Homozygote
Heterozygote
Erblichkeit
Polymorphism, Genetic
Alleles
Gen-Rearrangement
Genetische Prädisposition für eine Krankheit
Polymorphism, Single Nucleotide
Hämochromatose
Mutation
Phenotype
Haplotypes
Heterozygotenerkennung
Genetic Variation
Fall-Kontroll-Studien
Genes, Lethal
Mäuse, Mutantenstämme
Apolipoprotein E-2
Genes, Recessive
Catechol-o-Methyltransferase
Albinismus
Hypercholesterinämie, familiäre
Stammbaum
Polymerase-Kettenreaktion
Polymorphism, Restriction Fragment Length
Kreuzungen, genetische
Genetic Association Studies
Point Mutation
Base Sequence
Risikofaktoren
Gruppe asiatischer Abstammung
Genetic Testing
Linkage Disequilibrium
Gruppe europäischer Abstammung
Molekülsequenzdaten
Genetische Marker
Exons
Mutation, Missense
Genes, Dominant
DNA-Mutationsanalyse
Hyperlipoproteinämie Typ III
Ataxia teleangiectatica
Aminosäuresubstitution
Histokompatibilitätsantigene Klasse I
Chromosomenkartierung
Genetic Linkage
Ferritin
Gaucher-Krankheit
Apolipoproteine E
Models, Genetic
Xanthomatose
Hämoglobin E
Hair Color
Membranproteine
Ethylnitrosoharnstoff
Thalassämie
Amin-Oxidoreductasen
Juden
Alpha-1-Antitrypsinmangel
Hämoglobine, abnorme
DNA
Apolipoproteine A
Promoter Regions, Genetic
Genetik, Populations-
Kohortenstudien
DNA-Primer
Valin
Odds Ratio
Serotonin-Plasmamembran-Transportproteine
Reference Values
Transferrin
Genom
Peptidyl-Dipeptidase A
Apolipoprotein E4
Apolipoprotein C-III
Genes
Polyzythämie
Codon
Hypolipoproteinämie
Alpha-Thalassämie
Cholesterol
Embryonenverlust
Eisen
Tschechische Republik
Gene Deletion
Drosophila melanogaster
Mäuse, neurologische Mutanten
Manifestationssalter
Risiko
Gilbert-Krankheit
Arylkohlenwasserstoff-Hydroxylasen
Genotyping Techniques
Zystische Fibrose
Apolipoproteine B
Homocystein
Inzucht
China
Penetrance
Apolipoprotein A-I
Elektrophorese, Stärkegel-
Mäuse, Inzuchtstamm C57BL-
Ethnische Gruppen
Lipide
Prolin
Mäuse, Knockout-
Lipoproteine
Hämoglobinopathien
Polydaktylie
Sitosterole
Apolipoproteine C
Familiäres Mittelmeerfieber
Triglyceride
Fetales Hämoglobin
Selection, Genetic
Lipoproteine, HDL-Cholesterol-
Gen-Targeting
Publikationsbias
Stoffwechsel, angeborene Störungen
Sequenzanalyse, DNA-
Cholesterol-Ester-Transfer-Proteine
Neuralrohrdefekte
Lipidstoffwechsel, angeborene Störungen
Desoxyribonucleasen, Typ-II-, regionspezifische
Apolipoprotein E-3
Wolman-Krankheit
Folsäure
Siderosis
Spitzmäuse
Chromosomen
Lezithin-Azyltransferasemangel
Amino Acid Sequence
Cystic Fibrosis Transmembrane Conductance Regulator
Carrierproteine
Varianzanalyse
MNSs-Blutgruppensystem
Introns
Embryo
RNA, Messenger-
Faktor V
Globins
Lipoproteine, LDL-
Epistasis, Genetic
Mäuse, transgene
Fetaltod
Homozygotie ist ein Begriff aus der Genetik und beschreibt die Situation, in der ein Individuum zwei identische Allele eines Gens besitzt. Allele sind verschiedene Versionen desselben Gens, die an denselben genetischen Lokus auf einem Chromosomenpaar lokalisiert sind.
Wenn ein Mensch ein Gen in homozygoter Form besitzt, bedeutet das, dass beide Kopien dieses Gens (eine vom Vater geerbt und eine von der Mutter geerbt) identisch sind. Dies kann entweder passieren, wenn beide Elternteile dasselbe Allel weitergeben (z.B. AA x AA) oder wenn ein reinerbiges Merkmal vorliegt, bei dem das Gen nur in einer Form vorkommt (z.B. bb x bb).
Homozygote Individuen exprimieren das entsprechende Merkmal in der Regel in seiner reinsten Form, da beide Allele dieselben Informationen tragen. Dies kann sowohl vorteilhafte als auch nachteilige Auswirkungen haben, je nachdem, ob das Gen dominant oder rezessiv ist und welche Eigenschaften es kodiert.
In der Genetik, ein Heterozygoter Organismus ist eine Person oder ein Lebewesen, das zwei verschiedene Allele eines Gens hat, d.h. es trägt eine unterschiedliche Version des Gens auf jedem Chromosom in einem homologen Chromosomenpaar. Dies steht im Gegensatz zu Homozygotie, bei der beide Allele eines Gens identisch sind.
Heterozygote können sich klinisch manifestieren (manifeste Heterozygote) oder asymptomatisch sein (latente Heterozygote), abhängig von der Art des Gens und der Art der genetischen Erkrankung. Manchmal kann das Vorhandensein einer heterozygoten Genvariante auch mit Vorteilen einhergehen, wie z.B. bei der Resistenz gegen bestimmte Krankheiten.
Es ist wichtig zu beachten, dass Heterozygote nicht notwendigerweise die Hälfte der Merkmale ihrer Homozygoten Gegenstücke aufweisen müssen. Die Manifestation von Genvarianten wird durch komplexe genetische und umweltbedingte Faktoren beeinflusst, was zu einer Vielzahl von Phänotypen führen kann, selbst bei Individuen mit derselben Genvariante.
Erblichkeit bezieht sich in der Genetik auf die Übertragung von genetischen Merkmalen oder Krankheiten von Eltern auf ihre Nachkommen über die Vererbung von Genen. Sie beschreibt das Ausmaß, in dem ein bestimmtes Merkmal oder eine Erkrankung durch Unterschiede in den Genen beeinflusst wird.
Erblichkeit wird in der Regel als ein Wahrscheinlichkeitswert ausgedrückt und gibt an, wie hoch die Chance ist, dass ein Merkmal oder eine Krankheit auftritt, wenn man die Gene einer Person betrachtet. Eine Erblichkeit von 100% würde bedeuten, dass das Merkmal oder die Krankheit sicher vererbt wird, während eine Erblichkeit von 0% bedeutet, dass es nicht vererbt wird. In der Realität liegen die meisten Erblichkeitswerte irgendwo dazwischen.
Es ist wichtig zu beachten, dass Erblichkeit nur einen Teilaspekt der Entstehung von Merkmalen und Krankheiten darstellt. Umweltfaktoren und Wechselwirkungen zwischen Genen und Umwelt spielen oft ebenfalls eine Rolle bei der Entwicklung von Merkmalen und Krankheiten.
Allele sind verschiedene Varianten eines Gens, die an der gleichen Position auf einem Chromosomenpaar zu finden sind und ein bestimmtes Merkmal oder eine Eigenschaft codieren. Jeder Mensch erbt zwei Kopien jedes Gens - eine von jedem Elternteil. Wenn diese beiden Kopien des Gens unterschiedlich sind, werden sie als "Allele" bezeichnet.
Allele können kleine Unterschiede in ihrer DNA-Sequenz aufweisen, die zu verschiedenen Ausprägungen eines Merkmals führen können. Ein Beispiel ist das Gen, das für die Augenfarbe codiert. Es gibt mehrere verschiedene Allele dieses Gens, die jeweils leicht unterschiedliche DNA-Sequenzen aufweisen und zu verschiedenen Augenfarben führen können, wie beispielsweise braune, grüne oder blaue Augen.
Es ist wichtig zu beachten, dass nicht alle Gene mehrere Allele haben - einige Gene haben nur eine einzige Kopie, die bei allen Menschen gleich ist. Andere Gene können hunderte oder sogar tausende verschiedene Allele aufweisen. Die Gesamtheit aller Allele eines Individuums wird als sein Genotyp bezeichnet, während die Ausprägung der Merkmale, die durch diese Allele codiert werden, als Phänotyp bezeichnet wird.
Ein Gen-Rearrangement, auch genetisches Rearrangement genannt, ist eine Veränderung in der Anordnung von Genen auf einer Chromosomensequenz. Dies tritt auf, wenn zwei nicht-homologe Chromosomen oder zwei verschiedene Abschnitte des gleichen Chromosoms durch Kreuzungsübertragung während der Meiose ausgetauscht werden. Ein Gen-Rearrangement kann auch auftreten, wenn ein Stück Chromosomen durch eine Translokation, Deletion, Inversion oder Duplikation verloren geht oder hinzugefügt wird.
In der Immunologie spielen genetische Rearrangements eine wichtige Rolle bei der Entwicklung des adaptiven Immunsystems von Wirbeltieren. Durch V(D)J-Rearrangement werden die variablen Regionen der Antikörper und T-Zell-Rezeptor-Gene neu angeordnet, wodurch eine enorme Vielfalt an Rezeptoren entsteht, mit denen das Immunsystem verschiedene Antigene erkennen kann. Diese Rearrangements tragen dazu bei, dass jedes Individuum über ein einzigartiges Repertoire an Rezeptoren verfügt und somit in der Lage ist, eine breite Palette von Krankheitserregern zu bekämpfen.
Eine genetische Prädisposition für eine Krankheit bezieht sich auf die Erhöhung der Wahrscheinlichkeit, an einer bestimmten Erkrankung zu erkranken, aufgrund von genetischen Faktoren. Es bedeutet nicht, dass eine Person definitiv die Krankheit entwickeln wird, sondern dass sie ein erhöhtes Risiko im Vergleich zur Allgemeinbevölkerung hat.
Diese Prädisposition resultiert aus bestimmten Genvarianten oder Mutationen, die in den Genen einer Person vorhanden sind und die Funktion von Proteinen beeinflussen können, die an Krankheitsprozessen beteiligt sind. Manche dieser genetischen Faktoren werden autosomal-dominant vererbt, was bedeutet, dass eine Kopie des mutierten Gens ausreicht, um das Erkrankungsrisiko zu erhöhen. Andere Fälle können autosomal-rezessiv sein, bei denen zwei Kopien des mutierten Gens erforderlich sind, damit die Krankheit zum Ausbruch kommt.
Es ist wichtig anzumerken, dass genetische Prädispositionen oft in Kombination mit umweltbedingten Faktoren auftreten, wie beispielsweise Rauchen, Alkoholkonsum, Ernährung und Exposition gegenüber bestimmten Chemikalien. Diese Faktoren können das Erkrankungsrisiko weiter erhöhen oder abschwächen.
In der medizinischen Praxis kann die Kenntnis einer genetischen Prädisposition dazu beitragen, präventive Maßnahmen zu ergreifen, Früherkennungstests durchzuführen und individuelle Behandlungspläne zu entwickeln.
Hämochromatose ist eine genetisch bedingte Erkrankung, bei der es zu einer übermäßigen Aufnahme und Speicherung von Eisen im Körpergewebe, insbesondere in Leber, Bauchspeicheldrüse, Herz und Gelenken kommt. Dies geschieht durch eine Störung im Stoffwechsel des Eisens, die zu einer Anhäufung von Eisen in den Organen führt.
Die häufigste Form der Hämochromatose wird als HFE-Hämochromatose bezeichnet und ist autosomal-rezessiv vererbt. Sie betrifft vor allem Menschen mit nordeuropäischer Abstammung. Durch die Anhäufung von Eisen in den Organen kann es zu verschiedenen Symptomen kommen, wie beispielsweise Lebererkrankungen, Diabetes mellitus, Herzrhythmusstörungen, Bronzedermatitis (bräunliche Verfärbung der Haut) und Gelenkbeschwerden.
Die Behandlung besteht in der Regel in einer regelmäßigen Entfernung von Blut (Phlebotomie), um den Eisenspiegel im Körper zu reduzieren. Eine frühzeitige Diagnose und Behandlung können die Entstehung von Organschäden verhindern oder zumindest verlangsamen.
Eine Mutation ist eine dauerhafte, zufällige Veränderung der DNA-Sequenz in den Genen eines Organismus. Diese Veränderungen können spontan während des normalen Wachstums und Entwicklungsprozesses auftreten oder durch äußere Einflüsse wie ionisierende Strahlung, chemische Substanzen oder Viren hervorgerufen werden.
Mutationen können verschiedene Formen annehmen, wie z.B. Punktmutationen (Einzelnukleotidänderungen), Deletionen (Entfernung eines Teilstücks der DNA-Sequenz), Insertionen (Einfügung zusätzlicher Nukleotide) oder Chromosomenaberrationen (größere Veränderungen, die ganze Gene oder Chromosomen betreffen).
Die Auswirkungen von Mutationen auf den Organismus können sehr unterschiedlich sein. Manche Mutationen haben keinen Einfluss auf die Funktion des Gens und werden daher als neutral bezeichnet. Andere Mutationen können dazu führen, dass das Gen nicht mehr oder nur noch eingeschränkt funktioniert, was zu Krankheiten oder Behinderungen führen kann. Es gibt jedoch auch Mutationen, die einen Vorteil für den Organismus darstellen und zu einer verbesserten Anpassungsfähigkeit beitragen können.
Insgesamt spielen Mutationen eine wichtige Rolle bei der Evolution von Arten, da sie zur genetischen Vielfalt beitragen und so die Grundlage für natürliche Selektion bilden.
Haplotypen sind eine Reihe von Varianten (Allele) eines Gens oder nahegelegener Marker, die auf einem einzigen Chromosom vererbt werden. Sie repräsentieren ein charakteristisches Muster von Variationen in einem bestimmten Abschnitt des Genoms und werden oft als Einheit vererbt, da sie eng beieinander liegen und eine geringe Rekombinationsrate aufweisen.
Haplotypen sind nützlich in der Genetik, um Verwandtschaftsbeziehungen zu untersuchen, Krankheitsrisiken abzuschätzen und pharmakogenetische Studien durchzuführen. Durch die Analyse von Haplotypen kann man Rückschlüsse auf gemeinsame Vorfahren ziehen und die Evolution von Genen und Populationen besser verstehen.
Heterozygotenidentifikation bezieht sich auf die Fähigkeit, Individuen zu identifizieren, die heterozygote Merkmalsträger sind, d.h. sie haben zwei verschiedene Allele für ein bestimmtes Gen. Diese Identifikation ist in der Genetik und in der Humangenetik von großer Bedeutung, insbesondere bei genetischen Erkrankungen, die durch rezessive Allele verursacht werden.
Eine Person, die ein rezessives Allel für eine bestimmte Krankheit besitzt, zeigt in der Regel keine Symptome, wenn sie das dominante Allel für dieses Gen erbt. Wenn jedoch beide Elternteile Träger des rezessiven Allels sind, besteht eine 25%ige Chance, dass ihr Kind zwei Kopien des rezessiven Allels erhält und an der Krankheit erkrankt.
Die Heterozygotenidentifikation kann durch verschiedene Methoden wie Genotypisierung, Familienstudien oder prädiktive Tests durchgeführt werden. Die Identifizierung von Heterozygoten ist wichtig, um das Risiko von genetischen Erkrankungen bei zukünftigen Generationen abzuschätzen und mögliche präventive Maßnahmen zu ergreifen.
Genetic Variation bezieht sich auf die Unterschiede in der DNA-Sequenz oder der Anzahl der Kopien bestimmter Gene zwischen verschiedenen Individuen derselben Art. Diese Variationen entstehen durch Mutationen, Gen-Kreuzungen und Rekombination während der sexuellen Fortpflanzung.
Es gibt drei Hauptarten von genetischen Variationen:
1. Einzelnukleotidische Polymorphismen (SNPs): Dies sind die häufigsten Formen der genetischen Variation, bei denen ein einzelner Nukleotid (DNA-Baustein) in der DNA-Sequenz eines Individuums von dem eines anderen Individuums abweicht.
2. Insertionen/Deletionen (INDELs): Hierbei handelt es sich um kleine Abschnitte der DNA, die bei einigen Individuen vorhanden sind und bei anderen fehlen.
3. Kopienzahlvariationen (CNVs): Bei diesen Variationen liegt eine Abweichung in der Anzahl der Kopien bestimmter Gene oder Segmente der DNA vor.
Genetische Variationen können natürliche Unterschiede zwischen Individuen erklären, wie zum Beispiel die verschiedenen Reaktionen auf Medikamente oder das unterschiedliche Risiko für bestimmte Krankheiten. Einige genetische Variationen sind neutral und haben keinen Einfluss auf die Funktion des Organismus, während andere mit bestimmten Merkmalen oder Erkrankungen assoziiert sein können.
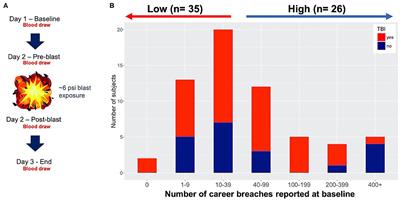
Eine Fall-Kontroll-Studie ist eine beobachtende Studie in der Epidemiologie, bei der die Exposition gegenüber einem potenziellen Risikofaktor für eine bestimmte Erkrankung zwischen den „Fällen“ (Personen mit der Erkrankung) und einer Kontrollgruppe ohne die Erkrankung verglichen wird. Die Kontrollgruppen werden üblicherweise so ausgewählt, dass sie dem Fall-Kollektiv hinsichtlich Alter, Geschlecht und anderen potentiell konfundierenden Variablen ähnlich sind. Anschließend wird die Häufigkeit der Exposition zu dem potenziellen Risikofaktor in beiden Gruppen verglichen. Fall-Kontroll-Studien eignen sich besonders gut, um seltene Erkrankungen zu untersuchen oder wenn eine langfristige Beobachtung nicht möglich ist.
In medical terms, "genes" refers to the basic units of heredity that are passed down from parents to offspring. They are made up of DNA and are located on chromosomes in the nucleus of cells. Each gene provides instructions for the production of a specific protein or set of proteins that play a crucial role in the development, functioning, and reproduction of an organism.
"Lethal" refers to something that causes death. In genetics, a lethal gene is one that results in the death of an organism before it can reach reproductive age or produce viable offspring. A lethal gene may cause embryonic lethality, meaning that the developing embryo dies before birth, or postnatal lethality, meaning that the organism dies shortly after birth or during early development.
Therefore, a "lethal gene" can be defined as a genetic mutation or variant that results in the death of an organism before it can reproduce, either due to embryonic or postnatal lethality.
Mutante Mausstämme sind genetisch veränderte Labortiere, die gezielt zur Erforschung von Krankheiten und zum Testen neuer Medikamente eingesetzt werden. Dabei wird das Erbgut der Mäuse durch verschiedene Methoden so verändert, dass sie bestimmte genetische Merkmale aufweisen, die denen von menschlichen Erkrankungen ähneln.
Diese Mutationen können spontan auftreten oder gezielt herbeigeführt werden, beispielsweise durch die Verwendung von Gentechnik oder Bestrahlung. Durch die Veränderung des Erbguts können Forscher untersuchen, wie sich die Genmutation auf das Verhalten, Wachstum und die Entwicklung der Mäuse auswirkt und ob sie anfälliger für bestimmte Krankheiten sind.
Mutante Mausstämme werden in der biomedizinischen Forschung häufig eingesetzt, um das Verständnis von Krankheitsprozessen zu verbessern und neue Behandlungsmethoden zu entwickeln. Ein bekanntes Beispiel ist die Knockout-Maus, bei der ein bestimmtes Gen gezielt deaktiviert wird, um die Funktion dieses Gens im Körper zu untersuchen.
Es ist wichtig zu beachten, dass Mutante Mausstämme zwar nützliche Modelle für die Erforschung menschlicher Krankheiten sein können, aber nicht immer ein perfektes Abbild der menschlichen Erkrankung darstellen. Daher müssen Forscher sorgfältig abwägen, ob und wie die Ergebnisse aus Tierversuchen auf den Menschen übertragbar sind.
Apolipoprotein E-2 (ApoE-2) ist eine Variante des Apolipoproteins E, das ein wichtiger Bestandteil der Lipoproteinkomplexe im Blukreislauf ist und bei der Cholesterintransport von den Geweben zum Leber verbunden mit der high-density lipoprotein (HDL) und low-density lipoprotein (LDL) Fraktionen spielt.
Es gibt drei Hauptformen von Apolipoprotein E, die durch Genvarianten am APOE-Gen auf Chromosom 19 codiert werden: E2, E3 und E4. ApoE-2 ist ein genetischer Variant, bei dem Cystein anstelle von Arginin an Position 112 und 158 der Aminosäuresequenz vorhanden ist.
Interessanterweise ist ApoE-2 assoziiert mit einem verminderten Risiko für die Entwicklung von Alzheimer-Krankheit, aber auch mit einem erhöhten Risiko für die Entwicklung von Typ-III-Hyperlipoproteinämie (auch bekannt als Remnant-Hyperlipidämie), eine seltene Erkrankung, die durch hohe Cholesterin- und Triglyceridspiegel im Blut gekennzeichnet ist.
"Recessive Genes" sind ein Konzept in der Genetik, bei dem die Merkmale eines Gens nur dann auftreten, wenn das Gen in beiden Kopien eines Chromosomenpaars vorhanden ist. Jeder Mensch hat zwei Kopien jedes Gens - eine von jedem Elternteil. Wenn ein Gen dominant ist, reicht es aus, es in einer Kopie zu haben, um das Merkmal zu exprimieren. Bei rezessiven Genen muss das Gen jedoch in beiden Kopien vorhanden sein, damit das Merkmal sichtbar wird.
Wenn ein Individuum ein rezessives Gen von einem Elternteil erbt und ein dominantes oder ein anderes Allel des Gens vom anderen Elternteil erbt, wird das rezessive Gen maskiert und das dominante Gen wird exprimiert. Dieses Individuum ist dann ein Träger des rezessiven Gens, zeigt aber keine Anzeichen dafür.
Rezessive Gene spielen eine wichtige Rolle in der Vererbung von erblichen Krankheiten und Merkmalen. Wenn beide Elternteile Träger eines rezessiven Gens sind, besteht für jedes Kind ein 25%iges Risiko, beide Kopien des Gens zu erben und die mit dem Gen verbundene Erkrankung oder das Merkmal auszudrücken.
Catechol-O-Methyltransferase (COMT) ist ein Enzym, das im menschlichen Körper vorkommt und an der Inaktivierung von Katecholaminen und Katecholestanolen beteiligt ist. Kommt ist hauptsächlich im Zytoplasma von Nervenzellen, Leber, Niere und Darm lokalisiert.
Das Enzym katalysiert den Transfer einer Methylgruppe von S-Adenosylmethionin (SAM) auf eine Hydroxygruppe in der catechol (dihydroxyphenyl) Gruppe von Substraten wie Dopamin, Noradrenalin und Adrenalin. Dies führt zur Bildung von methoxygruppenhaltigen Metaboliten, die weniger aktiv oder inaktiv sind als die ursprünglichen Substanzen.
Die Aktivität des COMT-Enzyms kann genetisch variieren, was Auswirkungen auf die Neurotransmission und den Stoffwechsel von Katecholaminen haben kann. Einige genetische Varianten des COMT-Gens wurden mit verschiedenen neurologischen Erkrankungen in Verbindung gebracht, wie z.B. Schizophrenie, Aufmerksamkeitsdefizit-Hyperaktivitätsstörung (ADHS) und Parkinson-Krankheit.
Albinismus ist eine genetisch bedingte Erkrankung, die durch einen Mangel oder vollständiges Fehlen des Enzyms Tyrosinase verursacht wird, das für die Produktion des Pigments Melanin in Haut, Haaren und Augen notwendig ist. Dies führt zu einer stark verminderten oder völlig fehlenden Pigmentierung der Haut, Haare und Augen. Menschen mit Albinismus haben oft weiße Haare, sehr helle Haut und hellblaue oder rosafarbene Augen. Darüber hinaus können sie eine verminderte Sehschärfe aufweisen, da die reduzierte Pigmentierung in der Augenrückseite (Retina) die normale Funktion des Sehnervs beeinträchtigen kann. Albinismus wird autosomal rezessiv vererbt, was bedeutet, dass ein Betroffener das defekte Gen von beiden Elternteilen erben muss, um an der Erkrankung zu leiden.
Familiäre Hypercholesterinämie ist ein genetisch bedingtes Lipidstoffwechselstörung, welches durch hohe Konzentrationen des Gesamtcholesterins und LDL-Cholesterins („schlechtes Cholesterin“) im Blut charakterisiert ist. Diese Erkrankung wird autosomal-dominant vererbt, was bedeutet dass eine Kopie des mutierten Gens ausreicht um die Krankheit zu manifestieren.
Die Mutationen betreffen in der Regel das Apolipoprotein B oder den LDL-Rezeptor Gen, wodurch die Fähigkeit des Körpers vermindert ist, LDL-Cholesterin aus dem Blutkreislauf zu entfernen. Dies führt zu einer Anhäufung von Cholesterin in den Blutgefäßen und steigert das Risiko für die Entwicklung von Atherosklerose und kardiovaskulären Erkrankungen, wie koronare Herzkrankheit, Herzinfarkt oder Schlaganfall.
Die Diagnose wird in der Regel durch eine Kombination aus Anamnese, klinischen Befunden, Lipidprofil und genetischer Testung gestellt. Die Behandlung umfasst in der Regel lifestyle Änderungen (wie Ernährungsumstellung und körperliche Aktivität) sowie medikamentöse Therapie mit Cholesterinsenkern wie Statinen oder PCSK9-Inhibitoren. In einigen Fällen kann auch eine LDL-Apherese notwendig sein, um die LDL-Cholesterinspiegel zu senken.
Genetische Kreuzungen beziehen sich auf die Paarung und Fortpflanzung zwischen zwei Individuen verschiedener reinbred Linien oder Arten, um neue Pflanzen- oder Tierhybriden zu erzeugen. Dieser Prozess ermöglicht es, gewünschte Merkmale von jeder Elternlinie in der nachfolgenden Generation zu kombinieren und kann zu einer Erweiterung der genetischen Vielfalt führen.
In der Genforschung können genetische Kreuzungen auch verwendet werden, um verschiedene Arten von Organismen gezielt zu kreuzen, um neue Eigenschaften oder Merkmale in den Nachkommen zu erzeugen. Durch die Analyse des Erbguts und der resultierenden Phänotypen können Forscher das Vererbungsmuster bestimmter Gene untersuchen und wertvolle Informationen über ihre Funktion und Interaktion gewinnen.
Es ist jedoch wichtig zu beachten, dass nicht alle genetischen Kreuzungen erfolgreich sind oder die erwarteten Ergebnisse liefern, da verschiedene Faktoren wie Kompatibilität der Genome, epistatische Effekte und genetische Drift eine Rolle spielen können.
Genetic association studies are a type of epidemiological research that aims to identify statistical associations between genetic variations and particular diseases or traits in a population. These studies compare the frequency of specific genetic markers, such as single nucleotide polymorphisms (SNPs) or copy number variants (CNVs), in individuals with a given disease or trait to those without it.
By identifying these associations, researchers can gain insights into the underlying genetic architecture of complex diseases and traits, which may ultimately lead to a better understanding of disease mechanisms, improved diagnostics, and the development of novel therapeutic strategies. It is important to note that while genetic association studies can identify statistical associations between genetic markers and diseases or traits, they do not necessarily imply causation, and further functional validation studies are often required to confirm the role of these genetic variants in disease pathogenesis.
In molecular biology, a base sequence refers to the specific order of nucleotides in a DNA or RNA molecule. In DNA, these nucleotides are adenine (A), cytosine (C), guanine (G), and thymine (T), while in RNA, uracil (U) takes the place of thymine. The base sequence contains genetic information that is essential for the synthesis of proteins and the regulation of gene expression. It is determined by the unique combination of these nitrogenous bases along the sugar-phosphate backbone of the nucleic acid molecule.
A 'Base Sequence' in a medical context typically refers to the specific order of these genetic building blocks, which can be analyzed and compared to identify genetic variations, mutations, or polymorphisms that may have implications for an individual's health, disease susceptibility, or response to treatments.
Es ist wichtig zu klären, dass es keine medizinische Definition der Gruppe "asiatischer Abstammung" gibt, da Rasse ein soziokulturelles Konstrukt und kein biologisches Merkmal ist. Die Klassifizierung von Menschen nach rassischen Kategorien ist umstritten und wird von vielen Experten abgelehnt, weil sie dazu beitragen kann, diskriminierende Praktiken zu rechtfertigen und Ungleichheiten aufrechtzuerhalten.
Im Gesundheitswesen werden jedoch häufig bestimmte Bevölkerungsgruppen identifiziert, die auf der Grundlage gemeinsamer kultureller, sprachlicher, geografischer oder historischer Merkmale zusammengefasst sind. Diese Gruppen können als "Asiatische Amerikaner und Inselbewohner des Pazifik" bezeichnet werden, wie es in den USA üblich ist. Diese Kategorie umfasst eine sehr heterogene Gruppe von Menschen mit unterschiedlichen ethnischen Hintergründen, Sprachen, Kulturen und Erfahrungen.
Es ist wichtig zu beachten, dass die Verwendung dieser Kategorien nicht bedeutet, dass es biologische Unterschiede zwischen den Gruppen gibt, sondern dass sie verwendet werden, um bestimmte Bevölkerungsgruppen zu identifizieren, die aufgrund historischer und soziokultureller Faktoren möglicherweise unterschiedliche Erfahrungen mit dem Gesundheitssystem machen oder unterschiedlichen Risikofaktoren für bestimmte Krankheiten ausgesetzt sind.
Genetic testing is a type of medical test that identifies changes in chromosomes, genes, or proteins. The results of a genetic test can confirm or rule out a suspected genetic condition or help determine a person's chance of developing or passing on a genetic disorder. Genetic tests are performed on a sample of blood, hair, skin, amniotic fluid (the fluid that surrounds a fetus during pregnancy), or other tissue. For example, a particular test might be used to identify a specific genetic variant or mutation associated with a condition such as cystic fibrosis or Huntington's disease.
There are several different types of genetic tests, including:
* Diagnostic testing: This type of test is used to confirm or rule out a suspected genetic condition in an individual who has symptoms of the condition.
* Predictive testing: This type of test is used to identify people who are at risk of developing a genetic disorder before they have symptoms.
* Carrier testing: This type of test is used to identify people who carry one copy of a gene mutation that, when present in two copies, causes a genetic disorder.
* Prenatal testing: This type of test is used to detect changes in a fetus's genes or chromosomes before birth.
* Newborn screening: This type of test is used to identify genetic disorders in newborn babies so that treatment can be started as early as possible.
It is important to note that genetic testing has both benefits and limitations. While it can provide valuable information about a person's health, it can also have potential risks, such as psychological distress or discrimination in employment or insurance. It is important for individuals considering genetic testing to receive accurate and unbiased information about the test and its implications so that they can make an informed decision about whether or not to proceed with testing.
Linkage Disequilibrium (LD) ist ein Konzept in der Genetik, das die nicht zufällige Assoziation zwischen Allelen verschiedener Gene oder unterschiedlicher Varianten eines Gens beschreibt. Normalerweise würden man erwarten, dass sich Allele unabhängig voneinander vererben, aber in bestimmten Situationen können zwei Allele häufiger gemeinsam vorkommen als es durch Zufall zu erwarten wäre.
Dies tritt auf, wenn diese Allele nahe beieinander auf demselben Chromosom liegen und nicht unabhängig sortiert werden können, da sie während der Meiose gemeinsam vererbt werden. Die Stärke des Linkage Disequilibrium hängt von der genetischen Distanz zwischen den Allelen ab - je näher sie beieinander liegen, desto stärker ist die Assoziation.
Linkage Disequilibrium wird oft in populationsgenetischen Studien untersucht, um Informationen über die Lage von Genen auf Chromosomen zu gewinnen und um genetische Varianten zu identifizieren, die mit bestimmten Krankheiten assoziiert sind.
Es gibt keine allgemein anerkannte medizinische Definition der Bezeichnung "Gruppe europäischer Abstammung". Der Begriff wird manchmal in biomedizinischen Forschungen oder öffentlichen Gesundheitsdaten verwendet, um Menschen mit gemeinsamen geografischen und historischen Ursprüngen in Europa zu beschreiben. Es ist jedoch wichtig anzumerken, dass die Verwendung solcher ethnischen Kategorien komplexe soziale und historische Konstrukte sind und nicht unbedingt biologisch oder genetisch bestimmbare Eigenschaften widerspiegeln. Die Verwendung dieser Begriffe in der Forschung oder klinischen Praxis sollte daher sorgfältig geprüft werden, um sicherzustellen, dass sie angemessen, nützlich und respektvoll sind.
Molekülsequenzdaten beziehen sich auf die Reihenfolge der Bausteine in Biomolekülen wie DNA, RNA oder Proteinen. Jedes Molekül hat eine einzigartige Sequenz, die seine Funktion und Struktur bestimmt.
In Bezug auf DNA und RNA besteht die Sequenz aus vier verschiedenen Nukleotiden (Adenin, Thymin/Uracil, Guanin und Cytosin), während Proteine aus 20 verschiedenen Aminosäuren bestehen. Die Sequenzdaten werden durch Laborverfahren wie DNA-Sequenzierung oder Massenspektrometrie ermittelt und können für Anwendungen in der Genetik, Biochemie und Pharmakologie verwendet werden.
Die Analyse von Molekülsequenzdaten kann zur Identifizierung genetischer Variationen, zur Vorhersage von Proteinstrukturen und -funktionen sowie zur Entwicklung neuer Medikamente beitragen.
Genetische Marker sind bestimmte Abschnitte der DNA, die mit einer bekanntermaßen variablen Position in der Genomsequenz eines Individuums assoziiert werden. Sie können in Form von einzelnen Nukleotiden (SNPs - Single Nucleotide Polymorphisms), Variationen in der Wiederholungszahl kurzer Sequenzen (VNTRs - Variable Number Tandem Repeats) oder Insertionen/Deletionen (InDels) auftreten.
Genetische Marker haben keine bekannte Funktion in sich selbst, aber sie können eng mit Genen verbunden sein, die für bestimmte Krankheiten prädisponieren oder Merkmale kontrollieren. Daher werden genetische Marker häufig bei der Kartierung von Krankheitsgenen und zur Abstammungstracing eingesetzt.
Es ist wichtig anzumerken, dass die Entdeckung und Nutzung genetischer Marker ein aktives Feld der Genforschung ist und neue Technologien wie Next-Generation Sequencing zu einer Explosion des verfügbaren Datenmaterials und möglicher neuer Anwendungen führen.
Exons sind die Abschnitte der DNA, die nach der Transkription und folgenden Prozessen wie Spleißen in das endgültige mature mRNA-Molekül eingebaut werden und somit die codierende Region für Proteine darstellen. Sie entsprechen den Bereichen, die nach dem Entfernen der nichtcodierenden Introns-Abschnitte in der reifen, translationsfähigen mRNA verbleiben. Im Allgemeinen enthalten Exons kodierende Sequenzen, die für Aminosäuren in einem Protein stehen, können aber auch Regulationssequenzen oder nichtcodierende RNA-Abschnitte wie beispielsweise RNA-Elemente mit Funktionen in der RNA-Struktur oder -Funktion enthalten.
Eine Missense-Mutation ist ein spezifischer Typ von Genmutation, bei der ein einzelner Nukleotid (DNA-Basenpaar) ausgetauscht wird, was dazu führt, dass ein anderes Aminosäure-Restmolekül anstelle des ursprünglichen eingebaut wird. Dies kann zu einer Veränderung der Proteinstruktur und -funktion führen, die je nach Art und Ort der Mutation im Genom variieren kann. Manchmal können Missense-Mutationen die Proteinfunktion beeinträchtigen oder sogar vollständig aufheben, was zu verschiedenen Krankheiten oder Fehlbildungen führen kann. Es ist wichtig zu beachten, dass nicht alle Missense-Mutationen pathogen sind und einige von ihnen möglicherweise keine Auswirkungen auf die Proteinfunktion haben.
In der Genetik versteht man unter "genes, dominant" die Ausprägung eines bestimmten Merkmals, die auftritt, wenn ein dominantes Allel vorhanden ist. Ein Allel ist eine Variante eines Gens, das an einer bestimmten Position auf einem Chromosom liegt. Wenn ein Individuum zwei unterschiedliche Allele für ein Gen besitzt (heterozygot), wird in der Regel das dominante Allel ausgeprägt, während das andere Allel, das rezessive Allel genannt wird, nicht zum Ausdruck kommt.
Zum Beispiel bei der Erbkrankheit Chorea Huntington ist das Gen, welches für die Proteinproduktion des Huntingtins verantwortlich ist, mutiert. Wenn eine Person ein dominantes Allel dieser Mutation besitzt, wird sie unabhängig davon, ob das zweite Allel rezessiv oder nicht betroffen ist, an der Krankheit erkranken.
Es sei jedoch angemerkt, dass die Dominanz eines Gens relativ ist und von Kontext zu Kontext variieren kann. In manchen Fällen können auch mehrere Gene zusammenwirken, um ein Merkmal auszubilden, was als polygenetische Vererbung bezeichnet wird.
Die DNA-Mutationsanalyse ist ein Prozess der Genetik, bei dem die Veränderungen in der DNA-Sequenz untersucht werden, um genetisch bedingte Krankheiten oder Veranlagungen zu diagnostizieren, zu bestätigen oder auszuschließen. Eine Mutation ist eine dauerhafte und oft zufällige Veränderung in der DNA-Sequenz, die die Genstruktur und -funktion beeinflussen kann.
Die DNA-Mutationsanalyse umfasst verschiedene Techniken wie PCR (Polymerasekettenreaktion), DNA-Sequenzierung, MLPA (Multiplex-Ligation-dependent Probe Amplification) und Array-CGH (Array Comparative Genomic Hybridization). Diese Techniken ermöglichen es, kleinste Veränderungen in der DNA zu erkennen, wie z.B. Einzelnukleotidpolymorphismen (SNPs), Deletionen, Insertionen oder Chromosomenaberrationen.
Die Ergebnisse der DNA-Mutationsanalyse können wichtige Informationen für die klinische Diagnose und Therapie von genetisch bedingten Krankheiten liefern, wie z.B. Krebs, erbliche Herzkrankheiten, Stoffwechselstörungen oder neuromuskuläre Erkrankungen. Die DNA-Mutationsanalyse wird auch in der Forschung eingesetzt, um die genetischen Grundlagen von Krankheiten besser zu verstehen und neue Therapieansätze zu entwickeln.
Hyperlipoproteinämie Typ III, auch bekannt als Broad-Beta-Disease oder Dysbetalipoproteinämie, ist ein genetischer Zustand, der durch eine Anhäufung von Chylomikronenremnants und Intermediärlipoproteinen (IDL) im Blut gekennzeichnet ist. Dies führt zu einem erhöhten Spiegel an Gesamtcholesterin, Triglyceriden und Lipoprotein a (Lp(a)) im Blutserum. Die Erkrankung wird durch eine Apolipoprotein E2-Homozygotie verursacht, die zu einer Funktionsstörung des Apolipoprotein E-Moleküls führt und somit die Clearance von Chylomikronenremnants und IDL aus dem Blut behindert.
Hyperlipoproteinämie Typ III ist ein seltener Zustand, der mit einem erhöhten Risiko für kardiovaskuläre Erkrankungen verbunden ist. Symptome können Xanthome (gelbliche, erhabene Hautläsionen), Arcus corneae (ein grauer Ring um die Cornea) und eine Pancreatitis (Entzündung der Bauchspeicheldrüse) umfassen. Die Diagnose wird durch klinische Bewertung, Laboruntersuchungen und Genotypisierung gestellt. Die Behandlung umfasst in der Regel eine cholesterinsenkende Therapie, Ernährungsmodifikationen und Gewichtsmanagement.
Ataxia teleangiectatica ist eine seltene, autosomal-rezessiv vererbte Neurodegenerative Erkrankung, die durch Gendefekte im ATM-Gen (Ataxia Telangiectasia Mutated) verursacht wird. Die Krankheit manifestiert sich normalerweise in den ersten zwei Lebensjahren mit zunehmender Instabilität und Koordinationsverlust der Muskelbewegungen (Ataxie), was zu Schwierigkeiten bei Gehen, Stehen und Balancieren führt.
Andere häufige Symptome sind eine erweiterte Blutgefäßwachstum (Teleangiektasien) im Gesicht und anderen Körperteilen, eine erhöhte Anfälligkeit für Infektionen aufgrund von Immunschwäche, eine progressive neurologische Degeneration, Verzögerung der motorischen Entwicklung, Sprachstörungen, Oculomotorische Apraxie (Störung der Augenbewegungen), Schwierigkeiten beim Schlucken und Atemwegsinfektionen.
Betroffene Personen haben auch ein erhöhtes Risiko für die Entwicklung von Krebs, insbesondere Leukämien und Lymphomen. Es gibt keine Heilung für Ataxia teleangiectatica, aber Symptome können behandelt werden, um die Lebensqualität zu verbessern.
Aminosäuresubstitution bezieht sich auf den Prozess, bei dem ein anderes Aminosäurerestmolekül in einen Proteinstrukturstrang eingebaut wird, anstelle des ursprünglichen Aminosäurerests an einer bestimmten Position. Dies tritt auf, wenn es eine genetische Variante oder Mutation gibt, die dazu führt, dass ein anderes Aminosäure codiert wird, was zu einer Veränderung der Aminosäurensequenz im Protein führt. Die Fähigkeit eines Proteins, seine Funktion aufrechtzuerhalten, nachdem eine Aminosäuresubstitution stattgefunden hat, hängt von der Art und Position der substituierten Aminosäure ab. Manche Substitutionen können die Proteinstruktur und -funktion beeinträchtigen oder sogar zerstören, während andere möglicherweise keine Auswirkungen haben.
Histokompatibilitätsantigene Klasse I sind Proteinkomplexe, die sich auf der Oberfläche der Zellen aller nucleären Organismen befinden. Sie spielen eine entscheidende Rolle im Immunsystem von Wirbeltieren und bestehen aus drei Komponenten: einem großen, transmem membranösen Protein (heavy chain) und zwei kleineren, nicht kovalent gebundenen Proteinen (light chains). Diese Antigene werden in drei verschiedene Untergruppen eingeteilt: HLA-A, HLA-B und HLA-C beim Menschen.
Histokompatibilitätsantigene Klasse I sind von großer Bedeutung bei der Transplantationsmedizin, da sie eine entscheidende Rolle bei der Abstoßungsreaktion gegenüber transplantierten Organen spielen. Die Übereinstimmung der Histokompatibilitätsantigene zwischen Spender und Empfänger ist ein wichtiger Faktor für das Gelingen einer Transplantation. Unterschiede in den HLA-Antigenen können zu einer Abstoßungsreaktion führen, bei der das Immunsystem des Empfängers die transplantierten Zellen als fremd erkennt und angreift.
Chromosomenkartierung ist ein Verfahren in der Genetik und Molekularbiologie, bei dem die Position von Genen oder anderen DNA-Sequenzen auf Chromosomen genau bestimmt wird. Dabei werden verschiedene molekularbiologische Techniken eingesetzt, wie beispielsweise die FISH (Fluoreszenz-in-situ-Hybridisierung) oder die Gelelektrophorese nach restrictionemfraktionierter DNA (RFLP).
Durch Chromosomenkartierung können genetische Merkmale und Krankheiten, die mit bestimmten Chromosomenabschnitten assoziiert sind, identifiziert werden. Diese Informationen sind von großer Bedeutung für die Erforschung von Vererbungsmechanismen und der Entwicklung gentherapeutischer Ansätze.
Die Chromosomenkartierung hat in den letzten Jahren durch die Fortschritte in der Genomsequenzierung und Bioinformatik an Präzision gewonnen, was zu einer detaillierteren Darstellung der genetischen Struktur von Organismen geführt hat.
Genetic linkage refers to the phenomenon where two or more genes are located physically close to each other on a chromosome and tend to be inherited together during meiosis. This means that the transmission of these genes is not independent, but rather they are linked and co-transmitted because the probability of their recombination (i.e., exchange of genetic material between homologous chromosomes) is relatively low. The degree of linkage between genes is measured by the recombination frequency, which reflects the percentage of meiotic events resulting in a crossover between the linked genes. Genes with a high recombination frequency are considered to be loosely linked or unlinked, while those with a low recombination frequency are tightly linked. The concept of genetic linkage is fundamental in genetics and has important implications for understanding patterns of inheritance, mapping gene locations, and identifying genetic variations associated with diseases or traits.
Ferritin ist ein Proteinkomplex, der in allen Zellen des menschlichen Körpers vorkommt und als Speicherprotein für Eisen dient. Es ist vor allem in Leber, Knochenmark, Milz und Muskeln konzentriert. Ferritin kann im Blutserum gemessen werden und dient als Maß für den Gesamtkörper-Eisenspeicher. Niedrige Serumferritinspiegel können auf einen Eisenmangel hinweisen, während hohe Spiegel auf eine übermäßige Eisenakkumulation oder Entzündungen hindeuten können.
Die Gaucher-Krankheit ist eine seltene, genetisch bedingte lysosomale Speichererkrankung, die durch einen Mangel an dem Enzym Glukozerebrosidase gekennzeichnet ist. Dieser Enzymmangel führt zu einem Anstieg des Lipids Glukozerebrosid in den Lysosomen von bestimmten Zellen, vor allem Makrophagen, was zur Bildung von Gaucher-Zellen führt. Diese Zellen sammeln sich hauptsächlich in Knochenmark, Leber und Milz an, wodurch diese Organe vergrößert werden können.
Es gibt drei Hauptformen der Gaucher-Krankheit: Typ 1, Typ 2 und Typ 3. Typ 1 ist die häufigste Form und verläuft chronisch, mit Symptomen wie Leber- und Milzvergrößerung, Blutarmut, Knochenschmerzen und -brüchen. Typ 2 ist eine akute neuropathische Form, die bei Säuglingen oder Kleinkindern auftritt und oft tödlich verläuft. Typ 3 ist eine langsam fortschreitende neuropathische Form, die im Kindes- oder Jugendalter beginnt und zu neurologischen Symptomen wie Sprachstörungen, Ataxie und Epilepsie führt.
Die Gaucher-Krankheit wird autosomal rezessiv vererbt, was bedeutet, dass ein Betroffener zwei Kopien des defekten Gens haben muss, um die Krankheit auszubilden. Die Diagnose erfolgt durch Bestimmung der Aktivität der Glukozerebrosidase im Blut oder in Gewebeproben und kann durch genetische Tests bestätigt werden. Die Behandlung umfasst Enzymersatztherapie, Substratreduktionstherapie und symptomatische Therapien.
Apolipoproteine E (ApoE) sind eine Klasse von Proteinen, die in der Regulation des Lipidstoffwechsels eine wichtige Rolle spielen. Sie sind Hauptbestandteil der Lipoproteinpartikel, wie sehr-low-density-Lipoproteine (VLDL) und high-density-Lipoproteine (HDL), die an der Bindung und Aufnahme von Lipiden in Zellen beteiligt sind.
Es gibt drei verschiedene Isoformen des ApoE-Proteins, ApoE2, ApoE3 und ApoE4, die durch genetische Variationen im APOE-Gen codiert werden. Diese Isoformen unterscheiden sich in ihrer Fähigkeit, Lipide zu binden und zu transportieren, was Auswirkungen auf das Risiko für verschiedene Erkrankungen hat.
Zum Beispiel ist die ApoE4-Isoform mit einem erhöhten Risiko für Alzheimer-Krankheit assoziiert, während ApoE2 mit einem reduzierten Risiko verbunden ist. Darüber hinaus spielt ApoE eine Rolle bei der Entstehung und Progression von Herz-Kreislauf-Erkrankungen, da es an der Clearance von Cholesterin aus dem Blutkreislauf beteiligt ist.
Insgesamt sind Apolipoproteine E wichtige Proteine im Lipidstoffwechsel und haben Auswirkungen auf das Risiko für verschiedene Erkrankungen, insbesondere Herz-Kreislauf-Erkrankungen und Alzheimer-Krankheit.
Genetic models in a medical context refer to theoretical frameworks that describe the inheritance and expression of specific genes or genetic variations associated with certain diseases or traits. These models are used to understand the underlying genetic architecture of a particular condition and can help inform research, diagnosis, and treatment strategies. They may take into account factors such as the mode of inheritance (e.g., autosomal dominant, autosomal recessive, X-linked), penetrance (the likelihood that a person with a particular genetic variant will develop the associated condition), expressivity (the range of severity of the condition among individuals with the same genetic variant), and potential interactions with environmental factors.
Hämoglobin E (HbE) ist eine Variation des Hämoglobins, das den Sauerstofftransport im Blut ermöglicht. Es entsteht durch eine genetische Mutation auf dem beta-Globin-Gen, die eine Aminosäuresubstitution an Position 26 der beta-Globinkette verursacht (Beta26 Glu -> Lys). Diese Mutation kann zu einer abnormalen Struktur und Funktion des Hämoglobins führen.
HbE ist relativ häufig in Südostasien, insbesondere in Thailand, Kambodscha, Laos, Vietnam und Teilen Chinas. Menschen, die zwei Kopien dieses Allels (HbEE) besitzen, können eine mild bis mäßig ausgeprägte Hämolytische Anämie entwickeln. Die Kombination von HbE mit anderen Hämoglobinvarianten wie HbS (das die Sichelzellenanämie verursacht) kann zu schwerwiegenderen Krankheitsbildern führen.
Es ist wichtig zu beachten, dass Menschen, die nur eine Kopie des HbE-Allels tragen (HbA/E), im Allgemeinen keine Symptome entwickeln und ein normales Leben führen können.
In der Medizin bezieht sich die Haarfarbe auf die natürliche oder durch Färbemittel veränderte Farbpigmentierung der Haare, die durch zwei Arten von Pigmenten bestimmt wird: Eumelanin und Phäomelanin. Die Menge und Art dieser Pigmente in den Haaren determinieren die Haarfarbe eines Menschen.
Eumelanin ist das dunklere Pigment, das in braunen und schwarzen Haaren vorkommt. Wenn nur wenig Eumelanin vorhanden ist, erscheinen die Haare blond. Phäomelanin hingegen ist das hellere Pigment, das in roten Haaren am stärksten konzentriert ist, aber auch in braunen und schwarzen Haaren vorkommen kann.
Es ist wichtig zu beachten, dass die Haarfarbe eines Menschen im Laufe des Lebens aufgrund von Alterung, Hormonänderungen und anderen Faktoren variieren kann.
Membranproteine sind Proteine, die sich in der Lipidbilayer-Membran von Zellen oder intrazellulären Organellen befinden. Sie durchdringen oder sind mit der Hydrophobischen Membran verbunden und spielen eine wichtige Rolle bei zellulären Funktionen, wie dem Transport von Molekülen, Signaltransduktion, Zell-Zell-Kommunikation und Erkennung. Membranproteine können in integral (dauerhaft eingebettet) oder peripher (vorübergehend assoziiert) eingeteilt werden, je nachdem, ob sie die Membran direkt durch eine hydrophobe Domäne stabilisieren oder über Wechselwirkungen mit anderen Proteinen assoziiert sind.
Ethylnitrosoharnstoff ist ein chemisches Medikament, das früher zur Behandlung von Angina pectoris eingesetzt wurde. Es wirkt als Vasodilatator, der die glatte Muskulatur in den Blutgefäßen entspannt und so den Blutfluss verbessert. Ethylnitrosoharnstoff ist ein Ester von Nitrosoharnstoff und wird nach der oralen Einnahme schnell vom Körper metabolisiert.
Aufgrund seiner unerwünschten Wirkungen, wie zum Beispiel Kopfschmerzen, niedriger Blutdruck, Übelkeit und Erbrechen, wird Ethylnitrosoharnstoff heutzutage nur noch selten eingesetzt. Stattdessen werden andere Medikamente bevorzugt, die ähnliche Wirkungen haben, aber besser verträglich sind.
Aminoxidoreduktasen sind ein Teil der Enzymsuppe in lebenden Organismen und spielen eine wichtige Rolle im Stoffwechsel von Aminen. Genauer gesagt, katalysieren Aminoxidoreduktasen die Oxidation von primären Aminen zu Aldehyden, wobei zugleich das Coenzym Pyridoxalphosphat (PLP) reduziert wird.
Die medizinische Bedeutung von Aminoxidoreduktasen liegt darin, dass sie an der Entgiftung von exogenen Aminen beteiligt sind, die in den Körper gelangen und potentiell toxisch sein können. Darüber hinaus sind Aminoxidoreduktasen auch an der Synthese von Neurotransmittern wie Serotonin und Dopamin beteiligt, indem sie die Umwandlung von Aminosäuren in Aminoverbindungen katalysieren.
Es gibt verschiedene Arten von Aminoxidoreduktasen, die sich in ihrer Lokalisation, ihrem Substratspektrum und ihrer Regulation unterscheiden. Ein Beispiel ist die semikarbazide-sensitive Aminoxidase (SSAO), die auch als Vascular Adhesion Protein 1 (VAP-1) bezeichnet wird und an der Entzündungsreaktion beteiligt ist. Mutationen in den Genen, die für Aminoxidoreduktasen codieren, können zu Stoffwechselstörungen führen, wie beispielsweise bei der primären Hyperoxalurie Typ I, einer Erkrankung des Harnstein-Stoffwechsels.
Es ist nicht angemessen oder korrekt, eine medizinische Definition für 'Juden' zu geben, da Judentum eine ethnoreligiöse Identität und keine medizinische oder gesundheitsbezogene Kategorie ist. Die Religion des Judentums, die Kultur und die Traditionen werden von Menschen auf der ganzen Welt praktiziert und sind nicht mit bestimmten Krankheiten oder Gesundheitszuständen verbunden.
Die Angabe einer medizinischen Definition für 'Juden' wäre ungenau, irreführend und könnte zu Diskriminierung und Vorurteilen führen. Es ist wichtig, dass wir alle Menschen als Individuen respektieren und ihre ethnische, religiöse und kulturelle Identität schätzen, ohne sie mit medizinischen oder gesundheitsbezogenen Konzepten in Verbindung zu bringen.
Alpha-1-Antitrypsinmangel ist ein genetisch bedingter Mangel an dem Protein Alpha-1-Antitrypsin (AAT), das normalerweise in der Leber produziert wird und im Blut zirkuliert. Das Protein gehört zur Gruppe der Serpin-Proteine und hat die Funktion, andere Enzyme zu inhibieren, insbesondere das Enzym Elastase, das von weißen Blutkörperchen (Neutrophilen) produziert wird.
Elastase ist ein Protein, das normalerweise hilft, Bakterien im Körper zu zerstören. Wenn es jedoch nicht durch AAT gehemmt wird, kann es Gewebe schädigen, insbesondere in den Lungen. Bei Menschen mit Alpha-1-Antitrypsinmangel ist das Protein defekt und kann seine Funktion nicht richtig ausüben, was zu einem Übermaß an Elastase führt und die Lunge angreift.
Die Krankheit wird autosomal-rezessiv vererbt, was bedeutet, dass eine Person das Gen nur dann von ihren Eltern geerbt haben muss, wenn beide Elternteile Träger des Gens sind. Wenn ein Mensch zwei Kopien des defekten Gens hat, führt dies zu einem schweren Mangel an Alpha-1-Antitrypsin und einem erhöhten Risiko für Lungen- und Lebererkrankungen.
Die Symptome von Alpha-1-Antitrypsinmangel können variieren, aber die meisten Menschen mit dieser Krankheit entwickeln im Erwachsenenalter eine chronisch obstruktive Lungenerkrankung (COPD), insbesondere wenn sie rauchen oder anderen schädlichen Umweltfaktoren ausgesetzt sind. Andere Symptome können Lebererkrankungen, Hautausschläge und Blutgerinnungsstörungen sein.
Die Behandlung von Alpha-1-Antitrypsinmangel umfasst in der Regel die Verwendung von Ersatzpräparaten aus menschlichem Plasma, die das fehlende Protein ersetzen. Andere Behandlungen können auch die Einnahme von Medikamenten zur Linderung der Symptome und die Änderung des Lebensstils umfassen, wie zum Beispiel das Aufhören des Rauchens.
Abnorme Hämoglobine sind Varianten des Hämoglobins, die aufgrund genetischer Mutationen in der Struktur oder Menge des Hämoglobinmoleküls von dem normalfall (HbA) abweichen. Diese Abweichungen können zu verschiedenen klinischen Manifestationen führen, wie z.B. anormaler Sauerstofftransport, erhöhter Affinität des Hämoglobins für Sauerstoff oder veränderte Elektrophorese-Muster. Einige der bekanntesten Beispiele sind HbS (verursacht Sichelzellanämie), HbC und HbE, die jeweils durch eine einzelne Aminosäure-Substitution im Hämoglobinmolekül gekennzeichnet sind. Diese Varianten können zu milden bis schweren klinischen Symptomen führen, wie Anämie, Jaundice, Splenomegalie und einem erhöhten Risiko für Infektionen. Es ist wichtig zu beachten, dass nicht alle abnormen Hämoglobine mit Krankheiten assoziiert sind und manche asymptomatisch bleiben können.
DNA, oder Desoxyribonukleinsäure, ist ein Molekül, das die genetische Information in allen Lebewesen und vielen Viren enthält. Es besteht aus zwei langen, sich wiederholenden Ketten von Nukleotiden, die durch Wasserstoffbrückenbindungen miteinander verbunden sind und eine Doppelhelix bilden.
Jeder Nukleotidstrang in der DNA besteht aus einem Zucker (Desoxyribose), einem Phosphatmolekül und einer von vier Nukleobasen: Adenin, Thymin, Guanin oder Cytosin. Die Reihenfolge dieser Basen entlang des Moleküls bildet den genetischen Code, der für die Synthese von Proteinen und anderen wichtigen Molekülen in der Zelle verantwortlich ist.
DNA wird oft als "Blaupause des Lebens" bezeichnet, da sie die Anweisungen enthält, die für das Wachstum, die Entwicklung und die Funktion von Lebewesen erforderlich sind. Die DNA in den Zellen eines Organismus wird in Chromosomen organisiert, die sich im Zellkern befinden.
Apolipoproteine A sind ein Hauptbestandteil der High-Density-Lipoproteine (HDL) oder "guten Cholesterins" im Blutkreislauf. Es gibt mehrere Arten von Apolipoproteinen, aber Apolipoprotein A-1 ist das häufigste und wichtigste Protein in HDL-Partikeln.
Apolipoprotein A-1 spielt eine entscheidende Rolle bei der Umwandlung von Cholesterin in wasserlösliche Partikel, die dann vom Körper ausgeschieden werden können. Es hilft auch, entzündliche Prozesse im Körper zu reduzieren und wirkt als Antioxidans, indem es die Oxidation von Low-Density-Lipoproteinen (LDL) oder "schlechtem Cholesterin" verhindert.
Eine niedrige Konzentration von Apolipoprotein A-1 im Blutkreislauf ist ein Risikofaktor für die Entwicklung von Herz-Kreislauf-Erkrankungen, während höhere Konzentrationen mit einem geringeren Risiko verbunden sind.
Population Genetics ist ein Teilgebiet der Genetik, das sich mit der Verteilung und dem Vorkommen von Genen und Allelen in populationsbiologischen Einheiten beschäftigt. Es untersucht die genetische Variation zwischen Individuen einer Population und wie solche Variation durch verschiedene evolutionäre Kräfte wie Mutation, Genfluss, genetische Drift und Selektion beeinflusst wird.
Die Populationsgenetik liefert wichtige Erkenntnisse zur Entstehung und Verbreitung von Krankheiten in Populationen sowie zur Anpassung von Arten an ihre Umwelt. Sie ist daher ein zentrales Forschungsgebiet der theoretischen und angewandten Genetik, Evolutionsbiologie und medizinischen Genetik.
Eine Kohortenstudie ist eine beobachtende, longitudinale Studie, bei der eine definierte Gruppe von Menschen (die Kohorte), die ein gemeinsames Merkmal oder Erlebnis teilen (z.B. Geburtsjahrgang, Berufsgruppe, Krankheit), über einen längeren Zeitraum hinsichtlich des Auftretens bestimmter Ereignisse oder Erkrankungen untersucht wird. Die Exposition gegenüber einem potenziellen Risikofaktor wird meist zu Beginn der Studie erfasst und das Auftreten der Erkrankung wird dann im Verlauf beobachtet. Kohortenstudien ermöglichen die Bestimmung von Inzidenzraten, relativem Risiko und attributablem Risiko und sind damit gut geeignet, um kausale Zusammenhänge zwischen Exposition und Erkrankung zu untersuchen.
Ein DNA-Primer ist ein kurzes, einzelsträngiges Stück DNA oder RNA, das spezifisch an die Template-Stränge einer DNA-Sequenz bindet und die Replikation oder Amplifikation der DNA durch Polymerasen ermöglicht. Primers sind notwendig, da Polymerasen nur in 5'-3' Richtung synthetisieren können und deshalb an den Startpunkt der Synthese binden müssen. In der PCR (Polymerase Chain Reaction) sind DNA-Primer entscheidend, um die exakte Amplifikation bestimmter DNA-Sequenzen zu gewährleisten. Sie werden spezifisch an die Sequenz vor und nach der Zielregion designed und erlauben so eine gezielte Vermehrung des gewünschten DNA-Abschnitts.
Die Odds Ratio (OR) ist ein statistisches Maß, das zur Beschreibung der Stärke eines Zusammenhangs zwischen zwei binären Ereignissen in einer beobachteten Population verwendet wird. Dabei handelt es sich um das Verhältnis der Odds (Chancen) für das Auftreten eines Ereignisses in der Expositionsgruppe im Vergleich zur Nicht-Expositionsgruppe.
Die Odds Ratio wird oft in epidemiologischen und klinischen Studien, wie z. B. Fall-Kontroll- oder Kohortenstudien, verwendet, um die Wahrscheinlichkeit eines outcomes (z.B. Krankheit) basierend auf einer Exposition (z.B. Risikofaktor) zu quantifizieren.
Eine Odds Ratio von >1 deutet darauf hin, dass das Ereignis in der Expositionsgruppe wahrscheinlicher ist als in der Nicht-Expositionsgruppe. Umgekehrt bedeutet eine OR von
Das Genom ist die Gesamtheit der Erbinformationen (DNA), die in einer Zelle eines Lebewesens enthalten sind. Es umfasst alle Gene, also die Abschnitte der DNA, die für die Synthese von Proteinen verantwortlich sind, sowie nichtcodierende DNA-Sequenzen, die verschiedene Funktionen haben können, wie z.B. die Regulation der Genexpression oder die Strukturierung der Chromosomen. Das menschliche Genom besteht aus etwa 3 Milliarden Basenpaaren und enthält schätzungsweise 20.000-25.000 Gene. Die Entschlüsselung des menschlichen Genoms im Rahmen des Humangenomprojekts hat wichtige Fortschritte in der Biomedizin und den Naturwissenschaften im Allgemeinen ermöglicht.
Apolipoprotein E (APOE) ist ein Protein, das bei der Regulierung des Cholesterin-Stoffwechsels und der Aufrechterhaltung der Gehirnfunktion eine wichtige Rolle spielt. Es gibt drei verschiedene Allele (Formen) dieses Gens: E2, E3 und E4. Apolipoprotein E4 ist eine dieser Varianten des APOE-Gens, von denen bekannt ist, dass sie das Risiko für die Entwicklung der Alzheimer-Krankheit erhöht.
Die APOE4-Variante ist mit etwa 10-15% der Bevölkerung vertreten und macht etwa 25% der Menschen aus, die an Alzheimer erkranken. Das Vorhandensein von einem oder zwei Kopien des APOE4-Allels erhöht das Risiko für die Entwicklung der Alzheimer-Krankheit im Vergleich zu denen, die keine Kopie dieses Allels haben.
Es ist jedoch wichtig anzumerken, dass das Vorhandensein von APOE4 nicht bedeutet, dass eine Person sicher an Alzheimer erkranken wird. Viele Menschen mit APOE4 werden nie Alzheimer bekommen und viele Menschen ohne APOE4 können die Krankheit entwickeln. Das Vorhandensein von APOE4 ist nur ein Risikofaktor für die Erkrankung, und es gibt viele andere Faktoren, die das Alzheimer-Risiko beeinflussen können, einschließlich des Lebensstils, der Umwelt und der Genetik.
Apolipoprotein C-III (ApoC-III) ist ein kleines, wasserlösliches Protein, das hauptsächlich in den Leberzellen synthetisiert wird. Es ist ein wichtiger Bestandteil von Lipoproteinen, insbesondere sehr niedrig Dichte Lipoproteine (VLDL) und Low-Density-Lipoproteine (LDL), die für den Transport von Fetten im Körper verantwortlich sind.
ApoC-III spielt eine wichtige Rolle bei der Regulation des Fettstoffwechsels, indem es die Clearance von Lipoproteinen aus dem Blutkreislauf hemmt und die Lipaseaktivität inhibiert, was zu einem Anstieg des Plasmaspiegels von Triglyceriden führt. Hohe Konzentrationen von ApoC-III im Blut sind mit einem erhöhten Risiko für kardiovaskuläre Erkrankungen assoziiert.
Es gibt auch Hinweise darauf, dass ApoC-III an der Entwicklung von Insulinresistenz und Typ-2-Diabetes beteiligt sein könnte. Einige Studien haben gezeigt, dass niedrigere ApoC-III-Spiegel mit einem geringeren Risiko für kardiovaskuläre Erkrankungen und Diabetes einhergehen.
In der Medizin bezieht sich "Genes" auf den Prozess der Wiederherstellung der normalen Funktion und des Wohlbefindens nach einer Krankheit, Verletzung oder Operation. Dieser Begriff beschreibt den Zustand, in dem ein Patient die Symptome seiner Erkrankung überwunden hat und wieder in der Lage ist, seine täglichen Aktivitäten ohne Beeinträchtigung auszuführen.
Es ist wichtig zu beachten, dass "Genes" nicht immer bedeutet, dass eine Person vollständig geheilt ist oder keine Spuren der Erkrankung mehr aufweist. Manchmal kann es sich lediglich um eine Verbesserung des Zustands handeln, bei der die Symptome abgeklungen sind und das Risiko einer erneuten Verschlechterung minimiert wurde.
Die Dauer des Genesungsprozesses hängt von verschiedenen Faktoren ab, wie Art und Schwere der Erkrankung, dem Alter und Gesundheitszustand des Patienten sowie der Qualität der medizinischen Versorgung und Nachsorge. In einigen Fällen kann die Genesung schnell und vollständig sein, während sie in anderen Fällen langwierig und möglicherweise unvollständig sein kann.
Ein Codon ist ein dreibasiger Nukleotidabschnitt in der DNA oder RNA, der für die genetische Information zur Synthese eines spezifischen Aminosäurerestes in einem Protein kodiert. Es gibt 64 mögliche Codone (inklusive Start- und Stoppcodons), die sich aus den vier Nukleotiden Adenin, Thymin/Uracil, Guanin und Cytosin zusammensetzen. Jedes Codon wird von einem korrespondierenden tRNA-Molekül erkannt und decodiert, um die entsprechende Aminosäure während der Proteinsynthese zu transportieren.
Hypolipoproteinämie ist ein medizinischer Zustand, der durch niedrige Konzentrationen von Lipoproteinen im Blut gekennzeichnet ist. Lipoproteine sind komplexe Partikel, die Fette (Lipide) und Proteine umfassen und für den Transport von Fetten in unserem Körper wesentlich sind. Es gibt verschiedene Arten von Lipoproteinen, wie Chylomikronen, VLDL (very low-density lipoproteins), LDL (low-density lipoproteins) und HDL (high-density lipoproteins). Jede Art von Lipoprotein hat eine bestimmte Funktion im Transport von Fetten.
Eine Hypolipoproteinämie kann auftreten, wenn die Leber nicht in der Lage ist, ausreichende Mengen an Lipoproteinen zu produzieren oder wenn es genetische Störungen gibt, die den Abbau von Lipoproteinen beeinträchtigen. Diese Bedingung kann sich auf verschiedene Weise manifestieren, einschließlich niedriger Cholesterin- und Triglyceridspiegel im Blut.
Es ist wichtig zu beachten, dass eine niedrige Konzentration von Lipoproteinen im Blut nicht unbedingt schädlich sein muss. Einige Formen der Hypolipoproteinämie können jedoch mit einem erhöhten Risiko für neurologische Erkrankungen verbunden sein, wie beispielsweise spinale Muskelatrophie oder periphere Neuropathie. Darüber hinaus kann eine schwere Hypolipoproteinämie zu Mangelernährung führen, da Fette eine wichtige Energiequelle darstellen.
Die Alpha-Thalassämie ist ein genetisches Blutkrankheit, welches durch eine reduzierte Fähigkeit oder Unfähigkeit zur Synthese des Hämoglobin-Proteins im roten Blutfarbstoff (Hämoglobin) der roten Blutkörperchen (Erythrozyten) gekennzeichnet ist. Dies wird durch Mutationen im Gen verursacht, das für die Alpha-Kette des Hämoglobins kodiert.
Die Schwere der Erkrankung hängt von der Anzahl der fehlenden oder nicht funktionsfähigen Gene ab. Bei schweren Formen der Alpha-Thalassämie kann es zu einer Anämie kommen, die sich in Müdigkeit, Blässe, Kurzatmigkeit und im Wachstumsrückstand bei Kindern äußert. In extremen Fällen können intrauterine oder neonatale Tod auftreten.
Die Alpha-Thalassämie ist verbreitet in Bevölkerungsgruppen, die aus geografischen Gebieten mit hoher Malaria-Übertragungsrate stammen, wie zum Beispiel im Mittelmeerraum, in Afrika, im Nahen Osten und in Südostasien. Die Diagnose der Alpha-Thalassämie erfolgt durch Laboruntersuchungen, einschließlich Hämoglobin-Elektrophorese und Gentests. Die Behandlung hängt von der Schwere der Erkrankung ab und kann Bluttransfusionen, Eisenchelationstherapie und Stammzellentransplantation umfassen.
Cholesterin ist ein fettartiger, wachsartiger Alkohol, der in den Membranen von Zellen im Körper vorkommt und für die Produktion von Hormonen, Vitamin D und Gallensäuren unerlässlich ist. Es wird hauptsächlich vom Körper selbst produziert, aber es kann auch mit der Nahrung aufgenommen werden, insbesondere durch den Verzehr von tierischen Produkten.
Cholesterin wird im Blutkreislauf durch Lipoproteine transportiert, die als "gutes Cholesterin" (High-Density-Lipoprotein, HDL) und "schlechtes Cholesterin" (Low-Density-Lipoprotein, LDL) bezeichnet werden. Ein hoher Spiegel von LDL-Cholesterin im Blutkreislauf kann zu Ablagerungen in den Arterienwänden führen und das Risiko für Herzkrankheiten und Schlaganfälle erhöhen. Ein niedriger HDL-Spiegel ist ebenfalls mit einem höheren Risiko für Herzkrankheiten verbunden.
Es ist wichtig, einen ausgewogenen Cholesterinspiegel im Blut aufrechtzuerhalten, um das Risiko von Herz-Kreislauf-Erkrankungen zu minimieren. Eine cholesterinarme Ernährung, regelmäßige körperliche Aktivität und gegebenenfalls Medikamente können dazu beitragen, den Cholesterinspiegel im Blut zu kontrollieren.
Embryonenverlust ist ein medizinischer Begriff, der den Verlust einer Schwangerschaft in den frühen Stadien bezeichnet, bevor das Fötus ein Herz hat geschlagen oder eine Größe von mehr als 5 mm erreicht hat. Dies tritt normalerweise vor der 8. Schwangerschaftswoche auf und wird manchmal auch als "früher Fruchtverlust" oder "fehlgeschlagene Empfängnis" bezeichnet.
Die Ursachen für Embryonenverlust können vielfältig sein, dazu gehören Chromosomenanomalien, Hormonstörungen, anatomische Probleme der Gebärmutter oder andere Erkrankungen bei der Mutter. In vielen Fällen ist die Ursache jedoch unbekannt und der Verlust passiert spontan und unerwartet.
Embryonenverlust kann ein emotional belastendes Ereignis sein, und es wird empfohlen, dass Frauen, die einen solchen Verlust erleiden, medizinische und psychologische Unterstützung suchen.
Eisen ist ein essentielles Spurenelement, das für den Sauerstofftransport im Körper unerlässlich ist. Es ist ein Hauptbestandteil des Hämoglobins in den roten Blutkörperchen und des Myoglobins in den Muskeln. Hämoglobin bindet Eisen, um Sauerstoff aus der Lunge aufzunehmen und zu den Geweben des Körpers zu transportieren, während Myoglobin Eisen verwendet, um Sauerstoff in den Muskeln zu speichern.
'Gene Deletion' ist ein Begriff aus der Genetik und bezeichnet den Verlust eines bestimmten Abschnitts oder sogar eines gesamten Gens auf einer DNA-Molekülstrangseite. Diese Mutation kann auftreten, wenn ein Stück Chromosomenmaterial herausgeschnitten wird oder durch fehlerhafte DNA-Reparaturmechanismen während der Zellteilung.
Die Folgen einer Gendeletion hängen davon ab, welches Gen betroffen ist und wie groß der gelöschte Abschnitt ist. In einigen Fällen kann eine Gendeletion zu keinen oder nur sehr milden Symptomen führen, während sie in anderen Fällen schwerwiegende Entwicklungsstörungen, Erkrankungen oder Behinderungen verursachen kann.
Es ist wichtig zu beachten, dass Gendeletionen bei der genetischen Beratung und Diagnostik eine große Rolle spielen, insbesondere wenn es um erbliche Krankheiten geht. Durch die Analyse von Chromosomen und Genen können Ärzte und Forscher feststellen, ob ein bestimmtes Gen fehlt oder ob es Veränderungen in der DNA-Sequenz gibt, die mit einer Erkrankung verbunden sind.
'Drosophila melanogaster' ist keine medizinische Bezeichnung, sondern die wissenschaftliche Bezeichnung für die Taufliege oder Fruchtfliege. Es handelt sich um ein kleines Insekt, das häufig in der biologischen und genetischen Forschung eingesetzt wird, da es eine kurze Generationszeit hat, leicht zu züchten und zu manipulieren ist, und sein Genom gut erforscht und verstanden ist. Die Entschlüsselung des Genoms von Drosophila melanogaster hat wertvolle Einblicke in die Funktionsweise von Genen bei verschiedenen Tierarten geliefert, einschließlich Menschen.
Neurologische Mutantenmäuse sind genetisch veränderte Labortiere, die für die Erforschung von neurologischen Erkrankungen und Störungen eingesetzt werden. Dabei wird das Erbgut der Mäuse so manipuliert, dass sie Veränderungen aufweisen, die dem menschlichen Krankheitsbild ähneln. Ziel ist es, durch das Studium dieser Tiere mehr über die zugrundeliegenden Mechanismen von neurologischen Erkrankungen wie Parkinson, Alzheimer, Epilepsie oder multipler Sklerose herauszufinden und neue Therapien zu entwickeln.
Es gibt verschiedene Arten von neurologischen Mutantenmäusen, die sich in der Art der genetischen Veränderung unterscheiden. Manche Mäuse tragen zusätzliche Kopien eines Gens, während andere ein Gen gezielt ausschalten (knockout) oder verändern (knock-in). Auch können mehrere Gene gleichzeitig verändert werden, um komplexe Krankheitsbilder abzubilden.
Die Verwendung von neurologischen Mutantenmäusen hat in den letzten Jahren zu wichtigen Erkenntnissen im Bereich der Neurowissenschaften beigetragen und ermöglicht es Forschern, neue Behandlungsansätze für neurologische Erkrankungen zu entwickeln.
Das Manifestationsalter ist ein Begriff aus der Medizin, der das typische Alter beschreibt, in dem die Symptome (die „Manifestationen“) einer bestimmten Erkrankung erstmals auftreten. Es handelt sich also um das durchschnittliche Alter, in dem die Krankheit klinisch sichtbar wird und vom Patienten oder von einem Arzt bemerkt werden kann.
Das Manifestationsalter ist ein wichtiger Faktor bei der Diagnose vieler Krankheiten, insbesondere bei Entwicklungsstörungen und genetischen Erkrankungen. Zum Beispiel manifestiert sich die spinale Muskelatrophie (SMA) typischerweise im Säuglings- oder Kleinkindalter, während die Alzheimer-Krankheit üblicherweise erst im höheren Erwachsenenalter auftritt.
Es ist jedoch wichtig zu beachten, dass das Manifestationsalter variieren kann und nicht bei allen Patienten mit derselben Erkrankung gleich ist. Einige Menschen können die Symptome einer Krankheit früher oder später im Leben entwickeln als üblich, was als atypische Manifestation oder variable Manifestation bezeichnet wird.
Die Gilbert-Krankheit, auch bekannt als Meulengracht-Syndrom oder Hyperbilirubinämie III, ist eine genetisch bedingte, mild verlaufende Lebererkrankung. Sie wird durch eine Mutation im Gen für die Bilirubin-Uridindiphosphat-Glucuronosyltransferase (UGT1A1) verursacht, was zu einer verminderten Fähigkeit führt, Bilirubin zu konjugieren und auszuscheiden.
Bilirubin ist ein Abbauprodukt des roten Blutfarbstoffs Hämoglobin. Unkonjugiertes Bilirubin wird in der Leber durch die UGT1A1-Enzymaktivität zu konjugiertem, wasserlöslichem Bilirubin umgewandelt und über die Galle ausgeschieden. Bei Menschen mit Gilbert-Krankheit ist die Aktivität des UGT1A1-Enzyms vermindert, wodurch sich unkonjugiertes Bilirubin im Blut ansammelt und zu leicht erhöhten Bilirubinwerten führt.
Die Gilbert-Krankheit ist in der Regel asymptomatisch oder verursacht nur milde Symptome wie Gelbfärbung der Haut (Ikterus) und der Augen (Sklerenikterus), insbesondere nach Fasten, Infektionen, Stress oder körperlicher Anstrengung. Die Diagnose erfolgt meist durch die Bestimmung des Bilirubinspiegels im Blut und Ausschluss anderer Lebererkrankungen.
Die Gilbert-Krankheit ist nicht behandlungsbedürftig, und es gibt keine bekannten Komplikationen oder Folgeschäden. Betroffene Personen wird lediglich geraten, auf einen gesunden Lebensstil zu achten, einschließlich ausreichender Flüssigkeitszufuhr und einer ausgewogenen Ernährung. In seltenen Fällen kann eine medikamentöse Behandlung erforderlich sein, wenn die Symptome störend sind oder sich die Leberwerte deutlich verschlechtern.
Arylkohlenwasserstoff-Hydroxylasen sind Enzyme, die an der Entgiftung von aromatischen Verbindungen und polyzyklischen aromatischen Kohlenwasserstoffen (PAK) beteiligt sind. Diese Verbindungen können in unserer Umwelt durch verschiedene Quellen wie Autoabgase, Zigarettenrauch oder industrielle Prozesse vorkommen und können für den Menschen krebserregend sein.
Die Arylkohlenwasserstoff-Hydroxylasen katalysieren die Hinzufügung einer Hydroxygruppe an das aromatische System der Verbindungen, was zu einer wasserlöslichen Verbindung führt, die dann leichter aus dem Körper ausgeschieden werden kann. Diese Enzyme sind Teil des xenobiotischen Stoffwechselwegs und kommen hauptsächlich in der Leber vor. Es gibt mehrere Isoformen von Arylkohlenwasserstoff-Hydroxylasen, darunter die bekannteste ist das Enzym CYP1A1. Die Aktivität dieser Enzyme kann durch verschiedene Faktoren wie Genetik, Ernährung und Exposition gegenüber Umweltgiften beeinflusst werden.
Genotyping techniques are a group of laboratory methods used to identify and analyze the genetic makeup (genotype) of an individual, organism, or virus. These techniques can be used to detect differences or variations in the DNA sequence, gene structure, or number of chromosomes between individuals. Genotyping is used in various fields such as medical research, forensic science, and ancestry testing.
There are several types of genotyping techniques, including:
1. Polymerase Chain Reaction (PCR) based methods: These methods use PCR to amplify specific regions of the DNA and then analyze them for variations. Examples include Amplification Refractory Mutation System (ARMS), TaqMan assays, and High-Resolution Melting (HRM) analysis.
2. Sequencing-based methods: These methods determine the order of nucleotides in a specific region of DNA. Examples include Sanger sequencing, Next-Generation Sequencing (NGS), and Whole Genome Sequencing (WGS).
3. Microarray-based methods: These methods use microarrays to analyze multiple genetic markers simultaneously. Examples include Single Nucleotide Polymorphism (SNP) arrays and Comparative Genomic Hybridization (CGH) arrays.
4. Fragment analysis-based methods: These methods use capillary electrophoresis or gel electrophoresis to separate DNA fragments based on their size, followed by detection using fluorescence or radioactivity. Examples include Short Tandem Repeat (STR) analysis and Restriction Fragment Length Polymorphism (RFLP).
Genotyping techniques have numerous applications in medicine, including diagnosis of genetic disorders, identification of disease-associated genes, pharmacogenomics, and infectious disease surveillance.
Apolipoprotein B (ApoB) ist ein großes, essentielles Protein, das bei der Bildung und Transport von Lipoproteinen im Körper beteiligt ist. Es gibt zwei Hauptformen von ApoB: ApoB-48 und ApoB-100.
ApoB-48 wird hauptsächlich in der Darmschleimhaut produziert und ist ein wichtiger Bestandteil von Chylomikronen, den Lipoproteinen, die Lipide aus der Nahrung transportieren.
ApoB-100 hingegen wird in der Leber synthetisiert und ist das Strukturprotein der sehr niedrig density Lipoproteine (VLDL), die während des Fettstoffwechsels Triglyceride aus der Leber zu den peripheren Geweben transportieren. Im Laufe des Stoffwechsels werden VLDL in Low-Density-Lipoproteine (LDL) umgewandelt, die hauptsächlich Cholesterin transportieren. ApoB-100 ist der einzige Apolipoprotein-Bestandteil von LDL und wird daher manchmal auch als "LDL-Rezeptor-Ligand" bezeichnet.
Erhöhte Konzentrationen von ApoB im Blutplasma sind mit einem erhöhten Risiko für kardiovaskuläre Erkrankungen verbunden, da sie auf eine erhöhte Anzahl von atherogenen Lipoproteinpartikeln hinweisen.
Homocystein ist eine nicht-proteinogene, schwefelhaltige Aminosäure, die im Stoffwechsel des Menschen entsteht. Es ist ein Intermediat im Methionin-Stoffwechsel und wird durch die Enzyme CBS (Cystathion-Beta-Synthase) und MTR (Methionin-Synthase) in weiterführende Stoffwechselwege überführt.
Erhöhte Homocystein-Spiegel im Blutplasma können ein Risikofaktor für die Entwicklung von kardiovaskulären Erkrankungen, Schlaganfall und venösen Thrombosen sein. Des Weiteren wurde ein Zusammenhang zwischen Hyperhomocysteinämie (erhöhtem Homocystein-Spiegel) und neurologischen Erkrankungen wie Demenz oder Alzheimer-Krankheit diskutiert, ist aber nicht abschließend geklärt.
Erniedrigte Spiegel an Vitamin B12, Folsäure oder Vitamin B6 können zu einem Anstieg des Homocystein-Spiegels führen, weshalb eine ausreichende Versorgung mit diesen Vitaminen empfohlen wird.
Inzucht, auch Inbreeding genannt, ist ein Begriff aus der Genetik und beschreibt die Paarung oder Fortpflanzung zwischen eng miteinander verwandten Organismen wie nahen Verwandten (z.B. Eltern mit ihren Kindern, Geschwistern, Onkeln/Tanten mit Nichten/Neffen). In der Humangenetik wird Inzucht oft als Maß für den Grad der Verwandtschaft zweier Menschen verwendet und wird durch den sogenannten Inzuchtkoeffizienten ausgedrückt.
Inzucht kann zu einer Erhöhung der Wahrscheinlichkeit führen, dass reinerbige oder homozygote Allele (Versionen eines Gens) auftreten, was wiederum das Risiko für genetisch bedingte Krankheiten und Entwicklungsstörungen erhöhen kann. Dies liegt daran, dass bei enger Verwandtschaft die Wahrscheinlichkeit größer ist, dass beide Elternteilen dieselben Allele für ein Gen geerbt haben.
Es ist wichtig zu beachten, dass Inzucht in der menschlichen Population aufgrund sozialer und kultureller Normen selten vorkommt. In einigen Tierpopulationen hingegen kann Inzucht auftreten, wenn die Population klein ist oder wenn es an geeigneten Partnern mangelt.
Es tut mir leid für die Verwirrung, aber "China" ist kein medizinischer Begriff oder Konzept. China ist ein Land in Ostasien, das offiziell als Volksrepublik China bekannt ist. Wenn Sie an eine bestimmte medizinische Bezeichnung denken, die mit dem Wort "China" verbunden sein könnte, wie beispielsweise "Chinasyndrom", das sich auf eine Kontamination durch radioaktive Substanzen bezieht, lassen Sie es mich bitte wissen und ich werde versuchen, Ihre Frage entsprechend zu beantworten.
Apolipoprotein A-I (ApoA-I) ist ein Protein, das hauptsächlich in der Hülle von High-Density-Lipoproteinen (HDL) vorkommt, die allgemein als "gutes Cholesterin" bekannt sind. Es wird hauptsächlich in der Leber und im Dünndarm synthetisiert. ApoA-I spielt eine wichtige Rolle bei der Umwandlung von Cholesterin in HDL, was zur Bildung von so genanntem "umgekehrtem Cholesterintransport" führt, einem Prozess, bei dem überschüssiges Cholesterin aus Geweben (einschließlich Arterienwänden) zu Leber und Darm transportiert wird, wo es metabolisiert oder ausgeschieden werden kann. ApoA-I hat auch entzündungshemmende Eigenschaften und spielt möglicherweise eine Rolle bei der endothelialen Funktion und der Immunantwort.
Stärkegel-Elektrophorese ist ein Labortest, bei dem Proteine in einer Probe aufgrund ihrer elektrischen Ladung und Größe getrennt werden. Im Gegensatz zu anderen Elektrophorese-Methoden verwendet Stärkegel-Elektrophorese eine Gelmatrix mit bekannter Porengröße, um die Migration der Proteine während des Tests besser zu kontrollieren und zu standardisieren.
Das Gel wird in ein Elektrophoresegerät eingesetzt, das eine elektrische Spannung appliziert, wodurch die Proteine in Richtung des Gegenions migrieren. Die Proteine bewegen sich durch das Gel mit einer Geschwindigkeit, die von ihrer Ladung und Größe abhängt. Große oder stark geladene Proteine migrieren schneller als kleinere oder weniger geladene Proteine.
Nach Abschluss der Elektrophorese wird das Gel typischerweise gefärbt, um die Position der Proteine sichtbar zu machen. Die Färbung kann durch verschiedene Methoden erfolgen, wie z.B. mit Coomassie-Brillantblau oder Silbernitrat. Das resultierende Muster der Proteinbanden wird dann analysiert, um Informationen über die Art und Menge der vorhandenen Proteine zu gewinnen.
Stärkegel-Elektrophorese ist ein wichtiges Werkzeug in der klinischen Diagnostik und Forschung, insbesondere in der Proteomik und Klinischen Biochemie. Es wird häufig eingesetzt, um Veränderungen im Proteinmuster bei verschiedenen Krankheiten wie Krebs, Stoffwechselstörungen und neurologischen Erkrankungen zu identifizieren.
Der Inzuchtstamm C57BL (C57 Black 6) ist ein spezifischer Stamm von Labormäusen, der durch enge Verwandtschaftspaarungen über mehrere Generationen hinweg gezüchtet wurde. Dieser Prozess, bekannt als Inzucht, dient dazu, eine genetisch homogene Population zu schaffen, bei der die meisten Tiere nahezu identische Genotypen aufweisen.
Die Mäuse des C57BL-Stammes sind für biomedizinische Forschungen sehr beliebt, da sie eine Reihe von vorteilhaften Eigenschaften besitzen. Dazu gehören:
1. Genetische Homogenität: Die enge Verwandtschaftspaarung führt dazu, dass die Tiere des C57BL-Stammes ein sehr ähnliches genetisches Profil aufweisen. Dies erleichtert die Reproduzierbarkeit von Experimenten und die Interpretation der Ergebnisse.
2. Robuste Gesundheit: Die Tiere des C57BL-Stammes gelten als gesund und leben im Allgemeinen lange. Sie sind anfällig für bestimmte Krankheiten, was sie zu einem geeigneten Modell für die Erforschung dieser Krankheiten macht.
3. Anfälligkeit für Krankheiten: C57BL-Mäuse sind anfällig für eine Reihe von Krankheiten, wie zum Beispiel Diabetes, Krebs, neurologische Erkrankungen und Immunerkrankungen. Dies macht sie zu einem wertvollen Modellorganismus für die Erforschung dieser Krankheiten und zur Entwicklung neuer Therapeutika.
4. Verfügbarkeit von genetisch veränderten Linien: Da der C57BL-Stamm seit langem in der Forschung eingesetzt wird, stehen zahlreiche genetisch veränderte Linien zur Verfügung. Diese Linien können für die Untersuchung spezifischer biologischer Prozesse oder Krankheiten eingesetzt werden.
5. Eignung für verschiedene experimentelle Ansätze: C57BL-Mäuse sind aufgrund ihrer Größe, Lebensdauer und Robustheit für eine Vielzahl von experimentellen Ansätzen geeignet, wie zum Beispiel Verhaltensstudien, Biochemie, Zellbiologie, Genetik und Immunologie.
Es ist wichtig zu beachten, dass C57BL-Mäuse nicht für jede Art von Forschung geeignet sind. Ihre Anfälligkeit für bestimmte Krankheiten kann sie als Modellorganismus ungeeignet machen, wenn das Ziel der Studie die Untersuchung einer anderen Krankheit ist. Darüber hinaus können genetische und Umweltfaktoren die Ergebnisse von Experimenten beeinflussen, was die Notwendigkeit einer sorgfältigen Planung und Durchführung von Experimenten unterstreicht.
In der Medizin und Epidemiologie werden ethnische Gruppen oft auf der Grundlage gemeinsamer kultureller, linguistischer, nationaler oder geografischer Merkmale definiert. Diese Definitionen können jedoch je nach Kontext und Studie variieren. Es ist wichtig zu beachten, dass die Zugehörigkeit zu einer ethnischen Gruppe in der Regel selbstgewählt ist und sich auf individuelle Identität und Selbstverständnis bezieht.
Es gibt jedoch auch objektive Kriterien wie Abstammung oder Herkunft, die bei der Definition von ethnischen Gruppen herangezogen werden können. Diese Kriterien umfassen häufig gemeinsame genetische Merkmale, die auf eine gemeinsame Abstammung hinweisen.
Es ist wichtig zu beachten, dass die Zugehörigkeit zu einer ethnischen Gruppe nicht mit der Rasse gleichzusetzen ist. Während Rasse sich auf physische Merkmale wie Hautfarbe, Haartextur und Körperbau bezieht, geht es bei der Ethnie um kulturelle, linguistische und historische Faktoren.
Insgesamt gibt es keine einheitliche Definition von "ethnischen Gruppen" in der Medizin, aber sie werden üblicherweise als soziale Konstrukte definiert, die auf gemeinsamen kulturellen, linguistischen, nationalen oder geografischen Merkmalen beruhen und sich oft auf genetische Verwandtschaft beziehen.
Lipide sind in der Biochemie und Medizin eine Gruppe von Stoffen, die hauptsächlich aus Kohlenwasserstoffketten bestehen und fettlöslich sind. Sie spielen eine wichtige Rolle als Energiereservoir, Strukturkomponenten von Zellmembranen und als Signalmoleküle im Körper.
Lipide umfassen eine Vielzahl von Verbindungen wie Triglyceride (Neutralfette), Phospholipide, Cholesterin und Lipoproteine. Zu den Funktionen von Lipiden gehören die Bereitstellung von Energie, die Unterstützung der Aufnahme und des Transports fettlöslicher Vitamine, die Schutzfunktionen der Haut und die Regulierung von Stoffwechselprozessen.
Eine übermäßige Ansammlung von Lipiden in Blutgefäßen kann jedoch zu Atherosklerose und Herz-Kreislauf-Erkrankungen führen, während ein Mangel an bestimmten Lipiden wie Omega-3-Fettsäuren mit Erkrankungen wie Herzkrankheiten und Entzündungen in Verbindung gebracht wird.
Knockout-Mäuse sind gentechnisch veränderte Mäuse, bei denen ein bestimmtes Gen gezielt ausgeschaltet („geknockt“) wurde, um die Funktion dieses Gens zu untersuchen. Dazu wird in der Regel ein spezifisches Stück der DNA, das für das Gen codiert, durch ein anderes Stück DNA ersetzt, welches ein selektives Merkmal trägt und es ermöglicht, die knockout-Zellen zu identifizieren. Durch diesen Prozess können Forscher die Auswirkungen des Fehlens eines bestimmten Gens auf die Physiologie, Entwicklung und Verhaltensweisen der Maus untersuchen. Knockout-Mäuse sind ein wichtiges Werkzeug in der biomedizinischen Forschung, um Krankheitsmechanismen zu verstehen und neue Therapeutika zu entwickeln.
Lipoproteine sind komplexe Partikel, die sich im Blutplasma befinden und hauptsächlich aus Proteinen (Apolipoproteinen) und Lipiden (Fetten und Cholesterin) bestehen. Ihre Hauptfunktion ist der Transport von Lipiden zwischen den Zellen des Körpers.
Lipoproteine werden in verschiedene Klassen eingeteilt, je nach ihrer Dichte:
- Chylomikronen: die leichtesten und größten Lipoproteine, die Lipide aus der Nahrung transportieren
- VLDL (very low density lipoproteins): sie transportieren Triglyceride von der Leber zu den peripheren Geweben
- IDL (intermediate density lipoproteins): sie sind ein Zwischenprodukt bei der Umwandlung von VLDL in LDL
- LDL (low density lipoproteins): sie werden oft als "schlechtes Cholesterin" bezeichnet, da hohe Konzentrationen im Blutplasma mit einem erhöhten Risiko für Herzkrankheiten verbunden sind
- HDL (high density lipoproteins): sie werden oft als "gutes Cholesterin" bezeichnet, da sie Cholesterin von den Zellen zu Leber transportieren und so das Risiko von Herzkrankheiten verringern können.
Eine ausgewogene Ernährung und ein gesunder Lebensstil können dazu beitragen, die Konzentrationen der verschiedenen Lipoproteine im Blutplasma zu optimieren und so das Risiko von Herz-Kreislauf-Erkrankungen zu verringern.
Hämoglobinopathien sind erbliche Störungen der Hämoglobinsynthese, bei denen es zu einer strukturellen oder quantitativen Abweichung des Hämoglobins kommt. Das Hämoglobin ist ein Proteinkomplex in den roten Blutkörperchen (Erythrozyten), der für den Sauerstofftransport im Blut verantwortlich ist. Die beiden häufigsten und klinisch bedeutsamen Hämoglobinopathien sind die Sichelzellkrankheit (HbS) und die Thalassämie.
Die Sichelzellkrankheit entsteht durch eine Punktmutation im Gen, das für die β-Kette des Hämoglobins codiert. Dies führt zu einer substitutionellen Aminosäureänderung (Glutamat zu Valin) und zur Bildung von abnorm verformbaren und instabilen Hämoglobinmolekülen, die bei niedrigen Sauerstoffspiegeln zu Sichelzellen und Mikrozirkulationsstörungen führen.
Thalassämien sind eine Gruppe von Erkrankungen, die durch eine verminderte Synthese der α- oder β-Ketten des Hämoglobins gekennzeichnet sind. Die ungleiche Produktion der Ketten führt zur Bildung von instabilen Tetrameren und zu einer Hemmung der Erythropoese, was schließlich zu einer Anämie führt.
Die klinischen Manifestationen der Hämoglobinopathien reichen von milden bis hin zu lebensbedrohlichen Erkrankungen und können Hämolyse, Infarkte, Organschäden und Entwicklungsverzögerungen umfassen. Die Diagnose erfolgt meist durch Hämoglobinelektrophorese und Gentestung. Die Behandlung kann symptomatisch oder kausal sein und hängt von der Schwere der Erkrankung ab.
Apolipoproteine C sind eine Klasse von Proteinen, die sich mit Lipoproteinpartikeln verbinden und eine wichtige Rolle bei der Regulation des Lipidstoffwechsels spielen. Es gibt drei Haupttypen von Apolipoprotein C: ApoC-I, ApoC-II und ApoC-III.
ApoC-I ist ein inhibitorisches Protein, das die Aktivität der Lipoproteinlipase hemmt und so die Clearance von Triglyceriden aus dem Blutkreislauf verringert.
ApoC-II ist ein aktivierendes Protein, das die Aktivität der Lipoproteinlipase erhöht und so die Clearance von Triglyceriden aus dem Blutkreislauf fördert.
ApoC-III ist ein inhibitorisches Protein, das die Aktivität der Lipoproteinlipase hemmt und so die Clearance von Triglyceriden aus dem Blutkreislauf verringert. Es wurde auch gezeigt, dass ApoC-III die HDL-Cholesterinwerte im Blut senken und die Entstehung von atherosklerotischen Plaques fördern kann.
Störungen im Metabolismus der Apolipoproteine C können zu Dyslipidämien führen, die wiederum das Risiko für kardiovaskuläre Erkrankungen erhöhen können.
Familiäres Mittelmeerfieber ist eine autosomal-rezessiv vererbte autoinflammatory Krankheit, die gekennzeichnet ist durch periodische Episoden von Fieber, Bauchschmerzen, Pleuritis, Perikarditis, Arthralgien und Hautausschlägen. Die Krankheit wird durch Mutationen im MEFV-Gen verursacht, das für das Protein Pyrin kodiert. Das familiäre Mittelmeerfieber ist am häufigsten in mediterranen Bevölkerungsgruppen wie Türken, Arabern, Juden und Italienern zu finden. Die Diagnose wird durch charakteristische klinische Merkmale und genetische Tests gestellt. Die Behandlung umfasst kolchizinhaltige Medikamente wie Colchicin zur Vorbeugung von Krankheitsepisoden und Entzündungshemmer wie nicht-steroidale Antirheumatika (NSAIDs) oder Glukokortikoide zur Linderung der Symptome während einer Episode.
Fetales Hämoglobin (HbF) ist eine Form des Hämoglobins, das hauptsächlich während der fetalen Entwicklung im Blutkreislauf des Fötus vorkommt und seine Aufgabe in der Sauerstofftransportfunktion erfüllt. Es besteht aus zwei Alpha- und zwei Gamma-Ketten (α2γ2). Im Vergleich zu adultem Hämoglobin (HbA) hat HbF eine höhere Affinität zur Bindung an Sauerstoff, was bedeutet, dass es Sauerstoff effektiver aus der Plazenta aufnimmt und an den Fötus abgibt.
Die Umwandlung von fetalem Hämoglobin in adultes Hämoglobin beginnt normalerweise während der letzten Phase der fetalen Entwicklung und dauert bis zum Alter von etwa sechs Monaten nach der Geburt an. Bei einigen Erkrankungen, wie z.B. Sichelzellkrankheit und Thalassämie, bleibt jedoch ein höherer Anteil an HbF im Blut erhalten, was als fetale Hämoglobinopathie bezeichnet wird. Dies kann vorübergehend oder dauerhaft sein und kann die Symptome der Erkrankung mildern, indem es die Bildung von krankhaften Hämoglobinen verringert.
Lipoproteine sind komplexe Partikel, die aus Lipiden (Fette) und Proteinen bestehen und im Blutplasma zirkulieren. Sie spielen eine wichtige Rolle bei der Fetttransportierung im Körper. Es gibt verschiedene Arten von Lipoproteinen, die sich in ihrer Dichte und Zusammensetzung unterscheiden, wie zum Beispiel Low-Density-Lipoproteine (LDL), Very-Low-Density-Lipoproteine (VLDL) und High-Density-Lipoproteine (HDL).
HDL-Cholesterol, auch bekannt als "gutes Cholesterin", bezieht sich auf das Cholesterin, das mit HDL-Partikeln assoziiert ist. HDL-Partikel sind kleiner und dichter als LDL-Partikel und werden manchmal als "Müllabfuhr des Körpers" bezeichnet, weil sie überschüssiges Cholesterin von Zellen im Körper aufnehmen und zu Leber transportieren, wo es abgebaut oder ausgeschieden werden kann. Hohe HDL-Cholesterinspiegel sind mit einem verringerten Risiko für Herzkrankheiten verbunden, während niedrige HDL-Cholesterinspiegel mit einem erhöhten Risiko assoziiert sind.
Gen-Targeting bezieht sich auf die gezielte Manipulation oder Modulation spezifischer Gene innerhalb von Zellen zur Behandlung oder Erforschung von Krankheiten. Dies wird in der Regel durch Techniken wie Geneditierung (z.B. CRISPR-Cas9), RNA-Interferenz oder Antisense-Oligonukleotide erreicht. Das Ziel ist es, die Funktion eines defekten Gens zu korrigieren, die Expression eines überaktiven Gens zu reduzieren oder gezielt therapeutische Proteine in den Zellen zu produzieren. Diese Techniken haben das Potenzial, neue Behandlungsmöglichkeiten für eine Vielzahl von Erkrankungen wie genetisch bedingte Krankheiten, Krebs und Virusinfektionen zu eröffnen.
Cholesteryl Ester Transfer Proteins (CETP) sind spezielle Proteine, die am Transport von Lipiden zwischen Lipoproteinen im Blutkreislauf beteiligt sind. Genauer gesagt, ermöglichen sie den Transfer von Cholesterylesternen von HDL (High-Density-Lipoprotein) zu LDL (Low-Density-Lipoprotein) und VLDL (Very Low-Density-Lipoprotein). Dieser Prozess ist wichtig, um den Cholesterinstoffwechsel im Körper aufrechtzuerhalten.
Eine Erhöhung der Aktivität von CETP kann dazu führen, dass sich mehr Cholesterin in LDL-Partikeln ansammelt und weniger in HDL-Partikeln verbleibt. Da hohe Konzentrationen von LDL-Cholesterin mit einem erhöhten Risiko für kardiovaskuläre Erkrankungen verbunden sind, während HDL-Cholesterin als schützend gilt, kann eine übermäßige Aktivität von CETP das Herzinfarktrisiko erhöhen.
Es gibt auch Medikamente, sogenannte CETP-Inhibitoren, die entwickelt wurden, um die Aktivität von CETP zu hemmen und so den HDL-Spiegel im Blut zu erhöhen und das Herzinfarktrisiko zu reduzieren. Allerdings haben klinische Studien mit diesen Medikamenten bisher gemischte Ergebnisse gezeigt, und ihre Verwendung ist derzeit umstritten.
Neuralrohrdefekte sind Fehlbildungen des sich entwickelnden Nervensystems während der Embryonalentwicklung. Das Neuralrohr ist die Struktur, aus der das zentrale Nervensystem – das Gehirn und das Rückenmark – hervorgeht. Bei einer gestörten Entwicklung kann es zu Fehlbildungen kommen, bei denen sich das Neuralrohr nicht vollständig schließt.
Es gibt drei Hauptarten von Neuralrohrdefekten:
1. Anenzephalie: Dabei handelt es sich um die schwerwiegendste Form von Neuralrohrdefekten, bei der sich der vordere Teil des Neuralrohrs nicht schließt. Das Gehirn und andere Schädelstrukturen fehlen oder sind nur unvollständig ausgebildet. Diese Fehlbildung ist mit dem Leben nicht vereinbar.
2. Spina bifida occulta: Hierbei handelt es sich um eine leichtere Form von Neuralrohrdefekten, bei der sich der hintere Teil des Neuralrohrs nicht schließt. Meist ist nur ein kleiner Bereich des Rückenmarks betroffen, und die Fehlbildung kann ohne Symptome verlaufen. In manchen Fällen können jedoch neurologische Ausfälle auftreten, wie beispielsweise Lähmungen in den Beinen oder Blasen- und Darminkontinenz.
3. Meningomyelozele: Bei dieser Form von Neuralrohrdefekten ist ein größerer Bereich des Rückenmarks betroffen, der durch eine sackartige Ausbuchtung nach außen hervortritt. Die Fehlbildung kann mit neurologischen Ausfällen verbunden sein, wie Lähmungen in den Beinen, Blasen- und Darminkontinenz sowie Sensibilitätsstörungen.
Die Ursachen von Neuralrohrdefekten sind noch nicht vollständig geklärt, aber es wird angenommen, dass genetische Faktoren und Umweltfaktoren wie eine unzureichende Versorgung mit Folsäure während der Schwangerschaft eine Rolle spielen.
Angeborene Störungen des Lipidstoffwechsels sind eine Gruppe von Erkrankungen, die aufgrund genetischer Mutationen auftreten und zu einer Stoffwechselstörung von Fetten (Lipiden) führen. Diese Erkrankungen können in der Leber, im Gehirn oder in anderen Organen Schäden verursachen und sind oft mit erhöhten Konzentrationen von Cholesterin oder Triglyceriden im Blut verbunden.
Es gibt verschiedene Arten von angeborenen Störungen des Lipidstoffwechsels, wie zum Beispiel:
* Hypercholesterinämien: Hierbei handelt es sich um Erkrankungen, die zu einem Anstieg des Cholesterinspiegels im Blut führen. Eine bekannte Form ist die familiäre Hypercholesterinämie, bei der eine Genmutation dazu führt, dass der Körper nicht in der Lage ist, überschüssiges Cholesterin aus dem Blut zu entfernen.
* Hypertriglyceridämien: Diese Erkrankungen verursachen einen Anstieg des Triglyceridspiegels im Blut. Eine bekannte Form ist die familiäre Hypertriglyceridämie, bei der eine Genmutation dazu führt, dass der Körper nicht in der Lage ist, überschüssige Triglyceride aus dem Blut zu entfernen.
* Lipoproteinlipase-Mangel: Dies ist eine seltene Erkrankung, bei der ein Enzymmangel dazu führt, dass Fette aus der Nahrung nicht richtig verdaut und aufgenommen werden können.
* Abetalipoproteinämie: Hierbei handelt es sich um eine seltene Erkrankung, bei der der Körper nicht in der Lage ist, Lipoproteine zu bilden, die für den Transport von Fetten im Blut erforderlich sind.
Die Behandlung dieser Erkrankungen hängt von der Art und Schwere der Erkrankung ab. In einigen Fällen kann eine Ernährungsumstellung oder Medikamente helfen, den Cholesterin- oder Triglyceridspiegel im Blut zu senken. In schwereren Fällen kann eine Operation erforderlich sein. Es ist wichtig, dass Menschen mit diesen Erkrankungen regelmäßig von einem Arzt untersucht werden, um sicherzustellen, dass sie richtig behandelt werden und Komplikationen vermieden werden.
Desoxyribonukleasen vom Typ II sind Enzyme, die spezifisch die DNA-Stränge spalten können. Sie sind in der Lage, die Phosphodiesterbindungen zwischen den Nukleotiden zu hydrolysieren und somit die DNA in kleinere Bruchstücke aufzuteilen.
Regionalspezifische Desoxyribonukleasen II sind eine Untergruppe dieser Enzyme, die an bestimmte Sequenzen der DNA binden und diese gezielt schneiden können. Sie werden oft in biochemischen und molekularbiologischen Anwendungen eingesetzt, um die DNA gezielt zu modifizieren oder zu sequenzieren.
Ein Beispiel für eine regionalspezifische Desoxyribonuklease II ist das Restriktionsendonuklease-Enzym, das an bestimmte Nukleotidsequenzen in der DNA bindet und diese spezifisch schneidet. Diese Enzyme werden oft aus Bakterien oder Bakteriophagen isoliert und sind ein wichtiges Werkzeug in der Molekularbiologie.
Apolipoprotein E-3 (ApoE-3) ist eine der drei verschiedenen isoformen des Apolipoproteins E, das ein wichtiges Protein im Lipidstoffwechsel des menschlichen Körpers ist. Es wird hauptsächlich in Leberzellen produziert und spielt eine entscheidende Rolle bei der Bindung, Transport und Stoffwechsel von Lipoproteinen, insbesondere Chylomikronen und VLDL (very low-density lipoproteins).
ApoE-3 ist die häufigste isoform des Apolipoproteins E und wird als neutral angesehen, da es weder mit einem erhöhten noch erniedrigten Risiko für Herz-Kreislauf-Erkrankungen assoziiert ist. Im Gegensatz dazu sind die beiden anderen isoformen ApoE-2 und ApoE-4 mit einem erhöhten Risiko für Hyperlipidämie, Alzheimer-Krankheit und anderen Erkrankungen verbunden.
ApoE-3 ist ein wichtiger Faktor bei der Regulierung des Cholesterinspiegels im Blut und trägt dazu bei, das Gleichgewicht zwischen den verschiedenen Lipoproteinen aufrechtzuerhalten. Es ist auch an der Aufnahme von Cholesterin in die Leber beteiligt und spielt eine Rolle bei der Immunantwort des Körpers.
Folsäure, auch als Vitamin B9 bekannt, ist eine wasserlösliche organische Verbindung, die für zahlreiche Stoffwechselvorgänge im menschlichen Körper unerlässlich ist. Sie spielt eine entscheidende Rolle bei der Zellteilung und dem Wachstum, insbesondere während der Schwangerschaft für das Wachstum des Fötus.
Folsäure trägt zur Bildung und Reparatur von DNA bei, hilft bei der Synthese von Aminosäuren und ist an der Methylierung von Homocystein zu Methionin beteiligt. Ein Mangel an Folsäure kann zu verschiedenen Gesundheitsproblemen führen, wie beispielsweise Anämie, Geburtsfehlern (Neuralrohrdefekten) bei Ungeborenen und einem erhöhten Risiko für Herz-Kreislauf-Erkrankungen.
Folsäure kommt in vielen Nahrungsmitteln vor, wie grünem Blattgemüse, Obst, Orangensaft, Bohnen, Nüssen und Vollkornprodukten. Da der Körper Folsäure nicht selbst herstellen kann, ist es wichtig, sie über die Ernährung oder Nahrungsergänzungsmittel aufzunehmen.
Chromosomen sind im Zellkern befindliche Strukturen, die die Erbinformationen in Form von Desoxyribonukleinsäure (DNA) enthalten. Sie sind bei der Zellteilung und -vermehrung von großer Bedeutung, da sie sich verdoppeln und dann zwischen den Tochterzellen gleichmäßig verteilen, um so eine genetisch identische Kopie der Elternzelle zu erzeugen.
Ein Chromosom besteht aus zwei Chromatiden, die durch einen Zentromer miteinander verbunden sind. Die Chromatiden enthalten jeweils ein lineares DNA-Molekül, das mit Proteinen assoziiert ist und in bestimmten Abschnitten (den Genen) die Erbinformationen kodiert.
Im Menschen gibt es 23 verschiedene Chromosomenpaare, von denen 22 Paare als Autosomen bezeichnet werden und ein Paar Geschlechtschromosomen (XX bei Frauen, XY bei Männern) bildet. Die Gesamtzahl der Chromosomen in einer menschlichen Zelle beträgt daher 46.
Lezithin-Azyltransferasemangel ist ein seltener Stoffwechseldefekt, bei dem das Enzym Lezithin-Azyltransferase (LAT) nicht oder nur in geringen Mengen vorhanden ist. Dieses Enzym ist für die Umwandlung des Lipids Lezithin in andere Verbindungen notwendig, insbesondere in Plasmalogen, eine Art von Phospholipid, das ein wichtiger Bestandteil der Zellmembranen ist.
Der Mangel an LAT führt zu einem Anstieg des Lezithinspiegels im Blut und zu einem Rückgang des Plasmalogenspiegels in den Zellmembranen, insbesondere in denen des Gehirns. Dies kann zu einer Reihe von neurologischen Symptomen führen, wie z.B. verzögerte Entwicklung, Muskelhypotonie (schlaffe Muskeln), Ataxie (koordinationsstörungen), Epilepsie und in schweren Fällen geistiger Behinderung.
Der Lezithin-Azyltransferasemangel wird autosomal-rezessiv vererbt, was bedeutet, dass ein Betroffener zwei Kopien des defekten Gens haben muss, um die Krankheit zu entwickeln. Die Diagnose erfolgt meist durch die Bestimmung der Aktivität der Lezithin-Azyltransferase im Blut oder in Fibroblasten (Hautzellen) und kann durch genetische Tests bestätigt werden.
Eine Aminosäuresequenz ist die genau festgelegte Reihenfolge der verschiedenen Aminosäuren, aus denen ein Proteinmolekül aufgebaut ist. Sie wird direkt durch die Nukleotidsequenz des entsprechenden Gens bestimmt und spielt eine zentrale Rolle bei der Funktion eines Proteins.
Die Aminosäuren sind über Peptidbindungen miteinander verknüpft, wobei die Carboxylgruppe (-COOH) einer Aminosäure mit der Aminogruppe (-NH2) der nächsten reagiert, wodurch eine neue Peptidbindung entsteht und Wasser abgespalten wird. Diese Reaktion wiederholt sich, bis die gesamte Kette der Proteinsequenz synthetisiert ist.
Die Aminosäuresequenz eines Proteins ist einzigartig und dient als wichtiges Merkmal zur Klassifizierung und Identifizierung von Proteinen. Sie bestimmt auch die räumliche Struktur des Proteins, indem sie hydrophobe und hydrophile Bereiche voneinander trennt und so die Sekundär- und Tertiärstruktur beeinflusst.
Abweichungen in der Aminosäuresequenz können zu Veränderungen in der Proteinstruktur und -funktion führen, was wiederum mit verschiedenen Krankheiten assoziiert sein kann. Daher ist die Bestimmung der Aminosäuresequenz von großer Bedeutung für das Verständnis der Funktion von Proteinen und deren Rolle bei Erkrankungen.
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) ist ein Protein, das als Chloridkanal in der Membran von Epithelzellen dient. Es spielt eine wichtige Rolle bei der Flüssigkeitsregulation in verschiedenen Organen, wie beispielsweise den Atemwegen, dem Verdauungstrakt und den Schweißdrüsen. Mutationen in dem Gen, das für CFTR codiert, können zu cystischer Fibrose führen, einer erblich bedingten Krankheit, die durch eine übermäßige Produktion von zähem Schleim in verschiedenen Organen gekennzeichnet ist. Diese Mutationen können dazu führen, dass das CFTR-Protein nicht richtig funktioniert oder gar nicht erst gebildet wird, was zu Störungen der Chlorid- und Flüssigkeitstransportmechanismen führt.
Carrierproteine, auch als Transportproteine bekannt, sind Moleküle, die die Funktion haben, andere Moleküle oder Ionen durch Membranen zu transportieren. Sie spielen eine wichtige Rolle im Stoffwechsel von Zellen und im interzellulären Kommunikationsprozess. Carrierproteine sind in der Lage, Substanzen wie Zucker, Aminosäuren, Ionen und andere Moleküle selektiv zu binden und diese durch die Membran zu transportieren, indem sie einen Konformationswandel durchlaufen.
Es gibt zwei Arten von Carrierproteinen: uniporter und symporter/antiporter. Uniporter transportieren nur eine Art von Substanz in eine Richtung, während Symporter und Antiporter jeweils zwei verschiedene Arten von Substanzen gleichzeitig in die gleiche oder entgegengesetzte Richtung transportieren.
Carrierproteine sind von großer Bedeutung für den Transport von Molekülen durch Zellmembranen, da diese normalerweise nicht-polar und lipophil sind und somit nur unpolare oder lipophile Moleküle passiv durch Diffusion durch die Membran transportieren können. Carrierproteine ermöglichen es so, auch polare und hydrophile Moleküle aktiv zu transportieren.
Das MNSs-Blutgruppensystem ist ein relativ seltenes humanes Blutgruppensystem, das auf die Anwesenheit oder Abwesenheit von bestimmten Antigenen auf der Oberfläche der roten Blutkörperchen (Erythrozyten) basiert. Es wird durch drei Hauptgene, M, N und S, codiert, die jeweils mehrere Allele haben können. Diese Gene steuern die Synthese von Glykoproteinen auf der Oberfläche der Erythrozyten, die als M, N, oder S-Antigene bekannt sind.
Die Kombination dieser Antigene führt zu verschiedenen Phänotypen im MNSs-System: M-, N-, MN, MS, NS und MNS. Die meisten Menschen sind entweder M oder N positiv, während die anderen Phänotypen seltener vorkommen.
Das MNSs-Blutgruppensystem ist klinisch relevant, da Antikörper gegen diese Antigene in einigen Fällen auftreten können und bei Bluttransfusionen oder Schwangerschaft Komplikationen verursachen können. Zum Beispiel kann eine Person mit dem M-Phänotyp Antikörper gegen N-Antigene bilden, was zu einer transfusionsbedingten hämolytischen Reaktion führen kann, wenn ihr Blut mit N-positivem Blut transfundiert wird. Ähnlich können Antikörper gegen M- oder S-Antigene bei entsprechenden Phänotypen auftreten und Komplikationen verursachen.
Es ist wichtig zu beachten, dass das MNSs-Blutgruppensystem unabhängig vom ABO-Blutgruppensystem ist, obwohl es einige Überschneidungen in der Antigenstruktur und -funktion gibt.
Introns sind nicht-kodierende Sequenzen in einem DNA-Molekül, die während der Transkription in mRNA (messenger RNA) umgeschrieben werden. Sie werden als "inneres" Segment zwischen zwei kodierenden Abschnitten oder Exons definiert.
Ein Embryo ist in der Medizin und Biologie die Bezeichnung für die frühe Entwicklungsphase eines Organismus vom Zeitpunkt der Befruchtung bis zum Beginn der Ausbildung der Körperorgane (ca. 8. Woche beim Menschen). In dieser Phase finden die Hauptprozesse der Embryogenese statt, wie Zellteilungen, Differenzierungen, Migrationen und Interaktionen, die zur Bildung der drei Keimblätter und der sich daraus differenzierenden Organe führen.
Bei Menschen wird nach der 8. Entwicklungswoche auch vom Fötus gesprochen. Es ist wichtig zu beachten, dass verschiedene Definitionen des Begriffs 'Embryo' in unterschiedlichen Kontexten und Rechtssystemen variieren können, insbesondere im Hinblick auf ethische und rechtliche Fragen der Fortpflanzungsmedizin.
Faktor V, auch bekannt als Proakzelerin oder Labile Faktor, ist ein Protein das im menschlichen Plasma vorkommt und eine wichtige Rolle in der Blutgerinnung spielt. Es wird von Leberzellen produziert und dient als Co-Faktor für die Aktivierung von Faktor X zu Faktor Xa während der Gerinnungskaskade.
Wenn Faktor V durch die Thrombin-aktivierte Faktor V-Protease in seine aktive Form Faktor Va umgewandelt wird, beschleunigt es die Umwandlung von Prothrombin zu Thrombin, was wiederum zur Bildung von Fibrin führt und so zur Blutgerinnung beiträgt.
Eine Fehlfunktion des Faktors V kann zu Gerinnungsstörungen führen, wie zum Beispiel die vererbte Thromboembolie, bei der das Risiko für venöse Thromboembolien (Blutgerinnsel in den Venen) erhöht ist.
Globine sind ein Teil von Proteinen, die als Globuline bezeichnet werden und in den Erythrozyten (roten Blutkörperchen) vorkommen. Die bekannteste und am besten untersuchte Gruppe der Globine sind die Hämoglobine, welche den Sauerstofftransport im Blut ermöglichen.
Hämoglobin besteht aus vier Proteinketten: zwei identische alpha-Globine und zwei identische beta-Globine. Diese Globin-Ketten sind mit einem Häm-Gruppen verbunden, die jeweils ein Eisen-Ion enthalten. Das Eisen im Hämoglobin bindet reversibel an Sauerstoff, was es ermöglicht, Sauerstoff von den Lungen zu den Geweben des Körpers zu transportieren.
Abnormalitäten in der Struktur oder Funktion von Globinen können zu verschiedenen Erkrankungen führen, wie z.B. Sichelzellanämie (eine Krankheit, die durch eine Mutation im beta-Globin-Gen verursacht wird) und Thalassämien (eine Gruppe von Erbkrankheiten, die durch eine Störung in der alpha- oder beta-Globin-Produktion gekennzeichnet sind).
LDL (Low-Density Lipoprotein) ist ein Typ von Lipoprotein, der hauptsächlich Cholesterin und andere Fette an die Zellen in Ihrem Körper transportiert. Es wird oft als "schlechtes Cholesterin" bezeichnet, weil hohe LDL-Spiegel das Risiko für Herzkrankheiten und Schlaganfälle erhöhen können, wenn sie sich an den Innenwänden der Arterien ablagern und so die Blutgefäße verengen oder verstopfen. Es ist wichtig, einen normalen LDL-Spiegel aufrechtzuerhalten, um das Risiko von Herz-Kreislauf-Erkrankungen zu minimieren.
Epistasis ist ein Begriff aus der Genetik und beschreibt ein Phänomen, bei dem die Auswirkungen eines Gens durch die Variation eines oder mehrerer anderer Gene beeinflusst werden. In anderen Worten, das Erscheinungsbild oder die Expression eines Gens wird durch die Interaktion mit einem anderen Gen verändert.
Es gibt verschiedene Arten von Epistasis, aber eine häufige Form ist die sogenannte rezessive Epistasis. Hierbei maskiert ein rezessives Allel (die weniger aktive Variante) eines Gens die Wirkung eines dominanten Allels (die aktivere Variante) eines anderen Gens.
Epistasis spielt eine wichtige Rolle bei der Ausprägung komplexer Merkmale und Krankheiten, wie beispielsweise Stoffwechselstörungen oder Erkrankungen des Immunsystems. Daher ist ein Verständnis von Epistasis für die humane Genetik und personalisierte Medizin von großer Bedeutung.
Transgenic Mice sind gentechnisch veränderte Mauslinien, bei denen Fremd-DNA (auch Transgen) in ihr Genom eingebracht wurde, um das genetische Material der Mäuse gezielt zu verändern. Das Ziel ist es, das Verständnis von Genfunktionen und krankheitsverursachenden Genmutationen zu verbessern.
Die Einführung des Transgens kann durch verschiedene Techniken erfolgen, wie beispielsweise per Mikroinjektion in die Keimzellen (Eizelle oder Spermien), durch Nukleofugierung in embryonale Stammzellen oder mithilfe von Virenvektoren.
Die transgenen Mäuse exprimieren das fremde Gen und können so als Modellorganismus für die Erforschung menschlicher Krankheiten dienen, um beispielsweise Krankheitsmechanismen besser zu verstehen oder neue Therapien zu entwickeln. Die Veränderungen im Genom der Tiere werden oft so gestaltet, dass sie die humane Krankheit nachahmen und somit ein geeignetes Modell für Forschungszwecke darstellen.
Fetales Tod, auch als intrauteriner Fruchttod oder stiller Tod bekannt, ist die Beendigung einer Schwangerschaft nach der 20. Schwangerschaftswoche mit dem Nachweis eines toten Fetus. In den USA wird es häufig definiert als Tod eines Fetus nach der 20. Schwangerschaftswoche mit einem Gewicht von mehr als 500 Gramm oder einer Länge von mehr als 20 cm. Die Ursachen des fetalen Todes sind vielfältig und können auf Komplikationen während der Schwangerschaft, genetische Faktoren, Infektionen, Mangelernährung oder Umweltfaktoren zurückzuführen sein. Der Nachweis eines fetalen Todes erfolgt durch Ultraschall oder nach der Entbindung durch klinische Untersuchung des Fetus.
HLA-Antigene, auch bekannt als Human Leukocyte Antigens, sind eine Klasse von Proteinen, die auf der Oberfläche der meisten Zellen im menschlichen Körper vorkommen. Sie spielen eine wichtige Rolle im Immunsystem, indem sie dem Körper helfen, zwischen "selbst" und "nicht-selbst" zu unterscheiden.
HLA-Antigene präsentieren kurze Proteinstücke, die aus both inneren und äußeren Quellen stammen, an T-Zellen des Immunsystems. Auf dieser Basis entscheidet das Immunsystem, ob eine Zelle als "normal" oder "krankhaft" eingestuft wird. Wenn beispielsweise ein Virus in eine Zelle eindringt und sich dort vermehrt, werden virale Proteinstücke von HLA-Antigenen präsentiert, was dazu führt, dass das Immunsystem die infizierte Zelle zerstört.
Es gibt drei Hauptklassen von HLA-Antigenen: Klasse I (A, B und C), Klasse II (DP, DQ und DR) und Klasse III. Jede Klasse hat eine unterschiedliche Funktion im Immunsystem. Unterschiede in HLA-Genen können die Anfälligkeit für bestimmte Krankheiten beeinflussen, einschließlich Autoimmunerkrankungen und Infektionskrankheiten. Darüber hinaus werden HLA-Typen zur Übereinstimmung bei Organtransplantationen verwendet, um die Wahrscheinlichkeit von Abstoßungsreaktionen zu minimieren.
 Homozygote familiäre Hypercholesterinämie
Homozygote familiäre Hypercholesterinämie
Mesomele Dysplasie Typ Langer
Morbus Wilson
Juvenile myelomonozytäre Leukämie
Nucleolus
Hämoglobinopathie
X-chromosomaler Erbgang
Mikrodeletionssyndrom
Klein-Waardenburg-Syndrom
Hyperlipoproteinämie
Fellfarben der Pferde
Scheckung
Bombay-Blutgruppe
Aussterben
Liste der betroffenen Merkmale des Gutachtens zur Auslegung des Verbotes von Qualzüchtungen
Selektion (Evolution)
Hardy-Weinberg-Gleichgewicht
Brachyurie
Faktor-XI-Mangel
Okulärer Albinismus Typ 1
Sabino Overo
Willebrand-Jürgens-Syndrom
Leuzismus
Rezessiv
Klotho (Protein)
Upshaw-Schulman-Syndrom
Silver-Locus
Zystenniere
Chinesischer Schopfhund
Malaria
Homozygote familiäre Hypercholesterinämie - Wikipedia
 Nutzenbewertungsverfahren zum Wirkstoff Evolocumab (Erneute Nutzenbewertung § 14: primäre Hypercholesterinämie, gemischte...
Nutzenbewertungsverfahren zum Wirkstoff Evolocumab (Erneute Nutzenbewertung § 14: primäre Hypercholesterinämie, gemischte...
 Ontozry: Neues Antikonvulsivum bei Epilepsie | APOTHEKE ADHOC
Ontozry: Neues Antikonvulsivum bei Epilepsie | APOTHEKE ADHOC
 Die Rolle von Vitamin D-Mangel bei Schilddrüsenerkrankungen | Symptome, Ursachen von Krankheiten
Die Rolle von Vitamin D-Mangel bei Schilddrüsenerkrankungen | Symptome, Ursachen von Krankheiten
 absetzer fohlen - Tiere kaufen & verkaufen: Tiermarkt & Haustiere auf Quoka.de
absetzer fohlen - Tiere kaufen & verkaufen: Tiermarkt & Haustiere auf Quoka.de
 Nicht bösartige hämatologische Erkrankungen | Universitätsklinikum Freiburg
Nicht bösartige hämatologische Erkrankungen | Universitätsklinikum Freiburg
 Von der Theorie zur Praxis: das dritte Jahr des Molekularbiologie-Studiums | Universität Basel
Von der Theorie zur Praxis: das dritte Jahr des Molekularbiologie-Studiums | Universität Basel
 OPUS Würzburg | Search
OPUS Würzburg | Search
 Familiäre Hypercholesterinämie: Ursachen, Risiken und Diagnose
Familiäre Hypercholesterinämie: Ursachen, Risiken und Diagnose
 Galaktokinase | LADR
Galaktokinase | LADR
 Atorvastatin AXiromed 10 mg/ -20 mg/ -40 mg/ -80 mg Filmtabletten: Dosierung, Nebenwirkung & Wirkung
Atorvastatin AXiromed 10 mg/ -20 mg/ -40 mg/ -80 mg Filmtabletten: Dosierung, Nebenwirkung & Wirkung
 Ginkgo biloba
Ginkgo biloba
 Hilfe zu Resistenz gegenüber aktiviertem Protein C: Prävention
Hilfe zu Resistenz gegenüber aktiviertem Protein C: Prävention
 Pharmakogenetics/genomics - Universitätsinstitut für Medizinisch-Chemische Labordiagnostik - Zentrallabor LKH
Pharmakogenetics/genomics - Universitätsinstitut für Medizinisch-Chemische Labordiagnostik - Zentrallabor LKH
 IPK-Forschungsteam klassifiziert erstmals Schlüssel-Gen für die Zellteilung
IPK-Forschungsteam klassifiziert erstmals Schlüssel-Gen für die Zellteilung
 Glucose-6-Phosphat-Dehydrogenase-Defizienz (Favismus): Labor & Diagnostik
Glucose-6-Phosphat-Dehydrogenase-Defizienz (Favismus): Labor & Diagnostik
 Alpha-1-Antitrypsin-Mangel
Alpha-1-Antitrypsin-Mangel
OPUS 4 | Search
 Simvabeta 80mg 100 stk → bei versandApo.de
Simvabeta 80mg 100 stk → bei versandApo.de
 Von der Erbse bis zur modernen genetischen Medizin - myPoint
Von der Erbse bis zur modernen genetischen Medizin - myPoint
 Genomia: Prüfung von Hunden: ARVC
Genomia: Prüfung von Hunden: ARVC
 Infantile neuroaxonale Dystrophie Seitelberger und Cockayne-Syndrom - Pädiatrie - eMedpedia
Infantile neuroaxonale Dystrophie Seitelberger und Cockayne-Syndrom - Pädiatrie - eMedpedia
 Dissertation Stefanie Krüger | PDF
Dissertation Stefanie Krüger | PDF
Studienhandbuch | Diseases caused by gene-environment-interaction
 Orkambi bei Kleinkindern: Real-life-Studie zeigt Langzeitsicherheit - Deutsche CF-Hilfe
Orkambi bei Kleinkindern: Real-life-Studie zeigt Langzeitsicherheit - Deutsche CF-Hilfe
 Wahre Größe misst sich nicht in Zentimetern ...<...
Wahre Größe misst sich nicht in Zentimetern ...<...
Zystische Fibrose (Mukoviszidose) im Kindes- und Jugendalter - Pädiatrie - eMedpedia
 Tabbys - Tabbys
Tabbys - Tabbys
Die IQ-Falle: Rezensionen durch Prof. Dr. Helmar Frank (Paderborn) und Prof. Dr. Petra Netter (Giessen)
Heterozygote3
- Sie definieren für Patienten mit einer familiären Hypercholesterinämie, gleichwohl ob homozygote oder heterozygote Hypercholesterinämie, einen LDL-Cholesterin-Zielwert (wikipedia.org)
- ehemals Typ I Hyperlipidämie), die in der Kindheit oder Jugend auftritt und häufig durch homozygote oder kombiniert-heterozygote pathogene Varianten im LPL -Gen oder dessen Cofaktoren APOC2 , APOA5 , GPIHBP1 oder LMF1 verursacht wird. (medizinische-genetik.de)
- Ursache für eine schwere Hypertriglyzeridämie sind homozygote oder gemischt heterozygote pathogene Varianten im LPL -Gen ( LPL-Defizienz ). (medizinische-genetik.de)
Individuen3
- Uniformitätsregel: Kreuzt man zwei homozygote Individuen, dann sind alle Nachkommen gleich 2. (explain-it-arium.de)
- Kreuzt man zwei reinerbige ( homozygote ) Individuen miteinander, die sich in nur einem Merkmal voneinander unterscheiden, die sog. (hpage.com)
- Kreuzt man homozygote Individuen einer Art, die sich in einem einzigen Merkmal unterscheiden, so sind die Nachkommen heterozygot und unter sich gleich. (naturundbildung.at)
Fohlen1
- Homozygote graue Pferde sind genetisch verpflichtet, das Gen bei der Zucht an 100 % ihrer Nachkommen weiterzugeben, sodass alle Fohlen grau und verblassen werden. (equigerminal.shop)
HoFH1
- Die homozygote familiäre Hypercholesterinämie (HoFH) ist eine erbliche, seltene Krankheit. (wikipedia.org)
Allels2
- dagegen sind Homozygote des normalen dominanten Allels anfällig für Malaria . (spektrum.de)
- In etwa 25 % der Europäer sind homozygote Träger des geschädigten Allels und damit von dieser Form der hereditären Laktoseintoleranz betroffen. (praxis-blumenthal.de)
Nachkommen1
- Homozygote graue Exemplare sind ideal, da sie bei der Zucht immer das graue Gen weitergeben und somit eventuelle graue Nachkommen garantieren. (equigerminal.shop)
Erbkrankheit2
- an einer Erbkrankheit, genannt homozygote familiäre Hypercholesterinämie leiden, welche zu erhöhten Cholesterinwerten in Ihrem Blut führt. (patienteninfo-service.de)
- Erbkrankheit, die zu erhöhten Cholesterinwerten führt (homozygote familiäre Hypercholesterinämie): Die Gabe eines Statins alleine reicht nicht aus, um den Spiegel zu senken. (gokapsel.de)
Seltene1
- Die homozygote Form der familiären Hypercholesterinämie ist eine seltene Erkrankung. (uni-wuerzburg.de)
Risiko1
- So wurde ein Gen identifiziert, dessen homozygote Träger ein erhöhtes Risiko für diese Allergie aufweisen. (360gradpferd.de)
Patienten1
- Sie rekrutierten F508del-homozygote Mukoviszidose-Patienten, die entweder mit Glukose-Intoleranz oder Diabetes diagnostiziert waren und ein Jahr lang mit Orkambi behandelt wurden. (dcfh.de)
Regeln1
- Der jetzt homozygote Nachkomme vererbt das Allel erneut und setzt somit die klassischen Regeln der Vererbung außer Kraft. (freiepresse.space)
Form1
- Wenn beide Eltern einen solchen Gendefekt vererben („homozygote" Form), verstärkt sich die Wirkung. (evi.at)