22q11 Deletion Syndrome
DiGeorge Syndrome
Chromosomes, Human, Pair 22
Velopharyngeal Insufficiency
Gene Deletion
Abnormalities, Multiple
Craniofacial Abnormalities
Chromosome Disorders
Intellectual Disability
Jacobsen Distal 11q Deletion Syndrome
Hypoparathyroidism
Developmental Disabilities
Parietal Bone
Cri-du-Chat Syndrome
Phenotype
Hemizygote
In Situ Hybridization, Fluorescence
Facies
Chromosome Breakage
Hypocalcemia
Catechol O-Methyltransferase
WAGR Syndrome
Muscle Hypotonia
Schizophrenia
Heart Defects, Congenital
T-Box Domain Proteins
Stanford-Binet Test
Exostoses, Multiple Hereditary
Learning Disorders
Down Syndrome
Metabolic Syndrome X
Mutation
Molecular Sequence Data
Monosomy
Language Development Disorders
Comparative Genomic Hybridization
Chromosome Aberrations
Pedigree
Chromosome Banding
Chromosome Mapping
Chromosomes, Human, Pair 1
Staphylococcus lugdunensis
Chromosomes, Human, Pair 6
Branchial Region
Intelligence Tests
Genotype
Base Sequence
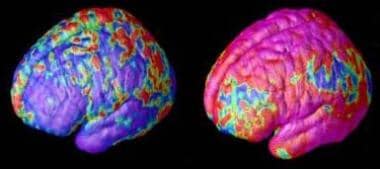
Brain
Gene Dosage
Nephrotic Syndrome
Magnetic Resonance Imaging
Siblings
Growth Disorders
Sjogren's Syndrome
Gene Rearrangement
Cognition Disorders
Translocation, Genetic
Case-Control Studies
Intelligence
Chromosomes, Human, Pair 18
Neuropsychological Tests
Seizures
Genetic Association Studies
Psychotic Disorders
Turner Syndrome
Psychiatric Status Rating Scales
Myelodysplastic Syndromes
Amino Acid Sequence
Genetic Predisposition to Disease
Williams Syndrome
Complement C1q
Polymorphism, Single Nucleotide
Prader-Willi Syndrome
Cushing Syndrome
Mice, Inbred C57BL
Mental Disorders
Analysis of Variance
Acute Coronary Syndrome
Polycystic Ovary Syndrome
Three phases of DiGeorge/22q11 deletion syndrome pathogenesis during brain development: patterning, proliferation, and mitochondrial functions of 22q11 genes. (1/16)
(+info)Dysregulation of presynaptic calcium and synaptic plasticity in a mouse model of 22q11 deletion syndrome. (2/16)
(+info)A patient with 22q11.2 deletion syndrome: case report. (3/16)
(+info)Atypical developmental trajectory of functionally significant cortical areas in children with chromosome 22q11.2 deletion syndrome. (4/16)
(+info)Hypoparathyroidism and autoimmunity in the 22q11.2 deletion syndrome. (5/16)
(+info)Proton magnetic resonance spectroscopy in 22q11 deletion syndrome. (6/16)
(+info)Genotype and cardiovascular phenotype correlations with TBX1 in 1,022 velo-cardio-facial/DiGeorge/22q11.2 deletion syndrome patients. (7/16)
(+info)Delayed-onset hypoparathyroidism in an adolescent with chromosome 22Q11 deletion syndrome. (8/16)
(+info)22q11 Deletion Syndrome, also known as DiGeorge Syndrome or Velocardiofacial Syndrome, is a genetic disorder caused by the deletion of a small piece of chromosome 22 at a specific location (q11.2). This deletion results in the poor development of several body systems, including the following:
* The third and fourth pharyngeal pouches, which give rise to various structures in the neck, such as the parathyroid glands and thymus. As a result, affected individuals often have hypocalcemia (low levels of calcium in the blood) due to decreased parathyroid hormone production, and may have immune deficiencies due to abnormal or missing thymus tissue.
* The fourth pharyngeal arch, which forms parts of the aortic arch, the cranial base, and the neck. This can lead to congenital heart defects, such as tetralogy of Fallot or interrupted aortic arch.
* The branchial arches, which contribute to the formation of the face and neck. This can result in distinctive facial features, such as a prominent nasal bridge, hooded eyelids, a small jaw, and low-set ears.
The severity of 22q11 Deletion Syndrome can vary widely, even among members of the same family. Common symptoms include heart defects, palate abnormalities, immune deficiencies, developmental delays, learning disabilities, behavioral problems, and hearing loss. Some individuals with this syndrome may also have psychiatric disorders, such as schizophrenia or anxiety disorders.
Treatment for 22q11 Deletion Syndrome typically involves a multidisciplinary approach, addressing each of the affected body systems. For example, heart defects may require surgical repair, while immune deficiencies may be managed with medications or thymus transplantation. Calcium supplements and vitamin D may be prescribed to treat hypocalcemia. Speech therapy, occupational therapy, and special education services can help address developmental delays and learning disabilities.
DiGeorge syndrome is a genetic disorder caused by the deletion of a small piece of chromosome 22. It is also known as 22q11.2 deletion syndrome. The symptoms and severity can vary widely among affected individuals, but often include birth defects such as congenital heart disease, poor immune system function, and palatal abnormalities. Characteristic facial features, learning disabilities, and behavioral problems are also common. Some people with DiGeorge syndrome may have mild symptoms while others may be more severely affected. The condition is typically diagnosed through genetic testing. Treatment is focused on managing the specific symptoms and may include surgery, medications, and therapy.
Human chromosome pair 22 consists of two rod-shaped structures present in the nucleus of each cell in the human body. Each chromosome is made up of DNA tightly coiled around histone proteins, forming a complex structure called a chromatin.
Chromosome pair 22 is one of the 22 autosomal pairs of human chromosomes, meaning they are not sex chromosomes (X or Y). Chromosome 22 is the second smallest human chromosome, with each arm of the chromosome designated as p and q. The short arm is labeled "p," and the long arm is labeled "q."
Chromosome 22 contains several genes that are associated with various genetic disorders, including DiGeorge syndrome, velocardiofacial syndrome, and cat-eye syndrome, which result from deletions or duplications of specific regions on the chromosome. Additionally, chromosome 22 is the location of the NRXN1 gene, which has been associated with an increased risk for autism spectrum disorder (ASD) and schizophrenia when deleted or disrupted.
Understanding the genetic makeup of human chromosome pair 22 can provide valuable insights into human genetics, evolution, and disease susceptibility, as well as inform medical diagnoses, treatments, and research.
A syndrome, in medical terms, is a set of symptoms that collectively indicate or characterize a disease, disorder, or underlying pathological process. It's essentially a collection of signs and/or symptoms that frequently occur together and can suggest a particular cause or condition, even though the exact physiological mechanisms might not be fully understood.
For example, Down syndrome is characterized by specific physical features, cognitive delays, and other developmental issues resulting from an extra copy of chromosome 21. Similarly, metabolic syndromes like diabetes mellitus type 2 involve a group of risk factors such as obesity, high blood pressure, high blood sugar, and abnormal cholesterol or triglyceride levels that collectively increase the risk of heart disease, stroke, and diabetes.
It's important to note that a syndrome is not a specific diagnosis; rather, it's a pattern of symptoms that can help guide further diagnostic evaluation and management.
A chromosome deletion is a type of genetic abnormality that occurs when a portion of a chromosome is missing or deleted. Chromosomes are thread-like structures located in the nucleus of cells that contain our genetic material, which is organized into genes.
Chromosome deletions can occur spontaneously during the formation of reproductive cells (eggs or sperm) or can be inherited from a parent. They can affect any chromosome and can vary in size, from a small segment to a large portion of the chromosome.
The severity of the symptoms associated with a chromosome deletion depends on the size and location of the deleted segment. In some cases, the deletion may be so small that it does not cause any noticeable symptoms. However, larger deletions can lead to developmental delays, intellectual disabilities, physical abnormalities, and various medical conditions.
Chromosome deletions are typically detected through a genetic test called karyotyping, which involves analyzing the number and structure of an individual's chromosomes. Other more precise tests, such as fluorescence in situ hybridization (FISH) or chromosomal microarray analysis (CMA), may also be used to confirm the diagnosis and identify the specific location and size of the deletion.
Velopharyngeal Insufficiency (VPI) is a medical condition that affects the proper functioning of the velopharyngeal valve, which is responsible for closing off the nasal cavity from the mouth during speech. This valve is made up of the soft palate (the back part of the roof of the mouth), the pharynx (the back of the throat), and the muscles that control their movement.
In VPI, the velopharyngeal valve does not close completely or properly during speech, causing air to escape through the nose and resulting in hypernasality, nasal emission, and/or articulation errors. This can lead to difficulties with speech clarity and understanding, as well as social and emotional challenges.
VPI can be present from birth (congenital) or acquired later in life due to factors such as cleft palate, neurological disorders, trauma, or surgery. Treatment for VPI may include speech therapy, surgical intervention, or a combination of both.
Gene deletion is a type of mutation where a segment of DNA, containing one or more genes, is permanently lost or removed from a chromosome. This can occur due to various genetic mechanisms such as homologous recombination, non-homologous end joining, or other types of genomic rearrangements.
The deletion of a gene can have varying effects on the organism, depending on the function of the deleted gene and its importance for normal physiological processes. If the deleted gene is essential for survival, the deletion may result in embryonic lethality or developmental abnormalities. However, if the gene is non-essential or has redundant functions, the deletion may not have any noticeable effects on the organism's phenotype.
Gene deletions can also be used as a tool in genetic research to study the function of specific genes and their role in various biological processes. For example, researchers may use gene deletion techniques to create genetically modified animal models to investigate the impact of gene deletion on disease progression or development.
'Abnormalities, Multiple' is a broad term that refers to the presence of two or more structural or functional anomalies in an individual. These abnormalities can be present at birth (congenital) or can develop later in life (acquired). They can affect various organs and systems of the body and can vary greatly in severity and impact on a person's health and well-being.
Multiple abnormalities can occur due to genetic factors, environmental influences, or a combination of both. Chromosomal abnormalities, gene mutations, exposure to teratogens (substances that cause birth defects), and maternal infections during pregnancy are some of the common causes of multiple congenital abnormalities.
Examples of multiple congenital abnormalities include Down syndrome, Turner syndrome, and VATER/VACTERL association. Acquired multiple abnormalities can result from conditions such as trauma, infection, degenerative diseases, or cancer.
The medical evaluation and management of individuals with multiple abnormalities depend on the specific abnormalities present and their impact on the individual's health and functioning. A multidisciplinary team of healthcare professionals is often involved in the care of these individuals to address their complex needs.
Craniofacial abnormalities refer to a group of birth defects that affect the development of the skull and face. These abnormalities can range from mild to severe and may involve differences in the shape and structure of the head, face, and jaws, as well as issues with the formation of facial features such as the eyes, nose, and mouth.
Craniofacial abnormalities can be caused by genetic factors, environmental influences, or a combination of both. Some common examples of craniofacial abnormalities include cleft lip and palate, craniosynostosis (premature fusion of the skull bones), and hemifacial microsomia (underdevelopment of one side of the face).
Treatment for craniofacial abnormalities may involve a team of healthcare professionals, including plastic surgeons, neurosurgeons, orthodontists, speech therapists, and other specialists. Treatment options may include surgery, bracing, therapy, and other interventions to help improve function and appearance.
Chromosome disorders are a group of genetic conditions caused by abnormalities in the number or structure of chromosomes. Chromosomes are thread-like structures located in the nucleus of cells that contain most of the body's genetic material, which is composed of DNA and proteins. Normally, humans have 23 pairs of chromosomes, for a total of 46 chromosomes.
Chromosome disorders can result from changes in the number of chromosomes (aneuploidy) or structural abnormalities in one or more chromosomes. Some common examples of chromosome disorders include:
1. Down syndrome: a condition caused by an extra copy of chromosome 21, resulting in intellectual disability, developmental delays, and distinctive physical features.
2. Turner syndrome: a condition that affects only females and is caused by the absence of all or part of one X chromosome, resulting in short stature, lack of sexual development, and other symptoms.
3. Klinefelter syndrome: a condition that affects only males and is caused by an extra copy of the X chromosome, resulting in tall stature, infertility, and other symptoms.
4. Cri-du-chat syndrome: a condition caused by a deletion of part of the short arm of chromosome 5, resulting in intellectual disability, developmental delays, and a distinctive cat-like cry.
5. Fragile X syndrome: a condition caused by a mutation in the FMR1 gene on the X chromosome, resulting in intellectual disability, behavioral problems, and physical symptoms.
Chromosome disorders can be diagnosed through various genetic tests, such as karyotyping, chromosomal microarray analysis (CMA), or fluorescence in situ hybridization (FISH). Treatment for these conditions depends on the specific disorder and its associated symptoms and may include medical interventions, therapies, and educational support.
A sequence deletion in a genetic context refers to the removal or absence of one or more nucleotides (the building blocks of DNA or RNA) from a specific region in a DNA or RNA molecule. This type of mutation can lead to the loss of genetic information, potentially resulting in changes in the function or expression of a gene. If the deletion involves a critical portion of the gene, it can cause diseases, depending on the role of that gene in the body. The size of the deleted sequence can vary, ranging from a single nucleotide to a large segment of DNA.
Intellectual disability (ID) is a term used when there are significant limitations in both intellectual functioning and adaptive behavior, which covers many everyday social and practical skills. This disability originates before the age of 18.
Intellectual functioning, also known as intelligence, refers to general mental capacity, such as learning, reasoning, problem-solving, and other cognitive skills. Adaptive behavior includes skills needed for day-to-day life, such as communication, self-care, social skills, safety judgement, and basic academic skills.
Intellectual disability is characterized by below-average intelligence or mental ability and a lack of skills necessary for day-to-day living. It can be mild, moderate, severe, or profound, depending on the degree of limitation in intellectual functioning and adaptive behavior.
It's important to note that people with intellectual disabilities have unique strengths and limitations, just like everyone else. With appropriate support and education, they can lead fulfilling lives and contribute to their communities in many ways.
Jacobsen Distal 11q Deletion Syndrome, also known as Jacobsen Syndrome or 11q terminal deletion disorder, is a rare genetic condition caused by a deletion of the distal portion of the long arm (q) of chromosome 11. The size of the deleted segment can vary significantly among individuals with this syndrome, which results in a range of symptoms and severity.
The medical definition of Jacobsen Distal 11q Deletion Syndrome is:
A contiguous gene deletion syndrome resulting from a chromosomal deletion of the distal region of the long arm of chromosome 11 (11q). The typical deletion size varies from 7 to 20 megabases, with breakpoints usually located between q23 and q25. Characteristic features include developmental delay, intellectual disability, distinctive facial dysmorphisms, growth retardation, congenital heart defects, skeletal abnormalities, gastrointestinal issues, and thrombocytopenia (low platelet count). The severity of the symptoms depends on the extent and location of the deletion. In some cases, additional chromosomal abnormalities or variants may contribute to the phenotype.
It is essential to note that this medical definition is a general guideline, and individual presentations can vary significantly. For an accurate diagnosis and personalized prognosis, consult with a certified medical professional or genetic counselor.
Hypoparathyroidism is a medical condition characterized by decreased levels or insufficient function of parathyroid hormone (PTH), which is produced and released by the parathyroid glands. These glands are located in the neck, near the thyroid gland, and play a crucial role in regulating calcium and phosphorus levels in the body.
In hypoparathyroidism, low PTH levels result in decreased absorption of calcium from the gut, increased excretion of calcium through the kidneys, and impaired regulation of bone metabolism. This leads to low serum calcium levels (hypocalcemia) and high serum phosphorus levels (hyperphosphatemia).
Symptoms of hypoparathyroidism can include muscle cramps, spasms, or tetany (involuntary muscle contractions), numbness or tingling sensations in the fingers, toes, and around the mouth, fatigue, weakness, anxiety, cognitive impairment, and in severe cases, seizures. Hypoparathyroidism can be caused by various factors, including surgical removal or damage to the parathyroid glands, autoimmune disorders, radiation therapy, genetic defects, or low magnesium levels. Treatment typically involves calcium and vitamin D supplementation to maintain normal serum calcium levels and alleviate symptoms. In some cases, recombinant PTH (Natpara) may be prescribed as well.
Developmental disabilities are a group of conditions that arise in childhood and are characterized by significant impairments in cognitive functioning, physical development, or both. These disabilities can affect various areas of an individual's life, including their ability to learn, communicate, socialize, and take care of themselves.
Examples of developmental disabilities include intellectual disabilities, cerebral palsy, autism spectrum disorder, Down syndrome, and fetal alcohol spectrum disorders. These conditions are typically diagnosed in childhood and can persist throughout an individual's life.
The causes of developmental disabilities are varied and can include genetic factors, environmental influences, and complications during pregnancy or childbirth. In some cases, the exact cause may be unknown.
It is important to note that individuals with developmental disabilities have unique strengths and abilities, as well as challenges. With appropriate support and services, they can lead fulfilling lives and participate actively in their communities.
The parietal bone is one of the four flat bones that form the skull's cranial vault, which protects the brain. There are two parietal bones in the skull, one on each side, located posterior to the frontal bone and temporal bone, and anterior to the occipital bone. Each parietal bone has a squamous part, which forms the roof and sides of the skull, and a smaller, wing-like portion called the mastoid process. The parietal bones contribute to the formation of the coronal and lambdoid sutures, which are fibrous joints that connect the bones in the skull.
Cri-du-chat syndrome is a genetic disorder caused by a deletion of part of chromosome 5. The name "Cri-du-chat" means "cry of the cat" in French, and refers to the characteristic high-pitched, distinctive cry of affected infants, which sounds similar to the meow of a cat.
The symptoms of Cri-du-chat syndrome can vary widely in severity, but typically include intellectual disability, developmental delays, speech and language difficulties, low muscle tone, and distinctive facial features such as wide-set eyes, a shortened jaw, and a rounded nose. Affected individuals may also have hearing and vision problems, heart defects, and gastrointestinal issues.
Cri-du-chat syndrome is usually not inherited and occurs randomly during the formation of the egg or sperm. It affects approximately 1 in 20,000 to 50,000 newborns worldwide. There is no cure for Cri-du-chat syndrome, but early intervention with therapies such as speech and language therapy, physical therapy, and occupational therapy can help improve outcomes and quality of life for affected individuals.
A phenotype is the physical or biochemical expression of an organism's genes, or the observable traits and characteristics resulting from the interaction of its genetic constitution (genotype) with environmental factors. These characteristics can include appearance, development, behavior, and resistance to disease, among others. Phenotypes can vary widely, even among individuals with identical genotypes, due to differences in environmental influences, gene expression, and genetic interactions.
A hemizygote is an individual or a cell that has only one copy of a particular gene, as opposed to the usual two copies (one from each parent) in a diploid organism. This condition typically occurs when the gene is located on a sex chromosome (X or Y). For example, males in humans are hemizygous for all genes located on the X chromosome since they have only one X chromosome and one Y chromosome. If a recessive allele is present on the X chromosome of a male, he will express that trait because there is no corresponding allele to mask its effect. In contrast, females have two X chromosomes and would need to inherit two copies of the recessive allele to express the trait.
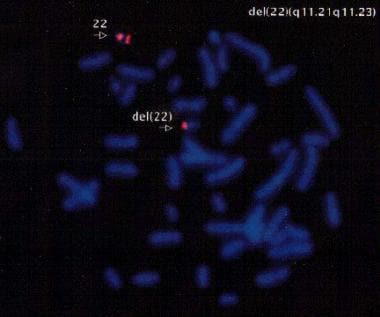
In situ hybridization, fluorescence (FISH) is a type of molecular cytogenetic technique used to detect and localize the presence or absence of specific DNA sequences on chromosomes through the use of fluorescent probes. This technique allows for the direct visualization of genetic material at a cellular level, making it possible to identify chromosomal abnormalities such as deletions, duplications, translocations, and other rearrangements.
The process involves denaturing the DNA in the sample to separate the double-stranded molecules into single strands, then adding fluorescently labeled probes that are complementary to the target DNA sequence. The probe hybridizes to the complementary sequence in the sample, and the location of the probe is detected by fluorescence microscopy.
FISH has a wide range of applications in both clinical and research settings, including prenatal diagnosis, cancer diagnosis and monitoring, and the study of gene expression and regulation. It is a powerful tool for identifying genetic abnormalities and understanding their role in human disease.
"Facies" is a medical term that refers to the typical appearance of a person or part of the body, particularly the face, which may provide clues about their underlying medical condition or genetic background. A specific facies is often associated with certain syndromes or disorders. For example, a "downsyndrome facies" refers to the distinctive facial features commonly found in individuals with Down syndrome, such as a flattened nasal bridge, almond-shaped eyes, and an upward slant to the eyelids.
It's important to note that while facies can provide valuable diagnostic information, it should be used in conjunction with other clinical findings and genetic testing to make a definitive diagnosis. Additionally, facies should be described objectively and without judgment, as they are simply physical characteristics associated with certain medical conditions.
Chromosome breakage is a medical term that refers to the breaking or fragmentation of chromosomes, which are thread-like structures located in the nucleus of cells that carry genetic information. Normally, chromosomes are tightly coiled and consist of two strands called chromatids, joined together at a central point called the centromere.
Chromosome breakage can occur spontaneously or be caused by environmental factors such as radiation or chemicals, or inherited genetic disorders. When a chromosome breaks, it can result in various genetic abnormalities, depending on the location and severity of the break.
For instance, if the break occurs in a region containing important genes, it can lead to the loss or alteration of those genes, causing genetic diseases or birth defects. In some cases, the broken ends of the chromosome may rejoin incorrectly, leading to chromosomal rearrangements such as translocations, deletions, or inversions. These rearrangements can also result in genetic disorders or cancer.
Chromosome breakage is commonly observed in individuals with certain inherited genetic conditions, such as Bloom syndrome, Fanconi anemia, and ataxia-telangiectasia, which are characterized by an increased susceptibility to chromosome breakage due to defects in DNA repair mechanisms.
Hypocalcemia is a medical condition characterized by an abnormally low level of calcium in the blood. Calcium is a vital mineral that plays a crucial role in various bodily functions, including muscle contraction, nerve impulse transmission, and bone formation. Normal calcium levels in the blood usually range from 8.5 to 10.2 milligrams per deciliter (mg/dL). Hypocalcemia is typically defined as a serum calcium level below 8.5 mg/dL or, when adjusted for albumin (a protein that binds to calcium), below 8.4 mg/dL (ionized calcium).
Hypocalcemia can result from several factors, such as vitamin D deficiency, hypoparathyroidism (underactive parathyroid glands), kidney dysfunction, certain medications, and severe magnesium deficiency. Symptoms of hypocalcemia may include numbness or tingling in the fingers, toes, or lips; muscle cramps or spasms; seizures; and, in severe cases, cognitive impairment or cardiac arrhythmias. Treatment typically involves correcting the underlying cause and administering calcium and vitamin D supplements to restore normal calcium levels in the blood.
Catechol-O-methyltransferase (COMT) is an enzyme that plays a role in the metabolism of catecholamines, which are neurotransmitters and hormones such as dopamine, norepinephrine, and epinephrine. COMT mediates the transfer of a methyl group from S-adenosylmethionine (SAM) to a catechol functional group in these molecules, resulting in the formation of methylated products that are subsequently excreted.
The methylation of catecholamines by COMT regulates their concentration and activity in the body, and genetic variations in the COMT gene can affect enzyme function and contribute to individual differences in the metabolism of these neurotransmitters. This has been implicated in various neurological and psychiatric conditions, including Parkinson's disease, schizophrenia, and attention deficit hyperactivity disorder (ADHD).
WAGR syndrome is a genetic disorder that stands for four main features: Wilms' tumor (a type of kidney cancer), aniridia (absence of the iris in the eye), genitourinary anomalies, and mental retardation. It is caused by a deletion of genetic material on chromosome 11, which includes the WAFT gene. This syndrome is rare and occurs in approximately 1 in 500,000 individuals.
The Wilms' tumor in WAGR syndrome typically develops during childhood, with about half of affected children developing this type of cancer by age 7. Aniridia is usually present at birth and can cause decreased vision or sensitivity to light. Genitourinary anomalies can include abnormalities of the reproductive and urinary systems, such as undescended testicles in males or structural abnormalities of the kidneys or urinary tract. Mental retardation ranges from mild to severe and is often accompanied by developmental delays and behavioral problems.
Early diagnosis and treatment of WAGR syndrome can improve outcomes for affected individuals. Treatment typically includes surveillance for Wilms' tumor, management of aniridia and genitourinary anomalies, and special education and therapy services for mental retardation.
Karyotyping is a medical laboratory test used to study the chromosomes in a cell. It involves obtaining a sample of cells from a patient, usually from blood or bone marrow, and then staining the chromosomes so they can be easily seen under a microscope. The chromosomes are then arranged in pairs based on their size, shape, and other features to create a karyotype. This visual representation allows for the identification and analysis of any chromosomal abnormalities, such as extra or missing chromosomes, or structural changes like translocations or inversions. These abnormalities can provide important information about genetic disorders, diseases, and developmental problems.
Muscle hypotonia, also known as decreased muscle tone, refers to a condition where the muscles appear to be flaccid or lacking in tension and stiffness. This results in reduced resistance to passive movements, making the limbs feel "floppy" or "like a rag doll." It can affect any muscle group in the body and can be caused by various medical conditions, including neurological disorders, genetic diseases, and injuries to the nervous system. Hypotonia should not be confused with muscle weakness, which refers to the inability to generate normal muscle strength.
Schizophrenia is a severe mental disorder characterized by disturbances in thought, perception, emotion, and behavior. It often includes hallucinations (usually hearing voices), delusions, paranoia, and disorganized speech and behavior. The onset of symptoms typically occurs in late adolescence or early adulthood. Schizophrenia is a complex, chronic condition that requires ongoing treatment and management. It significantly impairs social and occupational functioning, and it's often associated with reduced life expectancy due to comorbid medical conditions. The exact causes of schizophrenia are not fully understood, but research suggests that genetic, environmental, and neurodevelopmental factors play a role in its development.
Congenital heart defects (CHDs) are structural abnormalities in the heart that are present at birth. They can affect any part of the heart's structure, including the walls of the heart, the valves inside the heart, and the major blood vessels that lead to and from the heart.
Congenital heart defects can range from mild to severe and can cause various symptoms depending on the type and severity of the defect. Some common symptoms of CHDs include cyanosis (a bluish tint to the skin, lips, and fingernails), shortness of breath, fatigue, poor feeding, and slow growth in infants and children.
There are many different types of congenital heart defects, including:
1. Septal defects: These are holes in the walls that separate the four chambers of the heart. The two most common septal defects are atrial septal defect (ASD) and ventricular septal defect (VSD).
2. Valve abnormalities: These include narrowed or leaky valves, which can affect blood flow through the heart.
3. Obstruction defects: These occur when blood flow is blocked or restricted due to narrowing or absence of a part of the heart's structure. Examples include pulmonary stenosis and coarctation of the aorta.
4. Cyanotic heart defects: These cause a lack of oxygen in the blood, leading to cyanosis. Examples include tetralogy of Fallot and transposition of the great arteries.
The causes of congenital heart defects are not fully understood, but genetic factors and environmental influences during pregnancy may play a role. Some CHDs can be detected before birth through prenatal testing, while others may not be diagnosed until after birth or later in childhood. Treatment for CHDs may include medication, surgery, or other interventions to improve blood flow and oxygenation of the body's tissues.
Eye abnormalities refer to any structural or functional anomalies that affect the eye or its surrounding tissues. These abnormalities can be present at birth (congenital) or acquired later in life due to various factors such as injury, disease, or aging. Some examples of eye abnormalities include:
1. Strabismus: Also known as crossed eyes, strabismus is a condition where the eyes are misaligned and point in different directions.
2. Nystagmus: This is an involuntary movement of the eyes that can be horizontal, vertical, or rotatory.
3. Cataracts: A cataract is a clouding of the lens inside the eye that can cause vision loss.
4. Glaucoma: This is a group of eye conditions that damage the optic nerve and can lead to vision loss.
5. Retinal disorders: These include conditions such as retinal detachment, macular degeneration, and diabetic retinopathy.
6. Corneal abnormalities: These include conditions such as keratoconus, corneal ulcers, and Fuchs' dystrophy.
7. Orbital abnormalities: These include conditions such as orbital tumors, thyroid eye disease, and Graves' ophthalmopathy.
8. Ptosis: This is a condition where the upper eyelid droops over the eye.
9. Color blindness: A condition where a person has difficulty distinguishing between certain colors.
10. Microphthalmia: A condition where one or both eyes are abnormally small.
These are just a few examples of eye abnormalities, and there are many others that can affect the eye and its functioning. If you suspect that you have an eye abnormality, it is important to consult with an ophthalmologist for proper diagnosis and treatment.
T-box domain proteins are a family of transcription factors that share a highly conserved DNA-binding domain, known as the T-box. The T-box domain is a DNA-binding motif that specifically recognizes and binds to T-box binding elements (TBEs) in the regulatory regions of target genes. These proteins play crucial roles during embryonic development, particularly in the formation of specific tissues and organs, such as the heart, limbs, and brain. Mutations in T-box domain proteins can lead to various congenital defects and developmental disorders. Some examples of T-box domain proteins include TBX1, TBX5, and TBX20.
The Stanford-Binet Test is a widely used, individually administered intelligence test that was revised from the original Binet-Simon Scale by Lewis Terman at Stanford University in 1916. It is designed to measure various cognitive abilities and intelligence across a broad age range, from early childhood to adulthood. The test assesses five factors of cognitive ability: fluid reasoning, knowledge, quantitative reasoning, visual-spatial processing, and working memory.
The Stanford-Binet Test consists of several subtests that measure different skills and abilities. It yields a composite score, called the Intelligence Quotient (IQ), which is a ratio of mental age to chronological age, multiplied by 100. The test also provides detailed information about an individual's strengths and weaknesses in various areas of cognitive functioning.
Over the years, the Stanford-Binet Test has undergone several revisions to improve its psychometric properties, update its content, and reflect current theories of intelligence. The most recent version, the Stanford-Binet Fifth Edition (SB5), was published in 2003 and includes updated norms, a broader age range (2-85+ years), and a more comprehensive assessment of cognitive abilities.
The Stanford-Binet Test is used for various purposes, including identifying individuals who may have intellectual disabilities or giftedness, educational planning, career counseling, and research. It is considered a reliable and valid measure of intelligence, but like all psychological tests, it should be administered and interpreted by trained professionals who are aware of its limitations and potential sources of bias.
Multiple hereditary exostoses (MHE) is a genetic condition characterized by the growth of multiple benign tumors known as osteochondromas. These tumors typically develop at the ends of long bones near the growth plates and can cause various skeletal deformities, limitations in mobility, and other health issues.
MHE is usually inherited in an autosomal dominant pattern, meaning that a child has a 50% chance of inheriting the condition if one parent has it. However, some cases may result from spontaneous mutations. The condition typically becomes apparent during childhood or adolescence and can affect both sexes equally.
The primary diagnostic feature of MHE is the presence of multiple osteochondromas, which are made up of bone and cartilage. These growths can cause a range of symptoms, including pain, swelling, decreased mobility, and an increased risk of fractures. In some cases, they may also lead to complications such as nerve compression or vascular damage.
Treatment for MHE typically involves surgical removal of the osteochondromas, particularly if they are causing significant symptoms or complications. Regular monitoring is also important to detect any new growths and assess their potential impact on health. In addition, physical therapy and other supportive measures may be recommended to help manage symptoms and maintain mobility.
A learning disorder is a neurodevelopmental disorder that affects an individual's ability to acquire, process, and use information in one or more academic areas despite normal intelligence and adequate instruction. It can manifest as difficulties with reading (dyslexia), writing (dysgraphia), mathematics (dyscalculia), or other academic skills. Learning disorders are not the result of low intelligence, lack of motivation, or environmental factors alone, but rather reflect a significant discrepancy between an individual's cognitive abilities and their academic achievement. They can significantly impact a person's ability to perform in school, at work, and in daily life, making it important to diagnose and manage these disorders effectively.
Down syndrome is a genetic disorder caused by the presence of all or part of a third copy of chromosome 21. It is characterized by intellectual and developmental disabilities, distinctive facial features, and sometimes physical growth delays and health problems. The condition affects approximately one in every 700 babies born in the United States.
Individuals with Down syndrome have varying degrees of cognitive impairment, ranging from mild to moderate or severe. They may also have delayed development, including late walking and talking, and may require additional support and education services throughout their lives.
People with Down syndrome are at increased risk for certain health conditions, such as congenital heart defects, respiratory infections, hearing loss, vision problems, gastrointestinal issues, and thyroid disorders. However, many individuals with Down syndrome live healthy and fulfilling lives with appropriate medical care and support.
The condition is named after John Langdon Down, an English physician who first described the syndrome in 1866.
Metabolic syndrome, also known as Syndrome X, is a cluster of conditions that increase the risk of heart disease, stroke, and diabetes. It is not a single disease but a group of risk factors that often co-occur. According to the American Heart Association and the National Heart, Lung, and Blood Institute, a person has metabolic syndrome if they have any three of the following five conditions:
1. Abdominal obesity (waist circumference of 40 inches or more in men, and 35 inches or more in women)
2. Triglyceride level of 150 milligrams per deciliter of blood (mg/dL) or greater
3. HDL cholesterol level of less than 40 mg/dL in men or less than 50 mg/dL in women
4. Systolic blood pressure of 130 millimeters of mercury (mmHg) or greater, or diastolic blood pressure of 85 mmHg or greater
5. Fasting glucose level of 100 mg/dL or greater
Metabolic syndrome is thought to be caused by a combination of genetic and lifestyle factors, such as physical inactivity and a diet high in refined carbohydrates and unhealthy fats. Treatment typically involves making lifestyle changes, such as eating a healthy diet, getting regular exercise, and losing weight if necessary. In some cases, medication may also be needed to manage individual components of the syndrome, such as high blood pressure or high cholesterol.
A mutation is a permanent change in the DNA sequence of an organism's genome. Mutations can occur spontaneously or be caused by environmental factors such as exposure to radiation, chemicals, or viruses. They may have various effects on the organism, ranging from benign to harmful, depending on where they occur and whether they alter the function of essential proteins. In some cases, mutations can increase an individual's susceptibility to certain diseases or disorders, while in others, they may confer a survival advantage. Mutations are the driving force behind evolution, as they introduce new genetic variability into populations, which can then be acted upon by natural selection.
Molecular sequence data refers to the specific arrangement of molecules, most commonly nucleotides in DNA or RNA, or amino acids in proteins, that make up a biological macromolecule. This data is generated through laboratory techniques such as sequencing, and provides information about the exact order of the constituent molecules. This data is crucial in various fields of biology, including genetics, evolution, and molecular biology, allowing for comparisons between different organisms, identification of genetic variations, and studies of gene function and regulation.
Monosomy is a type of chromosomal abnormality in which there is only one copy of a particular chromosome instead of the usual pair in a diploid cell. In monosomy, an individual has one less chromosome than the normal diploid number (46 chromosomes) due to the absence of one member of a chromosome pair. This condition arises from the loss of one chromosome in an egg or sperm during gamete formation or at conception.
Examples of monosomy include Turner syndrome, which is characterized by the presence of only one X chromosome (45,X), and Cri du Chat syndrome, which results from a deletion of a portion of the short arm of chromosome 5 (46,del(5)(p15.2)). Monosomy can lead to developmental abnormalities, physical defects, intellectual disabilities, and various health issues depending on the chromosome involved.
Language development disorders, also known as language impairments or communication disorders, refer to a group of conditions that affect an individual's ability to understand and/or use spoken or written language in a typical manner. These disorders can manifest as difficulties with grammar, vocabulary, sentence structure, word finding, following directions, and/or conversational skills.
Language development disorders can be receptive (difficulty understanding language), expressive (difficulty using language to communicate), or mixed (a combination of both). They can occur in isolation or as part of a broader neurodevelopmental disorder, such as autism spectrum disorder or intellectual disability.
The causes of language development disorders are varied and may include genetic factors, environmental influences, neurological conditions, hearing loss, or other medical conditions. It is important to note that language development disorders are not the result of low intelligence or lack of motivation; rather, they reflect a specific impairment in the brain's language processing systems.
Early identification and intervention for language development disorders can significantly improve outcomes and help individuals develop effective communication skills. Treatment typically involves speech-language therapy, which may be provided individually or in a group setting, and may involve strategies such as modeling correct language use, practicing targeted language skills, and using visual aids to support comprehension.
Comparative genomic hybridization (CGH) is a molecular cytogenetic technique used to detect and measure changes in the DNA content of an individual's genome. It is a type of microarray-based analysis that compares the DNA of two samples, typically a test sample and a reference sample, to identify copy number variations (CNVs), including gains or losses of genetic material.
In CGH, the DNA from both samples is labeled with different fluorescent dyes, typically one sample with a green fluorophore and the other with a red fluorophore. The labeled DNAs are then co-hybridized to a microarray, which contains thousands of DNA probes representing specific genomic regions. The intensity of each spot on the array reflects the amount of DNA from each sample that has hybridized to the probe.
By comparing the ratio of green to red fluorescence intensities for each probe, CGH can detect gains or losses of genetic material in the test sample relative to the reference sample. A ratio of 1 indicates no difference in copy number between the two samples, while a ratio greater than 1 suggests a gain of genetic material, and a ratio less than 1 suggests a loss.
CGH is a powerful tool for detecting genomic imbalances associated with various genetic disorders, including cancer, developmental delay, intellectual disability, and congenital abnormalities. It can also be used to study the genomics of organisms in evolutionary biology and ecological studies.
Chromosome aberrations refer to structural and numerical changes in the chromosomes that can occur spontaneously or as a result of exposure to mutagenic agents. These changes can affect the genetic material encoded in the chromosomes, leading to various consequences such as developmental abnormalities, cancer, or infertility.
Structural aberrations include deletions, duplications, inversions, translocations, and rings, which result from breaks and rearrangements of chromosome segments. Numerical aberrations involve changes in the number of chromosomes, such as aneuploidy (extra or missing chromosomes) or polyploidy (multiples of a complete set of chromosomes).
Chromosome aberrations can be detected and analyzed using various cytogenetic techniques, including karyotyping, fluorescence in situ hybridization (FISH), and comparative genomic hybridization (CGH). These methods allow for the identification and characterization of chromosomal changes at the molecular level, providing valuable information for genetic counseling, diagnosis, and research.
I must clarify that the term "pedigree" is not typically used in medical definitions. Instead, it is often employed in genetics and breeding, where it refers to the recorded ancestry of an individual or a family, tracing the inheritance of specific traits or diseases. In human genetics, a pedigree can help illustrate the pattern of genetic inheritance in families over multiple generations. However, it is not a medical term with a specific clinical definition.
Chromosome banding is a technique used in cytogenetics to identify and describe the physical structure and organization of chromosomes. This method involves staining the chromosomes with specific dyes that bind differently to the DNA and proteins in various regions of the chromosome, resulting in a distinct pattern of light and dark bands when viewed under a microscope.
The most commonly used banding techniques are G-banding (Giemsa banding) and R-banding (reverse banding). In G-banding, the chromosomes are stained with Giemsa dye, which preferentially binds to the AT-rich regions, creating a characteristic banding pattern. The bands are numbered from the centromere (the constriction point where the chromatids join) outwards, with the darker bands (rich in A-T base pairs and histone proteins) labeled as "q" arms and the lighter bands (rich in G-C base pairs and arginine-rich proteins) labeled as "p" arms.
R-banding, on the other hand, uses a different staining procedure that results in a reversed banding pattern compared to G-banding. The darker R-bands correspond to the lighter G-bands, and vice versa. This technique is particularly useful for identifying and analyzing specific regions of chromosomes that may be difficult to visualize with G-banding alone.
Chromosome banding plays a crucial role in diagnosing genetic disorders, identifying chromosomal abnormalities, and studying the structure and function of chromosomes in both clinical and research settings.
Chromosome mapping, also known as physical mapping, is the process of determining the location and order of specific genes or genetic markers on a chromosome. This is typically done by using various laboratory techniques to identify landmarks along the chromosome, such as restriction enzyme cutting sites or patterns of DNA sequence repeats. The resulting map provides important information about the organization and structure of the genome, and can be used for a variety of purposes, including identifying the location of genes associated with genetic diseases, studying evolutionary relationships between organisms, and developing genetic markers for use in breeding or forensic applications.
Human chromosome pair 1 refers to the first pair of chromosomes in a set of 23 pairs found in the cells of the human body, excluding sex cells (sperm and eggs). Each cell in the human body, except for the gametes, contains 46 chromosomes arranged in 23 pairs. These chromosomes are rod-shaped structures that contain genetic information in the form of DNA.
Chromosome pair 1 is the largest pair, making up about 8% of the total DNA in a cell. Each chromosome in the pair consists of two arms - a shorter p arm and a longer q arm - connected at a centromere. Chromosome 1 carries an estimated 2,000-2,500 genes, which are segments of DNA that contain instructions for making proteins or regulating gene expression.
Defects or mutations in the genes located on chromosome 1 can lead to various genetic disorders and diseases, such as Charcot-Marie-Tooth disease type 1A, Huntington's disease, and certain types of cancer.
Staphylococcus lugdunensis is a type of Gram-positive, coagulase-negative bacterium that is part of the Staphylococcus genus. It is a facultative anaerobe, which means it can grow in the presence or absence of oxygen. This bacterium is commonly found on the skin and mucous membranes of humans and other animals.
While S. lugdunensis is generally considered to be a commensal organism, it has been increasingly recognized as an important cause of invasive infections, particularly in patients with pre-existing conditions or compromised immune systems. Infections caused by S. lugdunensis can range from skin and soft tissue infections to more serious conditions such as endocarditis, osteomyelitis, and bacteremia.
One notable feature of S. lugdunensis is its ability to produce a clumping factor, which is similar to the clumping factor produced by Staphylococcus aureus, a more well-known pathogenic species within the same genus. However, unlike S. aureus, S. lugdunensis is typically susceptible to many antibiotics and can be effectively treated with a variety of antimicrobial agents.
Human chromosome pair 6 consists of two rod-shaped structures present in the nucleus of each human cell. They are identical in size and shape and contain genetic material, made up of DNA and proteins, that is essential for the development and function of the human body.
Chromosome pair 6 is one of the 23 pairs of chromosomes found in humans, with one chromosome inherited from each parent. Each chromosome contains thousands of genes that provide instructions for the production of proteins and regulate various cellular processes.
Chromosome pair 6 contains several important genes, including those involved in the development and function of the immune system, such as the major histocompatibility complex (MHC) genes. It also contains genes associated with certain genetic disorders, such as hereditary neuropathy with liability to pressure palsies (HNPP), a condition that affects the nerves, and Waardenburg syndrome, a disorder that affects pigmentation and hearing.
Abnormalities in chromosome pair 6 can lead to various genetic disorders, including numerical abnormalities such as trisomy 6 (three copies of chromosome 6) or monosomy 6 (only one copy of chromosome 6), as well as structural abnormalities such as deletions, duplications, or translocations of parts of the chromosome.
The branchial region, also known as the pharyngeal region or viscerocranium, is a term used in human anatomy to refer to the area of the developing embryo that gives rise to structures derived from the branchial (or pharyngeal) arches. The branchial arches are a series of paired, rod-like structures that appear early in embryonic development and give rise to various head and neck structures, including the bones and muscles of the face, jaws, and neck, as well as the associated nerves, blood vessels, and connective tissues.
The branchial region is divided into several subregions, each corresponding to a specific branchial arch. The first branchial arch gives rise to structures such as the mandible (lower jaw), maxilla (upper jaw), and muscles of mastication (chewing). The second branchial arch forms the stapes and styloid process in the ear, as well as some neck muscles. The third and fourth branchial arches contribute to the formation of the larynx, thyroid cartilage, and other structures in the neck.
Abnormalities in the development of the branchial region can lead to a variety of congenital defects, such as cleft palate, micrognathia (small jaw), and branchial cysts or sinuses. These conditions may require surgical intervention to correct.
Cleft palate is a congenital birth defect that affects the roof of the mouth (palate). It occurs when the tissues that form the palate do not fuse together properly during fetal development, resulting in an opening or split in the palate. This can range from a small cleft at the back of the soft palate to a complete cleft that extends through the hard and soft palates, and sometimes into the nasal cavity.
A cleft palate can cause various problems such as difficulty with feeding, speaking, hearing, and ear infections. It may also affect the appearance of the face and mouth. Treatment typically involves surgical repair of the cleft palate, often performed during infancy or early childhood. Speech therapy, dental care, and other supportive treatments may also be necessary to address related issues.
Intelligence tests are standardized procedures used to assess various aspects of an individual's cognitive abilities, such as their problem-solving skills, logical reasoning, verbal comprehension, and spatial relations. These tests provide a quantitative measurement of intelligence, often reported as an Intelligence Quotient (IQ) score. It is important to note that intelligence is a multifaceted concept, and intelligence tests measure only certain aspects of it. They should not be considered the sole determinant of an individual's overall intellectual capabilities or potential.
Genotype, in genetics, refers to the complete heritable genetic makeup of an individual organism, including all of its genes. It is the set of instructions contained in an organism's DNA for the development and function of that organism. The genotype is the basis for an individual's inherited traits, and it can be contrasted with an individual's phenotype, which refers to the observable physical or biochemical characteristics of an organism that result from the expression of its genes in combination with environmental influences.
It is important to note that an individual's genotype is not necessarily identical to their genetic sequence. Some genes have multiple forms called alleles, and an individual may inherit different alleles for a given gene from each parent. The combination of alleles that an individual inherits for a particular gene is known as their genotype for that gene.
Understanding an individual's genotype can provide important information about their susceptibility to certain diseases, their response to drugs and other treatments, and their risk of passing on inherited genetic disorders to their offspring.
A base sequence in the context of molecular biology refers to the specific order of nucleotides in a DNA or RNA molecule. In DNA, these nucleotides are adenine (A), guanine (G), cytosine (C), and thymine (T). In RNA, uracil (U) takes the place of thymine. The base sequence contains genetic information that is transcribed into RNA and ultimately translated into proteins. It is the exact order of these bases that determines the genetic code and thus the function of the DNA or RNA molecule.
The brain is the central organ of the nervous system, responsible for receiving and processing sensory information, regulating vital functions, and controlling behavior, movement, and cognition. It is divided into several distinct regions, each with specific functions:
1. Cerebrum: The largest part of the brain, responsible for higher cognitive functions such as thinking, learning, memory, language, and perception. It is divided into two hemispheres, each controlling the opposite side of the body.
2. Cerebellum: Located at the back of the brain, it is responsible for coordinating muscle movements, maintaining balance, and fine-tuning motor skills.
3. Brainstem: Connects the cerebrum and cerebellum to the spinal cord, controlling vital functions such as breathing, heart rate, and blood pressure. It also serves as a relay center for sensory information and motor commands between the brain and the rest of the body.
4. Diencephalon: A region that includes the thalamus (a major sensory relay station) and hypothalamus (regulates hormones, temperature, hunger, thirst, and sleep).
5. Limbic system: A group of structures involved in emotional processing, memory formation, and motivation, including the hippocampus, amygdala, and cingulate gyrus.
The brain is composed of billions of interconnected neurons that communicate through electrical and chemical signals. It is protected by the skull and surrounded by three layers of membranes called meninges, as well as cerebrospinal fluid that provides cushioning and nutrients.
A newborn infant is a baby who is within the first 28 days of life. This period is also referred to as the neonatal period. Newborns require specialized care and attention due to their immature bodily systems and increased vulnerability to various health issues. They are closely monitored for signs of well-being, growth, and development during this critical time.
Gene dosage, in genetic terms, refers to the number of copies of a particular gene present in an organism's genome. Each gene usually has two copies (alleles) in diploid organisms, one inherited from each parent. An increase or decrease in the number of copies of a specific gene can lead to changes in the amount of protein it encodes, which can subsequently affect various biological processes and phenotypic traits.
For example, gene dosage imbalances have been associated with several genetic disorders, such as Down syndrome (trisomy 21), where an individual has three copies of chromosome 21 instead of the typical two copies, leading to developmental delays and intellectual disabilities. Similarly, in certain cases of cancer, gene amplification (an increase in the number of copies of a particular gene) can result in overexpression of oncogenes, contributing to tumor growth and progression.
Nephrotic syndrome is a group of symptoms that indicate kidney damage, specifically damage to the glomeruli—the tiny blood vessel clusters in the kidneys that filter waste and excess fluids from the blood. The main features of nephrotic syndrome are:
1. Proteinuria (excess protein in urine): Large amounts of a protein called albumin leak into the urine due to damaged glomeruli, which can't properly filter proteins. This leads to low levels of albumin in the blood, causing fluid buildup and swelling.
2. Hypoalbuminemia (low blood albumin levels): As albumin leaks into the urine, the concentration of albumin in the blood decreases, leading to hypoalbuminemia. This can cause edema (swelling), particularly in the legs, ankles, and feet.
3. Edema (fluid retention and swelling): With low levels of albumin in the blood, fluids move into the surrounding tissues, causing swelling or puffiness. The swelling is most noticeable around the eyes, face, hands, feet, and abdomen.
4. Hyperlipidemia (high lipid/cholesterol levels): The kidneys play a role in regulating lipid metabolism. Damage to the glomeruli can lead to increased lipid production and high cholesterol levels in the blood.
Nephrotic syndrome can result from various underlying kidney diseases, such as minimal change disease, membranous nephropathy, or focal segmental glomerulosclerosis. Treatment depends on the underlying cause and may include medications to control inflammation, manage high blood pressure, and reduce proteinuria. In some cases, dietary modifications and lifestyle changes are also recommended.
Medical Definition:
Magnetic Resonance Imaging (MRI) is a non-invasive diagnostic imaging technique that uses a strong magnetic field and radio waves to create detailed cross-sectional or three-dimensional images of the internal structures of the body. The patient lies within a large, cylindrical magnet, and the scanner detects changes in the direction of the magnetic field caused by protons in the body. These changes are then converted into detailed images that help medical professionals to diagnose and monitor various medical conditions, such as tumors, injuries, or diseases affecting the brain, spinal cord, heart, blood vessels, joints, and other internal organs. MRI does not use radiation like computed tomography (CT) scans.
I'm sorry for any confusion, but "siblings" is not a medical term. It is a term used in genealogy and sociology to refer to the brothers and sisters that someone has. Sibling relationships can have medical implications, such as when inherited genetic disorders are present in a family, but the term "siblings" itself does not have a specific medical definition.
Growth disorders are medical conditions that affect a person's growth and development, leading to shorter or taller stature than expected for their age, sex, and ethnic group. These disorders can be caused by various factors, including genetic abnormalities, hormonal imbalances, chronic illnesses, malnutrition, and psychosocial issues.
There are two main types of growth disorders:
1. Short stature: This refers to a height that is significantly below average for a person's age, sex, and ethnic group. Short stature can be caused by various factors, including genetic conditions such as Turner syndrome or dwarfism, hormonal deficiencies, chronic illnesses, malnutrition, and psychosocial issues.
2. Tall stature: This refers to a height that is significantly above average for a person's age, sex, and ethnic group. Tall stature can be caused by various factors, including genetic conditions such as Marfan syndrome or Klinefelter syndrome, hormonal imbalances, and certain medical conditions like acromegaly.
Growth disorders can have significant impacts on a person's physical, emotional, and social well-being. Therefore, it is essential to diagnose and manage these conditions early to optimize growth and development and improve overall quality of life. Treatment options for growth disorders may include medication, nutrition therapy, surgery, or a combination of these approaches.
Sjögren's syndrome is a chronic autoimmune disorder in which the body's immune system mistakenly attacks its own moisture-producing glands, particularly the tear and salivary glands. This can lead to symptoms such as dry eyes, dry mouth, and dryness in other areas of the body. In some cases, it may also affect other organs, leading to a variety of complications.
There are two types of Sjögren's syndrome: primary and secondary. Primary Sjögren's syndrome occurs when the condition develops on its own, while secondary Sjögren's syndrome occurs when it develops in conjunction with another autoimmune disease, such as rheumatoid arthritis or lupus.
The exact cause of Sjögren's syndrome is not fully understood, but it is believed to involve a combination of genetic and environmental factors. Treatment typically focuses on relieving symptoms and may include artificial tears, saliva substitutes, medications to stimulate saliva production, and immunosuppressive drugs in more severe cases.
"Gene rearrangement" is a process that involves the alteration of the order, orientation, or copy number of genes or gene segments within an organism's genome. This natural mechanism plays a crucial role in generating diversity and specificity in the immune system, particularly in vertebrates.
In the context of the immune system, gene rearrangement occurs during the development of B-cells and T-cells, which are responsible for adaptive immunity. The process involves breaking and rejoining DNA segments that encode antigen recognition sites, resulting in a unique combination of gene segments and creating a vast array of possible antigen receptors.
There are two main types of gene rearrangement:
1. V(D)J recombination: This process occurs in both B-cells and T-cells. It involves the recombination of variable (V), diversity (D), and joining (J) gene segments to form a functional antigen receptor gene. In humans, there are multiple copies of V, D, and J segments for each antigen receptor gene, allowing for a vast number of possible combinations.
2. Class switch recombination: This process occurs only in mature B-cells after antigen exposure. It involves the replacement of the constant (C) region of the immunoglobulin heavy chain gene with another C region, resulting in the production of different isotypes of antibodies (IgG, IgA, or IgE) that have distinct effector functions while maintaining the same antigen specificity.
These processes contribute to the generation of a diverse repertoire of antigen receptors, allowing the immune system to recognize and respond effectively to a wide range of pathogens.
Cognitive disorders are a category of mental health disorders that primarily affect cognitive abilities including learning, memory, perception, and problem-solving. These disorders can be caused by various factors such as brain injury, degenerative diseases, infection, substance abuse, or developmental disabilities. Examples of cognitive disorders include dementia, amnesia, delirium, and intellectual disability. It's important to note that the specific definition and diagnostic criteria for cognitive disorders may vary depending on the medical source or classification system being used.
Translocation, genetic, refers to a type of chromosomal abnormality in which a segment of a chromosome is transferred from one chromosome to another, resulting in an altered genome. This can occur between two non-homologous chromosomes (non-reciprocal translocation) or between two homologous chromosomes (reciprocal translocation). Genetic translocations can lead to various clinical consequences, depending on the genes involved and the location of the translocation. Some translocations may result in no apparent effects, while others can cause developmental abnormalities, cancer, or other genetic disorders. In some cases, translocations can also increase the risk of having offspring with genetic conditions.
A case-control study is an observational research design used to identify risk factors or causes of a disease or health outcome. In this type of study, individuals with the disease or condition (cases) are compared with similar individuals who do not have the disease or condition (controls). The exposure history or other characteristics of interest are then compared between the two groups to determine if there is an association between the exposure and the disease.
Case-control studies are often used when it is not feasible or ethical to conduct a randomized controlled trial, as they can provide valuable insights into potential causes of diseases or health outcomes in a relatively short period of time and at a lower cost than other study designs. However, because case-control studies rely on retrospective data collection, they are subject to biases such as recall bias and selection bias, which can affect the validity of the results. Therefore, it is important to carefully design and conduct case-control studies to minimize these potential sources of bias.
I must clarify that I cannot provide a "medical definition" of intelligence, as intelligence is not a concept that is typically defined within the field of medicine. Intelligence is a term used to describe the ability to learn, understand, and make judgments or decisions based on reason, experience, and information. It is often measured through various cognitive abilities such as problem-solving, critical thinking, creativity, and knowledge acquisition.
The concept of intelligence is studied in many fields, including psychology, neuroscience, and education. In medicine, healthcare professionals may assess a person's cognitive abilities to better understand their health status or develop treatment plans. However, there is no specific "medical definition" for intelligence. Instead, it is a multifaceted concept that can be influenced by various genetic, environmental, and experiential factors.
Human chromosome pair 18 consists of two rod-shaped structures present in the nucleus of each cell of the human body. Chromosomes are made up of DNA, protein, and RNA, and they carry genetic information that determines an individual's physical characteristics, biochemical processes, and susceptibility to disease.
Chromosome pair 18 is one of the 23 pairs of chromosomes that make up the human genome. Each member of chromosome pair 18 has a length of about 75 million base pairs and contains around 600 genes. Chromosome pair 18 is also known as the "smart chromosome" because it contains many genes involved in brain development, function, and cognition.
Abnormalities in chromosome pair 18 can lead to genetic disorders such as Edwards syndrome (trisomy 18), in which there is an extra copy of chromosome 18, or deletion of a portion of the chromosome, leading to various developmental and cognitive impairments.
Neuropsychological tests are a type of psychological assessment that measures cognitive functions, such as attention, memory, language, problem-solving, and perception. These tests are used to help diagnose and understand the cognitive impact of neurological conditions, including dementia, traumatic brain injury, stroke, Parkinson's disease, and other disorders that affect the brain.
The tests are typically administered by a trained neuropsychologist and can take several hours to complete. They may involve paper-and-pencil tasks, computerized tasks, or interactive activities. The results of the tests are compared to normative data to help identify any areas of cognitive weakness or strength.
Neuropsychological testing can provide valuable information for treatment planning, rehabilitation, and assessing response to treatment. It can also be used in research to better understand the neural basis of cognition and the impact of neurological conditions on cognitive function.
A seizure is an uncontrolled, abnormal firing of neurons (brain cells) that can cause various symptoms such as convulsions, loss of consciousness, altered awareness, or changes in behavior. Seizures can be caused by a variety of factors including epilepsy, brain injury, infection, toxic substances, or genetic disorders. They can also occur without any identifiable cause, known as idiopathic seizures. Seizures are a medical emergency and require immediate attention.
Genetic association studies are a type of epidemiological research that aims to identify statistical associations between genetic variations and particular traits or diseases. These studies typically compare the frequency of specific genetic markers, such as single nucleotide polymorphisms (SNPs), in individuals with a given trait or disease to those without it.
The goal of genetic association studies is to identify genetic factors that contribute to the risk of developing common complex diseases, such as diabetes, heart disease, or cancer. By identifying these genetic associations, researchers hope to gain insights into the underlying biological mechanisms of these diseases and develop new strategies for prevention, diagnosis, and treatment.
It's important to note that while genetic association studies can identify statistical associations between genetic markers and traits or diseases, they cannot prove causality. Further research is needed to confirm and validate these findings and to understand the functional consequences of the identified genetic variants.
Psychotic disorders are a group of severe mental health conditions characterized by distorted perceptions, thoughts, and emotions that lead to an inability to recognize reality. The two most common symptoms of psychotic disorders are hallucinations and delusions. Hallucinations are when a person sees, hears, or feels things that aren't there, while delusions are fixed, false beliefs that are not based on reality.
Other symptoms may include disorganized speech, disorganized behavior, catatonic behavior, and negative symptoms such as apathy and lack of emotional expression. Schizophrenia is the most well-known psychotic disorder, but other types include schizoaffective disorder, delusional disorder, brief psychotic disorder, shared psychotic disorder, and substance-induced psychotic disorder.
Psychotic disorders can be caused by a variety of factors, including genetics, brain chemistry imbalances, trauma, and substance abuse. Treatment typically involves a combination of medication, therapy, and support services to help manage symptoms and improve quality of life.
Turner Syndrome is a genetic disorder that affects females, caused by complete or partial absence of one X chromosome. The typical karyotype is 45,X0 instead of the normal 46,XX in women. This condition leads to distinctive physical features and medical issues in growth, development, and fertility. Characteristic features include short stature, webbed neck, low-set ears, and swelling of the hands and feet. Other potential symptoms can include heart defects, hearing and vision problems, skeletal abnormalities, kidney issues, and learning disabilities. Not all individuals with Turner Syndrome will have every symptom, but most will require medical interventions and monitoring throughout their lives to address various health concerns associated with the condition.
Psychiatric Status Rating Scales are standardized assessment tools used by mental health professionals to evaluate and rate the severity of a person's psychiatric symptoms and functioning. These scales provide a systematic and structured approach to measuring various aspects of an individual's mental health, such as mood, anxiety, psychosis, behavior, and cognitive abilities.
The purpose of using Psychiatric Status Rating Scales is to:
1. Assess the severity and improvement of psychiatric symptoms over time.
2. Aid in diagnostic decision-making and treatment planning.
3. Monitor treatment response and adjust interventions accordingly.
4. Facilitate communication among mental health professionals about a patient's status.
5. Provide an objective basis for research and epidemiological studies.
Examples of Psychiatric Status Rating Scales include:
1. Clinical Global Impression (CGI): A brief, subjective rating scale that measures overall illness severity, treatment response, and improvement.
2. Positive and Negative Syndrome Scale (PANSS): A comprehensive scale used to assess the symptoms of psychosis, including positive, negative, and general psychopathology domains.
3. Hamilton Rating Scale for Depression (HRSD) or Montgomery-Åsberg Depression Rating Scale (MADRS): Scales used to evaluate the severity of depressive symptoms.
4. Young Mania Rating Scale (YMRS): A scale used to assess the severity of manic or hypomanic symptoms.
5. Brief Psychiatric Rating Scale (BPRS) or Symptom Checklist-90 Revised (SCL-90-R): Scales that measure a broad range of psychiatric symptoms and psychopathology.
6. Global Assessment of Functioning (GAF): A scale used to rate an individual's overall psychological, social, and occupational functioning on a hypothetical continuum of mental health-illness.
It is important to note that Psychiatric Status Rating Scales should be administered by trained mental health professionals to ensure accurate and reliable results.
Myelodysplastic syndromes (MDS) are a group of diverse bone marrow disorders characterized by dysplasia (abnormal development or maturation) of one or more types of blood cells or by ineffective hematopoiesis, resulting in cytopenias (lower than normal levels of one or more types of blood cells). MDS can be classified into various subtypes based on the number and type of cytopenias, the degree of dysplasia, the presence of ring sideroblasts, and cytogenetic abnormalities.
The condition primarily affects older adults, with a median age at diagnosis of around 70 years. MDS can evolve into acute myeloid leukemia (AML) in approximately 30-40% of cases. The pathophysiology of MDS involves genetic mutations and chromosomal abnormalities that lead to impaired differentiation and increased apoptosis of hematopoietic stem and progenitor cells, ultimately resulting in cytopenias and an increased risk of developing AML.
The diagnosis of MDS typically requires a bone marrow aspiration and biopsy, along with cytogenetic and molecular analyses to identify specific genetic mutations and chromosomal abnormalities. Treatment options for MDS depend on the subtype, severity of cytopenias, and individual patient factors. These may include supportive care measures, such as transfusions and growth factor therapy, or more aggressive treatments, such as chemotherapy and stem cell transplantation.
An amino acid sequence is the specific order of amino acids in a protein or peptide molecule, formed by the linking of the amino group (-NH2) of one amino acid to the carboxyl group (-COOH) of another amino acid through a peptide bond. The sequence is determined by the genetic code and is unique to each type of protein or peptide. It plays a crucial role in determining the three-dimensional structure and function of proteins.
Genetic predisposition to disease refers to an increased susceptibility or vulnerability to develop a particular illness or condition due to inheriting specific genetic variations or mutations from one's parents. These genetic factors can make it more likely for an individual to develop a certain disease, but it does not guarantee that the person will definitely get the disease. Environmental factors, lifestyle choices, and interactions between genes also play crucial roles in determining if a genetically predisposed person will actually develop the disease. It is essential to understand that having a genetic predisposition only implies a higher risk, not an inevitable outcome.
Williams Syndrome is a rare genetic disorder caused by the deletion of a small portion of chromosome 7. This results in various developmental and medical problems, which can include:
1. Distinctive facial features such as a broad forehead, wide-set eyes, short nose, and full lips.
2. Cardiovascular disease, particularly narrowed or missing blood vessels near the heart.
3. Developmental delays and learning disabilities, although most people with Williams Syndrome have an IQ in the mild to moderate range of intellectual disability.
4. A unique pattern of strengths and weaknesses in cognitive skills, such as strong language skills but significant difficulty with visual-spatial tasks.
5. Overly friendly or sociable personality, often displaying a lack of fear or wariness around strangers.
6. Increased risk of anxiety and depression.
7. Sensitive hearing and poor depth perception.
8. Short stature in adulthood.
Williams Syndrome affects about 1 in every 10,000 people worldwide, regardless of race or ethnic background. It is not an inherited disorder, but rather a spontaneous genetic mutation.
Complement C1q is a protein that is part of the complement system, which is a group of proteins in the blood that help to eliminate pathogens and damaged cells from the body. C1q is the first component of the classical complement pathway, which is activated by the binding of C1q to antibodies that are attached to the surface of a pathogen or damaged cell.
C1q is composed of six identical polypeptide chains, each containing a collagen-like region and a globular head region. The globular heads can bind to various structures, including the Fc regions of certain antibodies, immune complexes, and some types of cells. When C1q binds to an activating surface, it triggers a series of proteolytic reactions that lead to the activation of other complement components and the formation of the membrane attack complex (MAC), which can punch holes in the membranes of pathogens or damaged cells, leading to their destruction.
In addition to its role in the immune system, C1q has also been found to have roles in various physiological processes, including tissue remodeling, angiogenesis, and the clearance of apoptotic cells. Dysregulation of the complement system, including abnormalities in C1q function, has been implicated in a variety of diseases, including autoimmune disorders, inflammatory diseases, and neurodegenerative conditions.
Single Nucleotide Polymorphism (SNP) is a type of genetic variation that occurs when a single nucleotide (A, T, C, or G) in the DNA sequence is altered. This alteration must occur in at least 1% of the population to be considered a SNP. These variations can help explain why some people are more susceptible to certain diseases than others and can also influence how an individual responds to certain medications. SNPs can serve as biological markers, helping scientists locate genes that are associated with disease. They can also provide information about an individual's ancestry and ethnic background.
Prader-Willi Syndrome (PWS) is a genetic disorder that affects several parts of the body and is characterized by a range of symptoms including:
1. Developmental delays and intellectual disability.
2. Hypotonia (low muscle tone) at birth, which can lead to feeding difficulties in infancy.
3. Excessive appetite and obesity, typically beginning around age 2, due to a persistent hunger drive and decreased satiety.
4. Behavioral problems such as temper tantrums, stubbornness, and compulsive behaviors.
5. Hormonal imbalances leading to short stature, small hands and feet, incomplete sexual development, and decreased bone density.
6. Distinctive facial features including a thin upper lip, almond-shaped eyes, and a narrowed forehead.
7. Sleep disturbances such as sleep apnea or excessive daytime sleepiness.
PWS is caused by the absence of certain genetic material on chromosome 15, which results in abnormal gene function. It affects both males and females equally and has an estimated incidence of 1 in 10,000 to 30,000 live births. Early diagnosis and management can help improve outcomes for individuals with PWS.
Cushing syndrome is a hormonal disorder that occurs when your body is exposed to high levels of the hormone cortisol for a long time. This can happen due to various reasons such as taking high doses of corticosteroid medications or tumors that produce cortisol or adrenocorticotropic hormone (ACTH).
The symptoms of Cushing syndrome may include:
* Obesity, particularly around the trunk and upper body
* Thinning of the skin, easy bruising, and purple or red stretch marks on the abdomen, thighs, breasts, and arms
* Weakened bones, leading to fractures
* High blood pressure
* High blood sugar
* Mental changes such as depression, anxiety, and irritability
* Increased fatigue and weakness
* Menstrual irregularities in women
* Decreased fertility in men
Cushing syndrome can be diagnosed through various tests, including urine and blood tests to measure cortisol levels, saliva tests, and imaging tests to locate any tumors. Treatment depends on the cause of the condition but may include surgery, radiation therapy, chemotherapy, or adjusting medication dosages.
C57BL/6 (C57 Black 6) is an inbred strain of laboratory mouse that is widely used in biomedical research. The term "inbred" refers to a strain of animals where matings have been carried out between siblings or other closely related individuals for many generations, resulting in a population that is highly homozygous at most genetic loci.
The C57BL/6 strain was established in 1920 by crossing a female mouse from the dilute brown (DBA) strain with a male mouse from the black strain. The resulting offspring were then interbred for many generations to create the inbred C57BL/6 strain.
C57BL/6 mice are known for their robust health, longevity, and ease of handling, making them a popular choice for researchers. They have been used in a wide range of biomedical research areas, including studies of cancer, immunology, neuroscience, cardiovascular disease, and metabolism.
One of the most notable features of the C57BL/6 strain is its sensitivity to certain genetic modifications, such as the introduction of mutations that lead to obesity or impaired glucose tolerance. This has made it a valuable tool for studying the genetic basis of complex diseases and traits.
Overall, the C57BL/6 inbred mouse strain is an important model organism in biomedical research, providing a valuable resource for understanding the genetic and molecular mechanisms underlying human health and disease.
A mental disorder is a syndrome characterized by clinically significant disturbance in an individual's cognition, emotion regulation, or behavior. It's associated with distress and/or impaired functioning in social, occupational, or other important areas of life, often leading to a decrease in quality of life. These disorders are typically persistent and can be severe and disabling. They may be related to factors such as genetics, early childhood experiences, or trauma. Examples include depression, anxiety disorders, bipolar disorder, schizophrenia, and personality disorders. It's important to note that a diagnosis should be made by a qualified mental health professional.
Analysis of Variance (ANOVA) is a statistical technique used to compare the means of two or more groups and determine whether there are any significant differences between them. It is a way to analyze the variance in a dataset to determine whether the variability between groups is greater than the variability within groups, which can indicate that the groups are significantly different from one another.
ANOVA is based on the concept of partitioning the total variance in a dataset into two components: variance due to differences between group means (also known as "between-group variance") and variance due to differences within each group (also known as "within-group variance"). By comparing these two sources of variance, ANOVA can help researchers determine whether any observed differences between groups are statistically significant, or whether they could have occurred by chance.
ANOVA is a widely used technique in many areas of research, including biology, psychology, engineering, and business. It is often used to compare the means of two or more experimental groups, such as a treatment group and a control group, to determine whether the treatment had a significant effect. ANOVA can also be used to compare the means of different populations or subgroups within a population, to identify any differences that may exist between them.
DNA Mutational Analysis is a laboratory test used to identify genetic variations or changes (mutations) in the DNA sequence of a gene. This type of analysis can be used to diagnose genetic disorders, predict the risk of developing certain diseases, determine the most effective treatment for cancer, or assess the likelihood of passing on an inherited condition to offspring.
The test involves extracting DNA from a patient's sample (such as blood, saliva, or tissue), amplifying specific regions of interest using polymerase chain reaction (PCR), and then sequencing those regions to determine the precise order of nucleotide bases in the DNA molecule. The resulting sequence is then compared to reference sequences to identify any variations or mutations that may be present.
DNA Mutational Analysis can detect a wide range of genetic changes, including single-nucleotide polymorphisms (SNPs), insertions, deletions, duplications, and rearrangements. The test is often used in conjunction with other diagnostic tests and clinical evaluations to provide a comprehensive assessment of a patient's genetic profile.
It is important to note that not all mutations are pathogenic or associated with disease, and the interpretation of DNA Mutational Analysis results requires careful consideration of the patient's medical history, family history, and other relevant factors.
Acute Coronary Syndrome (ACS) is a term used to describe a range of conditions associated with sudden, reduced blood flow to the heart muscle. This reduction in blood flow, commonly caused by blood clots forming in coronary arteries, can lead to damage or death of the heart muscle and is often characterized by symptoms such as chest pain, shortness of breath, and fatigue.
There are three main types of ACS:
1. Unstable Angina: This occurs when there is reduced blood flow to the heart muscle, causing chest pain or discomfort, but the heart muscle is not damaged. It can be a warning sign for a possible future heart attack.
2. Non-ST Segment Elevation Myocardial Infarction (NSTEMI): This type of heart attack occurs when there is reduced blood flow to the heart muscle, causing damage or death of some of the muscle cells. However, the electrical activity of the heart remains relatively normal.
3. ST Segment Elevation Myocardial Infarction (STEMI): This is a serious and life-threatening type of heart attack that occurs when there is a complete blockage in one or more of the coronary arteries, causing extensive damage to the heart muscle. The electrical activity of the heart is significantly altered, which can lead to dangerous heart rhythms and even cardiac arrest.
Immediate medical attention is required for anyone experiencing symptoms of ACS, as prompt treatment can help prevent further damage to the heart muscle and reduce the risk of complications or death. Treatment options may include medications, lifestyle changes, and procedures such as angioplasty or bypass surgery.
Polycyctic Ovary Syndrome (PCOS) is a complex endocrine-metabolic disorder characterized by the presence of hyperandrogenism (excess male hormones), ovulatory dysfunction, and polycystic ovaries. The Rotterdam criteria are commonly used for diagnosis, which require at least two of the following three features:
1. Oligo- or anovulation (irregular menstrual cycles)
2. Clinical and/or biochemical signs of hyperandrogenism (e.g., hirsutism, acne, or high levels of androgens in the blood)
3. Polycystic ovaries on ultrasound examination (presence of 12 or more follicles measuring 2-9 mm in diameter, or increased ovarian volume >10 mL)
The exact cause of PCOS remains unclear, but it is believed to involve a combination of genetic and environmental factors. Insulin resistance and obesity are common findings in women with PCOS, which can contribute to the development of metabolic complications such as type 2 diabetes, dyslipidemia, and cardiovascular disease.
Management of PCOS typically involves a multidisciplinary approach that includes lifestyle modifications (diet, exercise, weight loss), medications to regulate menstrual cycles and reduce hyperandrogenism (e.g., oral contraceptives, metformin, anti-androgens), and fertility treatments if desired. Regular monitoring of metabolic parameters and long-term follow-up are essential for optimal management and prevention of complications.
 TANGO2
TANGO2
Neural crest
Imprinted brain hypothesis
Asymmetric crying facies
TBX1
22q11.2 distal deletion syndrome
ARVCF
Cardiac neural crest
Childhood schizophrenia
1968 in science
Trisomy 22
DiGeorge syndrome
Persistent truncus arteriosus
Double aortic arch
ZDHHC8
22q11.2 duplication syndrome
Right-sided aortic arch
Carrie Bearden
Angelo DiGeorge
Tetralogy of Fallot
Sensory gating
Cédric Blanpain
Post-transcriptional regulation
Microcephaly
Parkinson's disease
Chromosome 22
VACTERL association
Mitochondrial ribosomal protein L40
Malignant rhabdoid tumour
HIRA
 22q11 Deletion Syndrome | Harvard Catalyst Profiles | Harvard Catalyst
22q11 Deletion Syndrome | Harvard Catalyst Profiles | Harvard Catalyst
 22q11 Deletion Syndrome
22q11 Deletion Syndrome
Young adult outcomes for children With 22q11 deletion syndrome and comorbid ADHD<...
TANGO2 - Wikipedia
Otologic and audiologic outcomes in pediatric patients with velo-cardio-facial (22q11 deletion) syndrome<...
 Frontiers | Endocrine Diseases of Newborn: Epidemiology, Pathogenesis, Therapeutic Options, and Outcome "Current Insights Into...
Frontiers | Endocrine Diseases of Newborn: Epidemiology, Pathogenesis, Therapeutic Options, and Outcome "Current Insights Into...
 Velocardiofacial Syndrome: Background, Pathophysiology, Etiology
Velocardiofacial Syndrome: Background, Pathophysiology, Etiology
 4.5b Common Truncus (Q20.0) | NCBDDD | CDC
4.5b Common Truncus (Q20.0) | NCBDDD | CDC
Childhood-Onset Schizophrenia Workup: Approach Considerations, MRI or CT scanning, EEG
 Psychosis: Symptoms, Causes, and Risk Factors
Psychosis: Symptoms, Causes, and Risk Factors
 Clinical Practice Guidelines : Primary immunodeficiencies
Clinical Practice Guidelines : Primary immunodeficiencies
 Both rare and common genetic variants contribute to autism in the Faroe Islands | npj Genomic Medicine
Both rare and common genetic variants contribute to autism in the Faroe Islands | npj Genomic Medicine
Pediatric Hypocalcemia: Practice Essentials, Background, Pathophysiology
 Radoslava Jönsson | Göteborgs universitet
Radoslava Jönsson | Göteborgs universitet
 Variation in prevalence of chromosome 22q11 deletion in subtypes of conotruncal defect in 254 children - Amrita Vishwa...
Variation in prevalence of chromosome 22q11 deletion in subtypes of conotruncal defect in 254 children - Amrita Vishwa...
 DiGeorge Syndrome Demystifed | ScienceBlogs
DiGeorge Syndrome Demystifed | ScienceBlogs
 Type a title for your page here) - Velo-Cardio-Facial Syndrome Educational Foundation
Type a title for your page here) - Velo-Cardio-Facial Syndrome Educational Foundation
 Adolescence is the game-changer for schizophrenia - Neuroscience News
Adolescence is the game-changer for schizophrenia - Neuroscience News
 Chromosomal and cardiovascular anomalies associated with congenital laryngeal web
Chromosomal and cardiovascular anomalies associated with congenital laryngeal web
 COMT gene: MedlinePlus Genetics
COMT gene: MedlinePlus Genetics
 William's Syndrome - Physiopedia
William's Syndrome - Physiopedia
 Imelda Coyne'Trinity Research - Trinity College Dublin
Imelda Coyne'Trinity Research - Trinity College Dublin
 Meta-Analysis Finds No Support for Dopamine Hypothesis of Schizophrenia
Meta-Analysis Finds No Support for Dopamine Hypothesis of Schizophrenia
 ype Elgersma - Publications
ype Elgersma - Publications
Renal Agenesis/Hypoplasia | NCBDDD | CDC
Publications - Department of Neuroscience
Orphanet: Distal deletion 10p
 Publications - DNF UNIL
Publications - DNF UNIL
 RDNZ | Find a support group
RDNZ | Find a support groupDiGeorge22
- It encompasses several syndromes with overlapping abnormalities including the DIGEORGE SYNDROME, VELOCARDIOFACIAL SYNDROME, and CONOTRUNCAL AMOMALY FACE SYNDROME. (harvard.edu)
- There is a 1.5-3.0 Mb deletion containing around 30-40 genes, spanning this region that causes the most survivable genetic deletion disorder known as 22q11.2 deletion syndrome, which is most commonly known as DiGeorge syndrome or Velocaridofacial syndrome. (wikipedia.org)
- The vast phenotypes arise from deletions of not only DiGeorge Syndrome Critical Region (DGCR) genes and disease genes but other unidentified genes as well. (wikipedia.org)
- C22orf25 is in close proximity to DGCR8 as well as other genes known to play a part in DiGeorge Syndrome such as armadillo repeat gene deleted in Velocardiofacial syndrome (ARVCF), Cathechol-O-methyltransferase (COMT) and T-box 1 (TBX1). (wikipedia.org)
- Patients: Pediatric patients in AudGenDB with a diagnosis of velo-cardio-facial syndrome or DiGeorge syndrome. (nebraska.edu)
- Take, for instance, a common group of birth defects - forms of a disorder called DiGeorge syndrome. (scienceblogs.com)
- And yes, they revealed that, at least for those with DiGeorge syndrome, a face can tell something about the heart. (scienceblogs.com)
- DiGeorge Syndrome is named after my father doctor Angelo DiGeorge who died three years ago. (scienceblogs.com)
- I enjoy following the tremendous research being done all over the world in connection with DiGeorge Syndrome, which is by all accounts the most common chromosomal genetic deletion syndrome. (scienceblogs.com)
- I am also pleased that you correctly refer to it as "DiGeorge Syndrome" rather than as 22 q 11 deletion syndrome or by one of its many other monikers. (scienceblogs.com)
- For more than forty years it has been known is "DiGeorge Syndrome" in all of the medical text books and literature, but lately there seems to be a mighty effort by various institutions to rename it under there own flag. (scienceblogs.com)
- Accordingly, I urge all DiGeorge researchers, "DiGeorge families", and the entire medical community to respect and honor the remarkable man who first described the syndrome and who discovered the role of the thymus gland in human function by continuing to call it what it is, DiGeorge Syndrome. (scienceblogs.com)
- The reason for this is that the symptoms of DiGeorge syndrome vary considerably. (scienceblogs.com)
- All are now considered to be included within DiGeorge syndrome. (scienceblogs.com)
- 1. A hemizygous deletion of chromosome 22q11.2 can result in a spectrum of clinical disease including velocardiofacial syndrome (VCFS) and DiGeorge syndrome (DGS). (immunodeficiencysearch.com)
- Deletion of chromosome 22q11.2 can result in a spectrum of clinical disease ranging from velocardiofacial syndrome (VCFS) to DiGeorge syndrome (DGS). (immunodeficiencysearch.com)
- 22q11.2 Deletion Syndrome, also known as DiGeorge Syndrome, is a genetic disorder that affects approximately 1 in 4,000 live births. (mellaly.com)
- DiGeorge syndrome is thymic and parathyroid hypoplasia or aplasia leading to T-cell immunodeficiency and hypoparathyroidism. (msdmanuals.com)
- It results from gene deletions in the DiGeorge chromosomal region at 22q11, mutations in genes at chromosome 10p13, and mutations in other unknown genes, which cause dysembryogenesis of structures that develop from pharyngeal pouches during the 8th week of gestation. (msdmanuals.com)
- DiGeorge syndrome (ie, hypoparathyroidism, absence of thymus gland [T-cell abnormalities], cardiac anomalies) is associated with abnormal development of the third and fourth pharyngeal pouches from which the parathyroids derive embryologically and represents an example of a defect in parathyroid gland development. (medscape.com)
- DiGeorge syndrome and velocardiofacial syndrome are variants of the chromosome arm 22q11 microdeletion syndrome. (medscape.com)
- Several cases of chromosome 10p deletion have also been reported in which affected individuals have some features of DiGeorge syndrome. (medscape.com)
Velocardiofacial7
- Velocardiofacial syndrome (VCFS) is a genetic condition characterized by abnormal pharyngeal arch development that results in defective development of the parathyroid glands, thymus, and the conotruncal region of the heart. (medscape.com)
- Ophthalmologic abnormalities are seen in 70% of patients with velocardiofacial syndrome, such as posterior embryotoxon, bilateral cataracts, tortuous retinal vessels, and small optic disks. (medscape.com)
- Reports indicate that some patients with velocardiofacial syndrome may be mistakenly categorized as having CHARGE syndrome (ie, coloboma, heart defect, atresia choanae, retarded growth and development, and/or CNS anomalies, genital hypoplasia, and ear anomalies and/or deafness). (medscape.com)
- Velocardiofacial syndrome (VCFS) is caused by a deletion (microdeletion) at the q11.2 band, which is located on the long arm (q) of chromosome 22 (see the images below). (medscape.com)
- One example is the 22q11 deletion syndrome, or velocardiofacial syndrome. (medscape.com)
- Anal anomalies: an uncommon feature of velocardiofacial (Shprintzen) syndrome? (bmj.com)
- We report three cases of velocardiofacial syndrome (VCFS) with anal anomalies who have deletions of the 22q11 region and a further case where the proband has VCFS clinically and her father has an anal anomaly. (bmj.com)
Fluorescence in situ hybri3
- Chromosomal fluorescence in situ hybridization (FISH) demonstrating the deletion of one chromosomal region 22q11 segment. (medscape.com)
- Twelve patients underwent cytogenetic evaluation, including seven that were tested for a chromosome 22q11 deletion by fluorescence in situ hybridization. (nih.gov)
- [1] Diagnosis of the syndrome can be made at birth based on physical characteristics, but a true medical diagnosis is confirmed following a diagnostic test called fluorescence in situ hybridization (FISH). (physio-pedia.com)
VCFS4
- Objective: The focus of this study was to evaluate the prevalence, type, and severity of hearing impairment in patients with velo-cardio-facial syndrome (VCFS) and to compare these characteristics with patient demographics and other otologic factors. (nebraska.edu)
- VCFS is a genetic disorder associated with a deletion of chromosome #22q11. (vcfsef.org)
- The deletion that causes VCFS is found on the q-arm of chromosome #22. (vcfsef.org)
- It is important to consider VCFS in the differential diagnosis of children with anal anomalies and to look for other features of the syndrome, such as asymmetrical crying facies, submucous cleft of the palate, developmental delay, cardiac anomalies, and hypoparathyroidism. (bmj.com)
Children With 22q11 deletion1
- Frontal Hypoactivation During a Working Memory Task in Children With 22q11 Deletion Syndrome. (harvard.edu)
22q11.2 deletion37
- Bleeding Severity and Phenotype in 22q11.2 Deletion Syndrome-A Cross-Sectional Investigation. (harvard.edu)
- Abnormalities in gray matter microstructure in young adults with 22q11.2 deletion syndrome. (harvard.edu)
- Failed Progenitor Specification Underlies the Cardiopharyngeal Phenotypes in a Zebrafish Model of 22q11.2 Deletion Syndrome. (harvard.edu)
- Confined placental mosaicism for 22q11.2 deletion as the etiology for discordant positive NIPT results. (harvard.edu)
- Background 22q11.2 deletion syndrome (22q11DS) is a common microdeletion syndrome associated with a variety of negative health, cognitive, emotional, and behavioral outcomes. (syr.edu)
- The gene coding for C22orf25 is located on chromosome 22 and the location q11.21, so it is often associated with 22q11.2 deletion syndrome. (wikipedia.org)
- 22q11.2 deletion syndrome has a vast array of phenotypes and is not attributed to the loss of a single gene. (wikipedia.org)
- [ 5 ] The presence of an aortic arch anomaly increases the odds of having a 22q11.2 deletion, regardless of the intracardiac anatomy. (medscape.com)
- Patients with 22q11.2 Deletion Syndrome (22q11.2DS) have a 25-30% risk of developing schizophrenia, and also suffer frequent hearing loss. (biorxiv.org)
- These results reveal bottom-up neurobiological mechanisms through which peripheral hearing loss arising from the 22q11.2 deletion may promote the emergence of schizophrenia-relevant auditory brain and behavioral abnormalities, and also suggest a link between conductive hearing loss and reduced PV+ interneuron density in the auditory cortex. (biorxiv.org)
- In the Df1 /+ mouse model of human 22q11.2 Deletion Syndrome, we find that hearing loss shapes measures that are considered schizophrenia-relevant endophenotypes, such as central auditory gain and auditory sensorimotor gating. (biorxiv.org)
- These results suggest mechanisms through which hearing loss associated with the 22q11.2 deletion may promote emergence of schizophrenia-relevant auditory brain and behavioral abnormalities and indicate that conductive hearing loss may influence PV+ interneuron density in the auditory cortex. (biorxiv.org)
- The characteristic signs and symptoms of 22q11.2 deletion syndrome result from a deletion of a small piece of chromosome 22. (medlineplus.gov)
- Researchers believe that changes involving this enzyme in the prefrontal cortex may help explain the increased risk of behavioral problems and mental illness associated with 22q11.2 deletion syndrome. (medlineplus.gov)
- People with 22q11.2 deletion syndrome are much more likely than people without the condition to develop schizophrenia, depression , anxiety, and bipolar disorder . (medlineplus.gov)
- Variations in the COMT gene also may be associated with mental illness in people without 22q11.2 deletion syndrome. (medlineplus.gov)
- In this symposium, we will cover the topic of social impairments in different populations across the psychosis continuum: patients with attenuated psychotic symptoms (APS), patients affected by the 22q11.2 deletion syndrome (22q11DS) - a genetic condition associated with increased risk for the development of psychosis - and patients with a first psychotic episode (FEP). (escap.eu)
- Approximately 90% of patients with DGS have the 22q11.2 deletion. (immunodeficiencysearch.com)
- 4. Low T-cell numbers are present in 80% of patients with 22q11.2 deletion syndrome as a result of thymic hypoplasia. (immunodeficiencysearch.com)
- Of the patients who have a DGS phenotype, 90% have a 22q11.2 deletion (10% have other molecular etiologies such as 10p deletion, 17p13 deletion, 18q21 deletion, and CHD7 mutations). (immunodeficiencysearch.com)
- While often used interchangeably with DGS, patients with the confirmed chromosomal deletion should be referred to has having 22q11.2 deletion syndrome. (immunodeficiencysearch.com)
- Up to 80% of patients with 22q11.2 deletion will have decreased T-cell numbers as a result of thymic hypoplasia. (immunodeficiencysearch.com)
- Early detection of 22q11.2 Deletion Syndrome is crucial for the best possible outcomes. (mellaly.com)
- The symptoms of 22q11.2 Deletion Syndrome can be physical or behavioral. (mellaly.com)
- The greatest risk factor for 22q11.2 Deletion Syndrome is genetics. (mellaly.com)
- Early diagnosis of 22q11.2 Deletion Syndrome is critical for providing effective intervention. (mellaly.com)
- Recognizing the symptoms of 22q11.2 Deletion Syndrome is also crucial for diagnosis. (mellaly.com)
- Most intervention for 22q11.2 Deletion Syndrome is medical. (mellaly.com)
- Early detection is crucial for a better outcome in patients with 22q11.2 Deletion Syndrome. (mellaly.com)
- Early detection and intervention are crucial for patients with 22q11.2 Deletion Syndrome. (mellaly.com)
- 1. What's the life expectancy of patients affected by 22q11.2 Deletion Syndrome? (mellaly.com)
- It is vital to understand that life expectancy in patients with 22q11.2 Deletion Syndrome varies considerably. (mellaly.com)
- 2. Can 22q11.2 Deletion Syndrome be treated with medication? (mellaly.com)
- Yes, medication can be used to manage symptoms of 22q11.2 Deletion Syndrome, including anxiety and hyperactivity. (mellaly.com)
- 3. Can 22q11.2 Deletion Syndrome be detected during pregnancy? (mellaly.com)
- 5. What can be done to prevent the risk of 22q11.2 Deletion Syndrome? (mellaly.com)
- While it is not possible to prevent the risk of 22q11.2 Deletion Syndrome, genetic counseling and testing can help identify the risk for future children. (mellaly.com)
Congenital3
- Of the patients with cardiovascular anomalies, 55% also had chromosomal alterations, and 71% of patients with chromosomal alterations also had a cardiovascular defect, of which four had the triad of a congenital laryngeal web, a chromosome 22q11 deletion, and congenital cardiovascular anomalies. (nih.gov)
- Accordingly, patients with a congenital laryngeal web should undergo genetic screening, including evaluation for a chromosome 22q11 deletion, and a thorough cardiovascular evaluation, including imaging of the aortic arch. (nih.gov)
- Particular attention should be paid to identifying patients with the triad of a congenital laryngeal web, a chromosome 22q11 deletion, and cardiovascular anomalies, particularly a vascular ring. (nih.gov)
Chromosomes3
- Autosomal refers to the fact that the deletion is not found on the sex chromosomes and both males and females can be affected. (vcfsef.org)
- When a person has the deletion in one of their #22 chromosomes, there is a 50% or 1/2 chance that they will pass on that chromosome to their offspring. (vcfsef.org)
- Mosaic) aneuploidies and chromosomal rearrangements are a frequent cause of idiopathic MCA/MR. Starting in 1959 with the identification of trisomy-21 as the genetic basis of Down syndrome [ 10 ], microscopic observation of metaphase chromosomes has for several decades been the method of choice for detecting chromosome abnormalities in MCA/MR patients. (biomedcentral.com)
Patients16
- As many as 15-20% of patients have Pierre Robin syndrome, which includes small jaw, U-shaped cleft palate, and glossoptosis. (medscape.com)
- Df1 /+ mice have a multi-gene deletion analogous to the chromosomal microdeletion that causes human 22q11.2DS, and like human 22q11.2DS patients exhibit high rates of hearing loss arising primarily from susceptibility to middle ear inflammation. (biorxiv.org)
- His research interests pertain to non-inflammatory vasculopathies, particularly Kohlmeier-Degos disease, and he has spearheaded efforts at UCLA to develop multidisciplinary care for patients with the PANS and PANDAS syndromes -- conditions that contribute to OCD, tic, and behavioral problems in young children. (uclahealth.org)
- Researchers at the University of Geneva (UNIGE), Switzerland, have provided an initial answer after observing and analysing several years of patients with deletion syndrome. (neurosciencenews.com)
- The Geneva team has been following 275 patients aged 6 to 35 years for 18 years: a control groups of 135 individuals - i.e. individuals without genetic problems - and 140 people with deletion syndrome, including 53 with moderate to severe psychotic symptoms. (neurosciencenews.com)
- Chromosomal abnormalities were detected in seven of the 12 patients in whom cytogenetic evaluation was performed (28% of the entire cohort), including a chromosome 22q11 deletion in six and trisomy 21 in one. (nih.gov)
- [1] J.C.P. Williams observed in four patients an association between supravalvular aortic stenosis and the common physical and mental characteristics of this patient population and stated that it "may constitute a previously unrecognized syndrome" [1] . (physio-pedia.com)
- Later, A.J. Beuren described eleven new patients with the characteristics described by Williams and the disorder became known as Williams-Beuren Syndrome. (physio-pedia.com)
- The differential diagnosis for patients with distal monosomy 10p should include deletion 22q11 syndrome and other causes of hypoparathyroidism, depending on the phenotype. (orpha.net)
- The Effect of Hypocalcaemia and Hypoxia on Risk of Autism in Patients with 22q11 Deletion Syndrome. (emory.edu)
- Twelve copy number abnormalities were identified in 12 patients (24% of the total): seven deletions (six apparently de novo and one inherited from a phenotypically normal parent) and five duplications (one de novo and four inherited from phenotypically normal parents). (bmj.com)
- No recurrent deletion or duplication was identified within this cohort of patients. (bmj.com)
- FISH probes designed to detect subtelomeric rearrangements have given a significant diagnostic yield of around 6% in patients with unexplained learning disability and dysmorphic features, 1- 3 but interstitial chromosomal deletions and duplications are not detectable using this method. (bmj.com)
- 8. A severe form of this syndrome resulting from complete thymic aplasia (resulting in near total absence of T-cells) occurs in approximately 1% of patients. (immunodeficiencysearch.com)
- Patients typically develop erythroderma and rash similar to Omenns syndrome. (immunodeficiencysearch.com)
- In consecutive, unselected MCA/MR patients karyotyping enables the detection of pathogenic chromosomal abnormalities in about 4% of cases (excluding Down syndrome), despite its limited resolution to about 5-10 Mb. (biomedcentral.com)
Genes3
- Data from SNP array and whole exome sequencing revealed that individuals with autism had a higher burden of rare exonic copy-number variants altering autism associated genes (deletions ( p = 0.0352) or duplications ( p = 0.0352)), higher inbreeding status ( p = 0.023) and a higher load of rare homozygous deleterious variants ( p = 0.011) compared to controls. (nature.com)
- [1] The test reveals a recurring micro-deletion, with a size of 1,551,83 Mb, on chromosome band 7q11.23, which contains 24-28 genes. (physio-pedia.com)
- 2. Although the most common deletion includes a region containing more than 35 genes, the TBX1 gene has emerged as one of the most likely causes of the DGS phenotype. (immunodeficiencysearch.com)
Abnormalities1
- This psychiatric illness affects 0.5% of the general population, and it may be related to genetic abnormalities of chromosome 22, known as 22q11 deletion syndrome. (neurosciencenews.com)
Schizophrenia4
- Deletion of chromosomal locus 22q11.2 is associated with both schizophrenia and hearing loss in humans. (biorxiv.org)
- Thirty percent of people affected by the syndrome end up developing psychotic symptoms specific to schizophrenia, such as auditory hallucinations, memory problems, disorders affecting their perception of reality, and difficulties in social interactions characterised by strong paranoia. (neurosciencenews.com)
- individuals with copy number variants, such as the copy number deletion of 1.5-5 megabases at 22q11.2 - a genetic marker associated with a "~45% lifetime risk of developing psychosis and ~35% lifetime risk of developing schizophrenia. (madinamerica.com)
- Four of these examined relatives of individuals with "schizophrenia," while two reported individuals with the 22q11 deletion syndrome. (madinamerica.com)
Rare chromosomal1
- Distal monosomy 10p is a rare chromosomal disorder in which the tip of the short arm (p arm) of chromosome 10 is deleted resulting in a variable phenotype depending on the size of the deletion. (orpha.net)
Shprintzen2
- Shprintzen and colleagues first described the syndrome in 1978. (medscape.com)
- The original classifications included velo-cardio-facial syndrome, Shprintzen syndrome, 22q11 deletion syndrome, Sedlackova syndrome and conotruncal anomaly face syndrome. (scienceblogs.com)
Chromosome region1
- Karyotype of a patient with a deletion of chromosome region 22q11. (medscape.com)
Symptoms4
- However, not everyone who has the syndrome necessarily develops psychotic symptoms. (neurosciencenews.com)
- That's why we studied the development of this structure in detail," continues the UNIGE researcher, "so we could understand why some people affected by deletion syndrome eventually develop psychotic symptoms, while others don't. (neurosciencenews.com)
- The researchers then compared the developmental curves of the hippocampus in people with deletion syndrome but no psychotic symptoms with those who developed psychotic symptoms. (neurosciencenews.com)
- and (c) young male with deletion syndrome and psychotic symptoms. (neurosciencenews.com)
Disorder5
- 22q11 deletion syndrome is a neurogenetic disorder that targets chromosome 22. (neurosciencenews.com)
- As a result of the deletion, people with this disorder have only one copy of the COMT gene in each cell instead of the usual two copies. (medlineplus.gov)
- William's Syndrome was first recognized as a unique disorder in 1961. (physio-pedia.com)
- The video displays children with William's Syndrome and the common facial characteristics distinct to the genetic disorder. (physio-pedia.com)
- The above two pictures are from the following souce: Kaplan P, Wang P, Francke U. Williams (Williams Beuren) syndrome: a distinct neurobehavioral disorder. (physio-pedia.com)
Familial1
- Common truncus can occur in association with genetic conditions - especially deletion 22q11 - and can be familial. (cdc.gov)
Phenotype1
- Chromosome 10p deletions and CHD7 mutations can also result in a DGS clinical phenotype. (immunodeficiencysearch.com)
Developmental1
- In addition, variable developmental problems and schizoid features are also associated with this syndrome. (harvard.edu)
Phenotypes2
- Condition with a variable constellation of phenotypes due to deletion polymorphisms at chromosome location 22q11. (harvard.edu)
- Sonzogni M, Zhai P, Mientjes EJ, van Woerden GM , Elgersma Y . Assessing the requirements of prenatal UBE3A expression for rescue of behavioral phenotypes in a mouse model for Angelman syndrome. (neurotree.org)
Piece of chromosome 221
- This syndrome is caused by a missing piece of chromosome 22, and it affects multiple systems in the body. (mellaly.com)
Mutations1
- Aneurysm syndromes caused by mutations in TGF - beta receptor. (kauveryhospital.com)
Hypocalcemia1
- Hypocalcemia associated with a 22q11 microdeletion may be transiently present in infancy but recur later in life, particularly during periods of stress. (medscape.com)
Autoimmune1
- Genetically inherited forms arise from defects of parathyroid gland development, defects in the parathyroid hormone (PTH) gene, defects in the calcium-sensing receptor gene, defects in PTH action, defects in the autoimmune regulator gene, and genetic syndromes. (medscape.com)
Diagnosis4
- Diagnosis requires cytogenetic analysis and molecular characterization and should include a search for a translocation because deletion may be the result of transmission of a derivative chromosome. (orpha.net)
- Prenatal diagnosis is feasible and genetic counseling should be proposed and depends on the cytogenetic rearrangement responsible for the deletion ( de novo or translocation). (orpha.net)
- The diagnosis usually involves genetic testing, which can identify the deletion of chromosome 22. (mellaly.com)
- Early diagnosis and intervention provide the best possible outcome for those with the syndrome. (mellaly.com)
Occur3
- Common truncus can occur with genetic syndromes such as deletion 22q11, in which many external (e.g. cleft palate) as well as internal anomalies have been described. (cdc.gov)
- The mechanism of how and why a deletion occurs is not fully understood, but we do know that there is nothing that a parent does before or during a pregnancy that causes a deletion to occur. (vcfsef.org)
- This chromosomal deletion syndrome is estimated to occur in approximately 1 in 3000 children. (immunodeficiencysearch.com)
Williams-Beuren S1
- The above series of 3 pictures A, B, C was from the following journal article: Pober B, Johnson M, Urban Z. Mechanisms and treatment of cardiovascular disease in Williams-Beuren syndrome. (physio-pedia.com)
Clinical1
- Often, however, the combination of clinical features is not diagnostic of a particular syndrome but may nevertheless still suggest that a chromosomal imbalance is the likely underlying cause of the abnormality. (bmj.com)