feces. A portion of the urobilin produced in the liver is reabsorbed into the bloodstream and eventually excreted through the urine, giving it a yellow color. Therefore, urobilin can be detected in both urine and feces.
In the context of medical and biological sciences, a "binding site" refers to a specific location on a protein, molecule, or cell where another molecule can attach or bind. This binding interaction can lead to various functional changes in the original protein or molecule. The other molecule that binds to the binding site is often referred to as a ligand, which can be a small molecule, ion, or even another protein.
The binding between a ligand and its target binding site can be specific and selective, meaning that only certain ligands can bind to particular binding sites with high affinity. This specificity plays a crucial role in various biological processes, such as signal transduction, enzyme catalysis, or drug action.
In the case of drug development, understanding the location and properties of binding sites on target proteins is essential for designing drugs that can selectively bind to these sites and modulate protein function. This knowledge can help create more effective and safer therapeutic options for various diseases.
I'm sorry for any confusion, but "spectrum analysis" is not a commonly used medical term. Spectrum analysis is a term that is more frequently used in the fields of physics, mathematics, and engineering to describe the process of breaking down a signal or a wave into its different frequencies and amplitudes, creating a visual representation called a spectrum.
If you have any concerns about a medical issue, I would recommend consulting with a healthcare professional for accurate information and guidance.
Phosphatidylcholines (PtdCho) are a type of phospholipids that are essential components of cell membranes in living organisms. They are composed of a hydrophilic head group, which contains a choline moiety, and two hydrophobic fatty acid chains. Phosphatidylcholines are crucial for maintaining the structural integrity and function of cell membranes, and they also serve as important precursors for the synthesis of signaling molecules such as acetylcholine. They can be found in various tissues and biological fluids, including blood, and are abundant in foods such as soybeans, eggs, and meat. Phosphatidylcholines have been studied for their potential health benefits, including their role in maintaining healthy lipid metabolism and reducing the risk of cardiovascular disease.
Photosynthetic Reaction Center (RC) Complex Proteins are specialized protein-pigment structures that play a crucial role in the primary process of light-driven electron transport during photosynthesis. They are present in the thylakoid membranes of cyanobacteria, algae, and higher plants.
The Photosynthetic Reaction Center Complex Proteins are composed of two major components: the light-harvesting complex (LHC) and the reaction center (RC). The LHC contains antenna pigments like chlorophylls and carotenoids that absorb sunlight and transfer the excitation energy to the RC. The RC is a multi-subunit protein complex containing cofactors such as bacteriochlorophyll, pheophytin, quinones, and iron-sulfur clusters.
When a photon of light is absorbed by the antenna pigments in the LHC, the energy is transferred to the RC, where it initiates a charge separation event. This results in the transfer of an electron from a donor molecule to an acceptor molecule, creating a flow of electrical charge and generating a transmembrane electrochemical gradient. The energy stored in this gradient is then used to synthesize ATP and reduce NADP+, which are essential for carbon fixation and other metabolic processes in the cell.
In summary, Photosynthetic Reaction Center Complex Proteins are specialized protein structures involved in capturing light energy and converting it into chemical energy during photosynthesis, ultimately driving the synthesis of ATP and NADPH for use in carbon fixation and other metabolic processes.
Blood protein disorders refer to a group of medical conditions that affect the production or function of proteins in the blood. These proteins are crucial for maintaining the proper functioning of the body's immune system, transporting nutrients, and preventing excessive bleeding. Some examples of blood protein disorders include:
1. Hemophilia: A genetic disorder caused by a deficiency or absence of clotting factors in the blood, leading to prolonged bleeding and poor clot formation.
2. Von Willebrand disease: A genetic disorder characterized by abnormal or deficient von Willebrand factor, which is necessary for platelet function and proper clotting.
3. Dysproteinemias: Abnormal levels of certain proteins in the blood, such as immunoglobulins (antibodies) or paraproteins, which can indicate underlying conditions like multiple myeloma or macroglobulinemia.
4. Hypoproteinemia: Low levels of total protein in the blood, often caused by liver disease, malnutrition, or kidney disease.
5. Hyperproteinemia: Elevated levels of total protein in the blood, which can be caused by dehydration, inflammation, or certain types of cancer.
6. Hemoglobinopathies: Genetic disorders affecting the structure and function of hemoglobin, a protein found in red blood cells that carries oxygen throughout the body. Examples include sickle cell anemia and thalassemia.
7. Disorders of complement proteins: Abnormalities in the complement system, which is a group of proteins involved in the immune response, can lead to conditions like autoimmune disorders or recurrent infections.
Treatment for blood protein disorders varies depending on the specific condition and its severity but may include medications, transfusions, or other medical interventions.
Oxidoreductases are a class of enzymes that catalyze oxidation-reduction reactions, where a electron is transferred from one molecule to another. N-Demethylating oxidoreductases are a specific subclass of these enzymes that catalyze the removal of a methyl group (-CH3) from a nitrogen atom (-N) in a molecule, which is typically a xenobiotic compound (a foreign chemical substance found within an living organism). This process often involves the transfer of electrons and the formation of water as a byproduct.
The reaction catalyzed by N-demethylating oxidoreductases can be represented as follows:
R-N-CH3 + O2 + H2O → R-N-H + CH3OH + H2O2
where R represents the rest of the molecule. The removal of the methyl group is often an important step in the metabolism and detoxification of xenobiotic compounds, as it can make them more water soluble and facilitate their excretion from the body.
Chyle is a milky, slightly opaque fluid that is present in the lymphatic system. It is formed in the small intestine during the digestion of food, particularly fats. Chyle consists of emulsified fat droplets (chylomicrons), proteins, electrolytes, and lymphocytes suspended in a watery solution. It is transported through the lacteals in the villi of the small intestine into the cisterna chyli and then to the thoracic duct, where it empties into the left subclavian vein. From there, it mixes with blood and circulates throughout the body. Chyle formation plays a crucial role in fat absorption and transportation in the human body.
Oxidation-Reduction (redox) reactions are a type of chemical reaction involving a transfer of electrons between two species. The substance that loses electrons in the reaction is oxidized, and the substance that gains electrons is reduced. Oxidation and reduction always occur together in a redox reaction, hence the term "oxidation-reduction."
In biological systems, redox reactions play a crucial role in many cellular processes, including energy production, metabolism, and signaling. The transfer of electrons in these reactions is often facilitated by specialized molecules called electron carriers, such as nicotinamide adenine dinucleotide (NAD+/NADH) and flavin adenine dinucleotide (FAD/FADH2).
The oxidation state of an element in a compound is a measure of the number of electrons that have been gained or lost relative to its neutral state. In redox reactions, the oxidation state of one or more elements changes as they gain or lose electrons. The substance that is oxidized has a higher oxidation state, while the substance that is reduced has a lower oxidation state.
Overall, oxidation-reduction reactions are fundamental to the functioning of living organisms and are involved in many important biological processes.
Bromotrichloromethane is a type of halomethane, which is a class of chemicals containing carbon and halogen atoms. Specifically, bromotrichloromethane is a colorless liquid with the chemical formula CBrCl3. It has been used as a fire extinguishing agent, a refrigerant, and an intermediate in the production of other chemicals.
In medical terms, bromotrichloromethane may be encountered in the context of occupational health and safety or environmental exposure assessment. Exposure to high levels of this chemical can cause irritation to the eyes, skin, and respiratory tract, as well as potential neurological effects such as headache, dizziness, and loss of consciousness. Long-term exposure has been linked to liver and kidney damage in animal studies.
It is important to note that bromotrichloromethane is not used in medical treatments or procedures. Its use in industrial applications has been largely phased out due to its ozone-depleting properties and potential health hazards.
Unsaturated fats are a type of fat that are primarily found in liquid form at room temperature. They are called "unsaturated" because their chemical structure contains one or more double bonds between the carbon atoms, making them less saturated with hydrogen atoms than saturated fats.
There are two main types of unsaturated fats: monounsaturated and polyunsaturated. Monounsaturated fats contain a single double bond in their chemical structure, while polyunsaturated fats contain multiple double bonds.
Unsaturated fats are generally considered to be healthier than saturated fats because they can help lower levels of harmful cholesterol in the blood and reduce the risk of heart disease. Foods that are high in unsaturated fats include vegetable oils, nuts, seeds, avocados, and fish.
It's important to note that while unsaturated fats are generally healthier than saturated fats, they are still high in calories and should be consumed in moderation as part of a balanced diet. Additionally, some types of polyunsaturated fats, such as trans fats, can actually increase the risk of heart disease and other health problems, so it's important to choose sources of unsaturated fats carefully.
Dietary fats, also known as fatty acids, are a major nutrient that the body needs for energy and various functions. They are an essential component of cell membranes and hormones, and they help the body absorb certain vitamins. There are several types of dietary fats:
1. Saturated fats: These are typically solid at room temperature and are found in animal products such as meat, butter, and cheese, as well as tropical oils like coconut and palm oil. Consuming a high amount of saturated fats can raise levels of unhealthy LDL cholesterol and increase the risk of heart disease.
2. Unsaturated fats: These are typically liquid at room temperature and can be further divided into monounsaturated and polyunsaturated fats. Monounsaturated fats, found in foods such as olive oil, avocados, and nuts, can help lower levels of unhealthy LDL cholesterol while maintaining levels of healthy HDL cholesterol. Polyunsaturated fats, found in foods such as fatty fish, flaxseeds, and walnuts, have similar effects on cholesterol levels and also provide essential omega-3 and omega-6 fatty acids that the body cannot produce on its own.
3. Trans fats: These are unsaturated fats that have been chemically modified to be solid at room temperature. They are often found in processed foods such as baked goods, fried foods, and snack foods. Consuming trans fats can raise levels of unhealthy LDL cholesterol and lower levels of healthy HDL cholesterol, increasing the risk of heart disease.
It is recommended to limit intake of saturated and trans fats and to consume more unsaturated fats as part of a healthy diet.
'Escherichia coli' (E. coli) is a type of gram-negative, facultatively anaerobic, rod-shaped bacterium that commonly inhabits the intestinal tract of humans and warm-blooded animals. It is a member of the family Enterobacteriaceae and one of the most well-studied prokaryotic model organisms in molecular biology.
While most E. coli strains are harmless and even beneficial to their hosts, some serotypes can cause various forms of gastrointestinal and extraintestinal illnesses in humans and animals. These pathogenic strains possess virulence factors that enable them to colonize and damage host tissues, leading to diseases such as diarrhea, urinary tract infections, pneumonia, and sepsis.
E. coli is a versatile organism with remarkable genetic diversity, which allows it to adapt to various environmental niches. It can be found in water, soil, food, and various man-made environments, making it an essential indicator of fecal contamination and a common cause of foodborne illnesses. The study of E. coli has contributed significantly to our understanding of fundamental biological processes, including DNA replication, gene regulation, and protein synthesis.
Biliverdine is a greenish pigment that is a byproduct of the breakdown of heme, which is a component of hemoglobin in red blood cells. It is formed when bilirubin, another byproduct of heme degradation, is reduced in the liver. Biliverdine is then converted back to bilirubin and excreted from the body as part of bile.
Elevated levels of biliverdine in the blood can indicate liver dysfunction or other medical conditions that affect the breakdown of heme. It may also be present in high concentrations in certain types of hemolytic anemia, where there is excessive destruction of red blood cells and subsequent release of large amounts of heme into the circulation.
The Cytochrome P-450 (CYP450) enzyme system is a group of enzymes found primarily in the liver, but also in other organs such as the intestines, lungs, and skin. These enzymes play a crucial role in the metabolism and biotransformation of various substances, including drugs, environmental toxins, and endogenous compounds like hormones and fatty acids.
The name "Cytochrome P-450" refers to the unique property of these enzymes to bind to carbon monoxide (CO) and form a complex that absorbs light at a wavelength of 450 nm, which can be detected spectrophotometrically.
The CYP450 enzyme system is involved in Phase I metabolism of xenobiotics, where it catalyzes oxidation reactions such as hydroxylation, dealkylation, and epoxidation. These reactions introduce functional groups into the substrate molecule, which can then undergo further modifications by other enzymes during Phase II metabolism.
There are several families and subfamilies of CYP450 enzymes, each with distinct substrate specificities and functions. Some of the most important CYP450 enzymes include:
1. CYP3A4: This is the most abundant CYP450 enzyme in the human liver and is involved in the metabolism of approximately 50% of all drugs. It also metabolizes various endogenous compounds like steroids, bile acids, and vitamin D.
2. CYP2D6: This enzyme is responsible for the metabolism of many psychotropic drugs, including antidepressants, antipsychotics, and beta-blockers. It also metabolizes some endogenous compounds like dopamine and serotonin.
3. CYP2C9: This enzyme plays a significant role in the metabolism of warfarin, phenytoin, and nonsteroidal anti-inflammatory drugs (NSAIDs).
4. CYP2C19: This enzyme is involved in the metabolism of proton pump inhibitors, antidepressants, and clopidogrel.
5. CYP2E1: This enzyme metabolizes various xenobiotics like alcohol, acetaminophen, and carbon tetrachloride, as well as some endogenous compounds like fatty acids and prostaglandins.
Genetic polymorphisms in CYP450 enzymes can significantly affect drug metabolism and response, leading to interindividual variability in drug efficacy and toxicity. Understanding the role of CYP450 enzymes in drug metabolism is crucial for optimizing pharmacotherapy and minimizing adverse effects.
Proteolipids are a type of complex lipid-containing proteins that are insoluble in water and have a high content of hydrophobic amino acids. They are primarily found in the plasma membrane of cells, where they play important roles in maintaining the structural integrity and function of the membrane. Proteolipids are also found in various organelles, including mitochondria, lysosomes, and peroxisomes.
Proteolipids are composed of a hydrophobic protein core that is tightly associated with a lipid bilayer through non-covalent interactions. The protein component of proteolipids typically contains several transmembrane domains that span the lipid bilayer, as well as hydrophilic regions that face the cytoplasm or the lumen of organelles.
Proteolipids have been implicated in various cellular processes, including signal transduction, membrane trafficking, and ion transport. They are also associated with several neurological disorders, such as Alzheimer's disease, Parkinson's disease, and multiple sclerosis. The study of proteolipids is an active area of research in biochemistry and cell biology, with potential implications for the development of new therapies for neurological disorders.
Bile pigments are the yellow-brown colored end products of hemoglobin breakdown in the liver. Hemoglobin is a protein found in red blood cells that carries oxygen throughout the body. When these cells are broken down, heme (the non-protein part of hemoglobin) is converted into biliverdin, which is then converted into bilirubin. Bilirubin is further metabolized and excreted by the liver as a component of bile, a digestive fluid that helps break down fats in the small intestine.
Under normal conditions, the liver effectively removes and excretes bilirubin from the body through the bile ducts into the small intestine. However, when there is an overproduction of bilirubin or a problem with its elimination, it can accumulate in the blood, leading to jaundice (yellowing of the skin and eyes) and other symptoms associated with liver dysfunction.
In summary, bile pigments are the waste products formed during the breakdown of hemoglobin, primarily consisting of bilirubin, which is eliminated from the body via the liver and bile ducts.
Veillonellaceae is a family of Gram-negative, anaerobic bacteria found in various environments, including the human mouth and gut. The bacteria are known for their ability to produce acetic and lactic acid as end products of their metabolism. They are often part of the normal microbiota of the body, but they can also be associated with certain infections, particularly in individuals with weakened immune systems.
It's important to note that while Veillonellaceae bacteria are generally considered to be commensal organisms, meaning they exist harmoniously with their human hosts, they have been implicated in some disease states, such as periodontitis (gum disease) and bacterial pneumonia. However, more research is needed to fully understand the role of these bacteria in health and disease.
Spectrophotometry, Ultraviolet (UV-Vis) is a type of spectrophotometry that measures how much ultraviolet (UV) and visible light is absorbed or transmitted by a sample. It uses a device called a spectrophotometer to measure the intensity of light at different wavelengths as it passes through a sample. The resulting data can be used to determine the concentration of specific components within the sample, identify unknown substances, or evaluate the physical and chemical properties of materials.
UV-Vis spectroscopy is widely used in various fields such as chemistry, biology, pharmaceuticals, and environmental science. It can detect a wide range of substances including organic compounds, metal ions, proteins, nucleic acids, and dyes. The technique is non-destructive, meaning that the sample remains unchanged after the measurement.
In UV-Vis spectroscopy, the sample is placed in a cuvette or other container, and light from a source is directed through it. The light then passes through a monochromator, which separates it into its component wavelengths. The monochromatic light is then directed through the sample, and the intensity of the transmitted or absorbed light is measured by a detector.
The resulting absorption spectrum can provide information about the concentration and identity of the components in the sample. For example, if a compound has a known absorption maximum at a specific wavelength, its concentration can be determined by measuring the absorbance at that wavelength and comparing it to a standard curve.
Overall, UV-Vis spectrophotometry is a versatile and powerful analytical technique for quantitative and qualitative analysis of various samples in different fields.
Carrier proteins, also known as transport proteins, are a type of protein that facilitates the movement of molecules across cell membranes. They are responsible for the selective and active transport of ions, sugars, amino acids, and other molecules from one side of the membrane to the other, against their concentration gradient. This process requires energy, usually in the form of ATP (adenosine triphosphate).
Carrier proteins have a specific binding site for the molecule they transport, and undergo conformational changes upon binding, which allows them to move the molecule across the membrane. Once the molecule has been transported, the carrier protein returns to its original conformation, ready to bind and transport another molecule.
Carrier proteins play a crucial role in maintaining the balance of ions and other molecules inside and outside of cells, and are essential for many physiological processes, including nerve impulse transmission, muscle contraction, and nutrient uptake.
Centrifugation, Density Gradient is a medical laboratory technique used to separate and purify different components of a mixture based on their size, density, and shape. This method involves the use of a centrifuge and a density gradient medium, such as sucrose or cesium chloride, to create a stable density gradient within a column or tube.
The sample is carefully layered onto the top of the gradient and then subjected to high-speed centrifugation. During centrifugation, the particles in the sample move through the gradient based on their size, density, and shape, with heavier particles migrating faster and further than lighter ones. This results in the separation of different components of the mixture into distinct bands or zones within the gradient.
This technique is commonly used to purify and concentrate various types of biological materials, such as viruses, organelles, ribosomes, and subcellular fractions, from complex mixtures. It allows for the isolation of pure and intact particles, which can then be collected and analyzed for further study or use in downstream applications.
In summary, Centrifugation, Density Gradient is a medical laboratory technique used to separate and purify different components of a mixture based on their size, density, and shape using a centrifuge and a density gradient medium.
Flavins are a group of naturally occurring organic compounds that contain a characteristic isoalloxazine ring, which is a tricyclic aromatic structure. The most common and well-known flavin is flavin adenine dinucleotide (FAD), which plays a crucial role as a coenzyme in various biological oxidation-reduction reactions. FAD accepts electrons and hydrogens to form the reduced form, flavin adenine dinucleotide hydride (FADH2). Another important flavin is flavin mononucleotide (FMN), which is derived from FAD and functions similarly as a coenzyme. Flavins are yellow in color and can be found in various biological systems, including animals, plants, and microorganisms. They are involved in several metabolic pathways, such as the electron transport chain, where they contribute to energy production.
In the context of medical terminology, "light" doesn't have a specific or standardized definition on its own. However, it can be used in various medical terms and phrases. For example, it could refer to:
1. Visible light: The range of electromagnetic radiation that can be detected by the human eye, typically between wavelengths of 400-700 nanometers. This is relevant in fields such as ophthalmology and optometry.
2. Therapeutic use of light: In some therapies, light is used to treat certain conditions. An example is phototherapy, which uses various wavelengths of ultraviolet (UV) or visible light for conditions like newborn jaundice, skin disorders, or seasonal affective disorder.
3. Light anesthesia: A state of reduced consciousness in which the patient remains responsive to verbal commands and physical stimulation. This is different from general anesthesia where the patient is completely unconscious.
4. Pain relief using light: Certain devices like transcutaneous electrical nerve stimulation (TENS) units have a 'light' setting, indicating lower intensity or frequency of electrical impulses used for pain management.
Without more context, it's hard to provide a precise medical definition of 'light'.
In the context of medicine, iron is an essential micromineral and key component of various proteins and enzymes. It plays a crucial role in oxygen transport, DNA synthesis, and energy production within the body. Iron exists in two main forms: heme and non-heme. Heme iron is derived from hemoglobin and myoglobin in animal products, while non-heme iron comes from plant sources and supplements.
The recommended daily allowance (RDA) for iron varies depending on age, sex, and life stage:
* For men aged 19-50 years, the RDA is 8 mg/day
* For women aged 19-50 years, the RDA is 18 mg/day
* During pregnancy, the RDA increases to 27 mg/day
* During lactation, the RDA for breastfeeding mothers is 9 mg/day
Iron deficiency can lead to anemia, characterized by fatigue, weakness, and shortness of breath. Excessive iron intake may result in iron overload, causing damage to organs such as the liver and heart. Balanced iron levels are essential for maintaining optimal health.
Leghemoglobin is a type of protein known as a hemeprotein, found in the root nodules of leguminous plants (plants belonging to the family Fabaceae or Leguminosae). These root nodules are formed through a symbiotic relationship with nitrogen-fixing bacteria called Rhizobia.
The primary function of leghemoglobin is to facilitate the process of nitrogen fixation by maintaining an optimal oxygen concentration within the root nodule cells, where the Rhizobia reside. By binding and releasing oxygen reversibly, leghemoglobin protects the nitrogen-fixing enzyme, nitrogenase, from being inactivated by excess oxygen. This ensures that the Rhizobia can effectively convert atmospheric nitrogen gas (N2) into ammonia (NH3), which is then utilized by the plant for its growth and development.
In summary, leghemoglobin is a crucial protein in the process of biological nitrogen fixation, allowing leguminous plants to grow without the need for added nitrogen fertilizers.
Electron Spin Resonance (ESR) Spectroscopy, also known as Electron Paramagnetic Resonance (EPR) Spectroscopy, is a technique used to investigate materials with unpaired electrons. It is based on the principle of absorption of energy by the unpaired electrons when they are exposed to an external magnetic field and microwave radiation.
In this technique, a sample is placed in a magnetic field and microwave radiation is applied. The unpaired electrons in the sample absorb energy and change their spin state when the energy of the microwaves matches the energy difference between the spin states. This absorption of energy is recorded as a function of the magnetic field strength, producing an ESR spectrum.
ESR spectroscopy can provide information about the number, type, and behavior of unpaired electrons in a sample, as well as the local environment around the electron. It is widely used in physics, chemistry, and biology to study materials such as free radicals, transition metal ions, and defects in solids.
Copper is a chemical element with the symbol Cu (from Latin: *cuprum*) and atomic number 29. It is a soft, malleable, and ductile metal with very high thermal and electrical conductivity. Copper is found as a free element in nature, and it is also a constituent of many minerals such as chalcopyrite and bornite.
In the human body, copper is an essential trace element that plays a role in various physiological processes, including iron metabolism, energy production, antioxidant defense, and connective tissue synthesis. Copper is found in a variety of foods, such as shellfish, nuts, seeds, whole grains, and organ meats. The recommended daily intake of copper for adults is 900 micrograms (mcg) per day.
Copper deficiency can lead to anemia, neutropenia, impaired immune function, and abnormal bone development. Copper toxicity, on the other hand, can cause nausea, vomiting, abdominal pain, diarrhea, and in severe cases, liver damage and neurological symptoms. Therefore, it is important to maintain a balanced copper intake through diet and supplements if necessary.
I'm sorry for any confusion, but "Nickel" is not a medical term. It is a chemical element with the symbol Ni and atomic number 28. Nickel is a hard, silvery-white lustrous metal with a slight golden tinge. It is one of four elements that are ferromagnetic and is used as a common component in various alloys due to its properties such as resistance to corrosion and heat.
However, in a medical context, nickel may refer to:
* Nickel allergy: A type of allergic contact dermatitis caused by an immune system response to the presence of nickel in jewelry, clothing fasteners, or other items that come into contact with the skin. Symptoms can include redness, itching, and rash at the site of exposure.
* Nickel carbonyl: A highly toxic chemical compound (Ni(CO)4) that can cause respiratory and neurological problems if inhaled. It is produced during some industrial processes involving nickel and carbon monoxide and poses a health risk to workers if proper safety measures are not taken.
If you have any concerns about exposure to nickel or symptoms related to nickel allergy, it's best to consult with a healthcare professional for further evaluation and treatment.
Electron microscopy (EM) is a type of microscopy that uses a beam of electrons to create an image of the sample being examined, resulting in much higher magnification and resolution than light microscopy. There are several types of electron microscopy, including transmission electron microscopy (TEM), scanning electron microscopy (SEM), and reflection electron microscopy (REM).
In TEM, a beam of electrons is transmitted through a thin slice of the sample, and the electrons that pass through the sample are focused to form an image. This technique can provide detailed information about the internal structure of cells, viruses, and other biological specimens, as well as the composition and structure of materials at the atomic level.
In SEM, a beam of electrons is scanned across the surface of the sample, and the electrons that are scattered back from the surface are detected to create an image. This technique can provide information about the topography and composition of surfaces, as well as the structure of materials at the microscopic level.
REM is a variation of SEM in which the beam of electrons is reflected off the surface of the sample, rather than scattered back from it. This technique can provide information about the surface chemistry and composition of materials.
Electron microscopy has a wide range of applications in biology, medicine, and materials science, including the study of cellular structure and function, disease diagnosis, and the development of new materials and technologies.
LDL receptors (Low-Density Lipoprotein Receptors) are cell surface receptors that play a crucial role in the regulation of cholesterol homeostasis within the body. They are responsible for recognizing and binding to LDL particles, also known as "bad cholesterol," which are then internalized by the cell through endocytosis.
Once inside the cell, the LDL particles are broken down, releasing their cholesterol content, which can be used for various cellular processes such as membrane synthesis and hormone production. The LDL receptors themselves are recycled back to the cell surface, allowing for continued uptake of LDL particles.
Mutations in the LDL receptor gene can lead to a condition called familial hypercholesterolemia, which is characterized by high levels of LDL cholesterol in the blood and an increased risk of premature cardiovascular disease.
Bacterial proteins are a type of protein that are produced by bacteria as part of their structural or functional components. These proteins can be involved in various cellular processes, such as metabolism, DNA replication, transcription, and translation. They can also play a role in bacterial pathogenesis, helping the bacteria to evade the host's immune system, acquire nutrients, and multiply within the host.
Bacterial proteins can be classified into different categories based on their function, such as:
1. Enzymes: Proteins that catalyze chemical reactions in the bacterial cell.
2. Structural proteins: Proteins that provide structural support and maintain the shape of the bacterial cell.
3. Signaling proteins: Proteins that help bacteria to communicate with each other and coordinate their behavior.
4. Transport proteins: Proteins that facilitate the movement of molecules across the bacterial cell membrane.
5. Toxins: Proteins that are produced by pathogenic bacteria to damage host cells and promote infection.
6. Surface proteins: Proteins that are located on the surface of the bacterial cell and interact with the environment or host cells.
Understanding the structure and function of bacterial proteins is important for developing new antibiotics, vaccines, and other therapeutic strategies to combat bacterial infections.
Myoglobin is a protein found in the muscle tissue, particularly in red or skeletal muscles. It belongs to the globin family and has a similar structure to hemoglobin, another oxygen-binding protein found in red blood cells. Myoglobin's primary function is to store oxygen within the muscle cells, making it readily available for use during periods of increased oxygen demand, such as during physical exertion.
Myoglobin contains heme groups that bind to and release oxygen molecules. The protein has a higher affinity for oxygen than hemoglobin, allowing it to maintain its bound oxygen even in low-oxygen environments. When muscle cells are damaged or undergo necrosis (cell death), myoglobin is released into the bloodstream and can be detected in serum or urine samples. Elevated levels of myoglobin in the blood or urine may indicate muscle injury, trauma, or diseases affecting muscle integrity, such as rhabdomyolysis or muscular dystrophies.
Phycoerythrin is not a medical term, but a term used in biochemistry and cell biology. It refers to a type of protein found in certain algae and cyanobacteria that binds phycobilins, which are linear tetrapyrrole chromophores. Phycoerythrin is a light-harvesting pigment that absorbs light energy and transfers it to the photosynthetic reaction centers. It is often used in research and clinical settings as a fluorescent label for various applications, such as flow cytometry, immunohistochemistry, and microscopy.
Cytochromes are a type of hemeprotein found in the mitochondria and other cellular membranes of organisms. They contain a heme group, which is a prosthetic group composed of an iron atom surrounded by a porphyrin ring. This structure allows cytochromes to participate in redox reactions, acting as electron carriers in various biological processes.
There are several types of cytochromes, classified based on the type of heme they contain and their absorption spectra. Some of the most well-known cytochromes include:
* Cytochrome c: a small, mobile protein found in the inner mitochondrial membrane that plays a crucial role in the electron transport chain during cellular respiration.
* Cytochrome P450: a large family of enzymes involved in the metabolism of drugs, toxins, and other xenobiotics. They are found in various tissues, including the liver, lungs, and skin.
* Cytochrome b: a component of several electron transport chains, including those found in mitochondria, bacteria, and chloroplasts.
Cytochromes play essential roles in energy production, detoxification, and other metabolic processes, making them vital for the survival and function of living organisms.
D-Aspartate oxidase (DDO) is an enzyme that catalyzes the oxidation of D-aspartic acid to yield oxaloacetate and carbon dioxide. This reaction also results in the reduction of NAD+ to NADH. DDO plays a role in the metabolism of D-amino acids, which are less common than L-amino acids in biological systems. It is found in various organisms, including bacteria, plants, and animals. In humans, DDO is widely expressed in the body, with particularly high levels found in the testes, where it is involved in the regulation of D-aspartate and D-serine concentrations.
The egg yolk is the nutrient-rich, inner portion of an egg that is surrounded by a protective layer of egg white. It is typically yellowish-orange and has a creamy consistency. The egg yolk contains various essential nutrients such as proteins, fats, vitamins (like A, D, E, and K), minerals (such as calcium, phosphorus, zinc, and iron), and antioxidants (like lutein and zeaxanthin). It is also a significant source of cholesterol. The egg yolk plays an essential role in the development of embryos in birds and reptiles, providing them with necessary nutrients for growth and energy. In culinary applications, egg yolks are often used as emulsifiers, thickeners, and leavening agents in various dishes.
Biological pigments are substances produced by living organisms that absorb certain wavelengths of light and reflect others, resulting in the perception of color. These pigments play crucial roles in various biological processes such as photosynthesis, vision, and protection against harmful radiation. Some examples of biological pigments include melanin, hemoglobin, chlorophyll, carotenoids, and flavonoids.
Melanin is a pigment responsible for the color of skin, hair, and eyes in animals, including humans. Hemoglobin is a protein found in red blood cells that contains a porphyrin ring with an iron atom at its center, which gives blood its red color and facilitates oxygen transport. Chlorophyll is a green pigment found in plants, algae, and some bacteria that absorbs light during photosynthesis to convert carbon dioxide and water into glucose and oxygen. Carotenoids are orange, yellow, or red pigments found in fruits, vegetables, and some animals that protect against oxidative stress and help maintain membrane fluidity. Flavonoids are a class of plant pigments with antioxidant properties that have been linked to various health benefits.
Lipoprotein receptors are specialized proteins found on the surface of cells that play a crucial role in the metabolism of lipoproteins, which are complex particles composed of lipids and proteins. These receptors bind to specific lipoproteins in the bloodstream, facilitating their uptake into the cell for further processing.
There are several types of lipoprotein receptors, including:
1. LDL (Low-Density Lipoprotein) Receptor: This receptor is responsible for recognizing and internalizing LDL particles, which are rich in cholesterol. Once inside the cell, LDL particles release their cholesterol, which can then be used for various cellular functions or stored for later use. Defects in the LDL receptor can lead to elevated levels of LDL cholesterol in the blood and an increased risk of developing cardiovascular disease.
2. HDL (High-Density Lipoprotein) Receptor: This receptor is involved in the clearance of HDL particles from the bloodstream. HDL particles are responsible for transporting excess cholesterol from peripheral tissues to the liver, where it can be processed and eliminated from the body.
3. VLDL (Very Low-Density Lipoprotein) Receptor: This receptor recognizes and internalizes VLDL particles, which are produced by the liver and carry triglycerides and cholesterol to peripheral tissues. VLDL particles are subsequently converted into LDL particles in the bloodstream.
4. LRP (Low-Density Lipoprotein Receptor-Related Protein) Family: This family of receptors includes several members, such as LRP1 and LRP2, that play roles in various cellular processes, including lipid metabolism, protein trafficking, and cell signaling. They can bind to a variety of ligands, including lipoproteins, proteases, and extracellular matrix components.
In summary, lipoprotein receptors are essential for maintaining proper lipid metabolism and homeostasis by facilitating the uptake, processing, and elimination of lipoproteins in the body.
Lipoprotein lipase (LPL) is an enzyme that plays a crucial role in the metabolism of lipids. It is responsible for breaking down triglycerides, which are the main constituent of dietary fats and chylomicrons, into fatty acids and glycerol. These products are then taken up by cells for energy production or storage.
LPL is synthesized in various tissues, including muscle and fat, where it is attached to the inner lining of blood vessels (endothelium). The enzyme is activated when it comes into contact with lipoprotein particles, such as chylomicrons and very-low-density lipoproteins (VLDL), which transport triglycerides in the bloodstream.
Deficiencies or mutations in LPL can lead to various metabolic disorders, including hypertriglyceridemia, a condition characterized by high levels of triglycerides in the blood. Conversely, overexpression of LPL has been associated with increased risk of atherosclerosis due to excessive uptake of fatty acids by macrophages and their conversion into foam cells, which contribute to plaque formation in the arteries.
D-amino-acid oxidase (DAAO) is an enzyme that catalyzes the oxidative deamination of D-amino acids to their corresponding α-keto acids, ammonia, and hydrogen peroxide. This enzyme plays a crucial role in the metabolism of D-amino acids in various organisms, including humans. In humans, DAAO is primarily expressed in the brain and contributes to the regulation of neurotransmitter levels and other physiological processes. Genetic variations and dysregulation of DAAO have been implicated in several neurological disorders, such as schizophrenia and bipolar disorder.
Allylisopropylacetamide is not a term that has a widely accepted or established medical definition. It is a chemical compound with the formula (CH₂CHCH₂)N(C=O)CH(CH₃)₂, and it may have various chemical or industrial uses, but it is not a term that is commonly used in medical contexts.
If you have any specific questions about this compound or its potential uses or effects, I would recommend consulting with a relevant expert, such as a chemist or toxicologist, who can provide more detailed and accurate information based on their expertise and knowledge of the subject.
Two-dimensional immunoelectrophoresis (2DE) is a specialized laboratory technique used in the field of clinical pathology and immunology. This technique is a refined version of traditional immunoelectrophoresis that adds an additional electrophoretic separation step, enhancing its resolution and allowing for more detailed analysis of complex protein mixtures.
In two-dimensional immunoelectrophoresis, proteins are first separated based on their isoelectric points (pI) in the initial dimension using isoelectric focusing (IEF). This process involves applying an electric field to a protein mixture contained within a gel matrix, where proteins will migrate and stop migrating once they reach the pH that matches their own isoelectric point.
Following IEF, the separated proteins are then subjected to a second electrophoretic separation in the perpendicular direction (second dimension) based on their molecular weights using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). SDS is a negatively charged molecule that binds to proteins, giving them a uniform negative charge and allowing for separation based solely on size.
Once the two-dimensional separation is complete, the gel is then overlaid with specific antisera to detect and identify proteins of interest. The resulting precipitin arcs formed at the intersection of the antibody and antigen are compared to known standards or patterns to determine the identity and quantity of the separated proteins.
Two-dimensional immunoelectrophoresis is particularly useful in identifying and quantifying proteins in complex mixtures, such as those found in body fluids like serum, urine, or cerebrospinal fluid (CSF). It can be applied to various clinical scenarios, including diagnosis and monitoring of monoclonal gammopathies, autoimmune disorders, and certain infectious diseases.
"Inbred strains of rats" are genetically identical rodents that have been produced through many generations of brother-sister mating. This results in a high degree of homozygosity, where the genes at any particular locus in the genome are identical in all members of the strain.
Inbred strains of rats are widely used in biomedical research because they provide a consistent and reproducible genetic background for studying various biological phenomena, including the effects of drugs, environmental factors, and genetic mutations on health and disease. Additionally, inbred strains can be used to create genetically modified models of human diseases by introducing specific mutations into their genomes.
Some commonly used inbred strains of rats include the Wistar Kyoto (WKY), Sprague-Dawley (SD), and Fischer 344 (F344) rat strains. Each strain has its own unique genetic characteristics, making them suitable for different types of research.
A base sequence in the context of molecular biology refers to the specific order of nucleotides in a DNA or RNA molecule. In DNA, these nucleotides are adenine (A), guanine (G), cytosine (C), and thymine (T). In RNA, uracil (U) takes the place of thymine. The base sequence contains genetic information that is transcribed into RNA and ultimately translated into proteins. It is the exact order of these bases that determines the genetic code and thus the function of the DNA or RNA molecule.
Oleic acid is a monounsaturated fatty acid that is commonly found in various natural oils such as olive oil, sunflower oil, and grapeseed oil. Its chemical formula is cis-9-octadecenoic acid, and it is a colorless liquid at room temperature. Oleic acid is an important component of human diet and has been shown to have potential health benefits, including reducing the risk of heart disease and improving immune function. It is also used in the manufacture of soaps, cosmetics, and other personal care products.
Diatomaceous earth is not a medical term, but a natural product derived from the fossilized remains of diatoms, a type of algae. It is composed of silica and other minerals. While it has various industrial and agricultural uses, it is not typically used as a medication or treatment. However, some people may use food-grade diatomaceous earth for various health purposes, such as detoxification or improving digestive health, but these uses are not supported by scientific evidence and its safety and effectiveness for these purposes have not been established. As with any substance used for medicinal purposes, it is important to consult with a healthcare provider before using diatomaceous earth.
Apolipoprotein D (apoD) is a protein that is associated with high-density lipoprotein (HDL) particles in the blood. It is one of several apolipoproteins that are involved in the transport and metabolism of lipids, such as cholesterol and triglycerides, in the body.
ApoD is produced by the APOD gene and is found in various tissues, including the brain, where it is believed to play a role in protecting nerve cells from oxidative stress. It has also been studied for its potential role in Alzheimer's disease and other neurological disorders.
In addition to its role in lipid metabolism and neuroprotection, apoD has been shown to have anti-inflammatory properties and may be involved in the regulation of immune responses. However, more research is needed to fully understand the functions and mechanisms of action of this protein.
Recombinant proteins are artificially created proteins produced through the use of recombinant DNA technology. This process involves combining DNA molecules from different sources to create a new set of genes that encode for a specific protein. The resulting recombinant protein can then be expressed, purified, and used for various applications in research, medicine, and industry.
Recombinant proteins are widely used in biomedical research to study protein function, structure, and interactions. They are also used in the development of diagnostic tests, vaccines, and therapeutic drugs. For example, recombinant insulin is a common treatment for diabetes, while recombinant human growth hormone is used to treat growth disorders.
The production of recombinant proteins typically involves the use of host cells, such as bacteria, yeast, or mammalian cells, which are engineered to express the desired protein. The host cells are transformed with a plasmid vector containing the gene of interest, along with regulatory elements that control its expression. Once the host cells are cultured and the protein is expressed, it can be purified using various chromatography techniques.
Overall, recombinant proteins have revolutionized many areas of biology and medicine, enabling researchers to study and manipulate proteins in ways that were previously impossible.
Rhodopsin, also known as visual purple, is a light-sensitive protein found in the rods of the eye's retina. It is a type of opsin, a class of proteins that are activated by light and play a crucial role in vision. Rhodopsin is composed of two parts: an apoprotein called opsin and a chromophore called 11-cis-retinal. When light hits the retina, it changes the shape of the 11-cis-retinal, which in turn activates the rhodopsin protein. This activation triggers a series of chemical reactions that ultimately lead to the transmission of a visual signal to the brain. Rhodopsin is highly sensitive to light and allows for vision in low-light conditions.
Surface tension is not a term that has a specific medical definition. However, it is a physical chemistry concept that relates to the cohesive force between liquid molecules, causing the surface of the liquid to contract and have a higher intermolecular force than its bulk.
In a broader sense, surface tension can have implications in certain medical or biological contexts, such as the movement of liquids in the lungs or the stability of lipid bilayers in cell membranes. But it is not a term that is typically used to describe medical conditions or treatments.
Trypsin is a proteolytic enzyme, specifically a serine protease, that is secreted by the pancreas as an inactive precursor, trypsinogen. Trypsinogen is converted into its active form, trypsin, in the small intestine by enterokinase, which is produced by the intestinal mucosa.
Trypsin plays a crucial role in digestion by cleaving proteins into smaller peptides at specific arginine and lysine residues. This enzyme helps to break down dietary proteins into amino acids, allowing for their absorption and utilization by the body. Additionally, trypsin can activate other zymogenic pancreatic enzymes, such as chymotrypsinogen and procarboxypeptidases, thereby contributing to overall protein digestion.
Bacteriorhodopsins are a type of protein found in certain archaea, a group of single-celled microorganisms. They are most commonly found in the archaea of the genus Halobacterium, which live in extremely salty environments such as salt lakes and solar salterns.
Bacteriorhodopsins are embedded in the cell membrane of these archaea and contain a retinal molecule, which is a type of vitamin A derivative. When exposed to light, the retinal changes shape, which causes a conformational change in the bacteriorhodopsin protein. This leads to the pumping of protons (hydrogen ions) across the cell membrane, generating a proton gradient.
The proton gradient created by bacteriorhodopsins can be used to generate ATP, which is the main energy currency of the cell. Bacteriorhodopsins are therefore involved in energy production in these archaea and are often referred to as light-driven proton pumps. They have also been studied extensively for their potential applications in optoelectronics and biotechnology.
"Plant proteins" refer to the proteins that are derived from plant sources. These can include proteins from legumes such as beans, lentils, and peas, as well as proteins from grains like wheat, rice, and corn. Other sources of plant proteins include nuts, seeds, and vegetables.
Plant proteins are made up of individual amino acids, which are the building blocks of protein. While animal-based proteins typically contain all of the essential amino acids that the body needs to function properly, many plant-based proteins may be lacking in one or more of these essential amino acids. However, by consuming a variety of plant-based foods throughout the day, it is possible to get all of the essential amino acids that the body needs from plant sources alone.
Plant proteins are often lower in calories and saturated fat than animal proteins, making them a popular choice for those following a vegetarian or vegan diet, as well as those looking to maintain a healthy weight or reduce their risk of chronic diseases such as heart disease and cancer. Additionally, plant proteins have been shown to have a number of health benefits, including improving gut health, reducing inflammation, and supporting muscle growth and repair.
Molecular models are three-dimensional representations of molecular structures that are used in the field of molecular biology and chemistry to visualize and understand the spatial arrangement of atoms and bonds within a molecule. These models can be physical or computer-generated and allow researchers to study the shape, size, and behavior of molecules, which is crucial for understanding their function and interactions with other molecules.
Physical molecular models are often made up of balls (representing atoms) connected by rods or sticks (representing bonds). These models can be constructed manually using materials such as plastic or wooden balls and rods, or they can be created using 3D printing technology.
Computer-generated molecular models, on the other hand, are created using specialized software that allows researchers to visualize and manipulate molecular structures in three dimensions. These models can be used to simulate molecular interactions, predict molecular behavior, and design new drugs or chemicals with specific properties. Overall, molecular models play a critical role in advancing our understanding of molecular structures and their functions.
Chloroplasts are specialized organelles found in the cells of green plants, algae, and some protists. They are responsible for carrying out photosynthesis, which is the process by which these organisms convert light energy from the sun into chemical energy in the form of organic compounds, such as glucose.
Chloroplasts contain the pigment chlorophyll, which absorbs light energy from the sun. They also contain a system of membranes and enzymes that convert carbon dioxide and water into glucose and oxygen through a series of chemical reactions known as the Calvin cycle. This process not only provides energy for the organism but also releases oxygen as a byproduct, which is essential for the survival of most life forms on Earth.
Chloroplasts are believed to have originated from ancient cyanobacteria that were engulfed by early eukaryotic cells and eventually became integrated into their host's cellular machinery through a process called endosymbiosis. Over time, chloroplasts evolved to become an essential component of plant and algal cells, contributing to their ability to carry out photosynthesis and thrive in a wide range of environments.
I'm sorry for any confusion, but "Hordeum" is not a medical term. It is actually the genus name for barley in botany. If you have any medical terms or concepts that you would like me to explain, please let me know!
Electrophoresis, Agar Gel is a laboratory technique used to separate and analyze DNA, RNA, or proteins based on their size and electrical charge. In this method, the sample is mixed with agarose gel, a gelatinous substance derived from seaweed, and then solidified in a horizontal slab-like format. An electric field is applied to the gel, causing the negatively charged DNA or RNA molecules to migrate towards the positive electrode. The smaller molecules move faster through the gel than the larger ones, resulting in their separation based on size. This technique is widely used in molecular biology and genetics research, as well as in diagnostic testing for various genetic disorders.
Fluorescence spectrometry is a type of analytical technique used to investigate the fluorescent properties of a sample. It involves the measurement of the intensity of light emitted by a substance when it absorbs light at a specific wavelength and then re-emits it at a longer wavelength. This process, known as fluorescence, occurs because the absorbed energy excites electrons in the molecules of the substance to higher energy states, and when these electrons return to their ground state, they release the excess energy as light.
Fluorescence spectrometry typically measures the emission spectrum of a sample, which is a plot of the intensity of emitted light versus the wavelength of emission. This technique can be used to identify and quantify the presence of specific fluorescent molecules in a sample, as well as to study their photophysical properties.
Fluorescence spectrometry has many applications in fields such as biochemistry, environmental science, and materials science. For example, it can be used to detect and measure the concentration of pollutants in water samples, to analyze the composition of complex biological mixtures, or to study the properties of fluorescent nanomaterials.
Rhodopsin, also known as visual purple, is a light-sensitive pigment found in the rods of the vertebrate retina. It is a complex protein molecule made up of two major components: an opsin protein and retinal, a form of vitamin A. When light hits the retinal in rhodopsin, it changes shape, which initiates a series of chemical reactions leading to the activation of the visual pathway and ultimately results in vision. This process is known as phototransduction. Rhodopsin plays a crucial role in low-light vision or scotopic vision.
Radioimmunoassay (RIA) is a highly sensitive analytical technique used in clinical and research laboratories to measure concentrations of various substances, such as hormones, vitamins, drugs, or tumor markers, in biological samples like blood, urine, or tissues. The method relies on the specific interaction between an antibody and its corresponding antigen, combined with the use of radioisotopes to quantify the amount of bound antigen.
In a typical RIA procedure, a known quantity of a radiolabeled antigen (also called tracer) is added to a sample containing an unknown concentration of the same unlabeled antigen. The mixture is then incubated with a specific antibody that binds to the antigen. During the incubation period, the antibody forms complexes with both the radiolabeled and unlabeled antigens.
After the incubation, the unbound (free) radiolabeled antigen is separated from the antibody-antigen complexes, usually through a precipitation or separation step involving centrifugation, filtration, or chromatography. The amount of radioactivity in the pellet (containing the antibody-antigen complexes) is then measured using a gamma counter or other suitable radiation detection device.
The concentration of the unlabeled antigen in the sample can be determined by comparing the ratio of bound to free radiolabeled antigen in the sample to a standard curve generated from known concentrations of unlabeled antigen and their corresponding bound/free ratios. The higher the concentration of unlabeled antigen in the sample, the lower the amount of radiolabeled antigen that will bind to the antibody, resulting in a lower bound/free ratio.
Radioimmunoassays offer high sensitivity, specificity, and accuracy, making them valuable tools for detecting and quantifying low levels of various substances in biological samples. However, due to concerns about radiation safety and waste disposal, alternative non-isotopic immunoassay techniques like enzyme-linked immunosorbent assays (ELISAs) have become more popular in recent years.
Ethinyl estradiol is a synthetic form of the hormone estrogen that is often used in various forms of hormonal contraception, such as birth control pills. It works by preventing ovulation and thickening cervical mucus to make it more difficult for sperm to reach the egg. Ethinyl estradiol may also be used in combination with other hormones to treat menopausal symptoms or hormonal disorders.
It is important to note that while ethinyl estradiol can be an effective form of hormonal therapy, it can also carry risks and side effects, such as an increased risk of blood clots, stroke, and breast cancer. As with any medication, it should only be used under the guidance and supervision of a healthcare provider.
Heme proteins are a type of protein that contain a heme group, which is a prosthetic group composed of an iron atom contained in the center of a large organic ring called a porphyrin. The heme group gives these proteins their characteristic red color. Hemeproteins have various important functions in biological systems, including oxygen transport (e.g., hemoglobin), electron transfer (e.g., cytochromes), and enzymatic catalysis (e.g., peroxidases and catalases). The heme group can bind and release gases, such as oxygen and carbon monoxide, and can participate in redox reactions due to the ease with which iron can change its oxidation state.
Molecular cloning is a laboratory technique used to create multiple copies of a specific DNA sequence. This process involves several steps:
1. Isolation: The first step in molecular cloning is to isolate the DNA sequence of interest from the rest of the genomic DNA. This can be done using various methods such as PCR (polymerase chain reaction), restriction enzymes, or hybridization.
2. Vector construction: Once the DNA sequence of interest has been isolated, it must be inserted into a vector, which is a small circular DNA molecule that can replicate independently in a host cell. Common vectors used in molecular cloning include plasmids and phages.
3. Transformation: The constructed vector is then introduced into a host cell, usually a bacterial or yeast cell, through a process called transformation. This can be done using various methods such as electroporation or chemical transformation.
4. Selection: After transformation, the host cells are grown in selective media that allow only those cells containing the vector to grow. This ensures that the DNA sequence of interest has been successfully cloned into the vector.
5. Amplification: Once the host cells have been selected, they can be grown in large quantities to amplify the number of copies of the cloned DNA sequence.
Molecular cloning is a powerful tool in molecular biology and has numerous applications, including the production of recombinant proteins, gene therapy, functional analysis of genes, and genetic engineering.
Halobacterium is a genus of extremely halophilic archaea, which means they require a high salt concentration to grow. They are often found in salt lakes, salt pans, and other hypersaline environments. These microorganisms contain bacteriorhodopsin, a light-driven proton pump, which gives them a purple color and allows them to generate ATP using light energy, similar to photosynthesis in plants. Halobacteria are also known for their ability to survive under extreme conditions, such as high temperatures, radiation, and desiccation.
Rhodospirillales is an order of predominantly gram-negative, aerobic or anaerobic, motile bacteria that are found in various environments such as freshwater, marine habitats, and soil. Many species in this order are capable of photosynthesis, particularly those belonging to the family Rhodospirillaceae. These photosynthetic bacteria, called purple bacteria, use bacteriochlorophyll and can grow under anaerobic conditions using light as an energy source. The order Rhodospirillales belongs to the class Alphaproteobacteria within the phylum Proteobacteria.
It is important to note that medical definitions typically focus on bacteria, viruses, or other microorganisms of clinical relevance. While Rhodospirillales does include some species that can be pathogenic in certain circumstances, it is not primarily a medical term and is more commonly used in the context of environmental or general microbiology.
Chromatography, agarose is a type of chromatography technique that utilizes agarose gel as the stationary phase in the separation and analysis of biological molecules, such as DNA, RNA, and proteins. This method is commonly used in molecular biology for various applications, including DNA fragment separation, protein purification, and detection of specific nucleic acid sequences or proteins.
Agarose gel is a matrix made from agarose, a polysaccharide derived from seaweed. It has a porous structure with uniform pore size that allows for the size-based separation of molecules based on their ability to migrate through the gel under an electric field (in the case of electrophoresis) or by capillary action (in the case of capillary electrophoresis).
The charged molecules, such as DNA or proteins, interact with the agarose matrix and move through the gel at different rates depending on their size, charge, and shape. Smaller molecules can migrate more quickly through the pores of the gel, while larger molecules are retarded due to their inability to easily pass through the pores. This results in a separation of the molecules based on their physical properties, allowing for their analysis and characterization.
In summary, chromatography, agarose refers to the use of agarose gel as the stationary phase in the separation and analysis of biological molecules using various chromatography techniques, such as electrophoresis or capillary electrophoresis.
Ferredoxins are iron-sulfur proteins that play a crucial role in electron transfer reactions in various biological systems, particularly in photosynthesis and nitrogen fixation. They contain one or more clusters of iron and sulfur atoms (known as the iron-sulfur cluster) that facilitate the movement of electrons between different molecules during metabolic processes.
Ferredoxins have a relatively simple structure, consisting of a polypeptide chain that binds to the iron-sulfur cluster. This simple structure allows ferredoxins to participate in a wide range of redox reactions and makes them versatile electron carriers in biological systems. They can accept electrons from various donors and transfer them to different acceptors, depending on the needs of the cell.
In photosynthesis, ferredoxins play a critical role in the light-dependent reactions by accepting electrons from photosystem I and transferring them to NADP+, forming NADPH. This reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) is then used in the Calvin cycle for carbon fixation and the production of glucose.
In nitrogen fixation, ferredoxins help transfer electrons to the nitrogenase enzyme complex, which reduces atmospheric nitrogen gas (N2) into ammonia (NH3), making it available for assimilation by plants and other organisms.
Overall, ferredoxins are essential components of many metabolic pathways, facilitating electron transfer and energy conversion in various biological systems.
I believe there may be some confusion in your question. "Rabbits" is a common name used to refer to the Lagomorpha species, particularly members of the family Leporidae. They are small mammals known for their long ears, strong legs, and quick reproduction.
However, if you're referring to "rabbits" in a medical context, there is a term called "rabbit syndrome," which is a rare movement disorder characterized by repetitive, involuntary movements of the fingers, resembling those of a rabbit chewing. It is also known as "finger-chewing chorea." This condition is usually associated with certain medications, particularly antipsychotics, and typically resolves when the medication is stopped or adjusted.
Magnetic Resonance Spectroscopy (MRS) is a non-invasive diagnostic technique that provides information about the biochemical composition of tissues, including their metabolic state. It is often used in conjunction with Magnetic Resonance Imaging (MRI) to analyze various metabolites within body tissues, such as the brain, heart, liver, and muscles.
During MRS, a strong magnetic field, radio waves, and a computer are used to produce detailed images and data about the concentration of specific metabolites in the targeted tissue or organ. This technique can help detect abnormalities related to energy metabolism, neurotransmitter levels, pH balance, and other biochemical processes, which can be useful for diagnosing and monitoring various medical conditions, including cancer, neurological disorders, and metabolic diseases.
There are different types of MRS, such as Proton (^1^H) MRS, Phosphorus-31 (^31^P) MRS, and Carbon-13 (^13^C) MRS, each focusing on specific elements or metabolites within the body. The choice of MRS technique depends on the clinical question being addressed and the type of information needed for diagnosis or monitoring purposes.
"Cattle" is a term used in the agricultural and veterinary fields to refer to domesticated animals of the genus *Bos*, primarily *Bos taurus* (European cattle) and *Bos indicus* (Zebu). These animals are often raised for meat, milk, leather, and labor. They are also known as bovines or cows (for females), bulls (intact males), and steers/bullocks (castrated males). However, in a strict medical definition, "cattle" does not apply to humans or other animals.
The mesentery is a continuous fold of the peritoneum, the double-layered serous membrane that lines the abdominal cavity, which attaches the stomach, small intestine, large intestine (colon), and rectum to the posterior wall of the abdomen. It provides blood vessels, nerves, and lymphatic vessels to these organs.
Traditionally, the mesentery was thought to consist of separate and distinct sections along the length of the intestines. However, recent research has shown that the mesentery is a continuous organ, with a single continuous tethering point to the posterior abdominal wall. This new understanding of the anatomy of the mesentery has implications for the study of various gastrointestinal diseases and disorders.
Protein denaturation is a process in which the native structure of a protein is altered, leading to loss of its biological activity. This can be caused by various factors such as changes in temperature, pH, or exposure to chemicals or radiation. The three-dimensional shape of a protein is crucial for its function, and denaturation causes the protein to lose this shape, resulting in impaired or complete loss of function. Denaturation is often irreversible and can lead to the aggregation of proteins, which can have negative effects on cellular function and can contribute to diseases such as Alzheimer's and Parkinson's.
A holozyme is not a specific medical term, but rather a term used in biochemistry to refer to the complete, active form of an enzyme. An enzyme is a biological molecule that catalyzes chemical reactions in the body, and it is often made up of several different subunits or components.
The term "holozyme" comes from the Greek words "holos," meaning whole, and "enzyma," meaning in yeast. It was originally used to describe the active form of enzymes found in yeast cells, but it is now used more broadly to refer to any complete, active enzyme complex.
A holozyme typically consists of two types of subunits: a catalytic subunit, which contains the active site where the substrate binds and the reaction takes place, and one or more regulatory subunits, which control the activity of the enzyme under different conditions. The regulatory subunits may be activated or inhibited by various signals, such as hormones, metabolites, or other molecules, allowing the enzyme to respond to changes in the cellular environment.
In summary, a holozyme is the fully assembled and functional form of an enzyme, consisting of one or more catalytic subunits and one or more regulatory subunits that work together to carry out specific biochemical reactions in the body.
Pronase is not a medical term itself, but it is a proteolytic enzyme mixture derived from the bacterium Streptomyces griseus. The term "pronase" refers to a group of enzymes that can break down proteins into smaller peptides and individual amino acids by hydrolyzing their peptide bonds.
Pronase is used in various laboratory applications, including protein degradation, DNA and RNA isolation, and the removal of contaminating proteins from nucleic acid samples. It has also been used in some medical research contexts to study protein function and structure, as well as in certain therapeutic settings for its ability to break down proteins.
It is important to note that pronase is not a drug or a medical treatment itself but rather a laboratory reagent with potential applications in medical research and diagnostics.
A lyase is a type of enzyme that catalyzes the breaking of various chemical bonds in a molecule, often resulting in the formation of two new molecules. Lyases differ from other types of enzymes, such as hydrolases and oxidoreductases, because they create double bonds or rings as part of their reaction mechanism.
In the context of medical terminology, lyases are not typically discussed on their own, but rather as a type of enzyme that can be involved in various biochemical reactions within the body. For example, certain lyases play a role in the metabolism of carbohydrates, lipids, and amino acids, among other molecules.
One specific medical application of lyase enzymes is in the diagnosis of certain genetic disorders. For instance, individuals with hereditary fructose intolerance (HFI) lack the enzyme aldolase B, which is a type of lyase that helps break down fructose in the liver. By measuring the activity of aldolase B in a patient's blood or tissue sample, doctors can diagnose HFI and recommend appropriate dietary restrictions to manage the condition.
Overall, while lyases are not a medical diagnosis or condition themselves, they play important roles in various biochemical processes within the body and can be useful in the diagnosis of certain genetic disorders.
Hemin is defined as the iron(III) complex of protoporphyrin IX, which is a porphyrin derivative. It is a naturally occurring substance that is involved in various biological processes, most notably in the form of heme, which is a component of hemoglobin and other hemoproteins. Hemin is also used in medical research and therapy, such as in the treatment of methemoglobinemia and lead poisoning.
Carotenoids are a class of pigments that are naturally occurring in various plants and fruits. They are responsible for the vibrant colors of many vegetables and fruits, such as carrots, pumpkins, tomatoes, and leafy greens. There are over 600 different types of carotenoids, with beta-carotene, alpha-carotene, lycopene, lutein, and zeaxanthin being some of the most well-known.
Carotenoids have antioxidant properties, which means they can help protect the body's cells from damage caused by free radicals. Some carotenoids, such as beta-carotene, can be converted into vitamin A in the body, which is important for maintaining healthy vision, skin, and immune function. Other carotenoids, such as lycopene and lutein, have been studied for their potential role in preventing chronic diseases, including cancer and heart disease.
In addition to being found in plant-based foods, carotenoids can also be taken as dietary supplements. However, it is generally recommended to obtain nutrients from whole foods rather than supplements whenever possible, as food provides a variety of other beneficial compounds that work together to support health.
Zinc is an essential mineral that is vital for the functioning of over 300 enzymes and involved in various biological processes in the human body, including protein synthesis, DNA synthesis, immune function, wound healing, and cell division. It is a component of many proteins and participates in the maintenance of structural integrity and functionality of proteins. Zinc also plays a crucial role in maintaining the sense of taste and smell.
The recommended daily intake of zinc varies depending on age, sex, and life stage. Good dietary sources of zinc include red meat, poultry, seafood, beans, nuts, dairy products, and fortified cereals. Zinc deficiency can lead to various health problems, including impaired immune function, growth retardation, and developmental delays in children. On the other hand, excessive intake of zinc can also have adverse effects on health, such as nausea, vomiting, and impaired immune function.
Plastocyanin is a small, copper-containing protein that plays a crucial role in the photosynthetic electron transport chain. It functions as an electron carrier, facilitating the movement of electrons between two key protein complexes (cytochrome b6f and photosystem I) located in the thylakoid membrane of chloroplasts. Plastocyanin is a soluble protein found in the lumen of the thylakoids, and its copper ion serves as the site for electron transfer. The oxidized form of plastocyanin accepts an electron from cytochrome b6f and then donates it to photosystem I, helping to maintain the flow of electrons during light-dependent reactions in photosynthesis.
Phytochrome A is a type of phytochrome, which is a photoreceptor protein that plants use to detect and respond to different wavelengths of light. Specifically, phytochrome A is responsible for mediating the response to red light. It exists in two interconvertible forms: Pr (the inactive form, absorbing red light) and Pfr (the active form, absorbing far-red light). The conversion between these two forms triggers a range of physiological responses in plants, such as seed germination, stem elongation, leaf expansion, and flowering. Phytochrome A is the most sensitive phytochrome to changes in light quality and quantity, making it a crucial photoreceptor for plants' adaptation to their environment.
A Structure-Activity Relationship (SAR) in the context of medicinal chemistry and pharmacology refers to the relationship between the chemical structure of a drug or molecule and its biological activity or effect on a target protein, cell, or organism. SAR studies aim to identify patterns and correlations between structural features of a compound and its ability to interact with a specific biological target, leading to a desired therapeutic response or undesired side effects.
By analyzing the SAR, researchers can optimize the chemical structure of lead compounds to enhance their potency, selectivity, safety, and pharmacokinetic properties, ultimately guiding the design and development of novel drugs with improved efficacy and reduced toxicity.
Messenger RNA (mRNA) is a type of RNA (ribonucleic acid) that carries genetic information copied from DNA in the form of a series of three-base code "words," each of which specifies a particular amino acid. This information is used by the cell's machinery to construct proteins, a process known as translation. After being transcribed from DNA, mRNA travels out of the nucleus to the ribosomes in the cytoplasm where protein synthesis occurs. Once the protein has been synthesized, the mRNA may be degraded and recycled. Post-transcriptional modifications can also occur to mRNA, such as alternative splicing and addition of a 5' cap and a poly(A) tail, which can affect its stability, localization, and translation efficiency.
5-Aminolevulinate synthase (ALAS) is an enzyme that catalyzes the first step in heme biosynthesis, a metabolic pathway that produces heme, a porphyrin ring with an iron atom at its center. Heme is a crucial component of hemoglobin, cytochromes, and other important molecules in the body.
ALAS exists in two forms: ALAS1 and ALAS2. ALAS1 is expressed in all tissues, while ALAS2 is primarily expressed in erythroid cells (precursors to red blood cells). The reaction catalyzed by ALAS involves the condensation of glycine and succinyl-CoA to form 5-aminolevulinate.
Deficiencies or mutations in the ALAS2 gene can lead to a rare genetic disorder called X-linked sideroblastic anemia, which is characterized by abnormal red blood cell maturation and iron overload in mitochondria.
I am not aware of a medical definition for the term "darkness." In general, darkness refers to the absence of light. It is not a term that is commonly used in the medical field, and it does not have a specific clinical meaning. If you have a question about a specific medical term or concept, I would be happy to try to help you understand it.
HDL (High-Density Lipoprotein) cholesterol is often referred to as "good" cholesterol. It is a type of lipoprotein that helps remove excess cholesterol from cells and carry it back to the liver, where it can be broken down and removed from the body. High levels of HDL cholesterol have been associated with a lower risk of heart disease and stroke.
Affinity chromatography is a type of chromatography technique used in biochemistry and molecular biology to separate and purify proteins based on their biological characteristics, such as their ability to bind specifically to certain ligands or molecules. This method utilizes a stationary phase that is coated with a specific ligand (e.g., an antibody, antigen, receptor, or enzyme) that selectively interacts with the target protein in a sample.
The process typically involves the following steps:
1. Preparation of the affinity chromatography column: The stationary phase, usually a solid matrix such as agarose beads or magnetic beads, is modified by covalently attaching the ligand to its surface.
2. Application of the sample: The protein mixture is applied to the top of the affinity chromatography column, allowing it to flow through the stationary phase under gravity or pressure.
3. Binding and washing: As the sample flows through the column, the target protein selectively binds to the ligand on the stationary phase, while other proteins and impurities pass through. The column is then washed with a suitable buffer to remove any unbound proteins and contaminants.
4. Elution of the bound protein: The target protein can be eluted from the column using various methods, such as changing the pH, ionic strength, or polarity of the buffer, or by introducing a competitive ligand that displaces the bound protein.
5. Collection and analysis: The eluted protein fraction is collected and analyzed for purity and identity, often through techniques like SDS-PAGE or mass spectrometry.
Affinity chromatography is a powerful tool in biochemistry and molecular biology due to its high selectivity and specificity, enabling the efficient isolation of target proteins from complex mixtures. However, it requires careful consideration of the binding affinity between the ligand and the protein, as well as optimization of the elution conditions to minimize potential damage or denaturation of the purified protein.
High-performance liquid chromatography (HPLC) is a type of chromatography that separates and analyzes compounds based on their interactions with a stationary phase and a mobile phase under high pressure. The mobile phase, which can be a gas or liquid, carries the sample mixture through a column containing the stationary phase.
In HPLC, the mobile phase is a liquid, and it is pumped through the column at high pressures (up to several hundred atmospheres) to achieve faster separation times and better resolution than other types of liquid chromatography. The stationary phase can be a solid or a liquid supported on a solid, and it interacts differently with each component in the sample mixture, causing them to separate as they travel through the column.
HPLC is widely used in analytical chemistry, pharmaceuticals, biotechnology, and other fields to separate, identify, and quantify compounds present in complex mixtures. It can be used to analyze a wide range of substances, including drugs, hormones, vitamins, pigments, flavors, and pollutants. HPLC is also used in the preparation of pure samples for further study or use.
Corn oil is a type of vegetable oil that is extracted from the germ of corn (maize). It is rich in polyunsaturated fat, particularly linoleic acid, and contains about 25% saturated fat. Corn oil has a high smoke point, making it suitable for frying and baking. It is also used as an ingredient in margarine, salad dressings, and other food products. In addition to its use as a food product, corn oil is sometimes used topically on the skin as a moisturizer or emollient.
Apolipoprotein C-II (ApoC-II) is a type of apolipoprotein, which are proteins that bind to lipids to form lipoprotein complexes. ApoC-II is a component of several lipoproteins, including very low-density lipoproteins (VLDL) and chylomicrons, which are responsible for the transport of fat molecules, such as triglycerides and cholesterol, in the bloodstream.
ApoC-II plays a crucial role in the activation of lipoprotein lipase, an enzyme that breaks down triglycerides in VLDL and chylomicrons into fatty acids, which can then be taken up by cells for energy production or storage. Therefore, ApoC-II deficiency can lead to hypertriglyceridemia, a condition characterized by high levels of triglycerides in the blood.
In addition to its role in lipid metabolism, ApoC-II has been implicated in the development and progression of atherosclerosis, a chronic inflammatory disease that affects the arteries and can lead to serious cardiovascular complications, such as heart attack and stroke.
Hyperlipoproteinemia Type III, also known as Broad Beta Disease or Remnant Hyperlipidemia, is a genetic disorder characterized by an increased level of chylomicron remnants and intermediate-density lipoproteins (IDL) in the blood. This results in elevated levels of both low-density lipoprotein (LDL), or "bad" cholesterol, and triglycerides, and decreased levels of high-density lipoprotein (HDL), or "good" cholesterol. The condition can lead to premature atherosclerosis and an increased risk for cardiovascular disease. It is caused by mutations in the APOE gene, which encodes the apolipoprotein E protein, leading to abnormal clearance of lipoproteins from the blood.
Isoelectric focusing (IEF) is a technique used in electrophoresis, which is a method for separating proteins or other molecules based on their electrical charges. In IEF, a mixture of ampholytes (molecules that can carry both positive and negative charges) is used to create a pH gradient within a gel matrix. When an electric field is applied, the proteins or molecules migrate through the gel until they reach the point in the gradient where their net charge is zero, known as their isoelectric point (pI). At this point, they focus into a sharp band and stop moving, resulting in a highly resolved separation of the different components based on their pI. This technique is widely used in protein research for applications such as protein identification, characterization, and purification.
Perfusion, in medical terms, refers to the process of circulating blood through the body's organs and tissues to deliver oxygen and nutrients and remove waste products. It is a measure of the delivery of adequate blood flow to specific areas or tissues in the body. Perfusion can be assessed using various methods, including imaging techniques like computed tomography (CT) scans, magnetic resonance imaging (MRI), and perfusion scintigraphy.
Perfusion is critical for maintaining proper organ function and overall health. When perfusion is impaired or inadequate, it can lead to tissue hypoxia, acidosis, and cell death, which can result in organ dysfunction or failure. Conditions that can affect perfusion include cardiovascular disease, shock, trauma, and certain surgical procedures.
Lecithin:cholesterol acyltransferase (LCAT) deficiency is a genetic disorder that affects the metabolism of cholesterol in the body. LCAT is an enzyme that helps to convert cholesterol into a form that can be easily transported in the bloodstream.
In LCAT deficiency, the activity of this enzyme is reduced or absent, leading to an accumulation of cholesterol in various tissues and organs of the body. This can result in a range of symptoms, including corneal opacities (clouding of the clear outer layer of the eye), hemolytic anemia (destruction of red blood cells), proteinuria (excess protein in the urine), and kidney failure.
There are two main types of LCAT deficiency: a complete form, known as fish-eye disease, which is characterized by corneal opacities but few other symptoms; and an incomplete form, known as LCAT deficiency with systemic involvement, which can affect multiple organs and systems of the body.
LCAT deficiency is caused by mutations in the LCAT gene, which provides instructions for making the LCAT enzyme. Inheritance is autosomal recessive, meaning that an individual must inherit two copies of the mutated gene (one from each parent) to develop the disorder.
Coenzymes are small organic molecules that assist enzymes in catalyzing chemical reactions within cells. They typically act as carriers of specific atoms or groups of atoms during enzymatic reactions, facilitating the conversion of substrates into products. Coenzymes often bind temporarily to enzymes at the active site, forming an enzyme-coenzyme complex.
Coenzymes are usually derived from vitamins or minerals and are essential for maintaining proper metabolic functions in the body. Examples of coenzymes include nicotinamide adenine dinucleotide (NAD+), flavin adenine dinucleotide (FAD), and coenzyme A (CoA). When a coenzyme is used up in a reaction, it must be regenerated or replaced for the enzyme to continue functioning.
In summary, coenzymes are vital organic compounds that work closely with enzymes to facilitate biochemical reactions, ensuring the smooth operation of various metabolic processes within living organisms.
Photosystem II Protein Complex is a crucial component of the photosynthetic apparatus in plants, algae, and cyanobacteria. It is a multi-subunit protein complex located in the thylakoid membrane of the chloroplasts. Photosystem II plays a vital role in light-dependent reactions of photosynthesis, where it absorbs sunlight and uses its energy to drive the oxidation of water molecules into oxygen, electrons, and protons.
The protein complex consists of several subunits, including the D1 and D2 proteins, which form the reaction center, and several antenna proteins that capture light energy and transfer it to the reaction center. Photosystem II also contains various cofactors, such as pigments (chlorophylls and carotenoids), redox-active metal ions (manganese and calcium), and quinones, which facilitate the charge separation and electron transfer processes during photosynthesis.
Photosystem II Protein Complex is responsible for the initial charge separation event in photosynthesis, which sets off a series of redox reactions that ultimately lead to the reduction of NADP+ to NADPH and the synthesis of ATP, providing energy for the carbon fixation reactions in the Calvin cycle. Additionally, Photosystem II Protein Complex is involved in oxygen evolution, contributing to the Earth's atmosphere's oxygen levels and making it an essential component of global carbon fixation and oxygen production.
Cobalt is a chemical element with the symbol Co and atomic number 27. It is a hard, silver-white, lustrous, and brittle metal that is found naturally only in chemically combined form, except for small amounts found in meteorites. Cobalt is used primarily in the production of magnetic, wear-resistant, and high-strength alloys, as well as in the manufacture of batteries, magnets, and pigments.
In a medical context, cobalt is sometimes used in the form of cobalt-60, a radioactive isotope, for cancer treatment through radiation therapy. Cobalt-60 emits gamma rays that can be directed at tumors to destroy cancer cells. Additionally, small amounts of cobalt are present in some vitamin B12 supplements and fortified foods, as cobalt is an essential component of vitamin B12. However, exposure to high levels of cobalt can be harmful and may cause health effects such as allergic reactions, lung damage, heart problems, and neurological issues.
Aryl hydrocarbon hydroxylases (AHH) are a group of enzymes that play a crucial role in the metabolism of various aromatic and heterocyclic compounds, including potentially harmful substances such as polycyclic aromatic hydrocarbons (PAHs) and dioxins. These enzymes are primarily located in the endoplasmic reticulum of cells, particularly in the liver, but can also be found in other tissues.
The AHH enzymes catalyze the addition of a hydroxyl group (-OH) to the aromatic ring structure of these compounds, which is the first step in their biotransformation and eventual elimination from the body. This process can sometimes lead to the formation of metabolites that are more reactive and potentially toxic than the original compound. Therefore, the overall impact of AHH enzymes on human health is complex and depends on various factors, including the specific compounds being metabolized and individual genetic differences in enzyme activity.
LDL, or low-density lipoprotein, is often referred to as "bad" cholesterol. It is one of the lipoproteins that helps carry cholesterol throughout your body. High levels of LDL cholesterol can lead to a buildup of cholesterol in your arteries, which can increase the risk of heart disease and stroke.
Cholesterol is a type of fat (lipid) that is found in the cells of your body. Your body needs some cholesterol to function properly, but having too much can lead to health problems. LDL cholesterol is one of the two main types of cholesterol; the other is high-density lipoprotein (HDL), or "good" cholesterol.
It's important to keep your LDL cholesterol levels in a healthy range to reduce your risk of developing heart disease and stroke. A healthcare professional can help you determine what your target LDL cholesterol level should be based on your individual health status and risk factors.
A Schiff base is not a medical term per se, but rather a chemical concept that can be relevant in various scientific and medical fields. A Schiff base is a chemical compound that contains a carbon-nitrogen double bond with the nitrogen atom connected to an aryl or alkyl group, excluding hydrogen. This structure is also known as an azomethine.
The general formula for a Schiff base is R1R2C=NR3, where R1 and R2 are organic groups (aryl or alkyl), and R3 is a hydrogen atom or an organic group. These compounds can be synthesized by the condensation of a primary amine with a carbonyl compound, such as an aldehyde or ketone.
Schiff bases have been studied in various medical and biological contexts due to their potential bioactivities. Some Schiff bases exhibit antimicrobial, antifungal, anti-inflammatory, and anticancer properties. They can also serve as ligands for metal ions, forming complexes with potential applications in medicinal chemistry, such as in the development of new drugs or diagnostic agents.
Macromolecular substances, also known as macromolecules, are large, complex molecules made up of repeating subunits called monomers. These substances are formed through polymerization, a process in which many small molecules combine to form a larger one. Macromolecular substances can be naturally occurring, such as proteins, DNA, and carbohydrates, or synthetic, such as plastics and synthetic fibers.
In the context of medicine, macromolecular substances are often used in the development of drugs and medical devices. For example, some drugs are designed to bind to specific macromolecules in the body, such as proteins or DNA, in order to alter their function and produce a therapeutic effect. Additionally, macromolecular substances may be used in the creation of medical implants, such as artificial joints and heart valves, due to their strength and durability.
It is important for healthcare professionals to have an understanding of macromolecular substances and how they function in the body, as this knowledge can inform the development and use of medical treatments.
Oleic acid is a monounsaturated fatty acid that is commonly found in various natural oils such as olive oil, sunflower oil, and peanut oil. Its chemical formula is cis-9-octadecenoic acid, and it is a colorless liquid at room temperature with a slight odor. Oleic acid is an important component of human diet and has been shown to have various health benefits, including reducing the risk of heart disease and improving immune function. It is also used in the manufacture of soaps, cosmetics, and other industrial products.
Cysteine is a semi-essential amino acid, which means that it can be produced by the human body under normal circumstances, but may need to be obtained from external sources in certain conditions such as illness or stress. Its chemical formula is HO2CCH(NH2)CH2SH, and it contains a sulfhydryl group (-SH), which allows it to act as a powerful antioxidant and participate in various cellular processes.
Cysteine plays important roles in protein structure and function, detoxification, and the synthesis of other molecules such as glutathione, taurine, and coenzyme A. It is also involved in wound healing, immune response, and the maintenance of healthy skin, hair, and nails.
Cysteine can be found in a variety of foods, including meat, poultry, fish, dairy products, eggs, legumes, nuts, seeds, and some grains. It is also available as a dietary supplement and can be used in the treatment of various medical conditions such as liver disease, bronchitis, and heavy metal toxicity. However, excessive intake of cysteine may have adverse effects on health, including gastrointestinal disturbances, nausea, vomiting, and headaches.
A mutation is a permanent change in the DNA sequence of an organism's genome. Mutations can occur spontaneously or be caused by environmental factors such as exposure to radiation, chemicals, or viruses. They may have various effects on the organism, ranging from benign to harmful, depending on where they occur and whether they alter the function of essential proteins. In some cases, mutations can increase an individual's susceptibility to certain diseases or disorders, while in others, they may confer a survival advantage. Mutations are the driving force behind evolution, as they introduce new genetic variability into populations, which can then be acted upon by natural selection.
Hydrogen-ion concentration, also known as pH, is a measure of the acidity or basicity of a solution. It is defined as the negative logarithm (to the base 10) of the hydrogen ion activity in a solution. The standard unit of measurement is the pH unit. A pH of 7 is neutral, less than 7 is acidic, and greater than 7 is basic.
In medical terms, hydrogen-ion concentration is important for maintaining homeostasis within the body. For example, in the stomach, a high hydrogen-ion concentration (low pH) is necessary for the digestion of food. However, in other parts of the body such as blood, a high hydrogen-ion concentration can be harmful and lead to acidosis. Conversely, a low hydrogen-ion concentration (high pH) in the blood can lead to alkalosis. Both acidosis and alkalosis can have serious consequences on various organ systems if not corrected.
Molecular structure, in the context of biochemistry and molecular biology, refers to the arrangement and organization of atoms and chemical bonds within a molecule. It describes the three-dimensional layout of the constituent elements, including their spatial relationships, bond lengths, and angles. Understanding molecular structure is crucial for elucidating the functions and reactivities of biological macromolecules such as proteins, nucleic acids, lipids, and carbohydrates. Various experimental techniques, like X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and cryo-electron microscopy (cryo-EM), are employed to determine molecular structures at atomic resolution, providing valuable insights into their biological roles and potential therapeutic targets.
Temperature, in a medical context, is a measure of the degree of hotness or coldness of a body or environment. It is usually measured using a thermometer and reported in degrees Celsius (°C), degrees Fahrenheit (°F), or kelvin (K). In the human body, normal core temperature ranges from about 36.5-37.5°C (97.7-99.5°F) when measured rectally, and can vary slightly depending on factors such as time of day, physical activity, and menstrual cycle. Elevated body temperature is a common sign of infection or inflammation, while abnormally low body temperature can indicate hypothermia or other medical conditions.
Hydroxylamines are organic compounds that contain a hydroxy group (-OH) and an amino group (-NH2) in their structure. More specifically, they have the functional group R-N-OH, where R represents a carbon-containing radical. Hydroxylamines can be considered as derivatives of ammonia (NH3), where one hydrogen atom is replaced by a hydroxy group.
These compounds are important in organic chemistry and biochemistry due to their ability to act as reducing agents, nitrogen donors, and intermediates in various chemical reactions. They can be found in some natural substances and are also synthesized for use in pharmaceuticals, agrochemicals, and other industrial applications.
Examples of hydroxylamines include:
* Hydroxylamine (NH2OH) itself, which is a colorless liquid at room temperature with an odor similar to ammonia.
* N-Methylhydroxylamine (CH3NHOH), which is a solid that can be used as a reducing agent and a nucleophile in organic synthesis.
* Phenylhydroxylamine (C6H5NHOH), which is a solid used as an intermediate in the production of dyes, pharmaceuticals, and other chemicals.
It's important to note that hydroxylamines can be unstable and potentially hazardous, so they should be handled with care during laboratory work or industrial processes.
I apologize for any confusion, but "plants" is not a term that has a medical definition. The term "plants" refers to a large and diverse group of organisms that obtain their energy through photosynthesis, which is the process of converting sunlight into chemical energy. Plants are typically characterized by having cells with cell walls containing cellulose, chloroplasts containing the pigment chlorophyll, and the ability to synthesize their own food through photosynthesis.
In a medical or biological context, you might be thinking of "plant-based" or "phytomedicine," which refer to the use of plants or plant extracts as a form of medicine or treatment. Phytomedicines have been used for thousands of years in many traditional systems of medicine, and some plant-derived compounds have been found to have therapeutic benefits in modern medicine as well. However, "plants" itself does not have a medical definition.
Isotope labeling is a scientific technique used in the field of medicine, particularly in molecular biology, chemistry, and pharmacology. It involves replacing one or more atoms in a molecule with a radioactive or stable isotope of the same element. This modified molecule can then be traced and analyzed to study its structure, function, metabolism, or interaction with other molecules within biological systems.
Radioisotope labeling uses unstable radioactive isotopes that emit radiation, allowing for detection and quantification of the labeled molecule using various imaging techniques, such as positron emission tomography (PET) or single-photon emission computed tomography (SPECT). This approach is particularly useful in tracking the distribution and metabolism of drugs, hormones, or other biomolecules in living organisms.
Stable isotope labeling, on the other hand, employs non-radioactive isotopes that do not emit radiation. These isotopes have different atomic masses compared to their natural counterparts and can be detected using mass spectrometry. Stable isotope labeling is often used in metabolic studies, protein turnover analysis, or for identifying the origin of specific molecules within complex biological samples.
In summary, isotope labeling is a versatile tool in medical research that enables researchers to investigate various aspects of molecular behavior and interactions within biological systems.
Biological transport refers to the movement of molecules, ions, or solutes across biological membranes or through cells in living organisms. This process is essential for maintaining homeostasis, regulating cellular functions, and enabling communication between cells. There are two main types of biological transport: passive transport and active transport.
Passive transport does not require the input of energy and includes:
1. Diffusion: The random movement of molecules from an area of high concentration to an area of low concentration until equilibrium is reached.
2. Osmosis: The diffusion of solvent molecules (usually water) across a semi-permeable membrane from an area of lower solute concentration to an area of higher solute concentration.
3. Facilitated diffusion: The assisted passage of polar or charged substances through protein channels or carriers in the cell membrane, which increases the rate of diffusion without consuming energy.
Active transport requires the input of energy (in the form of ATP) and includes:
1. Primary active transport: The direct use of ATP to move molecules against their concentration gradient, often driven by specific transport proteins called pumps.
2. Secondary active transport: The coupling of the movement of one substance down its electrochemical gradient with the uphill transport of another substance, mediated by a shared transport protein. This process is also known as co-transport or counter-transport.
Thioctic acid is also known as alpha-lipoic acid. It is a vitamin-like chemical compound that is made naturally in the body and is found in small amounts in some foods like spinach, broccoli, and potatoes. Thioctic acid is an antioxidant that helps to protect cells from damage caused by free radicals. It also plays a role in energy production in the cells and has been studied for its potential benefits in the treatment of diabetes and nerve-related symptoms of diabetes such as pain, burning, itching, and numbness. Thioctic acid is available as a dietary supplement.
Medical Definition: Thioctic acid (also known as alpha-lipoic acid) is a vitamin-like antioxidant that is made naturally in the body and is found in small amounts in some foods. It plays a role in energy production in the cells, and has been studied for its potential benefits in the treatment of diabetes and nerve-related symptoms of diabetes such as pain, burning, itching, and numbness. Thioctic acid is also available as a dietary supplement.
Arteriosclerosis is a general term that describes the hardening and stiffening of the artery walls. It's a progressive condition that can occur as a result of aging, or it may be associated with certain risk factors such as high blood pressure, high cholesterol, diabetes, smoking, and a sedentary lifestyle.
The process of arteriosclerosis involves the buildup of plaque, made up of fat, cholesterol, calcium, and other substances, in the inner lining of the artery walls. Over time, this buildup can cause the artery walls to thicken and harden, reducing the flow of oxygen-rich blood to the body's organs and tissues.
Arteriosclerosis can affect any of the body's arteries, but it is most commonly found in the coronary arteries that supply blood to the heart, the cerebral arteries that supply blood to the brain, and the peripheral arteries that supply blood to the limbs. When arteriosclerosis affects the coronary arteries, it can lead to heart disease, angina, or heart attack. When it affects the cerebral arteries, it can lead to stroke or transient ischemic attack (TIA). When it affects the peripheral arteries, it can cause pain, numbness, or weakness in the limbs, and in severe cases, gangrene and amputation.
Hydroxylamine is not a medical term, but it is a chemical compound with the formula NH2OH. It's used in some industrial processes and can also be found as a byproduct of certain metabolic reactions in the body. In a medical context, exposure to high levels of hydroxylamine may cause irritation to the skin, eyes, and respiratory tract, and it may have harmful effects on the nervous system and blood if ingested or absorbed in large amounts. However, it is not a substance that is commonly encountered or monitored in medical settings.
Dithionitrobenzoic acid is not a medical term, as it is related to chemistry rather than medicine. It is an organic compound with the formula C6H4N2O4S2. This compound is a type of benzenediol that contains two sulfur atoms and two nitro groups. It is a white crystalline powder that is soluble in water and alcohol.
Dithionitrobenzoic acid is not used directly in medical applications, but it can be used as a reagent in chemical reactions that are relevant to medical research or analysis. For example, it can be used to determine the concentration of iron in biological samples through a reaction that produces a colored complex. However, if you have any specific questions related to its use or application in a medical context, I would recommend consulting with a medical professional or a researcher in the relevant field.
Pulmonary Alveolar Proteinosis (PAP) is a rare lung disorder characterized by the accumulation of surfactant, a lipoprotein complex that reduces surface tension within the alveoli, in the air sacs (alveoli) of the lungs. This accumulation can lead to difficulty breathing and reduced oxygen levels in the blood.
There are three types of PAP:
1. Congenital PAP: A very rare inherited form that affects infants and is caused by a genetic mutation that disrupts the production or function of granulocyte-macrophage colony-stimulating factor (GM-CSF), a protein important for the development and function of alveolar macrophages.
2. Secondary PAP: This form is associated with conditions that impair the clearance of surfactant by alveolar macrophages, such as hematologic disorders (e.g., leukemia), infections, exposure to inhaled irritants (e.g., silica dust), and certain medications.
3. Idiopathic PAP: The most common form, also known as autoimmune PAP, is caused by the development of autoantibodies against GM-CSF, which disrupts its function and leads to surfactant accumulation in the lungs.
Treatment for PAP may include whole lung lavage (WLL), a procedure where the affected lung is filled with saline solution and then drained to remove excess surfactant, as well as managing any underlying conditions. In some cases of idiopathic PAP, off-label use of inhaled GM-CSF has shown promise in improving symptoms and lung function.
Photosystem I Protein Complex, also known as PsaA/B-Protein or Photosystem I reaction center, is a large protein complex found in the thylakoid membrane of plant chloroplasts and cyanobacteria. It plays a crucial role in light-dependent reactions of photosynthesis, where it absorbs light energy and converts it into chemical energy in the form of NADPH.
The complex is composed of several subunits, including PsaA and PsaB, which are the core components that bind to chlorophyll a and bacteriochlorophyll a pigments. These pigments absorb light energy and transfer it to the reaction center, where it is used to drive the electron transport chain and generate a proton gradient across the membrane. This gradient is then used to produce ATP, which provides energy for the carbon fixation reactions in photosynthesis.
Photosystem I Protein Complex is also involved in cyclic electron flow, where electrons are recycled within the complex to generate additional ATP without producing NADPH. This process helps regulate the balance between ATP and NADPH production in the chloroplast and optimizes the efficiency of photosynthesis.
Transducin is a G protein found in the rod cells of the retina and plays a crucial role in the visual signal transduction pathway. It is responsible for converting the light-induced isomerization of rhodopsin into a biochemical signal, which ultimately leads to the activation of downstream effectors and the generation of a neural response.
Transducin has three subunits: alpha (Tα), beta (Tβ), and gamma (Tγ). When light activates rhodopsin, it interacts with the Tα subunit, causing it to exchange GDP for GTP and dissociate from the Tβγ complex. The activated Tα then interacts with a downstream effector called phosphodiesterase (PDE), which leads to the hydrolysis of cGMP and the closure of cGMP-gated ion channels in the plasma membrane. This results in the hyperpolarization of the rod cell, which is the initial step in the visual signal transduction pathway.
Overall, transducin is a key player in the conversion of light energy into neural signals, allowing us to see and perceive our visual world.
Cytochrome b is a type of cytochrome, which is a class of proteins that contain heme as a cofactor and are involved in electron transfer. Cytochromes are classified based on the type of heme they contain and their absorption spectra.
The cytochrome b group includes several subfamilies of cytochromes, including cytochrome b5, cytochrome b2, and cytochrome bc1 (also known as complex III). These cytochromes are involved in various biological processes, such as fatty acid desaturation, steroid metabolism, and the electron transport chain.
The electron transport chain is a series of protein complexes in the inner mitochondrial membrane that generates most of the ATP (adenosine triphosphate) required for cellular energy production. Cytochrome bc1 is a key component of the electron transport chain, where it functions as a dimer and catalyzes the transfer of electrons from ubiquinol to cytochrome c while simultaneously pumping protons across the membrane. This creates an electrochemical gradient that drives ATP synthesis.
Deficiencies or mutations in cytochrome b genes can lead to various diseases, such as mitochondrial disorders and cancer.
Thromboplastin is a substance that activates the coagulation cascade, leading to the formation of a clot (thrombus). It's primarily found in damaged or injured tissues and blood vessels, as well as in platelets (thrombocytes). There are two types of thromboplastin:
1. Extrinsic thromboplastin (also known as tissue factor): This is a transmembrane glycoprotein that is primarily found in subendothelial cells and released upon injury to the blood vessels. It initiates the extrinsic pathway of coagulation by binding to and activating Factor VII, ultimately leading to the formation of thrombin and fibrin clots.
2. Intrinsic thromboplastin (also known as plasma thromboplastin or factor III): This term is used less frequently and refers to a labile phospholipid component present in platelet membranes, which plays a role in the intrinsic pathway of coagulation.
In clinical settings, the term "thromboplastin" often refers to reagents used in laboratory tests like the prothrombin time (PT) and activated partial thromboplastin time (aPTT). These reagents contain a source of tissue factor and calcium ions to initiate and monitor the coagulation process.
Antibiotics are a type of medication used to treat infections caused by bacteria. They work by either killing the bacteria or inhibiting their growth.
Antineoplastics, also known as chemotherapeutic agents, are a class of drugs used to treat cancer. These medications target and destroy rapidly dividing cells, such as cancer cells, although they can also affect other quickly dividing cells in the body, such as those in the hair follicles or digestive tract, which can lead to side effects.
Antibiotics and antineoplastics are two different classes of drugs with distinct mechanisms of action and uses. It is important to use them appropriately and under the guidance of a healthcare professional.
Protein folding is the process by which a protein molecule naturally folds into its three-dimensional structure, following the synthesis of its amino acid chain. This complex process is determined by the sequence and properties of the amino acids, as well as various environmental factors such as temperature, pH, and the presence of molecular chaperones. The final folded conformation of a protein is crucial for its proper function, as it enables the formation of specific interactions between different parts of the molecule, which in turn define its biological activity. Protein misfolding can lead to various diseases, including neurodegenerative disorders such as Alzheimer's and Parkinson's disease.
"Chickens" is a common term used to refer to the domesticated bird, Gallus gallus domesticus, which is widely raised for its eggs and meat. However, in medical terms, "chickens" is not a standard term with a specific definition. If you have any specific medical concern or question related to chickens, such as food safety or allergies, please provide more details so I can give a more accurate answer.
"Cells, cultured" is a medical term that refers to cells that have been removed from an organism and grown in controlled laboratory conditions outside of the body. This process is called cell culture and it allows scientists to study cells in a more controlled and accessible environment than they would have inside the body. Cultured cells can be derived from a variety of sources, including tissues, organs, or fluids from humans, animals, or cell lines that have been previously established in the laboratory.
Cell culture involves several steps, including isolation of the cells from the tissue, purification and characterization of the cells, and maintenance of the cells in appropriate growth conditions. The cells are typically grown in specialized media that contain nutrients, growth factors, and other components necessary for their survival and proliferation. Cultured cells can be used for a variety of purposes, including basic research, drug development and testing, and production of biological products such as vaccines and gene therapies.
It is important to note that cultured cells may behave differently than they do in the body, and results obtained from cell culture studies may not always translate directly to human physiology or disease. Therefore, it is essential to validate findings from cell culture experiments using additional models and ultimately in clinical trials involving human subjects.
Phenobarbital is a barbiturate medication that is primarily used for the treatment of seizures and convulsions. It works by suppressing the abnormal electrical activity in the brain that leads to seizures. In addition to its anticonvulsant properties, phenobarbital also has sedative and hypnotic effects, which can be useful for treating anxiety, insomnia, and agitation.
Phenobarbital is available in various forms, including tablets, capsules, and elixirs, and it is typically taken orally. The medication works by binding to specific receptors in the brain called gamma-aminobutyric acid (GABA) receptors, which help to regulate nerve impulses in the brain. By increasing the activity of GABA, phenobarbital can help to reduce excessive neural activity and prevent seizures.
While phenobarbital is an effective medication for treating seizures and other conditions, it can also be habit-forming and carries a risk of dependence and addiction. Long-term use of the medication can lead to tolerance, meaning that higher doses may be needed to achieve the same effects. Abruptly stopping the medication can also lead to withdrawal symptoms, such as anxiety, restlessness, and seizures.
Like all medications, phenobarbital can have side effects, including dizziness, drowsiness, and impaired coordination. It can also interact with other medications, such as certain antidepressants and sedatives, so it is important to inform your healthcare provider of all medications you are taking before starting phenobarbital.
In summary, phenobarbital is a barbiturate medication used primarily for the treatment of seizures and convulsions. It works by binding to GABA receptors in the brain and increasing their activity, which helps to reduce excessive neural activity and prevent seizures. While phenobarbital can be effective, it carries a risk of dependence and addiction and can have side effects and drug interactions.
Iodine radioisotopes are radioactive isotopes of the element iodine, which decays and emits radiation in the form of gamma rays. Some commonly used iodine radioisotopes include I-123, I-125, I-131. These radioisotopes have various medical applications such as in diagnostic imaging, therapy for thyroid disorders, and cancer treatment.
For example, I-131 is commonly used to treat hyperthyroidism and differentiated thyroid cancer due to its ability to destroy thyroid tissue. On the other hand, I-123 is often used in nuclear medicine scans of the thyroid gland because it emits gamma rays that can be detected by a gamma camera, allowing for detailed images of the gland's structure and function.
It is important to note that handling and administering radioisotopes require specialized training and safety precautions due to their radiation-emitting properties.
Cycloheximide is an antibiotic that is primarily used in laboratory settings to inhibit protein synthesis in eukaryotic cells. It is derived from the actinobacteria species Streptomyces griseus. In medical terms, it is not used as a therapeutic drug in humans due to its significant side effects, including liver toxicity and potential neurotoxicity. However, it remains a valuable tool in research for studying protein function and cellular processes.
The antibiotic works by binding to the 60S subunit of the ribosome, thereby preventing the transfer RNA (tRNA) from delivering amino acids to the growing polypeptide chain during translation. This inhibition of protein synthesis can be lethal to cells, making cycloheximide a useful tool in studying cellular responses to protein depletion or misregulation.
In summary, while cycloheximide has significant research applications due to its ability to inhibit protein synthesis in eukaryotic cells, it is not used as a therapeutic drug in humans because of its toxic side effects.
Apoferritins are the protein shells or apoproteins of ferritin molecules that are devoid of iron. Ferritin is a protein in cells that stores iron and releases it in a form that can be used by the body. Apoferritin can bind with iron ions to form ferritin. It has a hollow, spherical structure and is often used as a model for studying protein folding and assembly.
A homozygote is an individual who has inherited the same allele (version of a gene) from both parents and therefore possesses two identical copies of that allele at a specific genetic locus. This can result in either having two dominant alleles (homozygous dominant) or two recessive alleles (homozygous recessive). In contrast, a heterozygote has inherited different alleles from each parent for a particular gene.
The term "homozygote" is used in genetics to describe the genetic makeup of an individual at a specific locus on their chromosomes. Homozygosity can play a significant role in determining an individual's phenotype (observable traits), as having two identical alleles can strengthen the expression of certain characteristics compared to having just one dominant and one recessive allele.
Peptides are short chains of amino acid residues linked by covalent bonds, known as peptide bonds. They are formed when two or more amino acids are joined together through a condensation reaction, which results in the elimination of a water molecule and the formation of an amide bond between the carboxyl group of one amino acid and the amino group of another.
Peptides can vary in length from two to about fifty amino acids, and they are often classified based on their size. For example, dipeptides contain two amino acids, tripeptides contain three, and so on. Oligopeptides typically contain up to ten amino acids, while polypeptides can contain dozens or even hundreds of amino acids.
Peptides play many important roles in the body, including serving as hormones, neurotransmitters, enzymes, and antibiotics. They are also used in medical research and therapeutic applications, such as drug delivery and tissue engineering.
"Physicochemical phenomena" is not a term that has a specific medical definition. However, in general terms, physicochemical phenomena refer to the physical and chemical interactions and processes that occur within living organisms or biological systems. These phenomena can include various properties and reactions such as pH levels, osmotic pressure, enzyme kinetics, and thermodynamics, among others.
In a broader context, physicochemical phenomena play an essential role in understanding the mechanisms of drug action, pharmacokinetics, and toxicity. For instance, the solubility, permeability, and stability of drugs are all physicochemical properties that can affect their absorption, distribution, metabolism, and excretion (ADME) within the body.
Therefore, while not a medical definition per se, an understanding of physicochemical phenomena is crucial to the study and practice of pharmacology, toxicology, and other related medical fields.
Hemolymph is not a term typically used in human medicine, but it is commonly used in the study of invertebrates, particularly arthropods such as insects and crustaceans. Hemolymph is the fluid that circulates within the open circulatory system of these animals, serving multiple functions similar to both blood and lymphatic systems in vertebrates.
In simpler terms, hemolymph is a combined fluid that performs the functions of both blood and lymph in invertebrates. It serves as a transport medium for nutrients, waste products, hormones, and immune cells (hemocytes) throughout the body. Hemolymph does not contain red and white blood cells like human blood; instead, hemocytes are the primary cellular components responsible for immune responses and wound healing in these animals.
The isoelectric point (pI) is a term used in biochemistry and molecular biology to describe the pH at which a molecule, such as a protein or peptide, carries no net electrical charge. At this pH, the positive and negative charges on the molecule are equal and balanced. The pI of a protein can be calculated based on its amino acid sequence and is an important property that affects its behavior in various chemical and biological environments. Proteins with different pIs may have different solubilities, stabilities, and interactions with other molecules, which can impact their function and role in the body.
Secondary protein structure refers to the local spatial arrangement of amino acid chains in a protein, typically described as regular repeating patterns held together by hydrogen bonds. The two most common types of secondary structures are the alpha-helix (α-helix) and the beta-pleated sheet (β-sheet). In an α-helix, the polypeptide chain twists around itself in a helical shape, with each backbone atom forming a hydrogen bond with the fourth amino acid residue along the chain. This forms a rigid rod-like structure that is resistant to bending or twisting forces. In β-sheets, adjacent segments of the polypeptide chain run parallel or antiparallel to each other and are connected by hydrogen bonds, forming a pleated sheet-like arrangement. These secondary structures provide the foundation for the formation of tertiary and quaternary protein structures, which determine the overall three-dimensional shape and function of the protein.
Physical chemistry is a branch of chemistry that deals with the fundamental principles and laws governing the behavior of matter and energy at the molecular and atomic levels. It combines elements of physics, chemistry, mathematics, and engineering to study the properties, composition, structure, and transformation of matter. Key areas of focus in physical chemistry include thermodynamics, kinetics, quantum mechanics, statistical mechanics, electrochemistry, and spectroscopy.
In essence, physical chemists aim to understand how and why chemical reactions occur, what drives them, and how they can be controlled or predicted. This knowledge is crucial for developing new materials, medicines, energy technologies, and other applications that benefit society.
Hydrolysis is a chemical process, not a medical one. However, it is relevant to medicine and biology.
Hydrolysis is the breakdown of a chemical compound due to its reaction with water, often resulting in the formation of two or more simpler compounds. In the context of physiology and medicine, hydrolysis is a crucial process in various biological reactions, such as the digestion of food molecules like proteins, carbohydrates, and fats. Enzymes called hydrolases catalyze these hydrolysis reactions to speed up the breakdown process in the body.
Mass spectrometry (MS) is an analytical technique used to identify and quantify the chemical components of a mixture or compound. It works by ionizing the sample, generating charged molecules or fragments, and then measuring their mass-to-charge ratio in a vacuum. The resulting mass spectrum provides information about the molecular weight and structure of the analytes, allowing for identification and characterization.
In simpler terms, mass spectrometry is a method used to determine what chemicals are present in a sample and in what quantities, by converting the chemicals into ions, measuring their masses, and generating a spectrum that shows the relative abundances of each ion type.
A peptide fragment is a short chain of amino acids that is derived from a larger peptide or protein through various biological or chemical processes. These fragments can result from the natural breakdown of proteins in the body during regular physiological processes, such as digestion, or they can be produced experimentally in a laboratory setting for research or therapeutic purposes.
Peptide fragments are often used in research to map the structure and function of larger peptides and proteins, as well as to study their interactions with other molecules. In some cases, peptide fragments may also have biological activity of their own and can be developed into drugs or diagnostic tools. For example, certain peptide fragments derived from hormones or neurotransmitters may bind to receptors in the body and mimic or block the effects of the full-length molecule.
Chromatiaceae is a family of bacteria that are primarily characterized by their ability to photosynthesize and store energy in the form of sulfur granules. These bacteria are often found in aquatic environments, such as in salt marshes, freshwater sediments, and marine ecosystems. They are capable of using reduced sulfur compounds as an electron donor during photosynthesis, which distinguishes them from other photosynthetic bacteria that use water as an electron donor.
Chromatiaceae bacteria are gram-negative rods or curved rods, and they typically form distinct layers in the environment where they live. They are often found in stratified water columns, where they can form a layer of purple or brown-colored cells that are visible to the naked eye. The pigmentation comes from bacteriochlorophylls and carotenoids, which are used in light absorption during photosynthesis.
These bacteria play an important role in the biogeochemical cycling of sulfur and carbon in aquatic environments. They can help to remove excess nutrients from the water column, and they can also serve as a food source for other organisms in the ecosystem. However, some species of Chromatiaceae can also be associated with harmful algal blooms or other environmental disturbances that can have negative impacts on water quality and aquatic life.
Site-directed mutagenesis is a molecular biology technique used to introduce specific and targeted changes to a specific DNA sequence. This process involves creating a new variant of a gene or a specific region of interest within a DNA molecule by introducing a planned, deliberate change, or mutation, at a predetermined site within the DNA sequence.
The methodology typically involves the use of molecular tools such as PCR (polymerase chain reaction), restriction enzymes, and/or ligases to introduce the desired mutation(s) into a plasmid or other vector containing the target DNA sequence. The resulting modified DNA molecule can then be used to transform host cells, allowing for the production of large quantities of the mutated gene or protein for further study.
Site-directed mutagenesis is a valuable tool in basic research, drug discovery, and biotechnology applications where specific changes to a DNA sequence are required to understand gene function, investigate protein structure/function relationships, or engineer novel biological properties into existing genes or proteins.
Leucine is an essential amino acid, meaning it cannot be produced by the human body and must be obtained through the diet. It is one of the three branched-chain amino acids (BCAAs), along with isoleucine and valine. Leucine is critical for protein synthesis and muscle growth, and it helps to regulate blood sugar levels, promote wound healing, and produce growth hormones.
Leucine is found in various food sources such as meat, dairy products, eggs, and certain plant-based proteins like soy and beans. It is also available as a dietary supplement for those looking to increase their intake for athletic performance or muscle recovery purposes. However, it's important to consult with a healthcare professional before starting any new supplement regimen.
Fasting is defined in medical terms as the abstinence from food or drink for a period of time. This practice is often recommended before certain medical tests or procedures, as it helps to ensure that the results are not affected by recent eating or drinking.
In some cases, fasting may also be used as a therapeutic intervention, such as in the management of seizures or other neurological conditions. Fasting can help to lower blood sugar and insulin levels, which can have a variety of health benefits. However, it is important to note that prolonged fasting can also have negative effects on the body, including malnutrition, dehydration, and electrolyte imbalances.
Fasting is also a spiritual practice in many religions, including Christianity, Islam, Buddhism, and Hinduism. In these contexts, fasting is often seen as a way to purify the mind and body, to focus on spiritual practices, or to express devotion or mourning.
Factor VII, also known as proconvertin, is a protein involved in the coagulation cascade, which is a series of chemical reactions that leads to the formation of a blood clot. Factor VII is synthesized in the liver and is activated when it comes into contact with tissue factor, which is exposed when blood vessels are damaged. Activated Factor VII then activates Factor X, leading to the formation of thrombin and ultimately a fibrin clot.
Inherited deficiencies or dysfunctions of Factor VII can lead to an increased risk of bleeding, while elevated levels of Factor VII have been associated with an increased risk of thrombosis (blood clots).
Azo compounds are organic compounds characterized by the presence of one or more azo groups (-N=N-) in their molecular structure. The term "azo" is derived from the Greek word "azō," meaning "to boil" or "to sparkle," which refers to the brightly colored nature of many azo compounds.
These compounds are synthesized by the reaction between aromatic amines and nitrous acid or its derivatives, resulting in the formation of diazonium salts, which then react with another aromatic compound containing an active methylene group to form azo compounds.
Azo compounds have diverse applications across various industries, including dyes, pigments, pharmaceuticals, and agrochemicals. They are known for their vibrant colors, making them widely used as colorants in textiles, leather, paper, and food products. In addition, some azo compounds exhibit unique chemical properties, such as solubility, stability, and reactivity, which make them valuable intermediates in the synthesis of various organic compounds.
However, certain azo compounds have been found to pose health risks due to their potential carcinogenicity and mutagenicity. As a result, regulations have been imposed on their use in consumer products, particularly those intended for oral consumption or direct skin contact.
Cyanobacteria, also known as blue-green algae, are a type of bacteria that obtain their energy through photosynthesis, similar to plants. They can produce oxygen and contain chlorophyll a, which gives them a greenish color. Some species of cyanobacteria can produce toxins that can be harmful to humans and animals if ingested or inhaled. They are found in various aquatic environments such as freshwater lakes, ponds, and oceans, as well as in damp soil and on rocks. Cyanobacteria are important contributors to the Earth's oxygen-rich atmosphere and play a significant role in the global carbon cycle.
Serum Amyloid A (SAA) protein is an acute phase protein produced primarily in the liver, although it can also be produced by other cells in response to inflammation. It is a member of the apolipoprotein family and is found in high-density lipoproteins (HDL) in the blood. SAA protein levels increase rapidly during the acute phase response to infection, trauma, or tissue damage, making it a useful biomarker for inflammation.
In addition to its role as an acute phase protein, SAA has been implicated in several disease processes, including atherosclerosis and amyloidosis. In amyloidosis, SAA can form insoluble fibrils that deposit in various tissues, leading to organ dysfunction. There are four subtypes of SAA in humans (SAA1, SAA2, SAA3, and SAA4), with SAA1 and SAA2 being the most responsive to inflammatory stimuli.
I'm sorry for any confusion, but "horses" is not a medical term. It is the common name given to the species *Equus ferus caballus*, which are large domesticated mammals used for transportation, work, and recreation. If you have any questions about horses or a related topic that you would like a medical perspective on, please let me know and I'd be happy to help!
An emulsion is a type of stable mixture of two immiscible liquids, such as oil and water, which are normally unable to mix together uniformly. In an emulsion, one liquid (the dispersed phase) is broken down into small droplets and distributed throughout the other liquid (the continuous phase), creating a stable, cloudy mixture.
In medical terms, emulsions can be used in various pharmaceutical and cosmetic applications. For example, certain medications may be formulated as oil-in-water or water-in-oil emulsions to improve their absorption, stability, or palatability. Similarly, some skincare products and makeup removers contain emulsifiers that help create stable mixtures of water and oils, allowing for effective cleansing and moisturizing.
Emulsions can also occur naturally in the body, such as in the digestion of fats. The bile salts produced by the liver help to form small droplets of dietary lipids (oil) within the watery environment of the small intestine, allowing for efficient absorption and metabolism of these nutrients.
Lysophosphatidylcholines (LPCs) are a type of glycerophospholipids, which are major components of cell membranes. They are formed by the hydrolysis of phosphatidylcholines, another type of glycerophospholipids, catalyzed by the enzyme phospholipase A2. LPCs contain a single fatty acid chain attached to a glycerol backbone and a choline headgroup.
In medical terms, LPCs have been implicated in various physiological and pathological processes, such as cell signaling, membrane remodeling, and inflammation. Elevated levels of LPCs have been found in several diseases, including cardiovascular disease, neurodegenerative disorders, and cancer. They can also serve as biomarkers for the diagnosis and prognosis of these conditions.
Hypercholesterolemia is a medical term that describes a condition characterized by high levels of cholesterol in the blood. Specifically, it refers to an abnormally elevated level of low-density lipoprotein (LDL) cholesterol, also known as "bad" cholesterol, which can contribute to the development of fatty deposits in the arteries called plaques. Over time, these plaques can narrow and harden the arteries, leading to atherosclerosis, a condition that increases the risk of heart disease, stroke, and other cardiovascular complications.
Hypercholesterolemia can be caused by various factors, including genetics, lifestyle choices, and underlying medical conditions. In some cases, it may not cause any symptoms until serious complications arise. Therefore, regular cholesterol screening is essential for early detection and management of hypercholesterolemia. Treatment typically involves lifestyle modifications, such as a healthy diet, regular exercise, and weight management, along with medication if necessary.
Spectrum analysis in the context of Raman spectroscopy refers to the measurement and interpretation of the Raman scattering spectrum of a material or sample. Raman spectroscopy is a non-destructive analytical technique that uses the inelastic scattering of light to examine the vibrational modes of molecules.
When a monochromatic light source, typically a laser, illuminates a sample, a small fraction of the scattered light undergoes a shift in frequency due to interactions with the molecular vibrations of the sample. This shift in frequency is known as the Raman shift and is unique to each chemical bond or functional group within a molecule.
In a Raman spectrum, the intensity of the scattered light is plotted against the Raman shift, which is expressed in wavenumbers (cm-1). The resulting spectrum provides a "fingerprint" of the sample's molecular structure and composition, allowing for the identification and characterization of various chemical components within the sample.
Spectrum analysis in Raman spectroscopy can reveal valuable information about the sample's crystallinity, phase transitions, polymorphism, molecular orientation, and other properties. This technique is widely used across various fields, including materials science, chemistry, biology, pharmaceuticals, and forensics, to analyze a diverse range of samples, from simple liquids and solids to complex biological tissues and nanomaterials.
Cytochrome P-450 CYP3A is a subfamily of the cytochrome P-450 enzyme superfamily, which are primarily involved in drug metabolism in the human body. These enzymes are found predominantly in the liver, but also in other tissues such as the small intestine, kidneys, and brain.
CYP3A enzymes are responsible for metabolizing a wide variety of drugs, including many statins, benzodiazepines, antidepressants, and opioids. They can also metabolize endogenous compounds such as steroids and bile acids. The activity of CYP3A enzymes can be influenced by various factors, including genetic polymorphisms, age, sex, pregnancy, and the presence of other drugs or diseases.
The name "cytochrome P-450" refers to the fact that these enzymes contain a heme group that absorbs light at a wavelength of 450 nanometers when it is complexed with carbon monoxide. The term "CYP3A" denotes the specific subfamily of cytochrome P-450 enzymes that share a high degree of sequence similarity and function.
Tritium is not a medical term, but it is a term used in the field of nuclear physics and chemistry. Tritium (symbol: T or 3H) is a radioactive isotope of hydrogen with two neutrons and one proton in its nucleus. It is also known as heavy hydrogen or superheavy hydrogen.
Tritium has a half-life of about 12.3 years, which means that it decays by emitting a low-energy beta particle (an electron) to become helium-3. Due to its radioactive nature and relatively short half-life, tritium is used in various applications, including nuclear weapons, fusion reactors, luminous paints, and medical research.
In the context of medicine, tritium may be used as a radioactive tracer in some scientific studies or medical research, but it is not a term commonly used to describe a medical condition or treatment.
Aminoglycosides are a class of antibiotics that are derived from bacteria and are used to treat various types of infections caused by gram-negative and some gram-positive bacteria. These antibiotics work by binding to the 30S subunit of the bacterial ribosome, which inhibits protein synthesis and ultimately leads to bacterial cell death.
Some examples of aminoglycosides include gentamicin, tobramycin, neomycin, and streptomycin. These antibiotics are often used in combination with other antibiotics to treat severe infections, such as sepsis, pneumonia, and urinary tract infections.
Aminoglycosides can have serious side effects, including kidney damage and hearing loss, so they are typically reserved for use in serious infections that cannot be treated with other antibiotics. They are also used topically to treat skin infections and prevent wound infections after surgery.
It's important to note that aminoglycosides should only be used under the supervision of a healthcare professional, as improper use can lead to antibiotic resistance and further health complications.
In the context of medical and health sciences, particle size generally refers to the diameter or dimension of particles, which can be in the form of solid particles, droplets, or aerosols. These particles may include airborne pollutants, pharmaceutical drugs, or medical devices such as nanoparticles used in drug delivery systems.
Particle size is an important factor to consider in various medical applications because it can affect the behavior and interactions of particles with biological systems. For example, smaller particle sizes can lead to greater absorption and distribution throughout the body, while larger particle sizes may be filtered out by the body's natural defense mechanisms. Therefore, understanding particle size and its implications is crucial for optimizing the safety and efficacy of medical treatments and interventions.
Oxidoreductases are a class of enzymes that catalyze oxidation-reduction reactions, which involve the transfer of electrons from one molecule (the reductant) to another (the oxidant). These enzymes play a crucial role in various biological processes, including energy production, metabolism, and detoxification.
The oxidoreductase-catalyzed reaction typically involves the donation of electrons from a reducing agent (donor) to an oxidizing agent (acceptor), often through the transfer of hydrogen atoms or hydride ions. The enzyme itself does not undergo any permanent chemical change during this process, but rather acts as a catalyst to lower the activation energy required for the reaction to occur.
Oxidoreductases are classified and named based on the type of electron donor or acceptor involved in the reaction. For example, oxidoreductases that act on the CH-OH group of donors are called dehydrogenases, while those that act on the aldehyde or ketone groups are called oxidases. Other examples include reductases, peroxidases, and catalases.
Understanding the function and regulation of oxidoreductases is important for understanding various physiological processes and developing therapeutic strategies for diseases associated with impaired redox homeostasis, such as cancer, neurodegenerative disorders, and cardiovascular disease.
NADH, NADPH oxidoreductases are a class of enzymes that catalyze the redox reaction between NADH or NADPH and various electron acceptors. These enzymes play a crucial role in cellular metabolism by transferring electrons from NADH or NADPH to other molecules, which is essential for many biochemical reactions.
NADH (nicotinamide adenine dinucleotide hydrogen) and NADPH (nicotinamide adenine dinucleotide phosphate hydrogen) are coenzymes that act as electron carriers in redox reactions. They consist of a nicotinamide ring, which undergoes reduction or oxidation by accepting or donating electrons and a proton (H+).
NADH, NADPH oxidoreductases are classified based on their structure and mechanism of action. Some examples include:
1. Dehydrogenases: These enzymes catalyze the oxidation of NADH or NADPH to NAD+ or NADP+ while reducing an organic substrate. Examples include lactate dehydrogenase, alcohol dehydrogenase, and malate dehydrogenase.
2. Oxidases: These enzymes catalyze the oxidation of NADH or NADPH to NAD+ or NADP+ while reducing molecular oxygen (O2) to water (H2O). Examples include NADH oxidase and NADPH oxidase.
3. Reductases: These enzymes catalyze the reduction of various electron acceptors using NADH or NADPH as a source of electrons. Examples include glutathione reductase, thioredoxin reductase, and nitrate reductase.
Overall, NADH, NADPH oxidoreductases are essential for maintaining the redox balance in cells and play a critical role in various metabolic pathways, including energy production, detoxification, and biosynthesis.
Calorimetry is the measurement and study of heat transfer, typically using a device called a calorimeter. In the context of medicine and physiology, calorimetry can be used to measure heat production or dissipation in the body, which can provide insight into various bodily functions and metabolic processes.
There are different types of calorimeters used for medical research and clinical applications, including direct and indirect calorimeters. Direct calorimetry measures the heat produced directly by the body, while indirect calorimetry estimates heat production based on oxygen consumption and carbon dioxide production rates. Indirect calorimetry is more commonly used in clinical settings to assess energy expenditure and metabolic rate in patients with various medical conditions or during specific treatments, such as critical illness, surgery, or weight management programs.
In summary, calorimetry in a medical context refers to the measurement of heat exchange within the body or between the body and its environment, which can offer valuable information for understanding metabolic processes and developing personalized treatment plans.
IDL, or intermediate-density lipoproteins, are a type of lipoprotein that is denser than low-density lipoproteins (LDL) but less dense than high-density lipoproteins (HDL). They are formed during the catabolism (breakdown) of VLDL (very low-density lipoproteins), another type of lipoprotein, by lipoprotein lipase, an enzyme that breaks down triglycerides in lipoproteins.
IDLs contain a higher proportion of cholesterol and apolipoprotein E (apoE) compared to VLDLs and LDLs. Some IDLs are taken up by the liver, while others are converted into LDL particles through the action of cholesteryl ester transfer protein (CETP), which exchanges triglycerides in LDL for cholesterol esters in IDL.
Elevated levels of IDLs in the blood may be a risk factor for cardiovascular disease, as they can contribute to the formation and accumulation of plaque in the arteries. However, IDLs are not typically measured in routine clinical testing, and their role in disease is not as well understood as that of LDL or HDL.
Electron Transport Complex III, also known as cytochrome bc1 complex or ubiquinol-cytochrome c reductase, is a protein complex located in the inner mitochondrial membrane of eukaryotic cells and the cytoplasmic membrane of prokaryotic cells. It plays a crucial role in the electron transport chain (ETC), a series of complexes that generate energy in the form of ATP through a process called oxidative phosphorylation.
In ETC, Electron Transport Complex III accepts electrons from ubiquinol and transfers them to cytochrome c. This electron transfer is coupled with the translocation of protons (H+ ions) across the membrane, creating an electrochemical gradient. The energy stored in this gradient drives the synthesis of ATP by ATP synthase.
Electron Transport Complex III consists of several subunits, including cytochrome b, cytochrome c1, and the Rieske iron-sulfur protein. These subunits work together to facilitate the electron transfer and proton translocation processes.
Microsomes, liver refers to a subcellular fraction of liver cells (hepatocytes) that are obtained during tissue homogenization and subsequent centrifugation. These microsomal fractions are rich in membranous structures known as the endoplasmic reticulum (ER), particularly the rough ER. They are involved in various important cellular processes, most notably the metabolism of xenobiotics (foreign substances) including drugs, toxins, and carcinogens.
The liver microsomes contain a variety of enzymes, such as cytochrome P450 monooxygenases, that are crucial for phase I drug metabolism. These enzymes help in the oxidation, reduction, or hydrolysis of xenobiotics, making them more water-soluble and facilitating their excretion from the body. Additionally, liver microsomes also host other enzymes involved in phase II conjugation reactions, where the metabolites from phase I are further modified by adding polar molecules like glucuronic acid, sulfate, or acetyl groups.
In summary, liver microsomes are a subcellular fraction of liver cells that play a significant role in the metabolism and detoxification of xenobiotics, contributing to the overall protection and maintenance of cellular homeostasis within the body.
A cell membrane, also known as the plasma membrane, is a thin semi-permeable phospholipid bilayer that surrounds all cells in animals, plants, and microorganisms. It functions as a barrier to control the movement of substances in and out of the cell, allowing necessary molecules such as nutrients, oxygen, and signaling molecules to enter while keeping out harmful substances and waste products. The cell membrane is composed mainly of phospholipids, which have hydrophilic (water-loving) heads and hydrophobic (water-fearing) tails. This unique structure allows the membrane to be flexible and fluid, yet selectively permeable. Additionally, various proteins are embedded in the membrane that serve as channels, pumps, receptors, and enzymes, contributing to the cell's overall functionality and communication with its environment.
Hypophysectomy is a surgical procedure that involves the removal or partial removal of the pituitary gland, also known as the hypophysis. The pituitary gland is a small endocrine gland located at the base of the brain, just above the nasal cavity, and is responsible for producing and secreting several important hormones that regulate various bodily functions.
Hypophysectomy may be performed for therapeutic or diagnostic purposes. In some cases, it may be used to treat pituitary tumors or other conditions that affect the function of the pituitary gland. It may also be performed as a research procedure in animal models to study the effects of pituitary hormone deficiency on various physiological processes.
The surgical approach for hypophysectomy may vary depending on the specific indication and the patient's individual anatomy. In general, however, the procedure involves making an incision in the skull and exposing the pituitary gland through a small opening in the bone. The gland is then carefully dissected and removed or partially removed as necessary.
Potential complications of hypophysectomy include damage to surrounding structures such as the optic nerves, which can lead to vision loss, and cerebrospinal fluid leaks. Additionally, removal of the pituitary gland can result in hormonal imbalances that may require long-term management with hormone replacement therapy.
Lysine is an essential amino acid, which means that it cannot be synthesized by the human body and must be obtained through the diet. Its chemical formula is (2S)-2,6-diaminohexanoic acid. Lysine is necessary for the growth and maintenance of tissues in the body, and it plays a crucial role in the production of enzymes, hormones, and antibodies. It is also essential for the absorption of calcium and the formation of collagen, which is an important component of bones and connective tissue. Foods that are good sources of lysine include meat, poultry, fish, eggs, and dairy products.
I'm sorry for any confusion, but "thermodynamics" is not a term that has a specific medical definition. It is a branch of physics that deals with the relationships between heat and other forms of energy. However, the principles of thermodynamics can be applied to biological systems, including those in the human body, such as in the study of metabolism or muscle function. But in a medical context, "thermodynamics" would not be a term used independently as a diagnosis, treatment, or any medical condition.
Sepharose is not a medical term itself, but it is a trade name for a type of gel that is often used in medical and laboratory settings. Sepharose is a type of cross-linked agarose gel, which is derived from seaweed. It is commonly used in chromatography, a technique used to separate and purify different components of a mixture based on their physical or chemical properties.
Sepharose gels are available in various forms, including beads and sheets, and they come in different sizes and degrees of cross-linking. These variations allow for the separation and purification of molecules with different sizes, charges, and other properties. Sepharose is known for its high porosity, mechanical stability, and low non-specific binding, making it a popular choice for many laboratory applications.
Sequence homology, amino acid, refers to the similarity in the order of amino acids in a protein or a portion of a protein between two or more species. This similarity can be used to infer evolutionary relationships and functional similarities between proteins. The higher the degree of sequence homology, the more likely it is that the proteins are related and have similar functions. Sequence homology can be determined through various methods such as pairwise alignment or multiple sequence alignment, which compare the sequences and calculate a score based on the number and type of matching amino acids.
Methionine is an essential amino acid, which means that it cannot be synthesized by the human body and must be obtained through the diet. It plays a crucial role in various biological processes, including:
1. Protein synthesis: Methionine is one of the building blocks of proteins, helping to create new proteins and maintain the structure and function of cells.
2. Methylation: Methionine serves as a methyl group donor in various biochemical reactions, which are essential for DNA synthesis, gene regulation, and neurotransmitter production.
3. Antioxidant defense: Methionine can be converted to cysteine, which is involved in the formation of glutathione, a potent antioxidant that helps protect cells from oxidative damage.
4. Homocysteine metabolism: Methionine is involved in the conversion of homocysteine back to methionine through a process called remethylation, which is essential for maintaining normal homocysteine levels and preventing cardiovascular disease.
5. Fat metabolism: Methionine helps facilitate the breakdown and metabolism of fats in the body.
Foods rich in methionine include meat, fish, dairy products, eggs, and some nuts and seeds.
An atherogenic diet is a type of eating pattern that can contribute to the development and progression of atherosclerosis, which is the hardening and narrowing of the arteries due to the buildup of fats, cholesterol, and other substances in the inner lining of the artery walls.
An atherogenic diet is typically high in saturated and trans fats, cholesterol, refined carbohydrates, and salt, and low in fiber, fruits, vegetables, and unsaturated fats. This type of diet can increase the levels of LDL (low-density lipoprotein) or "bad" cholesterol in the blood, which can lead to the formation of plaques in the arteries and increase the risk of cardiovascular disease, including heart attack and stroke.
Therefore, it is recommended to follow a heart-healthy diet that emphasizes fruits, vegetables, whole grains, lean proteins, and healthy fats to reduce the risk of atherosclerosis and other chronic diseases.
In the field of medicine, "time factors" refer to the duration of symptoms or time elapsed since the onset of a medical condition, which can have significant implications for diagnosis and treatment. Understanding time factors is crucial in determining the progression of a disease, evaluating the effectiveness of treatments, and making critical decisions regarding patient care.
For example, in stroke management, "time is brain," meaning that rapid intervention within a specific time frame (usually within 4.5 hours) is essential to administering tissue plasminogen activator (tPA), a clot-busting drug that can minimize brain damage and improve patient outcomes. Similarly, in trauma care, the "golden hour" concept emphasizes the importance of providing definitive care within the first 60 minutes after injury to increase survival rates and reduce morbidity.
Time factors also play a role in monitoring the progression of chronic conditions like diabetes or heart disease, where regular follow-ups and assessments help determine appropriate treatment adjustments and prevent complications. In infectious diseases, time factors are crucial for initiating antibiotic therapy and identifying potential outbreaks to control their spread.
Overall, "time factors" encompass the significance of recognizing and acting promptly in various medical scenarios to optimize patient outcomes and provide effective care.
Solubility is a fundamental concept in pharmaceutical sciences and medicine, which refers to the maximum amount of a substance (solute) that can be dissolved in a given quantity of solvent (usually water) at a specific temperature and pressure. Solubility is typically expressed as mass of solute per volume or mass of solvent (e.g., grams per liter, milligrams per milliliter). The process of dissolving a solute in a solvent results in a homogeneous solution where the solute particles are dispersed uniformly throughout the solvent.
Understanding the solubility of drugs is crucial for their formulation, administration, and therapeutic effectiveness. Drugs with low solubility may not dissolve sufficiently to produce the desired pharmacological effect, while those with high solubility might lead to rapid absorption and short duration of action. Therefore, optimizing drug solubility through various techniques like particle size reduction, salt formation, or solubilization is an essential aspect of drug development and delivery.
Fourier Transform Infrared (FTIR) spectroscopy is a type of infrared spectroscopy that uses the Fourier transform mathematical technique to convert the raw data obtained from an interferometer into a more interpretable spectrum. This technique allows for the simultaneous collection of a wide range of wavelengths, resulting in increased sensitivity and speed compared to traditional dispersive infrared spectroscopy.
FTIR spectroscopy measures the absorption or transmission of infrared radiation by a sample as a function of frequency, providing information about the vibrational modes of the molecules present in the sample. This can be used for identification and quantification of chemical compounds, analysis of molecular structure, and investigation of chemical interactions and reactions.
In summary, FTIR spectroscopy is a powerful analytical technique that uses infrared radiation to study the vibrational properties of molecules, with increased sensitivity and speed due to the use of Fourier transform mathematical techniques and an interferometer.
Retinol-binding proteins (RBPs) are a group of proteins found in the body that play a crucial role in transporting and delivering retinol (vitamin A alcohol) to various tissues and cells. RBPs are synthesized primarily in the liver and then secreted into the bloodstream, where they bind to retinol and form a complex called holo-RBP.
Cellular RBPs, also known as intracellular RBPs or CRBPs (cellular retinol-binding proteins), are a subclass of RBPs that function inside cells. They are responsible for transporting retinol within the cell and facilitating its conversion to retinal and then to retinoic acid, which are active forms of vitamin A involved in various physiological processes such as vision, immune function, and embryonic development.
CRBPs have a high affinity for retinol and help regulate its intracellular concentration by preventing its degradation and promoting its uptake into the cell. There are several isoforms of CRBPs, including CRBP-I, CRBP-II, CRBP-III, and CRBP-IV, each with distinct expression patterns and functions in different tissues and cells.
Overall, CRBPs play a critical role in maintaining the homeostasis of vitamin A metabolism and ensuring its proper utilization in various physiological processes.
Proteins are complex, large molecules that play critical roles in the body's functions. They are made up of amino acids, which are organic compounds that are the building blocks of proteins. Proteins are required for the structure, function, and regulation of the body's tissues and organs. They are essential for the growth, repair, and maintenance of body tissues, and they play a crucial role in many biological processes, including metabolism, immune response, and cellular signaling. Proteins can be classified into different types based on their structure and function, such as enzymes, hormones, antibodies, and structural proteins. They are found in various foods, especially animal-derived products like meat, dairy, and eggs, as well as plant-based sources like beans, nuts, and grains.
Heparin is defined as a highly sulfated glycosaminoglycan (a type of polysaccharide) that is widely present in many tissues, but is most commonly derived from the mucosal tissues of mammalian lungs or intestinal mucosa. It is an anticoagulant that acts as an inhibitor of several enzymes involved in the blood coagulation cascade, primarily by activating antithrombin III which then neutralizes thrombin and other clotting factors.
Heparin is used medically to prevent and treat thromboembolic disorders such as deep vein thrombosis, pulmonary embolism, and certain types of heart attacks. It can also be used during hemodialysis, cardiac bypass surgery, and other medical procedures to prevent the formation of blood clots.
It's important to note that while heparin is a powerful anticoagulant, it does not have any fibrinolytic activity, meaning it cannot dissolve existing blood clots. Instead, it prevents new clots from forming and stops existing clots from growing larger.
Lipid metabolism is the process by which the body breaks down and utilizes lipids (fats) for various functions, such as energy production, cell membrane formation, and hormone synthesis. This complex process involves several enzymes and pathways that regulate the digestion, absorption, transport, storage, and consumption of fats in the body.
The main types of lipids involved in metabolism include triglycerides, cholesterol, phospholipids, and fatty acids. The breakdown of these lipids begins in the digestive system, where enzymes called lipases break down dietary fats into smaller molecules called fatty acids and glycerol. These molecules are then absorbed into the bloodstream and transported to the liver, which is the main site of lipid metabolism.
In the liver, fatty acids may be further broken down for energy production or used to synthesize new lipids. Excess fatty acids may be stored as triglycerides in specialized cells called adipocytes (fat cells) for later use. Cholesterol is also metabolized in the liver, where it may be used to synthesize bile acids, steroid hormones, and other important molecules.
Disorders of lipid metabolism can lead to a range of health problems, including obesity, diabetes, cardiovascular disease, and non-alcoholic fatty liver disease (NAFLD). These conditions may be caused by genetic factors, lifestyle habits, or a combination of both. Proper diagnosis and management of lipid metabolism disorders typically involves a combination of dietary changes, exercise, and medication.
Monoclonal antibodies are a type of antibody that are identical because they are produced by a single clone of cells. They are laboratory-produced molecules that act like human antibodies in the immune system. They can be designed to attach to specific proteins found on the surface of cancer cells, making them useful for targeting and treating cancer. Monoclonal antibodies can also be used as a therapy for other diseases, such as autoimmune disorders and inflammatory conditions.
Monoclonal antibodies are produced by fusing a single type of immune cell, called a B cell, with a tumor cell to create a hybrid cell, or hybridoma. This hybrid cell is then able to replicate indefinitely, producing a large number of identical copies of the original antibody. These antibodies can be further modified and engineered to enhance their ability to bind to specific targets, increase their stability, and improve their effectiveness as therapeutic agents.
Monoclonal antibodies have several mechanisms of action in cancer therapy. They can directly kill cancer cells by binding to them and triggering an immune response. They can also block the signals that promote cancer growth and survival. Additionally, monoclonal antibodies can be used to deliver drugs or radiation directly to cancer cells, increasing the effectiveness of these treatments while minimizing their side effects on healthy tissues.
Monoclonal antibodies have become an important tool in modern medicine, with several approved for use in cancer therapy and other diseases. They are continuing to be studied and developed as a promising approach to treating a wide range of medical conditions.
Cyanogen bromide is a solid compound with the chemical formula (CN)Br. It is a highly reactive and toxic substance that is used in research and industrial settings for various purposes, such as the production of certain types of resins and gels. Cyanogen bromide is an alkyl halide, which means it contains a bromine atom bonded to a carbon atom that is also bonded to a cyano group (a nitrogen atom bonded to a carbon atom with a triple bond).
Cyanogen bromide is classified as a class B poison, which means it can cause harm or death if swallowed, inhaled, or absorbed through the skin. It can cause irritation and burns to the eyes, skin, and respiratory tract, and prolonged exposure can lead to more serious health effects, such as damage to the nervous system and kidneys. Therefore, it is important to handle cyanogen bromide with care and to use appropriate safety precautions when working with it.
Palmitic acid is a type of saturated fatty acid, which is a common component in many foods and also produced by the body. Its chemical formula is C16:0, indicating that it contains 16 carbon atoms and no double bonds. Palmitic acid is found in high concentrations in animal fats, such as butter, lard, and beef tallow, as well as in some vegetable oils, like palm kernel oil and coconut oil.
In the human body, palmitic acid can be synthesized from other substances or absorbed through the diet. It plays a crucial role in various biological processes, including energy storage, membrane structure formation, and signaling pathways regulation. However, high intake of palmitic acid has been linked to an increased risk of developing cardiovascular diseases due to its potential to raise low-density lipoprotein (LDL) cholesterol levels in the blood.
It is essential to maintain a balanced diet and consume palmitic acid-rich foods in moderation, along with regular exercise and a healthy lifestyle, to reduce the risk of chronic diseases.
Blood proteins, also known as serum proteins, are a group of complex molecules present in the blood that are essential for various physiological functions. These proteins include albumin, globulins (alpha, beta, and gamma), and fibrinogen. They play crucial roles in maintaining oncotic pressure, transporting hormones, enzymes, vitamins, and minerals, providing immune defense, and contributing to blood clotting.
Albumin is the most abundant protein in the blood, accounting for about 60% of the total protein mass. It functions as a transporter of various substances, such as hormones, fatty acids, and drugs, and helps maintain oncotic pressure, which is essential for fluid balance between the blood vessels and surrounding tissues.
Globulins are divided into three main categories: alpha, beta, and gamma globulins. Alpha and beta globulins consist of transport proteins like lipoproteins, hormone-binding proteins, and enzymes. Gamma globulins, also known as immunoglobulins or antibodies, are essential for the immune system's defense against pathogens.
Fibrinogen is a protein involved in blood clotting. When an injury occurs, fibrinogen is converted into fibrin, which forms a mesh to trap platelets and form a clot, preventing excessive bleeding.
Abnormal levels of these proteins can indicate various medical conditions, such as liver or kidney disease, malnutrition, infections, inflammation, or autoimmune disorders. Blood protein levels are typically measured through laboratory tests like serum protein electrophoresis (SPE) and immunoelectrophoresis (IEP).
Cell surface receptors, also known as membrane receptors, are proteins located on the cell membrane that bind to specific molecules outside the cell, known as ligands. These receptors play a crucial role in signal transduction, which is the process of converting an extracellular signal into an intracellular response.
Cell surface receptors can be classified into several categories based on their structure and mechanism of action, including:
1. Ion channel receptors: These receptors contain a pore that opens to allow ions to flow across the cell membrane when they bind to their ligands. This ion flux can directly activate or inhibit various cellular processes.
2. G protein-coupled receptors (GPCRs): These receptors consist of seven transmembrane domains and are associated with heterotrimeric G proteins that modulate intracellular signaling pathways upon ligand binding.
3. Enzyme-linked receptors: These receptors possess an intrinsic enzymatic activity or are linked to an enzyme, which becomes activated when the receptor binds to its ligand. This activation can lead to the initiation of various signaling cascades within the cell.
4. Receptor tyrosine kinases (RTKs): These receptors contain intracellular tyrosine kinase domains that become activated upon ligand binding, leading to the phosphorylation and activation of downstream signaling molecules.
5. Integrins: These receptors are transmembrane proteins that mediate cell-cell or cell-matrix interactions by binding to extracellular matrix proteins or counter-receptors on adjacent cells. They play essential roles in cell adhesion, migration, and survival.
Cell surface receptors are involved in various physiological processes, including neurotransmission, hormone signaling, immune response, and cell growth and differentiation. Dysregulation of these receptors can contribute to the development of numerous diseases, such as cancer, diabetes, and neurological disorders.
I'm sorry for any confusion, but "Molybdenum" is not a medical term. It is an element with the symbol Mo and atomic number 42 on the periodic table. Molybdenum is used in various industries, including medicine, for example in the production of surgical instruments and some prosthetics due to its strength and resistance to corrosion. However, it is not a term used to describe a medical condition or bodily process. If you have any questions related to elements and their uses in medicine, I'd be happy to help with those!
The peritoneal cavity is the potential space within the abdominal and pelvic regions, bounded by the parietal peritoneum lining the inner aspect of the abdominal and pelvic walls, and the visceral peritoneum covering the abdominal and pelvic organs. It contains a small amount of serous fluid that allows for the gliding of organs against each other during normal physiological activities such as digestion and movement. This cavity can become pathologically involved in various conditions, including inflammation, infection, hemorrhage, or neoplasia, leading to symptoms like abdominal pain, distention, or tenderness.
A phenotype is the physical or biochemical expression of an organism's genes, or the observable traits and characteristics resulting from the interaction of its genetic constitution (genotype) with environmental factors. These characteristics can include appearance, development, behavior, and resistance to disease, among others. Phenotypes can vary widely, even among individuals with identical genotypes, due to differences in environmental influences, gene expression, and genetic interactions.
Post-translational protein processing refers to the modifications and changes that proteins undergo after their synthesis on ribosomes, which are complex molecular machines responsible for protein synthesis. These modifications occur through various biochemical processes and play a crucial role in determining the final structure, function, and stability of the protein.
The process begins with the translation of messenger RNA (mRNA) into a linear polypeptide chain, which is then subjected to several post-translational modifications. These modifications can include:
1. Proteolytic cleavage: The removal of specific segments or domains from the polypeptide chain by proteases, resulting in the formation of mature, functional protein subunits.
2. Chemical modifications: Addition or modification of chemical groups to the side chains of amino acids, such as phosphorylation (addition of a phosphate group), glycosylation (addition of sugar moieties), methylation (addition of a methyl group), acetylation (addition of an acetyl group), and ubiquitination (addition of a ubiquitin protein).
3. Disulfide bond formation: The oxidation of specific cysteine residues within the polypeptide chain, leading to the formation of disulfide bonds between them. This process helps stabilize the three-dimensional structure of proteins, particularly in extracellular environments.
4. Folding and assembly: The acquisition of a specific three-dimensional conformation by the polypeptide chain, which is essential for its function. Chaperone proteins assist in this process to ensure proper folding and prevent aggregation.
5. Protein targeting: The directed transport of proteins to their appropriate cellular locations, such as the nucleus, mitochondria, endoplasmic reticulum, or plasma membrane. This is often facilitated by specific signal sequences within the protein that are recognized and bound by transport machinery.
Collectively, these post-translational modifications contribute to the functional diversity of proteins in living organisms, allowing them to perform a wide range of cellular processes, including signaling, catalysis, regulation, and structural support.
Protein biosynthesis is the process by which cells generate new proteins. It involves two major steps: transcription and translation. Transcription is the process of creating a complementary RNA copy of a sequence of DNA. This RNA copy, or messenger RNA (mRNA), carries the genetic information to the site of protein synthesis, the ribosome. During translation, the mRNA is read by transfer RNA (tRNA) molecules, which bring specific amino acids to the ribosome based on the sequence of nucleotides in the mRNA. The ribosome then links these amino acids together in the correct order to form a polypeptide chain, which may then fold into a functional protein. Protein biosynthesis is essential for the growth and maintenance of all living organisms.
Hyperlipoproteinemia Type II, also known as Fredrickson Type II or Familial Combined Hyperlipidemia, is a genetic disorder characterized by elevated levels of low-density lipoprotein (LDL) cholesterol and/or triglycerides in the blood. This condition can lead to an increased risk of developing cardiovascular diseases such as atherosclerosis and coronary artery disease.
The disorder is caused by mutations in several genes involved in lipid metabolism, including APOB, LDLR, PCSK9, and APOE. These genetic defects result in impaired clearance of LDL particles from the bloodstream, leading to their accumulation and increased risk of cardiovascular disease.
Individuals with Hyperlipoproteinemia Type II typically have elevated levels of both LDL cholesterol and triglycerides, although some may only have one or the other elevated. The disorder can present at any age, but it is often diagnosed in adulthood during routine cholesterol screening.
Treatment for Hyperlipoproteinemia Type II typically involves lifestyle modifications such as a heart-healthy diet, regular exercise, and weight loss. Medications such as statins, ezetimibe, and PCSK9 inhibitors may also be prescribed to lower LDL cholesterol levels and reduce the risk of cardiovascular disease.
Enzyme induction is a process by which the activity or expression of an enzyme is increased in response to some stimulus, such as a drug, hormone, or other environmental factor. This can occur through several mechanisms, including increasing the transcription of the enzyme's gene, stabilizing the mRNA that encodes the enzyme, or increasing the translation of the mRNA into protein.
In some cases, enzyme induction can be a beneficial process, such as when it helps the body to metabolize and clear drugs more quickly. However, in other cases, enzyme induction can have negative consequences, such as when it leads to the increased metabolism of important endogenous compounds or the activation of harmful procarcinogens.
Enzyme induction is an important concept in pharmacology and toxicology, as it can affect the efficacy and safety of drugs and other xenobiotics. It is also relevant to the study of drug interactions, as the induction of one enzyme by a drug can lead to altered metabolism and effects of another drug that is metabolized by the same enzyme.
I'm sorry for any confusion, but "photochemistry" is not a term typically used in medical definitions. Photochemistry is a branch of chemistry that deals with the chemical effects of light. It involves the absorption of light by a substance, which can lead to the promotion of an electron to a higher energy state, and subsequently result in various chemical reactions.
In a medical context, photochemical processes might be discussed in relation to certain therapies or diagnostic techniques, such as photodynamic therapy for cancer treatment, where a photosensitizing agent is used that reacts with light to produce singlet oxygen or other reactive species to destroy nearby cells. However, it's not a term used to define a specific medical condition or concept in the same way that one might define "inflammation" or "metabolism."
Cytochromes c are a group of small heme proteins found in the mitochondria of cells, involved in the electron transport chain and play a crucial role in cellular respiration. They accept and donate electrons during the process of oxidative phosphorylation, which generates ATP, the main energy currency of the cell. Cytochromes c contain a heme group, an organic compound that includes iron, which facilitates the transfer of electrons. The "c" in cytochromes c refers to the type of heme group they contain (cyt c has heme c). They are highly conserved across species and have been widely used as a molecular marker for evolutionary studies.
A lung is a pair of spongy, elastic organs in the chest that work together to enable breathing. They are responsible for taking in oxygen and expelling carbon dioxide through the process of respiration. The left lung has two lobes, while the right lung has three lobes. The lungs are protected by the ribcage and are covered by a double-layered membrane called the pleura. The trachea divides into two bronchi, which further divide into smaller bronchioles, leading to millions of tiny air sacs called alveoli, where the exchange of gases occurs.
Fatty acids are carboxylic acids with a long aliphatic chain, which are important components of lipids and are widely distributed in living organisms. They can be classified based on the length of their carbon chain, saturation level (presence or absence of double bonds), and other structural features.
The two main types of fatty acids are:
1. Saturated fatty acids: These have no double bonds in their carbon chain and are typically solid at room temperature. Examples include palmitic acid (C16:0) and stearic acid (C18:0).
2. Unsaturated fatty acids: These contain one or more double bonds in their carbon chain and can be further classified into monounsaturated (one double bond) and polyunsaturated (two or more double bonds) fatty acids. Examples of unsaturated fatty acids include oleic acid (C18:1, monounsaturated), linoleic acid (C18:2, polyunsaturated), and alpha-linolenic acid (C18:3, polyunsaturated).
Fatty acids play crucial roles in various biological processes, such as energy storage, membrane structure, and cell signaling. Some essential fatty acids cannot be synthesized by the human body and must be obtained through dietary sources.
"Saccharomyces cerevisiae" is not typically considered a medical term, but it is a scientific name used in the field of microbiology. It refers to a species of yeast that is commonly used in various industrial processes, such as baking and brewing. It's also widely used in scientific research due to its genetic tractability and eukaryotic cellular organization.
However, it does have some relevance to medical fields like medicine and nutrition. For example, certain strains of S. cerevisiae are used as probiotics, which can provide health benefits when consumed. They may help support gut health, enhance the immune system, and even assist in the digestion of certain nutrients.
In summary, "Saccharomyces cerevisiae" is a species of yeast with various industrial and potential medical applications.
"Swine" is a common term used to refer to even-toed ungulates of the family Suidae, including domestic pigs and wild boars. However, in a medical context, "swine" often appears in the phrase "swine flu," which is a strain of influenza virus that typically infects pigs but can also cause illness in humans. The 2009 H1N1 pandemic was caused by a new strain of swine-origin influenza A virus, which was commonly referred to as "swine flu." It's important to note that this virus is not transmitted through eating cooked pork products; it spreads from person to person, mainly through respiratory droplets produced when an infected person coughs or sneezes.
Metallothioneins (MTs) are a group of small, cysteine-rich, metal-binding proteins found in the cells of many organisms, including humans. They play important roles in various biological processes such as:
1. Metal homeostasis and detoxification: MTs can bind to various heavy metals like zinc, copper, cadmium, and mercury with high affinity. This binding helps regulate the concentration of these metals within cells and protects against metal toxicity.
2. Oxidative stress protection: Due to their high cysteine content, MTs act as antioxidants by scavenging reactive oxygen species (ROS) and free radicals, thus protecting cells from oxidative damage.
3. Immune response regulation: MTs are involved in the modulation of immune cell function and inflammatory responses. They can influence the activation and proliferation of immune cells, as well as the production of cytokines and chemokines.
4. Development and differentiation: MTs have been implicated in cell growth, differentiation, and embryonic development, particularly in tissues with high rates of metal turnover, such as the liver and kidneys.
5. Neuroprotection: In the brain, MTs play a role in protecting neurons from oxidative stress, excitotoxicity, and heavy metal toxicity. They have been implicated in various neurodegenerative disorders, including Alzheimer's and Parkinson's diseases.
There are four main isoforms of metallothioneins (MT-1, MT-2, MT-3, and MT-4) in humans, each with distinct tissue expression patterns and functions.
Nitrate reductases are a group of enzymes that catalyze the reduction of nitrate (NO3-) to nitrite (NO2-). This process is an essential part of the nitrogen cycle, where nitrate serves as a terminal electron acceptor in anaerobic respiration for many bacteria and archaea. In plants, this enzyme plays a crucial role in nitrogen assimilation by reducing nitrate to ammonium (NH4+), which can then be incorporated into organic compounds. Nitrate reductases require various cofactors, such as molybdenum, heme, and/or FAD, for their activity. There are three main types of nitrate reductases: membrane-bound (which use menaquinol as an electron donor), cytoplasmic (which use NADH or NADPH as an electron donor), and assimilatory (which also use NADH or NADPH as an electron donor).
Cytochrome P-450 CYP1A1 is an enzyme that is part of the cytochrome P450 family, which are a group of enzymes involved in the metabolism of drugs and other xenobiotics (foreign substances) in the body. Specifically, CYP1A1 is found primarily in the liver and lungs and plays a role in the metabolism of polycyclic aromatic hydrocarbons (PAHs), which are chemicals found in tobacco smoke and are produced by the burning of fossil fuels and other organic materials.
CYP1A1 also has the ability to activate certain procarcinogens, which are substances that can be converted into cancer-causing agents (carcinogens) within the body. Therefore, variations in the CYP1A1 gene may influence an individual's susceptibility to cancer and other diseases.
The term "P-450" refers to the fact that these enzymes absorb light at a wavelength of 450 nanometers when they are combined with carbon monoxide, giving them a characteristic pink color. The "CYP" stands for "cytochrome P," and the number and letter designations (e.g., 1A1) indicate the specific enzyme within the family.
Histidine is an essential amino acid, meaning it cannot be synthesized by the human body and must be obtained through dietary sources. Its chemical formula is C6H9N3O2. Histidine plays a crucial role in several physiological processes, including:
1. Protein synthesis: As an essential amino acid, histidine is required for the production of proteins, which are vital components of various tissues and organs in the body.
2. Hemoglobin synthesis: Histidine is a key component of hemoglobin, the protein in red blood cells responsible for carrying oxygen throughout the body. The imidazole side chain of histidine acts as a proton acceptor/donor, facilitating the release and uptake of oxygen by hemoglobin.
3. Acid-base balance: Histidine is involved in maintaining acid-base homeostasis through its role in the biosynthesis of histamine, which is a critical mediator of inflammatory responses and allergies. The decarboxylation of histidine results in the formation of histamine, which can increase vascular permeability and modulate immune responses.
4. Metal ion binding: Histidine has a high affinity for metal ions such as zinc, copper, and iron. This property allows histidine to participate in various enzymatic reactions and maintain the structural integrity of proteins.
5. Antioxidant defense: Histidine-containing dipeptides, like carnosine and anserine, have been shown to exhibit antioxidant properties by scavenging reactive oxygen species (ROS) and chelating metal ions. These compounds may contribute to the protection of proteins and DNA from oxidative damage.
Dietary sources of histidine include meat, poultry, fish, dairy products, and wheat germ. Histidine deficiency is rare but can lead to growth retardation, anemia, and impaired immune function.
Species specificity is a term used in the field of biology, including medicine, to refer to the characteristic of a biological entity (such as a virus, bacterium, or other microorganism) that allows it to interact exclusively or preferentially with a particular species. This means that the biological entity has a strong affinity for, or is only able to infect, a specific host species.
For example, HIV is specifically adapted to infect human cells and does not typically infect other animal species. Similarly, some bacterial toxins are species-specific and can only affect certain types of animals or humans. This concept is important in understanding the transmission dynamics and host range of various pathogens, as well as in developing targeted therapies and vaccines.
Isomerism is a term used in chemistry and biochemistry, including the field of medicine, to describe the existence of molecules that have the same molecular formula but different structural formulas. This means that although these isomers contain the same number and type of atoms, they differ in the arrangement of these atoms in space.
There are several types of isomerism, including constitutional isomerism (also known as structural isomerism) and stereoisomerism. Constitutional isomers have different arrangements of atoms, while stereoisomers have the same arrangement of atoms but differ in the spatial arrangement of their atoms in three-dimensional space.
Stereoisomerism can be further divided into subcategories such as enantiomers (mirror-image stereoisomers), diastereomers (non-mirror-image stereoisomers), and conformational isomers (stereoisomers that can interconvert by rotating around single bonds).
In the context of medicine, isomerism can be important because different isomers of a drug may have different pharmacological properties. For example, some drugs may exist as pairs of enantiomers, and one enantiomer may be responsible for the desired therapeutic effect while the other enantiomer may be inactive or even harmful. In such cases, it may be important to develop methods for producing pure enantiomers of the drug in order to maximize its efficacy and minimize its side effects.
Carbohydrates are a major nutrient class consisting of organic compounds that primarily contain carbon, hydrogen, and oxygen atoms. They are classified as saccharides, which include monosaccharides (simple sugars), disaccharides (double sugars), oligosaccharides (short-chain sugars), and polysaccharides (complex carbohydrates).
Monosaccharides, such as glucose, fructose, and galactose, are the simplest form of carbohydrates. They consist of a single sugar molecule that cannot be broken down further by hydrolysis. Disaccharides, like sucrose (table sugar), lactose (milk sugar), and maltose (malt sugar), are formed from two monosaccharide units joined together.
Oligosaccharides contain a small number of monosaccharide units, typically less than 20, while polysaccharides consist of long chains of hundreds to thousands of monosaccharide units. Polysaccharides can be further classified into starch (found in plants), glycogen (found in animals), and non-starchy polysaccharides like cellulose, chitin, and pectin.
Carbohydrates play a crucial role in providing energy to the body, with glucose being the primary source of energy for most cells. They also serve as structural components in plants (cellulose) and animals (chitin), participate in various metabolic processes, and contribute to the taste, texture, and preservation of foods.
A ligand, in the context of biochemistry and medicine, is a molecule that binds to a specific site on a protein or a larger biomolecule, such as an enzyme or a receptor. This binding interaction can modify the function or activity of the target protein, either activating it or inhibiting it. Ligands can be small molecules, like hormones or neurotransmitters, or larger structures, like antibodies. The study of ligand-protein interactions is crucial for understanding cellular processes and developing drugs, as many therapeutic compounds function by binding to specific targets within the body.
X-ray diffraction (XRD) is not strictly a medical definition, but it is a technique commonly used in the field of medical research and diagnostics. XRD is a form of analytical spectroscopy that uses the phenomenon of X-ray diffraction to investigate the crystallographic structure of materials. When a beam of X-rays strikes a crystal, it is scattered in specific directions and with specific intensities that are determined by the arrangement of atoms within the crystal. By measuring these diffraction patterns, researchers can determine the crystal structures of various materials, including biological macromolecules such as proteins and viruses.
In the medical field, XRD is often used to study the structure of drugs and drug candidates, as well as to analyze the composition and structure of tissues and other biological samples. For example, XRD can be used to investigate the crystal structures of calcium phosphate minerals in bone tissue, which can provide insights into the mechanisms of bone formation and disease. Additionally, XRD is sometimes used in the development of new medical imaging techniques, such as phase-contrast X-ray imaging, which has the potential to improve the resolution and contrast of traditional X-ray images.
A cell-free system is a biochemical environment in which biological reactions can occur outside of an intact living cell. These systems are often used to study specific cellular processes or pathways, as they allow researchers to control and manipulate the conditions in which the reactions take place. In a cell-free system, the necessary enzymes, substrates, and cofactors for a particular reaction are provided in a test tube or other container, rather than within a whole cell.
Cell-free systems can be derived from various sources, including bacteria, yeast, and mammalian cells. They can be used to study a wide range of cellular processes, such as transcription, translation, protein folding, and metabolism. For example, a cell-free system might be used to express and purify a specific protein, or to investigate the regulation of a particular metabolic pathway.
One advantage of using cell-free systems is that they can provide valuable insights into the mechanisms of cellular processes without the need for time-consuming and resource-intensive cell culture or genetic manipulation. Additionally, because cell-free systems are not constrained by the limitations of a whole cell, they offer greater flexibility in terms of reaction conditions and the ability to study complex or transient interactions between biological molecules.
Overall, cell-free systems are an important tool in molecular biology and biochemistry, providing researchers with a versatile and powerful means of investigating the fundamental processes that underlie life at the cellular level.
"Pyrroles" is not a medical term in and of itself, but "pyrrole" is an organic compound that contains one nitrogen atom and four carbon atoms in a ring structure. In the context of human health, "pyrroles" often refers to a group of compounds called pyrrol derivatives or pyrrole metabolites.
In clinical settings, "pyrroles" is sometimes used to refer to a urinary metabolite called "pyrrole-protein conjugate," which contains a pyrrole ring and is excreted in the urine. Elevated levels of this compound have been associated with certain psychiatric and behavioral disorders, such as schizophrenia and mood disorders. However, the relationship between pyrroles and these conditions is not well understood, and more research is needed to establish a clear medical definition or diagnostic criteria for "pyrrole disorder" or "pyroluria."
A plasmid is a small, circular, double-stranded DNA molecule that is separate from the chromosomal DNA of a bacterium or other organism. Plasmids are typically not essential for the survival of the organism, but they can confer beneficial traits such as antibiotic resistance or the ability to degrade certain types of pollutants.
Plasmids are capable of replicating independently of the chromosomal DNA and can be transferred between bacteria through a process called conjugation. They often contain genes that provide resistance to antibiotics, heavy metals, and other environmental stressors. Plasmids have also been engineered for use in molecular biology as cloning vectors, allowing scientists to replicate and manipulate specific DNA sequences.
Plasmids are important tools in genetic engineering and biotechnology because they can be easily manipulated and transferred between organisms. They have been used to produce vaccines, diagnostic tests, and genetically modified organisms (GMOs) for various applications, including agriculture, medicine, and industry.
Edetic acid, also known as ethylenediaminetetraacetic acid (EDTA), is not a medical term per se, but a chemical compound with various applications in medicine. EDTA is a synthetic amino acid that acts as a chelating agent, which means it can bind to metallic ions and form stable complexes.
In medicine, EDTA is primarily used in the treatment of heavy metal poisoning, such as lead or mercury toxicity. It works by binding to the toxic metal ions in the body, forming a stable compound that can be excreted through urine. This helps reduce the levels of harmful metals in the body and alleviate their toxic effects.
EDTA is also used in some diagnostic tests, such as the determination of calcium levels in blood. Additionally, it has been explored as a potential therapy for conditions like atherosclerosis and Alzheimer's disease, although its efficacy in these areas remains controversial and unproven.
It is important to note that EDTA should only be administered under medical supervision due to its potential side effects and the need for careful monitoring of its use.
Fibroblasts are specialized cells that play a critical role in the body's immune response and wound healing process. They are responsible for producing and maintaining the extracellular matrix (ECM), which is the non-cellular component present within all tissues and organs, providing structural support and biochemical signals for surrounding cells.
Fibroblasts produce various ECM proteins such as collagens, elastin, fibronectin, and laminins, forming a complex network of fibers that give tissues their strength and flexibility. They also help in the regulation of tissue homeostasis by controlling the turnover of ECM components through the process of remodeling.
In response to injury or infection, fibroblasts become activated and start to proliferate rapidly, migrating towards the site of damage. Here, they participate in the inflammatory response, releasing cytokines and chemokines that attract immune cells to the area. Additionally, they deposit new ECM components to help repair the damaged tissue and restore its functionality.
Dysregulation of fibroblast activity has been implicated in several pathological conditions, including fibrosis (excessive scarring), cancer (where they can contribute to tumor growth and progression), and autoimmune diseases (such as rheumatoid arthritis).
Carbon radioisotopes are radioactive isotopes of carbon, which is an naturally occurring chemical element with the atomic number 6. The most common and stable isotope of carbon is carbon-12 (^12C), but there are also several radioactive isotopes, including carbon-11 (^11C), carbon-14 (^14C), and carbon-13 (^13C). These radioisotopes have different numbers of neutrons in their nuclei, which makes them unstable and causes them to emit radiation.
Carbon-11 has a half-life of about 20 minutes and is used in medical imaging techniques such as positron emission tomography (PET) scans. It is produced by bombarding nitrogen-14 with protons in a cyclotron.
Carbon-14, also known as radiocarbon, has a half-life of about 5730 years and is used in archaeology and geology to date organic materials. It is produced naturally in the atmosphere by cosmic rays.
Carbon-13 is stable and has a natural abundance of about 1.1% in carbon. It is not radioactive, but it can be used as a tracer in medical research and in the study of metabolic processes.
Macrophages are a type of white blood cell that are an essential part of the immune system. They are large, specialized cells that engulf and destroy foreign substances, such as bacteria, viruses, parasites, and fungi, as well as damaged or dead cells. Macrophages are found throughout the body, including in the bloodstream, lymph nodes, spleen, liver, lungs, and connective tissues. They play a critical role in inflammation, immune response, and tissue repair and remodeling.
Macrophages originate from monocytes, which are a type of white blood cell produced in the bone marrow. When monocytes enter the tissues, they differentiate into macrophages, which have a larger size and more specialized functions than monocytes. Macrophages can change their shape and move through tissues to reach sites of infection or injury. They also produce cytokines, chemokines, and other signaling molecules that help coordinate the immune response and recruit other immune cells to the site of infection or injury.
Macrophages have a variety of surface receptors that allow them to recognize and respond to different types of foreign substances and signals from other cells. They can engulf and digest foreign particles, bacteria, and viruses through a process called phagocytosis. Macrophages also play a role in presenting antigens to T cells, which are another type of immune cell that helps coordinate the immune response.
Overall, macrophages are crucial for maintaining tissue homeostasis, defending against infection, and promoting wound healing and tissue repair. Dysregulation of macrophage function has been implicated in a variety of diseases, including cancer, autoimmune disorders, and chronic inflammatory conditions.
X-ray crystallography is a technique used in structural biology to determine the three-dimensional arrangement of atoms in a crystal lattice. In this method, a beam of X-rays is directed at a crystal and diffracts, or spreads out, into a pattern of spots called reflections. The intensity and angle of each reflection are measured and used to create an electron density map, which reveals the position and type of atoms in the crystal. This information can be used to determine the molecular structure of a compound, including its shape, size, and chemical bonds. X-ray crystallography is a powerful tool for understanding the structure and function of biological macromolecules such as proteins and nucleic acids.
Medical Definition of Vitamin A:
Vitamin A is a fat-soluble vitamin that is essential for normal vision, immune function, and cell growth. It is also an antioxidant that helps protect the body's cells from damage caused by free radicals. Vitamin A can be found in two main forms: preformed vitamin A, which is found in animal products such as dairy, fish, and meat, particularly liver; and provitamin A carotenoids, which are found in plant-based foods such as fruits, vegetables, and vegetable oils.
The most active form of vitamin A is retinoic acid, which plays a critical role in the development and maintenance of the heart, lungs, kidneys, and other organs. Vitamin A deficiency can lead to night blindness, dry skin, and increased susceptibility to infections. Chronic vitamin A toxicity can cause nausea, dizziness, headaches, coma, and even death.
Urea is not a medical condition but it is a medically relevant substance. Here's the definition:
Urea is a colorless, odorless solid that is the primary nitrogen-containing compound in the urine of mammals. It is a normal metabolic end product that is excreted by the kidneys and is also used as a fertilizer and in various industrial applications. Chemically, urea is a carbamide, consisting of two amino groups (NH2) joined by a carbon atom and having a hydrogen atom and a hydroxyl group (OH) attached to the carbon atom. Urea is produced in the liver as an end product of protein metabolism and is then eliminated from the body by the kidneys through urination. Abnormal levels of urea in the blood, known as uremia, can indicate impaired kidney function or other medical conditions.
Differential scanning calorimetry (DSC) is a thermoanalytical technique used to measure the difference in the amount of heat required to increase the temperature of a sample and a reference as a function of temperature. It is commonly used to study phase transitions, such as melting, crystallization, and glass transition, as well as chemical reactions, in a wide range of materials, including polymers, pharmaceuticals, and biological samples.
In DSC, the sample and reference are placed in separate pans and heated at a constant rate. The heat flow required to maintain this heating rate is continuously measured for both the sample and the reference. As the temperature of the sample changes during a phase transition or chemical reaction, the heat flow required to maintain the same heating rate will change relative to the reference. This allows for the measurement of the enthalpy change (ΔH) associated with the transition or reaction.
Differential scanning calorimetry is a powerful tool in materials science and research as it can provide information about the thermal behavior, stability, and composition of materials. It can also be used to study the kinetics of reactions and phase transitions, making it useful for optimizing processing conditions and developing new materials.
In the context of medicine, there is no specific medical definition for 'metals.' However, certain metals have significant roles in biological systems and are thus studied in physiology, pathology, and pharmacology. Some metals are essential to life, serving as cofactors for enzymatic reactions, while others are toxic and can cause harm at certain levels.
Examples of essential metals include:
1. Iron (Fe): It is a crucial component of hemoglobin, myoglobin, and various enzymes involved in energy production, DNA synthesis, and electron transport.
2. Zinc (Zn): This metal is vital for immune function, wound healing, protein synthesis, and DNA synthesis. It acts as a cofactor for over 300 enzymes.
3. Copper (Cu): Copper is essential for energy production, iron metabolism, antioxidant defense, and connective tissue formation. It serves as a cofactor for several enzymes.
4. Magnesium (Mg): Magnesium plays a crucial role in many biochemical reactions, including nerve and muscle function, protein synthesis, and blood pressure regulation.
5. Manganese (Mn): This metal is necessary for bone development, protein metabolism, and antioxidant defense. It acts as a cofactor for several enzymes.
6. Molybdenum (Mo): Molybdenum is essential for the function of certain enzymes involved in the metabolism of nucleic acids, proteins, and drugs.
7. Cobalt (Co): Cobalt is a component of vitamin B12, which plays a vital role in DNA synthesis, fatty acid metabolism, and nerve function.
Examples of toxic metals include:
1. Lead (Pb): Exposure to lead can cause neurological damage, anemia, kidney dysfunction, and developmental issues.
2. Mercury (Hg): Mercury is highly toxic and can cause neurological problems, kidney damage, and developmental issues.
3. Arsenic (As): Arsenic exposure can lead to skin lesions, cancer, neurological disorders, and cardiovascular diseases.
4. Cadmium (Cd): Cadmium is toxic and can cause kidney damage, bone demineralization, and lung irritation.
5. Chromium (Cr): Excessive exposure to chromium can lead to skin ulcers, respiratory issues, and kidney and liver damage.
Low-Density Lipoprotein Receptor-Related Protein 1 (LRP1) is a large transmembrane receptor protein that belongs to the low-density lipoprotein receptor family. It plays a crucial role in various biological processes, including cellular signaling, endocytosis, and intracellular trafficking of ligands. LRP1 is widely expressed in many tissues, particularly in the brain, liver, and vascular endothelial cells.
LRP1 interacts with a diverse array of ligands, such as extracellular matrix proteins, apolipoproteins, proteinases, proteinase inhibitors, and various pathogen-associated molecules. The receptor is involved in the clearance of these ligands from the extracellular space through endocytosis, followed by intracellular degradation or recycling.
In the context of lipid metabolism, LRP1 has been implicated in the cellular uptake and degradation of Apolipoprotein E (ApoE)-containing lipoproteins, which are involved in the reverse transport of cholesterol from peripheral tissues to the liver. Dysregulation of LRP1 function has been linked to several diseases, including atherosclerosis, Alzheimer's disease, and various neurological disorders.
In summary, Low-Density Lipoprotein Receptor-Related Protein 1 (LRP1) is a multifunctional transmembrane receptor that plays essential roles in cellular signaling, endocytosis, and intracellular trafficking of various ligands. Its dysfunction has been implicated in several diseases related to lipid metabolism, neurodegeneration, and neurological disorders.
Ion exchange chromatography is a type of chromatography technique used to separate and analyze charged molecules (ions) based on their ability to exchange bound ions in a solid resin or gel with ions of similar charge in the mobile phase. The stationary phase, often called an ion exchanger, contains fixed ated functional groups that can attract counter-ions of opposite charge from the sample mixture.
In this technique, the sample is loaded onto an ion exchange column containing the charged resin or gel. As the sample moves through the column, ions in the sample compete for binding sites on the stationary phase with ions already present in the column. The ions that bind most strongly to the stationary phase will elute (come off) slower than those that bind more weakly.
Ion exchange chromatography can be performed using either cation exchangers, which exchange positive ions (cations), or anion exchangers, which exchange negative ions (anions). The pH and ionic strength of the mobile phase can be adjusted to control the binding and elution of specific ions.
Ion exchange chromatography is widely used in various applications such as water treatment, protein purification, and chemical analysis.
Enzyme activation refers to the process by which an enzyme becomes biologically active and capable of carrying out its specific chemical or biological reaction. This is often achieved through various post-translational modifications, such as proteolytic cleavage, phosphorylation, or addition of cofactors or prosthetic groups to the enzyme molecule. These modifications can change the conformation or structure of the enzyme, exposing or creating a binding site for the substrate and allowing the enzymatic reaction to occur.
For example, in the case of proteolytic cleavage, an inactive precursor enzyme, known as a zymogen, is cleaved into its active form by a specific protease. This is seen in enzymes such as trypsin and chymotrypsin, which are initially produced in the pancreas as inactive precursors called trypsinogen and chymotrypsinogen, respectively. Once they reach the small intestine, they are activated by enteropeptidase, a protease that cleaves a specific peptide bond, releasing the active enzyme.
Phosphorylation is another common mechanism of enzyme activation, where a phosphate group is added to a specific serine, threonine, or tyrosine residue on the enzyme by a protein kinase. This modification can alter the conformation of the enzyme and create a binding site for the substrate, allowing the enzymatic reaction to occur.
Enzyme activation is a crucial process in many biological pathways, as it allows for precise control over when and where specific reactions take place. It also provides a mechanism for regulating enzyme activity in response to various signals and stimuli, such as hormones, neurotransmitters, or changes in the intracellular environment.
Substrate specificity in the context of medical biochemistry and enzymology refers to the ability of an enzyme to selectively bind and catalyze a chemical reaction with a particular substrate (or a group of similar substrates) while discriminating against other molecules that are not substrates. This specificity arises from the three-dimensional structure of the enzyme, which has evolved to match the shape, charge distribution, and functional groups of its physiological substrate(s).
Substrate specificity is a fundamental property of enzymes that enables them to carry out highly selective chemical transformations in the complex cellular environment. The active site of an enzyme, where the catalysis takes place, has a unique conformation that complements the shape and charge distribution of its substrate(s). This ensures efficient recognition, binding, and conversion of the substrate into the desired product while minimizing unwanted side reactions with other molecules.
Substrate specificity can be categorized as:
1. Absolute specificity: An enzyme that can only act on a single substrate or a very narrow group of structurally related substrates, showing no activity towards any other molecule.
2. Group specificity: An enzyme that prefers to act on a particular functional group or class of compounds but can still accommodate minor structural variations within the substrate.
3. Broad or promiscuous specificity: An enzyme that can act on a wide range of structurally diverse substrates, albeit with varying catalytic efficiencies.
Understanding substrate specificity is crucial for elucidating enzymatic mechanisms, designing drugs that target specific enzymes or pathways, and developing biotechnological applications that rely on the controlled manipulation of enzyme activities.
Membrane proteins are a type of protein that are embedded in the lipid bilayer of biological membranes, such as the plasma membrane of cells or the inner membrane of mitochondria. These proteins play crucial roles in various cellular processes, including:
1. Cell-cell recognition and signaling
2. Transport of molecules across the membrane (selective permeability)
3. Enzymatic reactions at the membrane surface
4. Energy transduction and conversion
5. Mechanosensation and signal transduction
Membrane proteins can be classified into two main categories: integral membrane proteins, which are permanently associated with the lipid bilayer, and peripheral membrane proteins, which are temporarily or loosely attached to the membrane surface. Integral membrane proteins can further be divided into three subcategories based on their topology:
1. Transmembrane proteins, which span the entire width of the lipid bilayer with one or more alpha-helices or beta-barrels.
2. Lipid-anchored proteins, which are covalently attached to lipids in the membrane via a glycosylphosphatidylinositol (GPI) anchor or other lipid modifications.
3. Monotopic proteins, which are partially embedded in the membrane and have one or more domains exposed to either side of the bilayer.
Membrane proteins are essential for maintaining cellular homeostasis and are targets for various therapeutic interventions, including drug development and gene therapy. However, their structural complexity and hydrophobicity make them challenging to study using traditional biochemical methods, requiring specialized techniques such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and single-particle cryo-electron microscopy (cryo-EM).
'Immune sera' refers to the serum fraction of blood that contains antibodies produced in response to an antigenic stimulus, such as a vaccine or an infection. These antibodies are proteins known as immunoglobulins, which are secreted by B cells (a type of white blood cell) and can recognize and bind to specific antigens. Immune sera can be collected from an immunized individual and used as a source of passive immunity to protect against infection or disease. It is often used in research and diagnostic settings to identify or measure the presence of specific antigens or antibodies.
"Competitive binding" is a term used in pharmacology and biochemistry to describe the behavior of two or more molecules (ligands) competing for the same binding site on a target protein or receptor. In this context, "binding" refers to the physical interaction between a ligand and its target.
When a ligand binds to a receptor, it can alter the receptor's function, either activating or inhibiting it. If multiple ligands compete for the same binding site, they will compete to bind to the receptor. The ability of each ligand to bind to the receptor is influenced by its affinity for the receptor, which is a measure of how strongly and specifically the ligand binds to the receptor.
In competitive binding, if one ligand is present in high concentrations, it can prevent other ligands with lower affinity from binding to the receptor. This is because the higher-affinity ligand will have a greater probability of occupying the binding site and blocking access to the other ligands. The competition between ligands can be described mathematically using equations such as the Langmuir isotherm, which describes the relationship between the concentration of ligand and the fraction of receptors that are occupied by the ligand.
Competitive binding is an important concept in drug development, as it can be used to predict how different drugs will interact with their targets and how they may affect each other's activity. By understanding the competitive binding properties of a drug, researchers can optimize its dosage and delivery to maximize its therapeutic effect while minimizing unwanted side effects.
Manganese is not a medical condition, but it's an essential trace element that is vital for human health. Here is the medical definition of Manganese:
Manganese (Mn) is a trace mineral that is present in tiny amounts in the body. It is found mainly in bones, the liver, kidneys, and pancreas. Manganese helps the body form connective tissue, bones, blood clotting factors, and sex hormones. It also plays a role in fat and carbohydrate metabolism, calcium absorption, and blood sugar regulation. Manganese is also necessary for normal brain and nerve function.
The recommended dietary allowance (RDA) for manganese is 2.3 mg per day for adult men and 1.8 mg per day for adult women. Good food sources of manganese include nuts, seeds, legumes, whole grains, green leafy vegetables, and tea.
In some cases, exposure to high levels of manganese can cause neurological symptoms similar to Parkinson's disease, a condition known as manganism. However, this is rare and usually occurs in people who are occupationally exposed to manganese dust or fumes, such as welders.
Glycoproteins are complex proteins that contain oligosaccharide chains (glycans) covalently attached to their polypeptide backbone. These glycans are linked to the protein through asparagine residues (N-linked) or serine/threonine residues (O-linked). Glycoproteins play crucial roles in various biological processes, including cell recognition, cell-cell interactions, cell adhesion, and signal transduction. They are widely distributed in nature and can be found on the outer surface of cell membranes, in extracellular fluids, and as components of the extracellular matrix. The structure and composition of glycoproteins can vary significantly depending on their function and location within an organism.
Reference values, also known as reference ranges or reference intervals, are the set of values that are considered normal or typical for a particular population or group of people. These values are often used in laboratory tests to help interpret test results and determine whether a patient's value falls within the expected range.
The process of establishing reference values typically involves measuring a particular biomarker or parameter in a large, healthy population and then calculating the mean and standard deviation of the measurements. Based on these statistics, a range is established that includes a certain percentage of the population (often 95%) and excludes extreme outliers.
It's important to note that reference values can vary depending on factors such as age, sex, race, and other demographic characteristics. Therefore, it's essential to use reference values that are specific to the relevant population when interpreting laboratory test results. Additionally, reference values may change over time due to advances in measurement technology or changes in the population being studied.
Dextrans are a type of complex glucose polymers that are formed by the action of certain bacteria on sucrose. They are branched polysaccharides consisting of linear chains of α-1,6 linked D-glucopyranosyl units with occasional α-1,3 branches.
Dextrans have a wide range of applications in medicine and industry. In medicine, dextrans are used as plasma substitutes, volume expanders, and anticoagulants. They are also used as carriers for drugs and diagnostic agents, and in the manufacture of immunoadsorbents for the removal of toxins and pathogens from blood.
Dextrans can be derived from various bacterial sources, but the most common commercial source is Leuconostoc mesenteroides B-512(F) or L. dextranicum. The molecular weight of dextrans can vary widely, ranging from a few thousand to several million Daltons, depending on the method of preparation and purification.
Dextrans are generally biocompatible and non-toxic, but they can cause allergic reactions in some individuals. Therefore, their use as medical products requires careful monitoring and testing for safety and efficacy.
Liposomes are artificially prepared, small, spherical vesicles composed of one or more lipid bilayers that enclose an aqueous compartment. They can encapsulate both hydrophilic and hydrophobic drugs, making them useful for drug delivery applications in the medical field. The lipid bilayer structure of liposomes is similar to that of biological membranes, which allows them to merge with and deliver their contents into cells. This property makes liposomes a valuable tool in delivering drugs directly to targeted sites within the body, improving drug efficacy while minimizing side effects.
"Klebsiella pneumoniae" is a medical term that refers to a type of bacteria belonging to the family Enterobacteriaceae. It's a gram-negative, encapsulated, non-motile, rod-shaped bacterium that can be found in various environments, including soil, water, and the gastrointestinal tracts of humans and animals.
"Klebsiella pneumoniae" is an opportunistic pathogen that can cause a range of infections, particularly in individuals with weakened immune systems or underlying medical conditions. It's a common cause of healthcare-associated infections, such as pneumonia, urinary tract infections, bloodstream infections, and wound infections.
The bacterium is known for its ability to produce a polysaccharide capsule that makes it resistant to phagocytosis by white blood cells, allowing it to evade the host's immune system. Additionally, "Klebsiella pneumoniae" has developed resistance to many antibiotics, making infections caused by this bacterium difficult to treat and a growing public health concern.
Enzyme stability refers to the ability of an enzyme to maintain its structure and function under various environmental conditions, such as temperature, pH, and the presence of denaturants or inhibitors. A stable enzyme retains its activity and conformation over time and across a range of conditions, making it more suitable for industrial and therapeutic applications.
Enzymes can be stabilized through various methods, including chemical modification, immobilization, and protein engineering. Understanding the factors that affect enzyme stability is crucial for optimizing their use in biotechnology, medicine, and research.
Molecular conformation, also known as spatial arrangement or configuration, refers to the specific three-dimensional shape and orientation of atoms that make up a molecule. It describes the precise manner in which bonds between atoms are arranged around a molecular framework, taking into account factors such as bond lengths, bond angles, and torsional angles.
Conformational isomers, or conformers, are different spatial arrangements of the same molecule that can interconvert without breaking chemical bonds. These isomers may have varying energies, stability, and reactivity, which can significantly impact a molecule's biological activity and function. Understanding molecular conformation is crucial in fields such as drug design, where small changes in conformation can lead to substantial differences in how a drug interacts with its target.
Mass spectrometry with electrospray ionization (ESI-MS) is an analytical technique used to identify and quantify chemical species in a sample based on the mass-to-charge ratio of charged particles. In ESI-MS, analytes are ionized through the use of an electrospray, where a liquid sample is introduced through a metal capillary needle at high voltage, creating an aerosol of charged droplets. As the solvent evaporates, the analyte molecules become charged and can be directed into a mass spectrometer for analysis.
ESI-MS is particularly useful for the analysis of large biomolecules such as proteins, peptides, and nucleic acids, due to its ability to gently ionize these species without fragmentation. The technique provides information about the molecular weight and charge state of the analytes, which can be used to infer their identity and structure. Additionally, ESI-MS can be interfaced with separation techniques such as liquid chromatography (LC) for further purification and characterization of complex samples.
Arginine is an α-amino acid that is classified as a semi-essential or conditionally essential amino acid, depending on the developmental stage and health status of the individual. The adult human body can normally synthesize sufficient amounts of arginine to meet its needs, but there are certain circumstances, such as periods of rapid growth or injury, where the dietary intake of arginine may become necessary.
The chemical formula for arginine is C6H14N4O2. It has a molecular weight of 174.20 g/mol and a pKa value of 12.48. Arginine is a basic amino acid, which means that it contains a side chain with a positive charge at physiological pH levels. The side chain of arginine is composed of a guanidino group, which is a functional group consisting of a nitrogen atom bonded to three methyl groups.
In the body, arginine plays several important roles. It is a precursor for the synthesis of nitric oxide, a molecule that helps regulate blood flow and immune function. Arginine is also involved in the detoxification of ammonia, a waste product produced by the breakdown of proteins. Additionally, arginine can be converted into other amino acids, such as ornithine and citrulline, which are involved in various metabolic processes.
Foods that are good sources of arginine include meat, poultry, fish, dairy products, nuts, seeds, and legumes. Arginine supplements are available and may be used for a variety of purposes, such as improving exercise performance, enhancing wound healing, and boosting immune function. However, it is important to consult with a healthcare provider before taking arginine supplements, as they can interact with certain medications and have potential side effects.
Polyethylene glycols (PEGs) are a family of synthetic, water-soluble polymers with a wide range of molecular weights. They are commonly used in the medical field as excipients in pharmaceutical formulations due to their ability to improve drug solubility, stability, and bioavailability. PEGs can also be used as laxatives to treat constipation or as bowel cleansing agents prior to colonoscopy examinations. Additionally, some PEG-conjugated drugs have been developed for use in targeted cancer therapies.
In a medical context, PEGs are often referred to by their average molecular weight, such as PEG 300, PEG 400, PEG 1500, and so on. Higher molecular weight PEGs tend to be more viscous and have longer-lasting effects in the body.
It's worth noting that while PEGs are generally considered safe for use in medical applications, some people may experience allergic reactions or hypersensitivity to these compounds. Prolonged exposure to high molecular weight PEGs has also been linked to potential adverse effects, such as decreased fertility and developmental toxicity in animal studies. However, more research is needed to fully understand the long-term safety of PEGs in humans.
I believe there might be a misunderstanding in your question. "Dogs" is not a medical term or condition. It is the common name for a domesticated carnivore of the family Canidae, specifically the genus Canis, which includes wolves, foxes, and other extant and extinct species of mammals. Dogs are often kept as pets and companions, and they have been bred in a wide variety of forms and sizes for different purposes, such as hunting, herding, guarding, assisting police and military forces, and providing companionship and emotional support.
If you meant to ask about a specific medical condition or term related to dogs, please provide more context so I can give you an accurate answer.
Sequence homology in nucleic acids refers to the similarity or identity between the nucleotide sequences of two or more DNA or RNA molecules. It is often used as a measure of biological relationship between genes, organisms, or populations. High sequence homology suggests a recent common ancestry or functional constraint, while low sequence homology may indicate a more distant relationship or different functions.
Nucleic acid sequence homology can be determined by various methods such as pairwise alignment, multiple sequence alignment, and statistical analysis. The degree of homology is typically expressed as a percentage of identical or similar nucleotides in a given window of comparison.
It's important to note that the interpretation of sequence homology depends on the biological context and the evolutionary distance between the sequences compared. Therefore, functional and experimental validation is often necessary to confirm the significance of sequence homology.
Nuclear Magnetic Resonance (NMR) Biomolecular is a research technique that uses magnetic fields and radio waves to study the structure and dynamics of biological molecules, such as proteins and nucleic acids. This technique measures the magnetic properties of atomic nuclei within these molecules, specifically their spin, which can be influenced by the application of an external magnetic field.
When a sample is placed in a strong magnetic field, the nuclei absorb and emit electromagnetic radiation at specific frequencies, known as resonance frequencies, which are determined by the molecular structure and environment of the nuclei. By analyzing these resonance frequencies and their interactions, researchers can obtain detailed information about the three-dimensional structure, dynamics, and interactions of biomolecules.
NMR spectroscopy is a non-destructive technique that allows for the study of biological molecules in solution, which makes it an important tool for understanding the function and behavior of these molecules in their natural environment. Additionally, NMR can be used to study the effects of drugs, ligands, and other small molecules on biomolecular structure and dynamics, making it a valuable tool in drug discovery and development.
Microsomes are subcellular membranous vesicles that are obtained as a byproduct during the preparation of cellular homogenates. They are not naturally occurring structures within the cell, but rather formed due to fragmentation of the endoplasmic reticulum (ER) during laboratory procedures. Microsomes are widely used in various research and scientific studies, particularly in the fields of biochemistry and pharmacology.
Microsomes are rich in enzymes, including the cytochrome P450 system, which is involved in the metabolism of drugs, toxins, and other xenobiotics. These enzymes play a crucial role in detoxifying foreign substances and eliminating them from the body. As such, microsomes serve as an essential tool for studying drug metabolism, toxicity, and interactions, allowing researchers to better understand and predict the effects of various compounds on living organisms.
A gene is a specific sequence of nucleotides in DNA that carries genetic information. Genes are the fundamental units of heredity and are responsible for the development and function of all living organisms. They code for proteins or RNA molecules, which carry out various functions within cells and are essential for the structure, function, and regulation of the body's tissues and organs.
Each gene has a specific location on a chromosome, and each person inherits two copies of every gene, one from each parent. Variations in the sequence of nucleotides in a gene can lead to differences in traits between individuals, including physical characteristics, susceptibility to disease, and responses to environmental factors.
Medical genetics is the study of genes and their role in health and disease. It involves understanding how genes contribute to the development and progression of various medical conditions, as well as identifying genetic risk factors and developing strategies for prevention, diagnosis, and treatment.
The small intestine is the portion of the gastrointestinal tract that extends from the pylorus of the stomach to the beginning of the large intestine (cecum). It plays a crucial role in the digestion and absorption of nutrients from food. The small intestine is divided into three parts: the duodenum, jejunum, and ileum.
1. Duodenum: This is the shortest and widest part of the small intestine, approximately 10 inches long. It receives chyme (partially digested food) from the stomach and begins the process of further digestion with the help of various enzymes and bile from the liver and pancreas.
2. Jejunum: The jejunum is the middle section, which measures about 8 feet in length. It has a large surface area due to the presence of circular folds (plicae circulares), finger-like projections called villi, and microvilli on the surface of the absorptive cells (enterocytes). These structures increase the intestinal surface area for efficient absorption of nutrients, electrolytes, and water.
3. Ileum: The ileum is the longest and final section of the small intestine, spanning about 12 feet. It continues the absorption process, mainly of vitamin B12, bile salts, and any remaining nutrients. At the end of the ileum, there is a valve called the ileocecal valve that prevents backflow of contents from the large intestine into the small intestine.
The primary function of the small intestine is to absorb the majority of nutrients, electrolytes, and water from ingested food. The mucosal lining of the small intestine contains numerous goblet cells that secrete mucus, which protects the epithelial surface and facilitates the movement of chyme through peristalsis. Additionally, the small intestine hosts a diverse community of microbiota, which contributes to various physiological functions, including digestion, immunity, and protection against pathogens.
Biological models, also known as physiological models or organismal models, are simplified representations of biological systems, processes, or mechanisms that are used to understand and explain the underlying principles and relationships. These models can be theoretical (conceptual or mathematical) or physical (such as anatomical models, cell cultures, or animal models). They are widely used in biomedical research to study various phenomena, including disease pathophysiology, drug action, and therapeutic interventions.
Examples of biological models include:
1. Mathematical models: These use mathematical equations and formulas to describe complex biological systems or processes, such as population dynamics, metabolic pathways, or gene regulation networks. They can help predict the behavior of these systems under different conditions and test hypotheses about their underlying mechanisms.
2. Cell cultures: These are collections of cells grown in a controlled environment, typically in a laboratory dish or flask. They can be used to study cellular processes, such as signal transduction, gene expression, or metabolism, and to test the effects of drugs or other treatments on these processes.
3. Animal models: These are living organisms, usually vertebrates like mice, rats, or non-human primates, that are used to study various aspects of human biology and disease. They can provide valuable insights into the pathophysiology of diseases, the mechanisms of drug action, and the safety and efficacy of new therapies.
4. Anatomical models: These are physical representations of biological structures or systems, such as plastic models of organs or tissues, that can be used for educational purposes or to plan surgical procedures. They can also serve as a basis for developing more sophisticated models, such as computer simulations or 3D-printed replicas.
Overall, biological models play a crucial role in advancing our understanding of biology and medicine, helping to identify new targets for therapeutic intervention, develop novel drugs and treatments, and improve human health.
A precipitin test is a type of immunodiagnostic test used to detect and measure the presence of specific antibodies or antigens in a patient's serum. The test is based on the principle of antigen-antibody interaction, where the addition of an antigen to a solution containing its corresponding antibody results in the formation of an insoluble immune complex known as a precipitin.
In this test, a small amount of the patient's serum is added to a solution containing a known antigen or antibody. If the patient has antibodies or antigens that correspond to the added reagent, they will bind and form a visible precipitate. The size and density of the precipitate can be used to quantify the amount of antibody or antigen present in the sample.
Precipitin tests are commonly used in the diagnosis of various infectious diseases, autoimmune disorders, and allergies. They can also be used in forensic science to identify biological samples. However, they have largely been replaced by more modern immunological techniques such as enzyme-linked immunosorbent assays (ELISAs) and radioimmunoassays (RIAs).
Streptomyces is a genus of Gram-positive, aerobic, saprophytic bacteria that are widely distributed in soil, water, and decaying organic matter. They are known for their complex morphology, forming branching filaments called hyphae that can differentiate into long chains of spores.
Streptomyces species are particularly notable for their ability to produce a wide variety of bioactive secondary metabolites, including antibiotics, antifungals, and other therapeutic compounds. In fact, many important antibiotics such as streptomycin, neomycin, tetracycline, and erythromycin are derived from Streptomyces species.
Because of their industrial importance in the production of antibiotics and other bioactive compounds, Streptomyces have been extensively studied and are considered model organisms for the study of bacterial genetics, biochemistry, and ecology.
Mitochondria are specialized structures located inside cells that convert the energy from food into ATP (adenosine triphosphate), which is the primary form of energy used by cells. They are often referred to as the "powerhouses" of the cell because they generate most of the cell's supply of chemical energy. Mitochondria are also involved in various other cellular processes, such as signaling, differentiation, and apoptosis (programmed cell death).
Mitochondria have their own DNA, known as mitochondrial DNA (mtDNA), which is inherited maternally. This means that mtDNA is passed down from the mother to her offspring through the egg cells. Mitochondrial dysfunction has been linked to a variety of diseases and conditions, including neurodegenerative disorders, diabetes, and aging.
Detergents are cleaning agents that are often used to remove dirt, grease, and stains from various surfaces. They contain one or more surfactants, which are compounds that lower the surface tension between two substances, such as water and oil, allowing them to mix more easily. This makes it possible for detergents to lift and suspend dirt particles in water so they can be rinsed away.
Detergents may also contain other ingredients, such as builders, which help to enhance the cleaning power of the surfactants by softening hard water or removing mineral deposits. Some detergents may also include fragrances, colorants, and other additives to improve their appearance or performance.
In a medical context, detergents are sometimes used as disinfectants or antiseptics, as they can help to kill bacteria, viruses, and other microorganisms on surfaces. However, it is important to note that not all detergents are effective against all types of microorganisms, and some may even be toxic or harmful if used improperly.
It is always important to follow the manufacturer's instructions when using any cleaning product, including detergents, to ensure that they are used safely and effectively.
Lipase is an enzyme that is produced by the pancreas and found in the digestive system of most organisms. Its primary function is to catalyze the hydrolysis of fats (triglycerides) into smaller molecules, such as fatty acids and glycerol, which can then be absorbed by the intestines and utilized for energy or stored for later use.
In medical terms, lipase levels in the blood are often measured to diagnose or monitor conditions that affect the pancreas, such as pancreatitis (inflammation of the pancreas), pancreatic cancer, or cystic fibrosis. Elevated lipase levels may indicate damage to the pancreas and its ability to produce digestive enzymes.
Serum albumin is the most abundant protein in human blood plasma, synthesized by the liver. It plays a crucial role in maintaining the oncotic pressure or colloid osmotic pressure of blood, which helps to regulate the fluid balance between the intravascular and extravascular spaces.
Serum albumin has a molecular weight of around 66 kDa and is composed of a single polypeptide chain. It contains several binding sites for various endogenous and exogenous substances, such as bilirubin, fatty acids, hormones, and drugs, facilitating their transport throughout the body. Additionally, albumin possesses antioxidant properties, protecting against oxidative damage.
Albumin levels in the blood are often used as a clinical indicator of liver function, nutritional status, and overall health. Low serum albumin levels may suggest liver disease, malnutrition, inflammation, or kidney dysfunction.
I apologize for the confusion, but "Bucladesine" is not a recognized medical term or a medication in current use in medicine. It's possible that there may be some mistake or typo in the spelling. If you have any more context about where you encountered this term, I might be able to provide a more accurate and helpful response.
Tertiary protein structure refers to the three-dimensional arrangement of all the elements (polypeptide chains) of a single protein molecule. It is the highest level of structural organization and results from interactions between various side chains (R groups) of the amino acids that make up the protein. These interactions, which include hydrogen bonds, ionic bonds, van der Waals forces, and disulfide bridges, give the protein its unique shape and stability, which in turn determines its function. The tertiary structure of a protein can be stabilized by various factors such as temperature, pH, and the presence of certain ions. Any changes in these factors can lead to denaturation, where the protein loses its tertiary structure and thus its function.
Liquid chromatography (LC) is a type of chromatography technique used to separate, identify, and quantify the components in a mixture. In this method, the sample mixture is dissolved in a liquid solvent (the mobile phase) and then passed through a stationary phase, which can be a solid or a liquid that is held in place by a solid support.
The components of the mixture interact differently with the stationary phase and the mobile phase, causing them to separate as they move through the system. The separated components are then detected and measured using various detection techniques, such as ultraviolet (UV) absorbance or mass spectrometry.
Liquid chromatography is widely used in many areas of science and medicine, including drug development, environmental analysis, food safety testing, and clinical diagnostics. It can be used to separate and analyze a wide range of compounds, from small molecules like drugs and metabolites to large biomolecules like proteins and nucleic acids.
Tryptophan is an essential amino acid, meaning it cannot be synthesized by the human body and must be obtained through dietary sources. Its chemical formula is C11H12N2O2. Tryptophan plays a crucial role in various biological processes as it serves as a precursor to several important molecules, including serotonin, melatonin, and niacin (vitamin B3). Serotonin is a neurotransmitter involved in mood regulation, appetite control, and sleep-wake cycles, while melatonin is a hormone that regulates sleep-wake patterns. Niacin is essential for energy production and DNA repair.
Foods rich in tryptophan include turkey, chicken, fish, eggs, cheese, milk, nuts, seeds, and whole grains. In some cases, tryptophan supplementation may be recommended to help manage conditions related to serotonin imbalances, such as depression or insomnia, but this should only be done under the guidance of a healthcare professional due to potential side effects and interactions with other medications.
I'm sorry for any confusion, but there seems to be a misunderstanding. Mathematics is not a medical term; it is a branch of science dedicated to the study of numbers, shapes, and structures. However, mathematics does have many applications in medicine, such as in modeling disease spread, analyzing medical images, or designing clinical trials. If you have any questions related to mathematics in a medical context, I'd be happy to help clarify those for you!
Endopeptidases are a type of enzyme that breaks down proteins by cleaving peptide bonds inside the polypeptide chain. They are also known as proteinases or endoproteinases. These enzymes work within the interior of the protein molecule, cutting it at specific points along its length, as opposed to exopeptidases, which remove individual amino acids from the ends of the protein chain.
Endopeptidases play a crucial role in various biological processes, such as digestion, blood coagulation, and programmed cell death (apoptosis). They are classified based on their catalytic mechanism and the structure of their active site. Some examples of endopeptidase families include serine proteases, cysteine proteases, aspartic proteases, and metalloproteases.
It is important to note that while endopeptidases are essential for normal physiological functions, they can also contribute to disease processes when their activity is unregulated or misdirected. For instance, excessive endopeptidase activity has been implicated in the pathogenesis of neurodegenerative disorders, cancer, and inflammatory conditions.
'Escherichia coli (E. coli) proteins' refer to the various types of proteins that are produced and expressed by the bacterium Escherichia coli. These proteins play a critical role in the growth, development, and survival of the organism. They are involved in various cellular processes such as metabolism, DNA replication, transcription, translation, repair, and regulation.
E. coli is a gram-negative, facultative anaerobe that is commonly found in the intestines of warm-blooded organisms. It is widely used as a model organism in scientific research due to its well-studied genetics, rapid growth, and ability to be easily manipulated in the laboratory. As a result, many E. coli proteins have been identified, characterized, and studied in great detail.
Some examples of E. coli proteins include enzymes involved in carbohydrate metabolism such as lactase, sucrase, and maltose; proteins involved in DNA replication such as the polymerases, single-stranded binding proteins, and helicases; proteins involved in transcription such as RNA polymerase and sigma factors; proteins involved in translation such as ribosomal proteins, tRNAs, and aminoacyl-tRNA synthetases; and regulatory proteins such as global regulators, two-component systems, and transcription factors.
Understanding the structure, function, and regulation of E. coli proteins is essential for understanding the basic biology of this important organism, as well as for developing new strategies for combating bacterial infections and improving industrial processes involving bacteria.
Immunoblotting, also known as western blotting, is a laboratory technique used in molecular biology and immunogenetics to detect and quantify specific proteins in a complex mixture. This technique combines the electrophoretic separation of proteins by gel electrophoresis with their detection using antibodies that recognize specific epitopes (protein fragments) on the target protein.
The process involves several steps: first, the protein sample is separated based on size through sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Next, the separated proteins are transferred onto a nitrocellulose or polyvinylidene fluoride (PVDF) membrane using an electric field. The membrane is then blocked with a blocking agent to prevent non-specific binding of antibodies.
After blocking, the membrane is incubated with a primary antibody that specifically recognizes the target protein. Following this, the membrane is washed to remove unbound primary antibodies and then incubated with a secondary antibody conjugated to an enzyme such as horseradish peroxidase (HRP) or alkaline phosphatase (AP). The enzyme catalyzes a colorimetric or chemiluminescent reaction that allows for the detection of the target protein.
Immunoblotting is widely used in research and clinical settings to study protein expression, post-translational modifications, protein-protein interactions, and disease biomarkers. It provides high specificity and sensitivity, making it a valuable tool for identifying and quantifying proteins in various biological samples.
Alanine is an alpha-amino acid that is used in the biosynthesis of proteins. The molecular formula for alanine is C3H7NO2. It is a non-essential amino acid, which means that it can be produced by the human body through the conversion of other nutrients, such as pyruvate, and does not need to be obtained directly from the diet.
Alanine is classified as an aliphatic amino acid because it contains a simple carbon side chain. It is also a non-polar amino acid, which means that it is hydrophobic and tends to repel water. Alanine plays a role in the metabolism of glucose and helps to regulate blood sugar levels. It is also involved in the transfer of nitrogen between tissues and helps to maintain the balance of nitrogen in the body.
In addition to its role as a building block of proteins, alanine is also used as a neurotransmitter in the brain and has been shown to have a calming effect on the nervous system. It is found in many foods, including meats, poultry, fish, eggs, dairy products, and legumes.
Bicarbonates, also known as sodium bicarbonate or baking soda, is a chemical compound with the formula NaHCO3. In the context of medical definitions, bicarbonates refer to the bicarbonate ion (HCO3-), which is an important buffer in the body that helps maintain normal pH levels in blood and other bodily fluids.
The balance of bicarbonate and carbonic acid in the body helps regulate the acidity or alkalinity of the blood, a condition known as pH balance. Bicarbonates are produced by the body and are also found in some foods and drinking water. They work to neutralize excess acid in the body and help maintain the normal pH range of 7.35 to 7.45.
In medical testing, bicarbonate levels may be measured as part of an electrolyte panel or as a component of arterial blood gas (ABG) analysis. Low bicarbonate levels can indicate metabolic acidosis, while high levels can indicate metabolic alkalosis. Both conditions can have serious consequences if not treated promptly and appropriately.
The endothelium is the thin, delicate tissue that lines the interior surface of blood vessels and lymphatic vessels. It is a single layer of cells called endothelial cells that are in contact with the blood or lymph fluid. The endothelium plays an essential role in maintaining vascular homeostasis by regulating blood flow, coagulation, platelet activation, immune function, and angiogenesis (the formation of new blood vessels). It also acts as a barrier between the vessel wall and the circulating blood or lymph fluid. Dysfunction of the endothelium has been implicated in various cardiovascular diseases, diabetes, inflammation, and cancer.
A cell line is a culture of cells that are grown in a laboratory for use in research. These cells are usually taken from a single cell or group of cells, and they are able to divide and grow continuously in the lab. Cell lines can come from many different sources, including animals, plants, and humans. They are often used in scientific research to study cellular processes, disease mechanisms, and to test new drugs or treatments. Some common types of human cell lines include HeLa cells (which come from a cancer patient named Henrietta Lacks), HEK293 cells (which come from embryonic kidney cells), and HUVEC cells (which come from umbilical vein endothelial cells). It is important to note that cell lines are not the same as primary cells, which are cells that are taken directly from a living organism and have not been grown in the lab.
Brain chemistry refers to the chemical processes that occur within the brain, particularly those involving neurotransmitters, neuromodulators, and neuropeptides. These chemicals are responsible for transmitting signals between neurons (nerve cells) in the brain, allowing for various cognitive, emotional, and physical functions.
Neurotransmitters are chemical messengers that transmit signals across the synapse (the tiny gap between two neurons). Examples of neurotransmitters include dopamine, serotonin, norepinephrine, GABA (gamma-aminobutyric acid), and glutamate. Each neurotransmitter has a specific role in brain function, such as regulating mood, motivation, attention, memory, and movement.
Neuromodulators are chemicals that modify the effects of neurotransmitters on neurons. They can enhance or inhibit the transmission of signals between neurons, thereby modulating brain activity. Examples of neuromodulators include acetylcholine, histamine, and substance P.
Neuropeptides are small protein-like molecules that act as neurotransmitters or neuromodulators. They play a role in various physiological functions, such as pain perception, stress response, and reward processing. Examples of neuropeptides include endorphins, enkephalins, and oxytocin.
Abnormalities in brain chemistry can lead to various neurological and psychiatric conditions, such as depression, anxiety disorders, schizophrenia, Parkinson's disease, and Alzheimer's disease. Understanding brain chemistry is crucial for developing effective treatments for these conditions.
Quaternary protein structure refers to the arrangement and interaction of multiple folded protein molecules in a multi-subunit complex. These subunits can be identical or different forms of the same protein or distinctly different proteins that associate to form a functional complex. The quaternary structure is held together by non-covalent interactions, such as hydrogen bonds, ionic bonds, and van der Waals forces. Understanding quaternary structure is crucial for comprehending the function, regulation, and assembly of many protein complexes involved in various cellular processes.
Deoxyribonucleic acid (DNA) is the genetic material present in the cells of organisms where it is responsible for the storage and transmission of hereditary information. DNA is a long molecule that consists of two strands coiled together to form a double helix. Each strand is made up of a series of four nucleotide bases - adenine (A), guanine (G), cytosine (C), and thymine (T) - that are linked together by phosphate and sugar groups. The sequence of these bases along the length of the molecule encodes genetic information, with A always pairing with T and C always pairing with G. This base-pairing allows for the replication and transcription of DNA, which are essential processes in the functioning and reproduction of all living organisms.
A heterozygote is an individual who has inherited two different alleles (versions) of a particular gene, one from each parent. This means that the individual's genotype for that gene contains both a dominant and a recessive allele. The dominant allele will be expressed phenotypically (outwardly visible), while the recessive allele may or may not have any effect on the individual's observable traits, depending on the specific gene and its function. Heterozygotes are often represented as 'Aa', where 'A' is the dominant allele and 'a' is the recessive allele.
Subcellular fractions refer to the separation and collection of specific parts or components of a cell, including organelles, membranes, and other structures, through various laboratory techniques such as centrifugation and ultracentrifugation. These fractions can be used in further biochemical and molecular analyses to study the structure, function, and interactions of individual cellular components. Examples of subcellular fractions include nuclear extracts, mitochondrial fractions, microsomal fractions (membrane vesicles), and cytosolic fractions (cytoplasmic extracts).
Chromatography, gas (GC) is a type of chromatographic technique used to separate, identify, and analyze volatile compounds or vapors. In this method, the sample mixture is vaporized and carried through a column packed with a stationary phase by an inert gas (carrier gas). The components of the mixture get separated based on their partitioning between the mobile and stationary phases due to differences in their adsorption/desorption rates or solubility.
The separated components elute at different times, depending on their interaction with the stationary phase, which can be detected and quantified by various detection systems like flame ionization detector (FID), thermal conductivity detector (TCD), electron capture detector (ECD), or mass spectrometer (MS). Gas chromatography is widely used in fields such as chemistry, biochemistry, environmental science, forensics, and food analysis.
Intracellular membranes refer to the membrane structures that exist within a eukaryotic cell (excluding bacteria and archaea, which are prokaryotic and do not have intracellular membranes). These membranes compartmentalize the cell, creating distinct organelles or functional regions with specific roles in various cellular processes.
Major types of intracellular membranes include:
1. Nuclear membrane (nuclear envelope): A double-membraned structure that surrounds and protects the genetic material within the nucleus. It consists of an outer and inner membrane, perforated by nuclear pores that regulate the transport of molecules between the nucleus and cytoplasm.
2. Endoplasmic reticulum (ER): An extensive network of interconnected tubules and sacs that serve as a major site for protein folding, modification, and lipid synthesis. The ER has two types: rough ER (with ribosomes on its surface) and smooth ER (without ribosomes).
3. Golgi apparatus/Golgi complex: A series of stacked membrane-bound compartments that process, sort, and modify proteins and lipids before they are transported to their final destinations within the cell or secreted out of the cell.
4. Lysosomes: Membrane-bound organelles containing hydrolytic enzymes for breaking down various biomolecules (proteins, carbohydrates, lipids, and nucleic acids) in the process called autophagy or from outside the cell via endocytosis.
5. Peroxisomes: Single-membrane organelles involved in various metabolic processes, such as fatty acid oxidation and detoxification of harmful substances like hydrogen peroxide.
6. Vacuoles: Membrane-bound compartments that store and transport various molecules, including nutrients, waste products, and enzymes. Plant cells have a large central vacuole for maintaining turgor pressure and storing metabolites.
7. Mitochondria: Double-membraned organelles responsible for generating energy (ATP) through oxidative phosphorylation and other metabolic processes, such as the citric acid cycle and fatty acid synthesis.
8. Chloroplasts: Double-membraned organelles found in plant cells that convert light energy into chemical energy during photosynthesis, producing oxygen and organic compounds (glucose) from carbon dioxide and water.
9. Endoplasmic reticulum (ER): A network of interconnected membrane-bound tubules involved in protein folding, modification, and transport; it is divided into two types: rough ER (with ribosomes on the surface) and smooth ER (without ribosomes).
10. Nucleus: Double-membraned organelle containing genetic material (DNA) and associated proteins involved in replication, transcription, RNA processing, and DNA repair. The nuclear membrane separates the nucleoplasm from the cytoplasm and contains nuclear pores for transporting molecules between the two compartments.
A bacterial gene is a segment of DNA (or RNA in some viruses) that contains the genetic information necessary for the synthesis of a functional bacterial protein or RNA molecule. These genes are responsible for encoding various characteristics and functions of bacteria such as metabolism, reproduction, and resistance to antibiotics. They can be transmitted between bacteria through horizontal gene transfer mechanisms like conjugation, transformation, and transduction. Bacterial genes are often organized into operons, which are clusters of genes that are transcribed together as a single mRNA molecule.
It's important to note that the term "bacterial gene" is used to describe genetic elements found in bacteria, but not all genetic elements in bacteria are considered genes. For example, some DNA sequences may not encode functional products and are therefore not considered genes. Additionally, some bacterial genes may be plasmid-borne or phage-borne, rather than being located on the bacterial chromosome.
Northern blotting is a laboratory technique used in molecular biology to detect and analyze specific RNA molecules (such as mRNA) in a mixture of total RNA extracted from cells or tissues. This technique is called "Northern" blotting because it is analogous to the Southern blotting method, which is used for DNA detection.
The Northern blotting procedure involves several steps:
1. Electrophoresis: The total RNA mixture is first separated based on size by running it through an agarose gel using electrical current. This separates the RNA molecules according to their length, with smaller RNA fragments migrating faster than larger ones.
2. Transfer: After electrophoresis, the RNA bands are denatured (made single-stranded) and transferred from the gel onto a nitrocellulose or nylon membrane using a technique called capillary transfer or vacuum blotting. This step ensures that the order and relative positions of the RNA fragments are preserved on the membrane, similar to how they appear in the gel.
3. Cross-linking: The RNA is then chemically cross-linked to the membrane using UV light or heat treatment, which helps to immobilize the RNA onto the membrane and prevent it from washing off during subsequent steps.
4. Prehybridization: Before adding the labeled probe, the membrane is prehybridized in a solution containing blocking agents (such as salmon sperm DNA or yeast tRNA) to minimize non-specific binding of the probe to the membrane.
5. Hybridization: A labeled nucleic acid probe, specific to the RNA of interest, is added to the prehybridization solution and allowed to hybridize (form base pairs) with its complementary RNA sequence on the membrane. The probe can be either a DNA or an RNA molecule, and it is typically labeled with a radioactive isotope (such as ³²P) or a non-radioactive label (such as digoxigenin).
6. Washing: After hybridization, the membrane is washed to remove unbound probe and reduce background noise. The washing conditions (temperature, salt concentration, and detergent concentration) are optimized based on the stringency required for specific hybridization.
7. Detection: The presence of the labeled probe is then detected using an appropriate method, depending on the type of label used. For radioactive probes, this typically involves exposing the membrane to X-ray film or a phosphorimager screen and analyzing the resulting image. For non-radioactive probes, detection can be performed using colorimetric, chemiluminescent, or fluorescent methods.
8. Data analysis: The intensity of the signal is quantified and compared to controls (such as housekeeping genes) to determine the relative expression level of the RNA of interest. This information can be used for various purposes, such as identifying differentially expressed genes in response to a specific treatment or comparing gene expression levels across different samples or conditions.
'Gene expression regulation' refers to the processes that control whether, when, and where a particular gene is expressed, meaning the production of a specific protein or functional RNA encoded by that gene. This complex mechanism can be influenced by various factors such as transcription factors, chromatin remodeling, DNA methylation, non-coding RNAs, and post-transcriptional modifications, among others. Proper regulation of gene expression is crucial for normal cellular function, development, and maintaining homeostasis in living organisms. Dysregulation of gene expression can lead to various diseases, including cancer and genetic disorders.
Western blotting is a laboratory technique used in molecular biology to detect and quantify specific proteins in a mixture of many different proteins. This technique is commonly used to confirm the expression of a protein of interest, determine its size, and investigate its post-translational modifications. The name "Western" blotting distinguishes this technique from Southern blotting (for DNA) and Northern blotting (for RNA).
The Western blotting procedure involves several steps:
1. Protein extraction: The sample containing the proteins of interest is first extracted, often by breaking open cells or tissues and using a buffer to extract the proteins.
2. Separation of proteins by electrophoresis: The extracted proteins are then separated based on their size by loading them onto a polyacrylamide gel and running an electric current through the gel (a process called sodium dodecyl sulfate-polyacrylamide gel electrophoresis or SDS-PAGE). This separates the proteins according to their molecular weight, with smaller proteins migrating faster than larger ones.
3. Transfer of proteins to a membrane: After separation, the proteins are transferred from the gel onto a nitrocellulose or polyvinylidene fluoride (PVDF) membrane using an electric current in a process called blotting. This creates a replica of the protein pattern on the gel but now immobilized on the membrane for further analysis.
4. Blocking: The membrane is then blocked with a blocking agent, such as non-fat dry milk or bovine serum albumin (BSA), to prevent non-specific binding of antibodies in subsequent steps.
5. Primary antibody incubation: A primary antibody that specifically recognizes the protein of interest is added and allowed to bind to its target protein on the membrane. This step may be performed at room temperature or 4°C overnight, depending on the antibody's properties.
6. Washing: The membrane is washed with a buffer to remove unbound primary antibodies.
7. Secondary antibody incubation: A secondary antibody that recognizes the primary antibody (often coupled to an enzyme or fluorophore) is added and allowed to bind to the primary antibody. This step may involve using a horseradish peroxidase (HRP)-conjugated or alkaline phosphatase (AP)-conjugated secondary antibody, depending on the detection method used later.
8. Washing: The membrane is washed again to remove unbound secondary antibodies.
9. Detection: A detection reagent is added to visualize the protein of interest by detecting the signal generated from the enzyme-conjugated or fluorophore-conjugated secondary antibody. This can be done using chemiluminescent, colorimetric, or fluorescent methods.
10. Analysis: The resulting image is analyzed to determine the presence and quantity of the protein of interest in the sample.
Western blotting is a powerful technique for identifying and quantifying specific proteins within complex mixtures. It can be used to study protein expression, post-translational modifications, protein-protein interactions, and more. However, it requires careful optimization and validation to ensure accurate and reproducible results.
Medical Definition:
Superoxide dismutase (SOD) is an enzyme that catalyzes the dismutation of superoxide radicals (O2-) into oxygen (O2) and hydrogen peroxide (H2O2). This essential antioxidant defense mechanism helps protect the body's cells from damage caused by reactive oxygen species (ROS), which are produced during normal metabolic processes and can lead to oxidative stress when their levels become too high.
There are three main types of superoxide dismutase found in different cellular locations:
1. Copper-zinc superoxide dismutase (CuZnSOD or SOD1) - Present mainly in the cytoplasm of cells.
2. Manganese superoxide dismutase (MnSOD or SOD2) - Located within the mitochondrial matrix.
3. Extracellular superoxide dismutase (EcSOD or SOD3) - Found in the extracellular spaces, such as blood vessels and connective tissues.
Imbalances in SOD levels or activity have been linked to various pathological conditions, including neurodegenerative diseases, cancer, and aging-related disorders.
A dose-response relationship in the context of drugs refers to the changes in the effects or symptoms that occur as the dose of a drug is increased or decreased. Generally, as the dose of a drug is increased, the severity or intensity of its effects also increases. Conversely, as the dose is decreased, the effects of the drug become less severe or may disappear altogether.
The dose-response relationship is an important concept in pharmacology and toxicology because it helps to establish the safe and effective dosage range for a drug. By understanding how changes in the dose of a drug affect its therapeutic and adverse effects, healthcare providers can optimize treatment plans for their patients while minimizing the risk of harm.
The dose-response relationship is typically depicted as a curve that shows the relationship between the dose of a drug and its effect. The shape of the curve may vary depending on the drug and the specific effect being measured. Some drugs may have a steep dose-response curve, meaning that small changes in the dose can result in large differences in the effect. Other drugs may have a more gradual dose-response curve, where larger changes in the dose are needed to produce significant effects.
In addition to helping establish safe and effective dosages, the dose-response relationship is also used to evaluate the potential therapeutic benefits and risks of new drugs during clinical trials. By systematically testing different doses of a drug in controlled studies, researchers can identify the optimal dosage range for the drug and assess its safety and efficacy.
Protein precursors, also known as proproteins or prohormones, are inactive forms of proteins that undergo post-translational modification to become active. These modifications typically include cleavage of the precursor protein by specific enzymes, resulting in the release of the active protein. This process allows for the regulation and control of protein activity within the body. Protein precursors can be found in various biological processes, including the endocrine system where they serve as inactive hormones that can be converted into their active forms when needed.
Thin-layer chromatography (TLC) is a type of chromatography used to separate, identify, and quantify the components of a mixture. In TLC, the sample is applied as a small spot onto a thin layer of adsorbent material, such as silica gel or alumina, which is coated on a flat, rigid support like a glass plate. The plate is then placed in a developing chamber containing a mobile phase, typically a mixture of solvents.
As the mobile phase moves up the plate by capillary action, it interacts with the stationary phase and the components of the sample. Different components of the mixture travel at different rates due to their varying interactions with the stationary and mobile phases, resulting in distinct spots on the plate. The distance each component travels can be measured and compared to known standards to identify and quantify the components of the mixture.
TLC is a simple, rapid, and cost-effective technique that is widely used in various fields, including forensics, pharmaceuticals, and research laboratories. It allows for the separation and analysis of complex mixtures with high resolution and sensitivity, making it an essential tool in many analytical applications.
Isoenzymes, also known as isoforms, are multiple forms of an enzyme that catalyze the same chemical reaction but differ in their amino acid sequence, structure, and/or kinetic properties. They are encoded by different genes or alternative splicing of the same gene. Isoenzymes can be found in various tissues and organs, and they play a crucial role in biological processes such as metabolism, detoxification, and cell signaling. Measurement of isoenzyme levels in body fluids (such as blood) can provide valuable diagnostic information for certain medical conditions, including tissue damage, inflammation, and various diseases.
Dimerization is a process in which two molecules, usually proteins or similar structures, bind together to form a larger complex. This can occur through various mechanisms, such as the formation of disulfide bonds, hydrogen bonding, or other non-covalent interactions. Dimerization can play important roles in cell signaling, enzyme function, and the regulation of gene expression.
In the context of medical research and therapy, dimerization is often studied in relation to specific proteins that are involved in diseases such as cancer. For example, some drugs have been developed to target and inhibit the dimerization of certain proteins, with the goal of disrupting their function and slowing or stopping the progression of the disease.
Gene expression is the process by which the information encoded in a gene is used to synthesize a functional gene product, such as a protein or RNA molecule. This process involves several steps: transcription, RNA processing, and translation. During transcription, the genetic information in DNA is copied into a complementary RNA molecule, known as messenger RNA (mRNA). The mRNA then undergoes RNA processing, which includes adding a cap and tail to the mRNA and splicing out non-coding regions called introns. The resulting mature mRNA is then translated into a protein on ribosomes in the cytoplasm through the process of translation.
The regulation of gene expression is a complex and highly controlled process that allows cells to respond to changes in their environment, such as growth factors, hormones, and stress signals. This regulation can occur at various stages of gene expression, including transcriptional activation or repression, RNA processing, mRNA stability, and translation. Dysregulation of gene expression has been implicated in many diseases, including cancer, genetic disorders, and neurological conditions.
Tissue distribution, in the context of pharmacology and toxicology, refers to the way that a drug or xenobiotic (a chemical substance found within an organism that is not naturally produced by or expected to be present within that organism) is distributed throughout the body's tissues after administration. It describes how much of the drug or xenobiotic can be found in various tissues and organs, and is influenced by factors such as blood flow, lipid solubility, protein binding, and the permeability of cell membranes. Understanding tissue distribution is important for predicting the potential effects of a drug or toxin on different parts of the body, and for designing drugs with improved safety and efficacy profiles.
An amino acid substitution is a type of mutation in which one amino acid in a protein is replaced by another. This occurs when there is a change in the DNA sequence that codes for a particular amino acid in a protein. The genetic code is redundant, meaning that most amino acids are encoded by more than one codon (a sequence of three nucleotides). As a result, a single base pair change in the DNA sequence may not necessarily lead to an amino acid substitution. However, if a change does occur, it can have a variety of effects on the protein's structure and function, depending on the nature of the substituted amino acids. Some substitutions may be harmless, while others may alter the protein's activity or stability, leading to disease.
RNA (Ribonucleic Acid) is a single-stranded, linear polymer of ribonucleotides. It is a nucleic acid present in the cells of all living organisms and some viruses. RNAs play crucial roles in various biological processes such as protein synthesis, gene regulation, and cellular signaling. There are several types of RNA including messenger RNA (mRNA), ribosomal RNA (rRNA), transfer RNA (tRNA), small nuclear RNA (snRNA), microRNA (miRNA), and long non-coding RNA (lncRNA). These RNAs differ in their structure, function, and location within the cell.
DNA restriction enzymes, also known as restriction endonucleases, are a type of enzyme that cut double-stranded DNA at specific recognition sites. These enzymes are produced by bacteria and archaea as a defense mechanism against foreign DNA, such as that found in bacteriophages (viruses that infect bacteria).
Restriction enzymes recognize specific sequences of nucleotides (the building blocks of DNA) and cleave the phosphodiester bonds between them. The recognition sites for these enzymes are usually palindromic, meaning that the sequence reads the same in both directions when facing the opposite strands of DNA.
Restriction enzymes are widely used in molecular biology research for various applications such as genetic engineering, genome mapping, and DNA fingerprinting. They allow scientists to cut DNA at specific sites, creating precise fragments that can be manipulated and analyzed. The use of restriction enzymes has been instrumental in the development of recombinant DNA technology and the Human Genome Project.
Complementary DNA (cDNA) is a type of DNA that is synthesized from a single-stranded RNA molecule through the process of reverse transcription. In this process, the enzyme reverse transcriptase uses an RNA molecule as a template to synthesize a complementary DNA strand. The resulting cDNA is therefore complementary to the original RNA molecule and is a copy of its coding sequence, but it does not contain non-coding regions such as introns that are present in genomic DNA.
Complementary DNA is often used in molecular biology research to study gene expression, protein function, and other genetic phenomena. For example, cDNA can be used to create cDNA libraries, which are collections of cloned cDNA fragments that represent the expressed genes in a particular cell type or tissue. These libraries can then be screened for specific genes or gene products of interest. Additionally, cDNA can be used to produce recombinant proteins in heterologous expression systems, allowing researchers to study the structure and function of proteins that may be difficult to express or purify from their native sources.
The endoplasmic reticulum (ER) is a network of interconnected tubules and sacs that are present in the cytoplasm of eukaryotic cells. It is a continuous membranous organelle that plays a crucial role in the synthesis, folding, modification, and transport of proteins and lipids.
The ER has two main types: rough endoplasmic reticulum (RER) and smooth endoplasmic reticulum (SER). RER is covered with ribosomes, which give it a rough appearance, and is responsible for protein synthesis. On the other hand, SER lacks ribosomes and is involved in lipid synthesis, drug detoxification, calcium homeostasis, and steroid hormone production.
In summary, the endoplasmic reticulum is a vital organelle that functions in various cellular processes, including protein and lipid metabolism, calcium regulation, and detoxification.
Bacterial DNA refers to the genetic material found in bacteria. It is composed of a double-stranded helix containing four nucleotide bases - adenine (A), thymine (T), guanine (G), and cytosine (C) - that are linked together by phosphodiester bonds. The sequence of these bases in the DNA molecule carries the genetic information necessary for the growth, development, and reproduction of bacteria.
Bacterial DNA is circular in most bacterial species, although some have linear chromosomes. In addition to the main chromosome, many bacteria also contain small circular pieces of DNA called plasmids that can carry additional genes and provide resistance to antibiotics or other environmental stressors.
Unlike eukaryotic cells, which have their DNA enclosed within a nucleus, bacterial DNA is present in the cytoplasm of the cell, where it is in direct contact with the cell's metabolic machinery. This allows for rapid gene expression and regulation in response to changing environmental conditions.
In a medical context, "hot temperature" is not a standard medical term with a specific definition. However, it is often used in relation to fever, which is a common symptom of illness. A fever is typically defined as a body temperature that is higher than normal, usually above 38°C (100.4°F) for adults and above 37.5-38°C (99.5-101.3°F) for children, depending on the source.
Therefore, when a medical professional talks about "hot temperature," they may be referring to a body temperature that is higher than normal due to fever or other causes. It's important to note that a high environmental temperature can also contribute to an elevated body temperature, so it's essential to consider both the body temperature and the environmental temperature when assessing a patient's condition.
In genetics, sequence alignment is the process of arranging two or more DNA, RNA, or protein sequences to identify regions of similarity or homology between them. This is often done using computational methods to compare the nucleotide or amino acid sequences and identify matching patterns, which can provide insight into evolutionary relationships, functional domains, or potential genetic disorders. The alignment process typically involves adjusting gaps and mismatches in the sequences to maximize the similarity between them, resulting in an aligned sequence that can be visually represented and analyzed.
Genetic polymorphism refers to the occurrence of multiple forms (called alleles) of a particular gene within a population. These variations in the DNA sequence do not generally affect the function or survival of the organism, but they can contribute to differences in traits among individuals. Genetic polymorphisms can be caused by single nucleotide changes (SNPs), insertions or deletions of DNA segments, or other types of genetic rearrangements. They are important for understanding genetic diversity and evolution, as well as for identifying genetic factors that may contribute to disease susceptibility in humans.
Immunoenzyme techniques are a group of laboratory methods used in immunology and clinical chemistry that combine the specificity of antibody-antigen reactions with the sensitivity and amplification capabilities of enzyme reactions. These techniques are primarily used for the detection, quantitation, or identification of various analytes (such as proteins, hormones, drugs, viruses, or bacteria) in biological samples.
In immunoenzyme techniques, an enzyme is linked to an antibody or antigen, creating a conjugate. This conjugate then interacts with the target analyte in the sample, forming an immune complex. The presence and amount of this immune complex can be visualized or measured by detecting the enzymatic activity associated with it.
There are several types of immunoenzyme techniques, including:
1. Enzyme-linked Immunosorbent Assay (ELISA): A widely used method for detecting and quantifying various analytes in a sample. In ELISA, an enzyme is attached to either the capture antibody or the detection antibody. After the immune complex formation, a substrate is added that reacts with the enzyme, producing a colored product that can be measured spectrophotometrically.
2. Immunoblotting (Western blot): A method used for detecting specific proteins in a complex mixture, such as a protein extract from cells or tissues. In this technique, proteins are separated by gel electrophoresis and transferred to a membrane, where they are probed with an enzyme-conjugated antibody directed against the target protein.
3. Immunohistochemistry (IHC): A method used for detecting specific antigens in tissue sections or cells. In IHC, an enzyme-conjugated primary or secondary antibody is applied to the sample, and the presence of the antigen is visualized using a chromogenic substrate that produces a colored product at the site of the antigen-antibody interaction.
4. Immunofluorescence (IF): A method used for detecting specific antigens in cells or tissues by employing fluorophore-conjugated antibodies. The presence of the antigen is visualized using a fluorescence microscope.
5. Enzyme-linked immunosorbent assay (ELISA): A method used for detecting and quantifying specific antigens or antibodies in liquid samples, such as serum or culture supernatants. In ELISA, an enzyme-conjugated detection antibody is added after the immune complex formation, and a substrate is added that reacts with the enzyme to produce a colored product that can be measured spectrophotometrically.
These techniques are widely used in research and diagnostic laboratories for various applications, including protein characterization, disease diagnosis, and monitoring treatment responses.
Anti-bacterial agents, also known as antibiotics, are a type of medication used to treat infections caused by bacteria. These agents work by either killing the bacteria or inhibiting their growth and reproduction. There are several different classes of anti-bacterial agents, including penicillins, cephalosporins, fluoroquinolones, macrolides, and tetracyclines, among others. Each class of antibiotic has a specific mechanism of action and is used to treat certain types of bacterial infections. It's important to note that anti-bacterial agents are not effective against viral infections, such as the common cold or flu. Misuse and overuse of antibiotics can lead to antibiotic resistance, which is a significant global health concern.
Crystallization is a process in which a substance transitions from a liquid or dissolved state to a solid state, forming a crystal lattice. In the medical context, crystallization can refer to the formation of crystals within the body, which can occur under certain conditions such as changes in pH, temperature, or concentration of solutes. These crystals can deposit in various tissues and organs, leading to the formation of crystal-induced diseases or disorders.
For example, in patients with gout, uric acid crystals can accumulate in joints, causing inflammation, pain, and swelling. Similarly, in nephrolithiasis (kidney stones), minerals in the urine can crystallize and form stones that can obstruct the urinary tract. Crystallization can also occur in other medical contexts, such as in the formation of dental calculus or plaque, and in the development of cataracts in the eye.
A chemical model is a simplified representation or description of a chemical system, based on the laws of chemistry and physics. It is used to explain and predict the behavior of chemicals and chemical reactions. Chemical models can take many forms, including mathematical equations, diagrams, and computer simulations. They are often used in research, education, and industry to understand complex chemical processes and develop new products and technologies.
For example, a chemical model might be used to describe the way that atoms and molecules interact in a particular reaction, or to predict the properties of a new material. Chemical models can also be used to study the behavior of chemicals at the molecular level, such as how they bind to each other or how they are affected by changes in temperature or pressure.
It is important to note that chemical models are simplifications of reality and may not always accurately represent every aspect of a chemical system. They should be used with caution and validated against experimental data whenever possible.
Multienzyme complexes are specialized protein structures that consist of multiple enzymes closely associated or bound together, often with other cofactors and regulatory subunits. These complexes facilitate the sequential transfer of substrates along a series of enzymatic reactions, also known as a metabolic pathway. By keeping the enzymes in close proximity, multienzyme complexes enhance reaction efficiency, improve substrate specificity, and maintain proper stoichiometry between different enzymes involved in the pathway. Examples of multienzyme complexes include the pyruvate dehydrogenase complex, the citrate synthase complex, and the fatty acid synthetase complex.
The intestinal mucosa is the innermost layer of the intestines, which comes into direct contact with digested food and microbes. It is a specialized epithelial tissue that plays crucial roles in nutrient absorption, barrier function, and immune defense. The intestinal mucosa is composed of several cell types, including absorptive enterocytes, mucus-secreting goblet cells, hormone-producing enteroendocrine cells, and immune cells such as lymphocytes and macrophages.
The surface of the intestinal mucosa is covered by a single layer of epithelial cells, which are joined together by tight junctions to form a protective barrier against harmful substances and microorganisms. This barrier also allows for the selective absorption of nutrients into the bloodstream. The intestinal mucosa also contains numerous lymphoid follicles, known as Peyer's patches, which are involved in immune surveillance and defense against pathogens.
In addition to its role in absorption and immunity, the intestinal mucosa is also capable of producing hormones that regulate digestion and metabolism. Dysfunction of the intestinal mucosa can lead to various gastrointestinal disorders, such as inflammatory bowel disease, celiac disease, and food allergies.
Recombinant fusion proteins are artificially created biomolecules that combine the functional domains or properties of two or more different proteins into a single protein entity. They are generated through recombinant DNA technology, where the genes encoding the desired protein domains are linked together and expressed as a single, chimeric gene in a host organism, such as bacteria, yeast, or mammalian cells.
The resulting fusion protein retains the functional properties of its individual constituent proteins, allowing for novel applications in research, diagnostics, and therapeutics. For instance, recombinant fusion proteins can be designed to enhance protein stability, solubility, or immunogenicity, making them valuable tools for studying protein-protein interactions, developing targeted therapies, or generating vaccines against infectious diseases or cancer.
Examples of recombinant fusion proteins include:
1. Etaglunatide (ABT-523): A soluble Fc fusion protein that combines the heavy chain fragment crystallizable region (Fc) of an immunoglobulin with the extracellular domain of the human interleukin-6 receptor (IL-6R). This fusion protein functions as a decoy receptor, neutralizing IL-6 and its downstream signaling pathways in rheumatoid arthritis.
2. Etanercept (Enbrel): A soluble TNF receptor p75 Fc fusion protein that binds to tumor necrosis factor-alpha (TNF-α) and inhibits its proinflammatory activity, making it a valuable therapeutic option for treating autoimmune diseases like rheumatoid arthritis, ankylosing spondylitis, and psoriasis.
3. Abatacept (Orencia): A fusion protein consisting of the extracellular domain of cytotoxic T-lymphocyte antigen 4 (CTLA-4) linked to the Fc region of an immunoglobulin, which downregulates T-cell activation and proliferation in autoimmune diseases like rheumatoid arthritis.
4. Belimumab (Benlysta): A monoclonal antibody that targets B-lymphocyte stimulator (BLyS) protein, preventing its interaction with the B-cell surface receptor and inhibiting B-cell activation in systemic lupus erythematosus (SLE).
5. Romiplostim (Nplate): A fusion protein consisting of a thrombopoietin receptor agonist peptide linked to an immunoglobulin Fc region, which stimulates platelet production in patients with chronic immune thrombocytopenia (ITP).
6. Darbepoetin alfa (Aranesp): A hyperglycosylated erythropoiesis-stimulating protein that functions as a longer-acting form of recombinant human erythropoietin, used to treat anemia in patients with chronic kidney disease or cancer.
7. Palivizumab (Synagis): A monoclonal antibody directed against the F protein of respiratory syncytial virus (RSV), which prevents RSV infection and is administered prophylactically to high-risk infants during the RSV season.
8. Ranibizumab (Lucentis): A recombinant humanized monoclonal antibody fragment that binds and inhibits vascular endothelial growth factor A (VEGF-A), used in the treatment of age-related macular degeneration, diabetic retinopathy, and other ocular disorders.
9. Cetuximab (Erbitux): A chimeric monoclonal antibody that binds to epidermal growth factor receptor (EGFR), used in the treatment of colorectal cancer and head and neck squamous cell carcinoma.
10. Adalimumab (Humira): A fully humanized monoclonal antibody that targets tumor necrosis factor-alpha (TNF-α), used in the treatment of various inflammatory diseases, including rheumatoid arthritis, psoriasis, and Crohn's disease.
11. Bevacizumab (Avastin): A recombinant humanized monoclonal antibody that binds to VEGF-A, used in the treatment of various cancers, including colorectal, lung, breast, and kidney cancer.
12. Trastuzumab (Herceptin): A humanized monoclonal antibody that targets HER2/neu receptor, used in the treatment of breast cancer.
13. Rituximab (Rituxan): A chimeric monoclonal antibody that binds to CD20 antigen on B cells, used in the treatment of non-Hodgkin's lymphoma and rheumatoid arthritis.
14. Palivizumab (Synagis): A humanized monoclonal antibody that binds to the F protein of respiratory syncytial virus, used in the prevention of respiratory syncytial virus infection in high-risk infants.
15. Infliximab (Remicade): A chimeric monoclonal antibody that targets TNF-α, used in the treatment of various inflammatory diseases, including Crohn's disease, ulcerative colitis, rheumatoid arthritis, and ankylosing spondylitis.
16. Natalizumab (Tysabri): A humanized monoclonal antibody that binds to α4β1 integrin, used in the treatment of multiple sclerosis and Crohn's disease.
17. Adalimumab (Humira): A fully human monoclonal antibody that targets TNF-α, used in the treatment of various inflammatory diseases, including rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn's disease, and ulcerative colitis.
18. Golimumab (Simponi): A fully human monoclonal antibody that targets TNF-α, used in the treatment of rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, and ulcerative colitis.
19. Certolizumab pegol (Cimzia): A PEGylated Fab' fragment of a humanized monoclonal antibody that targets TNF-α, used in the treatment of rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, and Crohn's disease.
20. Ustekinumab (Stelara): A fully human monoclonal antibody that targets IL-12 and IL-23, used in the treatment of psoriasis, psoriatic arthritis, and Crohn's disease.
21. Secukinumab (Cosentyx): A fully human monoclonal antibody that targets IL-17A, used in the treatment of psoriasis, psoriatic arthritis, and ankylosing spondylitis.
22. Ixekizumab (Taltz): A fully human monoclonal antibody that targets IL-17A, used in the treatment of psoriasis and psoriatic arthritis.
23. Brodalumab (Siliq): A fully human monoclonal antibody that targets IL-17 receptor A, used in the treatment of psoriasis.
24. Sarilumab (Kevzara): A fully human monoclonal antibody that targets the IL-6 receptor, used in the treatment of rheumatoid arthritis.
25. Tocilizumab (Actemra): A humanized monoclonal antibody that targets the IL-6 receptor, used in the treatment of rheumatoid arthritis, systemic juvenile idiopathic arthritis, polyarticular juvenile idiopathic arthritis, giant cell arteritis, and chimeric antigen receptor T-cell-induced cytokine release syndrome.
26. Siltuximab (Sylvant): A chimeric monoclonal antibody that targets IL-6, used in the treatment of multicentric Castleman disease.
27. Satralizumab (Enspryng): A humanized monoclonal antibody that targets IL-6 receptor alpha, used in the treatment of neuromyelitis optica spectrum disorder.
28. Sirukumab (Plivensia): A human monoclonal antibody that targets IL-6, used in the treatment
Gene expression regulation in bacteria refers to the complex cellular processes that control the production of proteins from specific genes. This regulation allows bacteria to adapt to changing environmental conditions and ensure the appropriate amount of protein is produced at the right time.
Bacteria have a variety of mechanisms for regulating gene expression, including:
1. Operon structure: Many bacterial genes are organized into operons, which are clusters of genes that are transcribed together as a single mRNA molecule. The expression of these genes can be coordinately regulated by controlling the transcription of the entire operon.
2. Promoter regulation: Transcription is initiated at promoter regions upstream of the gene or operon. Bacteria have regulatory proteins called sigma factors that bind to the promoter and recruit RNA polymerase, the enzyme responsible for transcribing DNA into RNA. The binding of sigma factors can be influenced by environmental signals, allowing for regulation of transcription.
3. Attenuation: Some operons have regulatory regions called attenuators that control transcription termination. These regions contain hairpin structures that can form in the mRNA and cause transcription to stop prematurely. The formation of these hairpins is influenced by the concentration of specific metabolites, allowing for regulation of gene expression based on the availability of those metabolites.
4. Riboswitches: Some bacterial mRNAs contain regulatory elements called riboswitches that bind small molecules directly. When a small molecule binds to the riboswitch, it changes conformation and affects transcription or translation of the associated gene.
5. CRISPR-Cas systems: Bacteria use CRISPR-Cas systems for adaptive immunity against viruses and plasmids. These systems incorporate short sequences from foreign DNA into their own genome, which can then be used to recognize and cleave similar sequences in invading genetic elements.
Overall, gene expression regulation in bacteria is a complex process that allows them to respond quickly and efficiently to changing environmental conditions. Understanding these regulatory mechanisms can provide insights into bacterial physiology and help inform strategies for controlling bacterial growth and behavior.
The Fluorescent Antibody Technique (FAT) is a type of immunofluorescence assay used in laboratory medicine and pathology for the detection and localization of specific antigens or antibodies in tissues, cells, or microorganisms. In this technique, a fluorescein-labeled antibody is used to selectively bind to the target antigen or antibody, forming an immune complex. When excited by light of a specific wavelength, the fluorescein label emits light at a longer wavelength, typically visualized as green fluorescence under a fluorescence microscope.
The FAT is widely used in diagnostic microbiology for the identification and characterization of various bacteria, viruses, fungi, and parasites. It has also been applied in the diagnosis of autoimmune diseases and certain cancers by detecting specific antibodies or antigens in patient samples. The main advantage of FAT is its high sensitivity and specificity, allowing for accurate detection and differentiation of various pathogens and disease markers. However, it requires specialized equipment and trained personnel to perform and interpret the results.
Estradiol is a type of estrogen, which is a female sex hormone. It is the most potent and dominant form of estrogen in humans. Estradiol plays a crucial role in the development and maintenance of secondary sexual characteristics in women, such as breast development and regulation of the menstrual cycle. It also helps maintain bone density, protect the lining of the uterus, and is involved in cognition and mood regulation.
Estradiol is produced primarily by the ovaries, but it can also be synthesized in smaller amounts by the adrenal glands and fat cells. In men, estradiol is produced from testosterone through a process called aromatization. Abnormal levels of estradiol can contribute to various health issues, such as hormonal imbalances, infertility, osteoporosis, and certain types of cancer.
The aorta is the largest artery in the human body, which originates from the left ventricle of the heart and carries oxygenated blood to the rest of the body. It can be divided into several parts, including the ascending aorta, aortic arch, and descending aorta. The ascending aorta gives rise to the coronary arteries that supply blood to the heart muscle. The aortic arch gives rise to the brachiocephalic, left common carotid, and left subclavian arteries, which supply blood to the head, neck, and upper extremities. The descending aorta travels through the thorax and abdomen, giving rise to various intercostal, visceral, and renal arteries that supply blood to the chest wall, organs, and kidneys.
 Apoprotein
Apoprotein
ApoA-I Milano
Aequorin
Andre Francis Palmer
Hemoprotein
Vertebrate visual opsin
Free radical damage to DNA
Cytochrome c family
Surfactant protein A2
Aplysioviolin
Surfactant protein A1
Apolipoprotein
David J. Galton
CYP2A6
Genetic association
Candidate gene
Light-harvesting complex
Succinate dehydrogenase
Retinal
Drosophila melanogaster
Rhodopsin
Opsin
Transferrin
Serum amyloid A1
Holoprotein
Lipid-lowering agent
Brian Andrew Hills
Lipoprotein lipase deficiency
Chylomicron retention disease
Autumn leaf color
Apoprotein - Wikipedia
 Apoproteins | Harvard Catalyst Profiles | Harvard Catalyst
Apoproteins | Harvard Catalyst Profiles | Harvard Catalyst
JCI - A-IMilano apoprotein. Decreased high density lipoprotein cholesterol levels with significant lipoprotein modifications...
 Photosystem I P700 chlorophyll a apoprotein A2 (Chlorella vulgaris) | Protein Target - PubChem
Photosystem I P700 chlorophyll a apoprotein A2 (Chlorella vulgaris) | Protein Target - PubChem
 Apoprotein E is synthesized and secreted by resident and thioglycollate-elicited macrophages but not by pyran copolymer- or...
Apoprotein E is synthesized and secreted by resident and thioglycollate-elicited macrophages but not by pyran copolymer- or...
 Molecular cloning of the gene sequences of a major apoprotein in avian very low density lipoproteins.
Molecular cloning of the gene sequences of a major apoprotein in avian very low density lipoproteins.
 Respiratory Distress Syndrome: Background, Etiology, Epidemiology
Respiratory Distress Syndrome: Background, Etiology, Epidemiology
 Apolipoprotein B100: MedlinePlus Medical Encyclopedia
Apolipoprotein B100: MedlinePlus Medical Encyclopedia
 "Endotoxin suppresses expression of apoprotein e by mouse macrophages i" by Z Werb and J R. Chin
"Endotoxin suppresses expression of apoprotein e by mouse macrophages i" by Z Werb and J R. Chin
The influence of particle size and multiple apoprotein E-receptor interactions on the endocytic targeting of beta-VLDL in mouse...
 A transplant model to study the long term effects of apoprotein AI overexpression on large, complex atherosclerotic lesions in...
A transplant model to study the long term effects of apoprotein AI overexpression on large, complex atherosclerotic lesions in...
 Paralogs of the C-Terminal Domain of the Cyanobacterial Orange Carotenoid Protein Are Carotenoid Donors to Helical Carotenoid...
Paralogs of the C-Terminal Domain of the Cyanobacterial Orange Carotenoid Protein Are Carotenoid Donors to Helical Carotenoid...
 RCSB PDB - 4UB8: Native structure of photosystem II (dataset-2) by a femtosecond X-ray laser
RCSB PDB - 4UB8: Native structure of photosystem II (dataset-2) by a femtosecond X-ray laser
The distinction between the exposed regions and the buried regions of apoproteins in high density lipoproteins by their...
 The Three Dimensional Structure of Proteins Flashcards
The Three Dimensional Structure of Proteins Flashcards
 Apolipoprotein E Polyclonal Antibody (BS-4892R)
Apolipoprotein E Polyclonal Antibody (BS-4892R)
 Structural basis for recognition of transcriptional terminator structures by ProQ/FinO domain RNA chaperones | Nature...
Structural basis for recognition of transcriptional terminator structures by ProQ/FinO domain RNA chaperones | Nature...
 Role of apolipoprotein E in neurodegenerative diseases | NDT
Role of apolipoprotein E in neurodegenerative diseases | NDT
 Apolipoprotein B100 | Multimedia Encyclopedia | Health Information | St. Luke's Hospital
Apolipoprotein B100 | Multimedia Encyclopedia | Health Information | St. Luke's Hospital
![txid205537[Organism:noexp] - Identical Protein Groups - NCBI](data:image/png;base64,iVBORw0KGgoAAAANSUhEUgAAABAAAAAQCAYAAAAf8/9hAAAB1ElEQVQ4jaWSPWgTcRjGf/ehuWhobO2JxGJRY3taTTRV2yoqSpW6iIWO4iAoUsRBioNDKUWKLU7i4KA4OfhVREQnETRia03k7IdiS0LaQYKJQg3mLtfc30GySNUDn/V5nx/vy/vAf0pqad3db2xquiBJku93s2Tb2eEHdw1rTcsxol23sObTjN7oIp9KVmaU9kMdTxcLAyiqGtA0bfms+XKQULSdQG2EmnUx0q9ughAA8p/CFW0IN3Sv0vUI5p2zIMpUrd5JeP/Jii//80ZJUlrb9lyV8qn3zI5dB8A4MoBWtcITAKBmZe3eRmPzccYf9uIUsyzx6zQd7fMMAIjFdgxpkuPy4clFANbu6qa6fouybXtznxeAoqoBn0/zz5kvBqVQ5DBasJ5gXaPnDQAWFpwCkiwLZekyAMp2wTPAsqy5d8nEZcIHThPQo7jlIua9854BibdvekqKX8PouARAOn6F+c8pT4Bc7svz6U8f77O1cwDVV439PcPU4yHw8AUhhDPyOn4OfWOMuuZfBZp41INTLACorhC2/Jc2zsxMX8vl8lMcPBUHFL5mnpEZGa748sS42esKYS8WLtl2NjE22s/6fScIhtr48W2S5O0zIFwvp3vST6Z+myCvkaonAAAAAElFTkSuQmCC) txid205537[Organism:noexp] - Identical Protein Groups - NCBI
txid205537[Organism:noexp] - Identical Protein Groups - NCBI
 Gunnar C Hansson | Göteborgs universitet
Gunnar C Hansson | Göteborgs universitet
 wheat germ versus cholesterol
wheat germ versus cholesterol
 The effect of different dietary fatty acids on lipoprotein metabolism: concentration-dependent effects of diets enriched in...
The effect of different dietary fatty acids on lipoprotein metabolism: concentration-dependent effects of diets enriched in...
Familial type 5 hyperlipoproteinemia AND humans[mesh] AND review[publication type] - Search Results - PubMed
US20120157377A1 - Methods to enhance night vision and treatment of night blindness - Google Patents
 Research | Osteopathic Medicine | Touro University California
Research | Osteopathic Medicine | Touro University California
 Green tea Information | Mount Sinai - New York
Green tea Information | Mount Sinai - New York
 Replacing Arginine 33 for Alanine in the Hemophore HasA from Pseudomonas aeruginosa Causes Closure of the H32 Loop in the Apo...
Replacing Arginine 33 for Alanine in the Hemophore HasA from Pseudomonas aeruginosa Causes Closure of the H32 Loop in the Apo...
 Guidelines for School and Community Programs to Promote Lifelong Physical Activity Among Young People
Guidelines for School and Community Programs to Promote Lifelong Physical Activity Among Young People
 Search Results
Search ResultsLipoproteins3
- We have found that apoprotein E (ApoE), a component of plasma lipoproteins, was synthesized and secreted by resident and nonspecifically stimulated macrophages elicited with thioglycollate broth, but not by activated macrophages obtained from mice treated with bacillus Calmette-Guerin, pyran copolymer, whole Corynebacterium parvum, or bacterial endotoxin. (rupress.org)
- Scholars@Duke publication: Molecular cloning of the gene sequences of a major apoprotein in avian very low density lipoproteins. (duke.edu)
- 1. By means of 2-dimensional gradient-gel electrophoresis, the very low density lipoproteins (VLDL) apoproteins E and C profiles from human and swine plasma were studied. (unh.edu)
Lipoprotein5
- Apoprotein may refer to: Apoenzyme, the protein part of an enzyme without its characteristic prosthetic group Apolipoprotein, a lipid-binding protein that is a constituent of the plasma lipoprotein This disambiguation page lists articles associated with the title Apoprotein. (wikipedia.org)
- The observation in otherwise clinically healthy subjects of hypertriglyceridemia, reduced HDL-cholesterol, and marked apoprotein abnormalities, without a significant incidence of atherosclerotic disease in the family suggests this is a new disease entity in the field of lipoprotein pathology, very probably related to an altered amino acid composition of the apo-A-I protein (see Weisgraber et al. (jci.org)
- Surfactant is a complex lipoprotein (see the image below) composed of six phospholipids and four apoproteins. (medscape.com)
- In addition, treatment with fenofibrate results in increases in high density lipoprotein (HDL) and apoproteins apoAI and apoAII. (nih.gov)
- Through this mechanism, fenofibrate increases lipolysis and elimination of triglyceride-rich particles from plasma by activating lipoprotein lipase and reducing production of apoprotein C-III (an inhibitor of lipoprotein lipase activity). (nih.gov)
Proteins2
- Among the four surfactant apoproteins identified, surfactant protein B (SP-B) and SP-C are two small hydrophobic proteins that make up 2-4% of the surfactant mass and are present in commercially available surfactant preparations. (medscape.com)
- Incontrast to the copper metallochaperone mechanism involving thiol ligandexchanges between structurally similar chaperones and target proteins, wepropose that the Hsp40-like module interacts with urease apoprotein and/orother urease accessory proteins, while the Atx1-like domain delivershistidyl-bound nickel to the urease active site. (embl-heidelberg.de)
Particle size1
- The influence of particle size and multiple apoprotein E-receptor interactions on the endocytic targeting of beta-VLDL in mouse peritoneal macrophages. (silverchair.com)
MeSH1
- Apoproteins" is a descriptor in the National Library of Medicine's controlled vocabulary thesaurus, MeSH (Medical Subject Headings) . (harvard.edu)
Isoelectric2
- Analytical isoelectric focusing of HDL apoproteins provided evidence for multiple isoproteins in the apoprotein(apo)-A-I range, with nine different bands being detected instead of the usual four bands observed in normal subjects. (jci.org)
- 2. The molecular properties (isoelectric point and molecular weight) of the VLDL apoproteins and their isoforms were determined and showed many similarities between species. (unh.edu)
Synthesis1
- Activation of PPARα also induces an increase in the synthesis of apoproteins A-I, A-II and HDL-cholesterol. (nih.gov)
Profiles2
- This graph shows the total number of publications written about "Apoproteins" by people in Harvard Catalyst Profiles by year, and whether "Apoproteins" was a major or minor topic of these publication. (harvard.edu)
- Below are the most recent publications written about "Apoproteins" by people in Profiles. (harvard.edu)
Large1
- Fluorescence microscopy experiments revealed that large, intestinally derived, apoprotein (Apo) E-rich beta-VLDL was targeted mostly to widely distributed vesicles, whereas small, hepatically derived beta-VLDL was targeted more centrally (like LDL). (silverchair.com)
Model1
- Hypotheses about the evolution of reaction centers and their evidences (Olson, Blankenship, Vermaas models, „export model", „redox switch" hypothesis, apoprotein early model). (elte.hu)
Expression2
- Endotoxin suppresses expression of apoprotein e by mouse macrophages in vivo and in culture. (jax.org)
- Here, we explored the diversity, ecological distribution, and expression of opsin genes that encode the apoproteins of Type I r. (researchgate.net)
Lipoprotein4
- Apoprotein may refer to: Apoenzyme, the protein part of an enzyme without its characteristic prosthetic group Apolipoprotein, a lipid-binding protein that is a constituent of the plasma lipoprotein This disambiguation page lists articles associated with the title Apoprotein. (wikipedia.org)
- Surfactant is a complex lipoprotein (see the image below) composed of six phospholipids and four apoproteins. (medscape.com)
- Dietary fish oil increases conversion of very low density lipoprotein apoprotein B to low density lipoprotein. (scialert.net)
- All subfractions competed equally well in binding iodinated total lipoproteins and individual subfractions, but apoproteins common to all subfractions were ineffective in inhibiting lipoprotein acquisition. (bmj.com)
ApoB1001
- When lipid synthesis is limited in HepG2 cells apoprotein B100 (apoB100) isn't secreted but quickly degraded from the ubiquitin-proteasome pathway. (biobender.com)
B1001
- What does it mean if your Apoprotein B100 (ApoB 100) result is too high? (healthmatters.io)
Surfactant4
- A micropapillary pattern is predictive of a poor prognosis in lung adenocarcinoma, and reduced surfactant apoprotein A expression in the micropapillary pattern is an excellent indicator of a poor prognosis. (nih.gov)
- It was noteworthy that the disease-free interval in patients with high surfactant apoprotein A expression was significantly better than in patients with low surfactant apoprotein A expression (P=0.03), and no recurrence or death occurred in patients with high surfactant apoprotein A expression. (nih.gov)
- High MUC1 expression on the surface is an important characteristic of a micropapillary pattern, and reduced surfactant apoprotein A expression in the micropapillary pattern may be an excellent indicator for poor prognosis in small-size lung adenocarcinoma. (nih.gov)
- Among the four surfactant apoproteins identified, surfactant protein B (SP-B) and SP-C are two small hydrophobic proteins that make up 2-4% of the surfactant mass and are present in commercially available surfactant preparations. (medscape.com)
Phospholipids1
- Opsin the rhodopsin apoprotein was recently shown to be an ATP-independent flippase (or scramblase) that equilibrates phospholipids across photoreceptor disc membranes in mammalian retina a process required for disc homeostasis. (healthyconnectionsinc.com)
Enzyme1
- The protein encoded by this gene is an enzyme that covalently links a heme group to the apoprotein of cytochrome c. (antikoerper-online.de)
Proteins1
- Research findings of Dr. Fredrickson and colleagues have also included the discovery of several previously unknown apolipo-proteins, and new knowledge including descriptions concerning the structure and function of various apoproteins. (nih.gov)
Particles2
Plasma1
- Diagnosis is made by the absence of apoprotein B (apo B) in plasma. (msdmanuals.com)
Transfer1
- FeS cluster assembly is a complex process involving the mobilisation of Fe and S atoms from storage sources, their assembly into [Fe-S] form, their transport to specific cellular locations, and their transfer to recipient apoproteins. (embl-heidelberg.de)
Patients2
- Nov. 4, 2003 - Infusion of the Milano apoprotein A rapidly causes regression of atherosclerosis in patients with acute coronary syndromes (ACS), according to the results of a preliminary randomized trial published in the Nov. 5 issue of The Journal of the American Medical Association. (medscape.com)
- Nous avons réalisé un essai en double aveugle contre placebo sur 50 patients atteints de diabète de type 2 randomisés pour recevoir 2 g/jour d'acides gras oméga 3 purifiés ou un placebo pendant 10 semaines. (who.int)