Heterozygote
Heterozygotenerkennung
Homozygote
Alleles
Erblichkeit
Mutation
Stammbaum
Phenotype
Polymorphism, Genetic
Gen-Rearrangement
Thalassämie
Genes, Recessive
Hyperlipoproteinämie Typ I
Hämoglobin A2
Genetic Variation
Genes, Dominant
DNA-Mutationsanalyse
Genes, Lethal
Kreuzungen, genetische
Base Sequence
Hämoglobine, abnorme
Hämochromatose
Polymerase-Kettenreaktion
Ataxia teleangiectatica
Genetische Prädisposition für eine Krankheit
Point Mutation
Haplotypes
Hypercholesterinämie, familiäre
Mutation, Missense
Molekülsequenzdaten
Zystinurie
Hypobetalipoproteinämie
Tay-Sachs-Krankheit
Exons
Hämoglobin E
Juden
Adenin-Phosphoribosyltransferase
Genetic Linkage
Mäuse, Mutantenstämme
Models, Genetic
Fetales Hämoglobin
Alpha-Thalassämie
Genetic Testing
Polymorphism, Single Nucleotide
Hämoglobinopathien
Chromosomenkartierung
Inzucht
Genetik, Populations-
Spitzmäuse
Mäuse, Knockout-
Sandhoff-Krankheit
Polymorphism, Restriction Fragment Length
Beta-Thalassämie
Glukosephosphat-Dehydrogenasemangel
DNA
Genetische Marker
Aminosäurenstoffwechsel, angeborene Störungen
Phenylketonurie
Microsatellite Repeats
Globins
Frameshift Mutation
Selection, Genetic
Lipidstoffwechsel, angeborene Störungen
Fall-Kontroll-Studien
Stoffwechsel, angeborene Störungen
Codon, Nonsens-
Fabry-Krankheit
Hypolipoproteinämie
DNA-Beschädigung
Hybrid Vigor
Hexosaminidase A
Gene Deletion
Homozystinurie
Mäuse, Inzuchtstamm C57BL-
Lipoidose
Lezithin-Azyltransferasemangel
Zystische Fibrose
Apolipoproteine B
Crossing Over, Genetic
Elektrophorese, Stärkegel-
Lesch-Nyhan-Syndrom
DNA-Primer
Lipoprotein-Lipase
Ethylnitrosoharnstoff
Apolipoprotein B-100
Genes
Sequenzanalyse, DNA-
Haploidy
Alpha-1-Antitrypsinmangel
Albinismus
Chromosome Inversion
Hämoglobin, Sichelzell-
Sequence Deletion
Beta-N-Acetylhexosaminidase
Fibroblasten
Hyperlipoproteinämie
2-Aminopurin
Amino Acid Sequence
Linkage Disequilibrium
Gruppe asiatischer Abstammung
Polymorphism, Single-Stranded Conformational
Faktor V
Aminosäuresubstitution
Hair Color
Hemizygote
Chromosomen
Diploidy
Purin-Pyrimidin-Stoffwechsel, angeborene Störungen
Glucosephosphat-Dehydrogenase
X-Chromosom
Xanthomatose
Founder Effect
Familiäres Mittelmeerfieber
Haar
Eisenüberlastung
Recombination, Genetic
Drosophila melanogaster
Gene Dosage
Italien
Zwergwuchs
Erythrozyten
Introns
Tangier-Krankheit
Geschlechtschromosomen
Syndrom
Membranproteine
Gen-Targeting
Zystinose
Penetrance
HLA-Antigene
Familie, Gesundheitszustand der
Adrenale Hyperplasie, kongenitale
Geschlechtschromosomale Aberrationen
Chromosomendeletion
Hyperargininämie
Pränatale Diagnostik
Sonderurlaub zur Angehörigenpflege
Elektroretinographie
Mukopolysaccharidose Typ II
Cystic Fibrosis Transmembrane Conductance Regulator
Alpha-Galactosidase
RNA, Messenger-
HLA-DRB3 Chains
Alpha-1-Antitrypsin
Ornithincarbamoyltransferase-Mangelkrankheit
Kind, neugeborenes
Risikofaktoren
Codon
Ferritin
Rezeptoren, LDL-
Leukozyten
Genotyping Techniques
Sichelzellanlage
Augenproteine
Genetischer Komplementaritätstest
Pregnancy
Glykogenspeicherkrankheit Typ V
Epistasis, Genetic
Augenabnormalitäten
Manifestationssalter
Mäuse, transgene
Apolipoprotein A-I
Embryonenverlust
Blotting, Southern
Mäuse, Inzuchtstämme
Heteroduplex-Analyse
Lipoproteine
Gaucher-Krankheit
Iduronat-Sulfatase
Anämie, Sichelzellen-
Hybridisierung, genetische
Eisen
Tschechische Republik
Cholesterol
Zellen, kultivierte
Mäuse, neurologische Mutanten
Sitosterole
Genetic Association Studies
Fetaltod
Gene Expression Regulation, Developmental
Serumproteinelektrophorese
Xeroderma pigmentosum
Lymphozyten
Gruppe afrikanischer Abstammung
Histokompatibilitätsantigene Klasse I
Schwei
Isoenzyme
Apolipoproteine E
Retinitis pigmentosa
Lipoproteine, HDL-Cholesterol-
Hexosaminidasen
Apolipoproteine A
Neuralrohrdefekte
Tyrosinämien
Anämie, hämolytische, kongenitale nichtsphärozytäre
Retinadegeneration
Hämoglobin C
Genetic Heterogeneity
Genetic Load
Reference Values
Embryo
Carrierproteine
In der Genetik, ein Heterozygoter Organismus ist eine Person oder ein Lebewesen, das zwei verschiedene Allele eines Gens hat, d.h. es trägt eine unterschiedliche Version des Gens auf jedem Chromosom in einem homologen Chromosomenpaar. Dies steht im Gegensatz zu Homozygotie, bei der beide Allele eines Gens identisch sind.
Heterozygote können sich klinisch manifestieren (manifeste Heterozygote) oder asymptomatisch sein (latente Heterozygote), abhängig von der Art des Gens und der Art der genetischen Erkrankung. Manchmal kann das Vorhandensein einer heterozygoten Genvariante auch mit Vorteilen einhergehen, wie z.B. bei der Resistenz gegen bestimmte Krankheiten.
Es ist wichtig zu beachten, dass Heterozygote nicht notwendigerweise die Hälfte der Merkmale ihrer Homozygoten Gegenstücke aufweisen müssen. Die Manifestation von Genvarianten wird durch komplexe genetische und umweltbedingte Faktoren beeinflusst, was zu einer Vielzahl von Phänotypen führen kann, selbst bei Individuen mit derselben Genvariante.
Heterozygotenidentifikation bezieht sich auf die Fähigkeit, Individuen zu identifizieren, die heterozygote Merkmalsträger sind, d.h. sie haben zwei verschiedene Allele für ein bestimmtes Gen. Diese Identifikation ist in der Genetik und in der Humangenetik von großer Bedeutung, insbesondere bei genetischen Erkrankungen, die durch rezessive Allele verursacht werden.
Eine Person, die ein rezessives Allel für eine bestimmte Krankheit besitzt, zeigt in der Regel keine Symptome, wenn sie das dominante Allel für dieses Gen erbt. Wenn jedoch beide Elternteile Träger des rezessiven Allels sind, besteht eine 25%ige Chance, dass ihr Kind zwei Kopien des rezessiven Allels erhält und an der Krankheit erkrankt.
Die Heterozygotenidentifikation kann durch verschiedene Methoden wie Genotypisierung, Familienstudien oder prädiktive Tests durchgeführt werden. Die Identifizierung von Heterozygoten ist wichtig, um das Risiko von genetischen Erkrankungen bei zukünftigen Generationen abzuschätzen und mögliche präventive Maßnahmen zu ergreifen.
Homozygotie ist ein Begriff aus der Genetik und beschreibt die Situation, in der ein Individuum zwei identische Allele eines Gens besitzt. Allele sind verschiedene Versionen desselben Gens, die an denselben genetischen Lokus auf einem Chromosomenpaar lokalisiert sind.
Wenn ein Mensch ein Gen in homozygoter Form besitzt, bedeutet das, dass beide Kopien dieses Gens (eine vom Vater geerbt und eine von der Mutter geerbt) identisch sind. Dies kann entweder passieren, wenn beide Elternteile dasselbe Allel weitergeben (z.B. AA x AA) oder wenn ein reinerbiges Merkmal vorliegt, bei dem das Gen nur in einer Form vorkommt (z.B. bb x bb).
Homozygote Individuen exprimieren das entsprechende Merkmal in der Regel in seiner reinsten Form, da beide Allele dieselben Informationen tragen. Dies kann sowohl vorteilhafte als auch nachteilige Auswirkungen haben, je nachdem, ob das Gen dominant oder rezessiv ist und welche Eigenschaften es kodiert.
Allele sind verschiedene Varianten eines Gens, die an der gleichen Position auf einem Chromosomenpaar zu finden sind und ein bestimmtes Merkmal oder eine Eigenschaft codieren. Jeder Mensch erbt zwei Kopien jedes Gens - eine von jedem Elternteil. Wenn diese beiden Kopien des Gens unterschiedlich sind, werden sie als "Allele" bezeichnet.
Allele können kleine Unterschiede in ihrer DNA-Sequenz aufweisen, die zu verschiedenen Ausprägungen eines Merkmals führen können. Ein Beispiel ist das Gen, das für die Augenfarbe codiert. Es gibt mehrere verschiedene Allele dieses Gens, die jeweils leicht unterschiedliche DNA-Sequenzen aufweisen und zu verschiedenen Augenfarben führen können, wie beispielsweise braune, grüne oder blaue Augen.
Es ist wichtig zu beachten, dass nicht alle Gene mehrere Allele haben - einige Gene haben nur eine einzige Kopie, die bei allen Menschen gleich ist. Andere Gene können hunderte oder sogar tausende verschiedene Allele aufweisen. Die Gesamtheit aller Allele eines Individuums wird als sein Genotyp bezeichnet, während die Ausprägung der Merkmale, die durch diese Allele codiert werden, als Phänotyp bezeichnet wird.
Erblichkeit bezieht sich in der Genetik auf die Übertragung von genetischen Merkmalen oder Krankheiten von Eltern auf ihre Nachkommen über die Vererbung von Genen. Sie beschreibt das Ausmaß, in dem ein bestimmtes Merkmal oder eine Erkrankung durch Unterschiede in den Genen beeinflusst wird.
Erblichkeit wird in der Regel als ein Wahrscheinlichkeitswert ausgedrückt und gibt an, wie hoch die Chance ist, dass ein Merkmal oder eine Krankheit auftritt, wenn man die Gene einer Person betrachtet. Eine Erblichkeit von 100% würde bedeuten, dass das Merkmal oder die Krankheit sicher vererbt wird, während eine Erblichkeit von 0% bedeutet, dass es nicht vererbt wird. In der Realität liegen die meisten Erblichkeitswerte irgendwo dazwischen.
Es ist wichtig zu beachten, dass Erblichkeit nur einen Teilaspekt der Entstehung von Merkmalen und Krankheiten darstellt. Umweltfaktoren und Wechselwirkungen zwischen Genen und Umwelt spielen oft ebenfalls eine Rolle bei der Entwicklung von Merkmalen und Krankheiten.
Eine Mutation ist eine dauerhafte, zufällige Veränderung der DNA-Sequenz in den Genen eines Organismus. Diese Veränderungen können spontan während des normalen Wachstums und Entwicklungsprozesses auftreten oder durch äußere Einflüsse wie ionisierende Strahlung, chemische Substanzen oder Viren hervorgerufen werden.
Mutationen können verschiedene Formen annehmen, wie z.B. Punktmutationen (Einzelnukleotidänderungen), Deletionen (Entfernung eines Teilstücks der DNA-Sequenz), Insertionen (Einfügung zusätzlicher Nukleotide) oder Chromosomenaberrationen (größere Veränderungen, die ganze Gene oder Chromosomen betreffen).
Die Auswirkungen von Mutationen auf den Organismus können sehr unterschiedlich sein. Manche Mutationen haben keinen Einfluss auf die Funktion des Gens und werden daher als neutral bezeichnet. Andere Mutationen können dazu führen, dass das Gen nicht mehr oder nur noch eingeschränkt funktioniert, was zu Krankheiten oder Behinderungen führen kann. Es gibt jedoch auch Mutationen, die einen Vorteil für den Organismus darstellen und zu einer verbesserten Anpassungsfähigkeit beitragen können.
Insgesamt spielen Mutationen eine wichtige Rolle bei der Evolution von Arten, da sie zur genetischen Vielfalt beitragen und so die Grundlage für natürliche Selektion bilden.
Ein Gen-Rearrangement, auch genetisches Rearrangement genannt, ist eine Veränderung in der Anordnung von Genen auf einer Chromosomensequenz. Dies tritt auf, wenn zwei nicht-homologe Chromosomen oder zwei verschiedene Abschnitte des gleichen Chromosoms durch Kreuzungsübertragung während der Meiose ausgetauscht werden. Ein Gen-Rearrangement kann auch auftreten, wenn ein Stück Chromosomen durch eine Translokation, Deletion, Inversion oder Duplikation verloren geht oder hinzugefügt wird.
In der Immunologie spielen genetische Rearrangements eine wichtige Rolle bei der Entwicklung des adaptiven Immunsystems von Wirbeltieren. Durch V(D)J-Rearrangement werden die variablen Regionen der Antikörper und T-Zell-Rezeptor-Gene neu angeordnet, wodurch eine enorme Vielfalt an Rezeptoren entsteht, mit denen das Immunsystem verschiedene Antigene erkennen kann. Diese Rearrangements tragen dazu bei, dass jedes Individuum über ein einzigartiges Repertoire an Rezeptoren verfügt und somit in der Lage ist, eine breite Palette von Krankheitserregern zu bekämpfen.
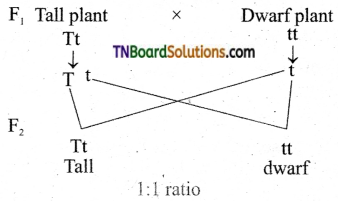
"Recessive Genes" sind ein Konzept in der Genetik, bei dem die Merkmale eines Gens nur dann auftreten, wenn das Gen in beiden Kopien eines Chromosomenpaars vorhanden ist. Jeder Mensch hat zwei Kopien jedes Gens - eine von jedem Elternteil. Wenn ein Gen dominant ist, reicht es aus, es in einer Kopie zu haben, um das Merkmal zu exprimieren. Bei rezessiven Genen muss das Gen jedoch in beiden Kopien vorhanden sein, damit das Merkmal sichtbar wird.
Wenn ein Individuum ein rezessives Gen von einem Elternteil erbt und ein dominantes oder ein anderes Allel des Gens vom anderen Elternteil erbt, wird das rezessive Gen maskiert und das dominante Gen wird exprimiert. Dieses Individuum ist dann ein Träger des rezessiven Gens, zeigt aber keine Anzeichen dafür.
Rezessive Gene spielen eine wichtige Rolle in der Vererbung von erblichen Krankheiten und Merkmalen. Wenn beide Elternteile Träger eines rezessiven Gens sind, besteht für jedes Kind ein 25%iges Risiko, beide Kopien des Gens zu erben und die mit dem Gen verbundene Erkrankung oder das Merkmal auszudrücken.
Hyperlipoproteinämie Typ I, auch bekannt als "Familiale Lipoprotein-Lipase-Defizienz", ist ein seltenes genetisches Störung des Fettstoffwechsels, welche durch einen Mangel an der Lipoproteinlipase (LPL) oder an deren Co-Faktoren verursacht wird. Dies führt zu einem Anstieg von Chylomikronen im Blut, sehr große Lipoproteinkomplexe, die Fette aus der Nahrung transportieren.
Diese Erkrankung geht mit extrem hohen Konzentrationen von Triglyceriden (>1000 mg/dL) einher und kann zu wiederkehrenden Akuten Pancreatitis (Bauchspeicheldrüsenentzündungen) führen. Die Diagnose wird durch Laboruntersuchungen gestellt, bei denen erhöhte Chylomikron-Spiegel und niedrige HDL-Cholesterinspiegel nachgewiesen werden können.
Die Behandlung von Hyperlipoproteinämie Typ I umfasst in der Regel eine fettarme Diät, Medikamente zur Senkung der Triglyceridwerte und gegebenenfalls Enzymersatztherapie mit Lipasen. Eine frühzeitige Diagnose und Behandlung sind wichtig, um das Risiko von Komplikationen wie Pankreatitis zu minimieren.
Hämoglobin A2 ist ein Typ von Hämoglobin, das hauptsächlich in den roten Blutkörperchen (Erythrozyten) vorkommt. Es besteht aus zwei Alpha- und zwei Delta-Ketten (α2δ2) und macht normalerweise weniger als 3% des gesamten Hämoglobins im Körper aus.
Hämoglobin A2 spielt im Allgemeinen keine bedeutende Rolle beim Sauerstofftransport im Körper, jedoch kann seine Messung und Analyse bei diagnostischen Zwecken nützlich sein. Zum Beispiel wird der Hämoglobin A2-Spiegel bei Menschen mit Beta-Thalassämien oft erhöht, was ein hilfreiches Indiz für die Diagnose und Überwachung dieser Erkrankungen sein kann.
Genetic Variation bezieht sich auf die Unterschiede in der DNA-Sequenz oder der Anzahl der Kopien bestimmter Gene zwischen verschiedenen Individuen derselben Art. Diese Variationen entstehen durch Mutationen, Gen-Kreuzungen und Rekombination während der sexuellen Fortpflanzung.
Es gibt drei Hauptarten von genetischen Variationen:
1. Einzelnukleotidische Polymorphismen (SNPs): Dies sind die häufigsten Formen der genetischen Variation, bei denen ein einzelner Nukleotid (DNA-Baustein) in der DNA-Sequenz eines Individuums von dem eines anderen Individuums abweicht.
2. Insertionen/Deletionen (INDELs): Hierbei handelt es sich um kleine Abschnitte der DNA, die bei einigen Individuen vorhanden sind und bei anderen fehlen.
3. Kopienzahlvariationen (CNVs): Bei diesen Variationen liegt eine Abweichung in der Anzahl der Kopien bestimmter Gene oder Segmente der DNA vor.
Genetische Variationen können natürliche Unterschiede zwischen Individuen erklären, wie zum Beispiel die verschiedenen Reaktionen auf Medikamente oder das unterschiedliche Risiko für bestimmte Krankheiten. Einige genetische Variationen sind neutral und haben keinen Einfluss auf die Funktion des Organismus, während andere mit bestimmten Merkmalen oder Erkrankungen assoziiert sein können.
In der Genetik versteht man unter "genes, dominant" die Ausprägung eines bestimmten Merkmals, die auftritt, wenn ein dominantes Allel vorhanden ist. Ein Allel ist eine Variante eines Gens, das an einer bestimmten Position auf einem Chromosom liegt. Wenn ein Individuum zwei unterschiedliche Allele für ein Gen besitzt (heterozygot), wird in der Regel das dominante Allel ausgeprägt, während das andere Allel, das rezessive Allel genannt wird, nicht zum Ausdruck kommt.
Zum Beispiel bei der Erbkrankheit Chorea Huntington ist das Gen, welches für die Proteinproduktion des Huntingtins verantwortlich ist, mutiert. Wenn eine Person ein dominantes Allel dieser Mutation besitzt, wird sie unabhängig davon, ob das zweite Allel rezessiv oder nicht betroffen ist, an der Krankheit erkranken.
Es sei jedoch angemerkt, dass die Dominanz eines Gens relativ ist und von Kontext zu Kontext variieren kann. In manchen Fällen können auch mehrere Gene zusammenwirken, um ein Merkmal auszubilden, was als polygenetische Vererbung bezeichnet wird.
Die DNA-Mutationsanalyse ist ein Prozess der Genetik, bei dem die Veränderungen in der DNA-Sequenz untersucht werden, um genetisch bedingte Krankheiten oder Veranlagungen zu diagnostizieren, zu bestätigen oder auszuschließen. Eine Mutation ist eine dauerhafte und oft zufällige Veränderung in der DNA-Sequenz, die die Genstruktur und -funktion beeinflussen kann.
Die DNA-Mutationsanalyse umfasst verschiedene Techniken wie PCR (Polymerasekettenreaktion), DNA-Sequenzierung, MLPA (Multiplex-Ligation-dependent Probe Amplification) und Array-CGH (Array Comparative Genomic Hybridization). Diese Techniken ermöglichen es, kleinste Veränderungen in der DNA zu erkennen, wie z.B. Einzelnukleotidpolymorphismen (SNPs), Deletionen, Insertionen oder Chromosomenaberrationen.
Die Ergebnisse der DNA-Mutationsanalyse können wichtige Informationen für die klinische Diagnose und Therapie von genetisch bedingten Krankheiten liefern, wie z.B. Krebs, erbliche Herzkrankheiten, Stoffwechselstörungen oder neuromuskuläre Erkrankungen. Die DNA-Mutationsanalyse wird auch in der Forschung eingesetzt, um die genetischen Grundlagen von Krankheiten besser zu verstehen und neue Therapieansätze zu entwickeln.
In medical terms, "genes" refers to the basic units of heredity that are passed down from parents to offspring. They are made up of DNA and are located on chromosomes in the nucleus of cells. Each gene provides instructions for the production of a specific protein or set of proteins that play a crucial role in the development, functioning, and reproduction of an organism.
"Lethal" refers to something that causes death. In genetics, a lethal gene is one that results in the death of an organism before it can reach reproductive age or produce viable offspring. A lethal gene may cause embryonic lethality, meaning that the developing embryo dies before birth, or postnatal lethality, meaning that the organism dies shortly after birth or during early development.
Therefore, a "lethal gene" can be defined as a genetic mutation or variant that results in the death of an organism before it can reproduce, either due to embryonic or postnatal lethality.
Genetische Kreuzungen beziehen sich auf die Paarung und Fortpflanzung zwischen zwei Individuen verschiedener reinbred Linien oder Arten, um neue Pflanzen- oder Tierhybriden zu erzeugen. Dieser Prozess ermöglicht es, gewünschte Merkmale von jeder Elternlinie in der nachfolgenden Generation zu kombinieren und kann zu einer Erweiterung der genetischen Vielfalt führen.
In der Genforschung können genetische Kreuzungen auch verwendet werden, um verschiedene Arten von Organismen gezielt zu kreuzen, um neue Eigenschaften oder Merkmale in den Nachkommen zu erzeugen. Durch die Analyse des Erbguts und der resultierenden Phänotypen können Forscher das Vererbungsmuster bestimmter Gene untersuchen und wertvolle Informationen über ihre Funktion und Interaktion gewinnen.
Es ist jedoch wichtig zu beachten, dass nicht alle genetischen Kreuzungen erfolgreich sind oder die erwarteten Ergebnisse liefern, da verschiedene Faktoren wie Kompatibilität der Genome, epistatische Effekte und genetische Drift eine Rolle spielen können.
In molecular biology, a base sequence refers to the specific order of nucleotides in a DNA or RNA molecule. In DNA, these nucleotides are adenine (A), cytosine (C), guanine (G), and thymine (T), while in RNA, uracil (U) takes the place of thymine. The base sequence contains genetic information that is essential for the synthesis of proteins and the regulation of gene expression. It is determined by the unique combination of these nitrogenous bases along the sugar-phosphate backbone of the nucleic acid molecule.
A 'Base Sequence' in a medical context typically refers to the specific order of these genetic building blocks, which can be analyzed and compared to identify genetic variations, mutations, or polymorphisms that may have implications for an individual's health, disease susceptibility, or response to treatments.
Abnorme Hämoglobine sind Varianten des Hämoglobins, die aufgrund genetischer Mutationen in der Struktur oder Menge des Hämoglobinmoleküls von dem normalfall (HbA) abweichen. Diese Abweichungen können zu verschiedenen klinischen Manifestationen führen, wie z.B. anormaler Sauerstofftransport, erhöhter Affinität des Hämoglobins für Sauerstoff oder veränderte Elektrophorese-Muster. Einige der bekanntesten Beispiele sind HbS (verursacht Sichelzellanämie), HbC und HbE, die jeweils durch eine einzelne Aminosäure-Substitution im Hämoglobinmolekül gekennzeichnet sind. Diese Varianten können zu milden bis schweren klinischen Symptomen führen, wie Anämie, Jaundice, Splenomegalie und einem erhöhten Risiko für Infektionen. Es ist wichtig zu beachten, dass nicht alle abnormen Hämoglobine mit Krankheiten assoziiert sind und manche asymptomatisch bleiben können.
Hämochromatose ist eine genetisch bedingte Erkrankung, bei der es zu einer übermäßigen Aufnahme und Speicherung von Eisen im Körpergewebe, insbesondere in Leber, Bauchspeicheldrüse, Herz und Gelenken kommt. Dies geschieht durch eine Störung im Stoffwechsel des Eisens, die zu einer Anhäufung von Eisen in den Organen führt.
Die häufigste Form der Hämochromatose wird als HFE-Hämochromatose bezeichnet und ist autosomal-rezessiv vererbt. Sie betrifft vor allem Menschen mit nordeuropäischer Abstammung. Durch die Anhäufung von Eisen in den Organen kann es zu verschiedenen Symptomen kommen, wie beispielsweise Lebererkrankungen, Diabetes mellitus, Herzrhythmusstörungen, Bronzedermatitis (bräunliche Verfärbung der Haut) und Gelenkbeschwerden.
Die Behandlung besteht in der Regel in einer regelmäßigen Entfernung von Blut (Phlebotomie), um den Eisenspiegel im Körper zu reduzieren. Eine frühzeitige Diagnose und Behandlung können die Entstehung von Organschäden verhindern oder zumindest verlangsamen.
Ataxia teleangiectatica ist eine seltene, autosomal-rezessiv vererbte Neurodegenerative Erkrankung, die durch Gendefekte im ATM-Gen (Ataxia Telangiectasia Mutated) verursacht wird. Die Krankheit manifestiert sich normalerweise in den ersten zwei Lebensjahren mit zunehmender Instabilität und Koordinationsverlust der Muskelbewegungen (Ataxie), was zu Schwierigkeiten bei Gehen, Stehen und Balancieren führt.
Andere häufige Symptome sind eine erweiterte Blutgefäßwachstum (Teleangiektasien) im Gesicht und anderen Körperteilen, eine erhöhte Anfälligkeit für Infektionen aufgrund von Immunschwäche, eine progressive neurologische Degeneration, Verzögerung der motorischen Entwicklung, Sprachstörungen, Oculomotorische Apraxie (Störung der Augenbewegungen), Schwierigkeiten beim Schlucken und Atemwegsinfektionen.
Betroffene Personen haben auch ein erhöhtes Risiko für die Entwicklung von Krebs, insbesondere Leukämien und Lymphomen. Es gibt keine Heilung für Ataxia teleangiectatica, aber Symptome können behandelt werden, um die Lebensqualität zu verbessern.
Eine genetische Prädisposition für eine Krankheit bezieht sich auf die Erhöhung der Wahrscheinlichkeit, an einer bestimmten Erkrankung zu erkranken, aufgrund von genetischen Faktoren. Es bedeutet nicht, dass eine Person definitiv die Krankheit entwickeln wird, sondern dass sie ein erhöhtes Risiko im Vergleich zur Allgemeinbevölkerung hat.
Diese Prädisposition resultiert aus bestimmten Genvarianten oder Mutationen, die in den Genen einer Person vorhanden sind und die Funktion von Proteinen beeinflussen können, die an Krankheitsprozessen beteiligt sind. Manche dieser genetischen Faktoren werden autosomal-dominant vererbt, was bedeutet, dass eine Kopie des mutierten Gens ausreicht, um das Erkrankungsrisiko zu erhöhen. Andere Fälle können autosomal-rezessiv sein, bei denen zwei Kopien des mutierten Gens erforderlich sind, damit die Krankheit zum Ausbruch kommt.
Es ist wichtig anzumerken, dass genetische Prädispositionen oft in Kombination mit umweltbedingten Faktoren auftreten, wie beispielsweise Rauchen, Alkoholkonsum, Ernährung und Exposition gegenüber bestimmten Chemikalien. Diese Faktoren können das Erkrankungsrisiko weiter erhöhen oder abschwächen.
In der medizinischen Praxis kann die Kenntnis einer genetischen Prädisposition dazu beitragen, präventive Maßnahmen zu ergreifen, Früherkennungstests durchzuführen und individuelle Behandlungspläne zu entwickeln.
Haplotypen sind eine Reihe von Varianten (Allele) eines Gens oder nahegelegener Marker, die auf einem einzigen Chromosom vererbt werden. Sie repräsentieren ein charakteristisches Muster von Variationen in einem bestimmten Abschnitt des Genoms und werden oft als Einheit vererbt, da sie eng beieinander liegen und eine geringe Rekombinationsrate aufweisen.
Haplotypen sind nützlich in der Genetik, um Verwandtschaftsbeziehungen zu untersuchen, Krankheitsrisiken abzuschätzen und pharmakogenetische Studien durchzuführen. Durch die Analyse von Haplotypen kann man Rückschlüsse auf gemeinsame Vorfahren ziehen und die Evolution von Genen und Populationen besser verstehen.
Familiäre Hypercholesterinämie ist ein genetisch bedingtes Lipidstoffwechselstörung, welches durch hohe Konzentrationen des Gesamtcholesterins und LDL-Cholesterins („schlechtes Cholesterin“) im Blut charakterisiert ist. Diese Erkrankung wird autosomal-dominant vererbt, was bedeutet dass eine Kopie des mutierten Gens ausreicht um die Krankheit zu manifestieren.
Die Mutationen betreffen in der Regel das Apolipoprotein B oder den LDL-Rezeptor Gen, wodurch die Fähigkeit des Körpers vermindert ist, LDL-Cholesterin aus dem Blutkreislauf zu entfernen. Dies führt zu einer Anhäufung von Cholesterin in den Blutgefäßen und steigert das Risiko für die Entwicklung von Atherosklerose und kardiovaskulären Erkrankungen, wie koronare Herzkrankheit, Herzinfarkt oder Schlaganfall.
Die Diagnose wird in der Regel durch eine Kombination aus Anamnese, klinischen Befunden, Lipidprofil und genetischer Testung gestellt. Die Behandlung umfasst in der Regel lifestyle Änderungen (wie Ernährungsumstellung und körperliche Aktivität) sowie medikamentöse Therapie mit Cholesterinsenkern wie Statinen oder PCSK9-Inhibitoren. In einigen Fällen kann auch eine LDL-Apherese notwendig sein, um die LDL-Cholesterinspiegel zu senken.
Eine Missense-Mutation ist ein spezifischer Typ von Genmutation, bei der ein einzelner Nukleotid (DNA-Basenpaar) ausgetauscht wird, was dazu führt, dass ein anderes Aminosäure-Restmolekül anstelle des ursprünglichen eingebaut wird. Dies kann zu einer Veränderung der Proteinstruktur und -funktion führen, die je nach Art und Ort der Mutation im Genom variieren kann. Manchmal können Missense-Mutationen die Proteinfunktion beeinträchtigen oder sogar vollständig aufheben, was zu verschiedenen Krankheiten oder Fehlbildungen führen kann. Es ist wichtig zu beachten, dass nicht alle Missense-Mutationen pathogen sind und einige von ihnen möglicherweise keine Auswirkungen auf die Proteinfunktion haben.
Molekülsequenzdaten beziehen sich auf die Reihenfolge der Bausteine in Biomolekülen wie DNA, RNA oder Proteinen. Jedes Molekül hat eine einzigartige Sequenz, die seine Funktion und Struktur bestimmt.
In Bezug auf DNA und RNA besteht die Sequenz aus vier verschiedenen Nukleotiden (Adenin, Thymin/Uracil, Guanin und Cytosin), während Proteine aus 20 verschiedenen Aminosäuren bestehen. Die Sequenzdaten werden durch Laborverfahren wie DNA-Sequenzierung oder Massenspektrometrie ermittelt und können für Anwendungen in der Genetik, Biochemie und Pharmakologie verwendet werden.
Die Analyse von Molekülsequenzdaten kann zur Identifizierung genetischer Variationen, zur Vorhersage von Proteinstrukturen und -funktionen sowie zur Entwicklung neuer Medikamente beitragen.
Hypobetalipoproteinämie ist eine genetisch bedingte Stoffwechselstörung, bei der die Synthese und Sekretion der Betalipoproteine (z.B. LDL oder "bad cholesterol") in der Leber eingeschränkt ist. Dies führt zu niedrigen Konzentrationen von Chylomikronen, VLDL und damit auch von LDL und Apolipoprotein B im Blutserum. Die Erkrankung kann sich klinisch durch Symptome wie Steatose (Fettleber), Ataxie (Störung der Bewegungskoordination) und Neuropathie (Nervenschädigung) manifestieren, aber auch asymptomatisch verlaufen. Die Diagnose wird meist durch Laboruntersuchungen gestellt, die niedrige Werte von Gesamtcholesterin, LDL-Cholesterin und Apolipoprotein B aufzeigen.
Exons sind die Abschnitte der DNA, die nach der Transkription und folgenden Prozessen wie Spleißen in das endgültige mature mRNA-Molekül eingebaut werden und somit die codierende Region für Proteine darstellen. Sie entsprechen den Bereichen, die nach dem Entfernen der nichtcodierenden Introns-Abschnitte in der reifen, translationsfähigen mRNA verbleiben. Im Allgemeinen enthalten Exons kodierende Sequenzen, die für Aminosäuren in einem Protein stehen, können aber auch Regulationssequenzen oder nichtcodierende RNA-Abschnitte wie beispielsweise RNA-Elemente mit Funktionen in der RNA-Struktur oder -Funktion enthalten.
Hämoglobin E (HbE) ist eine Variation des Hämoglobins, das den Sauerstofftransport im Blut ermöglicht. Es entsteht durch eine genetische Mutation auf dem beta-Globin-Gen, die eine Aminosäuresubstitution an Position 26 der beta-Globinkette verursacht (Beta26 Glu -> Lys). Diese Mutation kann zu einer abnormalen Struktur und Funktion des Hämoglobins führen.
HbE ist relativ häufig in Südostasien, insbesondere in Thailand, Kambodscha, Laos, Vietnam und Teilen Chinas. Menschen, die zwei Kopien dieses Allels (HbEE) besitzen, können eine mild bis mäßig ausgeprägte Hämolytische Anämie entwickeln. Die Kombination von HbE mit anderen Hämoglobinvarianten wie HbS (das die Sichelzellenanämie verursacht) kann zu schwerwiegenderen Krankheitsbildern führen.
Es ist wichtig zu beachten, dass Menschen, die nur eine Kopie des HbE-Allels tragen (HbA/E), im Allgemeinen keine Symptome entwickeln und ein normales Leben führen können.
Es ist nicht angemessen oder korrekt, eine medizinische Definition für 'Juden' zu geben, da Judentum eine ethnoreligiöse Identität und keine medizinische oder gesundheitsbezogene Kategorie ist. Die Religion des Judentums, die Kultur und die Traditionen werden von Menschen auf der ganzen Welt praktiziert und sind nicht mit bestimmten Krankheiten oder Gesundheitszuständen verbunden.
Die Angabe einer medizinischen Definition für 'Juden' wäre ungenau, irreführend und könnte zu Diskriminierung und Vorurteilen führen. Es ist wichtig, dass wir alle Menschen als Individuen respektieren und ihre ethnische, religiöse und kulturelle Identität schätzen, ohne sie mit medizinischen oder gesundheitsbezogenen Konzepten in Verbindung zu bringen.
Adenin-Phosphoribosyltransferase (APRT) ist ein enzymatisches Molekül, das in der menschlichen Biochemie eine wichtige Rolle bei dem Prozess der Nukleotidsynthese spielt. Genauer gesagt ist APRT an der Synthese von Adenin-Nukleotiden beteiligt, indem es Adenin mit Phosphoribosylpyrophosphat (PRPP) zu Adenosinmonophosphat (AMP) verbindet. Eine Fehlfunktion oder ein Mangel an diesem Enzym kann zu einer seltenen Stoffwechselstörung führen, die als Adenin-Phosphoribosyltransferase-Mangelkrankheit bekannt ist. Diese Krankheit kann zu einer Anhäufung von 2,8-Dihydroxyadenin in verschiedenen Körpergeweben führen, was wiederum Nierensteine und Nierenschäden verursachen kann.
Genetic linkage refers to the phenomenon where two or more genes are located physically close to each other on a chromosome and tend to be inherited together during meiosis. This means that the transmission of these genes is not independent, but rather they are linked and co-transmitted because the probability of their recombination (i.e., exchange of genetic material between homologous chromosomes) is relatively low. The degree of linkage between genes is measured by the recombination frequency, which reflects the percentage of meiotic events resulting in a crossover between the linked genes. Genes with a high recombination frequency are considered to be loosely linked or unlinked, while those with a low recombination frequency are tightly linked. The concept of genetic linkage is fundamental in genetics and has important implications for understanding patterns of inheritance, mapping gene locations, and identifying genetic variations associated with diseases or traits.
Mutante Mausstämme sind genetisch veränderte Labortiere, die gezielt zur Erforschung von Krankheiten und zum Testen neuer Medikamente eingesetzt werden. Dabei wird das Erbgut der Mäuse durch verschiedene Methoden so verändert, dass sie bestimmte genetische Merkmale aufweisen, die denen von menschlichen Erkrankungen ähneln.
Diese Mutationen können spontan auftreten oder gezielt herbeigeführt werden, beispielsweise durch die Verwendung von Gentechnik oder Bestrahlung. Durch die Veränderung des Erbguts können Forscher untersuchen, wie sich die Genmutation auf das Verhalten, Wachstum und die Entwicklung der Mäuse auswirkt und ob sie anfälliger für bestimmte Krankheiten sind.
Mutante Mausstämme werden in der biomedizinischen Forschung häufig eingesetzt, um das Verständnis von Krankheitsprozessen zu verbessern und neue Behandlungsmethoden zu entwickeln. Ein bekanntes Beispiel ist die Knockout-Maus, bei der ein bestimmtes Gen gezielt deaktiviert wird, um die Funktion dieses Gens im Körper zu untersuchen.
Es ist wichtig zu beachten, dass Mutante Mausstämme zwar nützliche Modelle für die Erforschung menschlicher Krankheiten sein können, aber nicht immer ein perfektes Abbild der menschlichen Erkrankung darstellen. Daher müssen Forscher sorgfältig abwägen, ob und wie die Ergebnisse aus Tierversuchen auf den Menschen übertragbar sind.
Genetic models in a medical context refer to theoretical frameworks that describe the inheritance and expression of specific genes or genetic variations associated with certain diseases or traits. These models are used to understand the underlying genetic architecture of a particular condition and can help inform research, diagnosis, and treatment strategies. They may take into account factors such as the mode of inheritance (e.g., autosomal dominant, autosomal recessive, X-linked), penetrance (the likelihood that a person with a particular genetic variant will develop the associated condition), expressivity (the range of severity of the condition among individuals with the same genetic variant), and potential interactions with environmental factors.
Fetales Hämoglobin (HbF) ist eine Form des Hämoglobins, das hauptsächlich während der fetalen Entwicklung im Blutkreislauf des Fötus vorkommt und seine Aufgabe in der Sauerstofftransportfunktion erfüllt. Es besteht aus zwei Alpha- und zwei Gamma-Ketten (α2γ2). Im Vergleich zu adultem Hämoglobin (HbA) hat HbF eine höhere Affinität zur Bindung an Sauerstoff, was bedeutet, dass es Sauerstoff effektiver aus der Plazenta aufnimmt und an den Fötus abgibt.
Die Umwandlung von fetalem Hämoglobin in adultes Hämoglobin beginnt normalerweise während der letzten Phase der fetalen Entwicklung und dauert bis zum Alter von etwa sechs Monaten nach der Geburt an. Bei einigen Erkrankungen, wie z.B. Sichelzellkrankheit und Thalassämie, bleibt jedoch ein höherer Anteil an HbF im Blut erhalten, was als fetale Hämoglobinopathie bezeichnet wird. Dies kann vorübergehend oder dauerhaft sein und kann die Symptome der Erkrankung mildern, indem es die Bildung von krankhaften Hämoglobinen verringert.
Die Alpha-Thalassämie ist ein genetisches Blutkrankheit, welches durch eine reduzierte Fähigkeit oder Unfähigkeit zur Synthese des Hämoglobin-Proteins im roten Blutfarbstoff (Hämoglobin) der roten Blutkörperchen (Erythrozyten) gekennzeichnet ist. Dies wird durch Mutationen im Gen verursacht, das für die Alpha-Kette des Hämoglobins kodiert.
Die Schwere der Erkrankung hängt von der Anzahl der fehlenden oder nicht funktionsfähigen Gene ab. Bei schweren Formen der Alpha-Thalassämie kann es zu einer Anämie kommen, die sich in Müdigkeit, Blässe, Kurzatmigkeit und im Wachstumsrückstand bei Kindern äußert. In extremen Fällen können intrauterine oder neonatale Tod auftreten.
Die Alpha-Thalassämie ist verbreitet in Bevölkerungsgruppen, die aus geografischen Gebieten mit hoher Malaria-Übertragungsrate stammen, wie zum Beispiel im Mittelmeerraum, in Afrika, im Nahen Osten und in Südostasien. Die Diagnose der Alpha-Thalassämie erfolgt durch Laboruntersuchungen, einschließlich Hämoglobin-Elektrophorese und Gentests. Die Behandlung hängt von der Schwere der Erkrankung ab und kann Bluttransfusionen, Eisenchelationstherapie und Stammzellentransplantation umfassen.
Genetic testing is a type of medical test that identifies changes in chromosomes, genes, or proteins. The results of a genetic test can confirm or rule out a suspected genetic condition or help determine a person's chance of developing or passing on a genetic disorder. Genetic tests are performed on a sample of blood, hair, skin, amniotic fluid (the fluid that surrounds a fetus during pregnancy), or other tissue. For example, a particular test might be used to identify a specific genetic variant or mutation associated with a condition such as cystic fibrosis or Huntington's disease.
There are several different types of genetic tests, including:
* Diagnostic testing: This type of test is used to confirm or rule out a suspected genetic condition in an individual who has symptoms of the condition.
* Predictive testing: This type of test is used to identify people who are at risk of developing a genetic disorder before they have symptoms.
* Carrier testing: This type of test is used to identify people who carry one copy of a gene mutation that, when present in two copies, causes a genetic disorder.
* Prenatal testing: This type of test is used to detect changes in a fetus's genes or chromosomes before birth.
* Newborn screening: This type of test is used to identify genetic disorders in newborn babies so that treatment can be started as early as possible.
It is important to note that genetic testing has both benefits and limitations. While it can provide valuable information about a person's health, it can also have potential risks, such as psychological distress or discrimination in employment or insurance. It is important for individuals considering genetic testing to receive accurate and unbiased information about the test and its implications so that they can make an informed decision about whether or not to proceed with testing.
Hämoglobinopathien sind erbliche Störungen der Hämoglobinsynthese, bei denen es zu einer strukturellen oder quantitativen Abweichung des Hämoglobins kommt. Das Hämoglobin ist ein Proteinkomplex in den roten Blutkörperchen (Erythrozyten), der für den Sauerstofftransport im Blut verantwortlich ist. Die beiden häufigsten und klinisch bedeutsamen Hämoglobinopathien sind die Sichelzellkrankheit (HbS) und die Thalassämie.
Die Sichelzellkrankheit entsteht durch eine Punktmutation im Gen, das für die β-Kette des Hämoglobins codiert. Dies führt zu einer substitutionellen Aminosäureänderung (Glutamat zu Valin) und zur Bildung von abnorm verformbaren und instabilen Hämoglobinmolekülen, die bei niedrigen Sauerstoffspiegeln zu Sichelzellen und Mikrozirkulationsstörungen führen.
Thalassämien sind eine Gruppe von Erkrankungen, die durch eine verminderte Synthese der α- oder β-Ketten des Hämoglobins gekennzeichnet sind. Die ungleiche Produktion der Ketten führt zur Bildung von instabilen Tetrameren und zu einer Hemmung der Erythropoese, was schließlich zu einer Anämie führt.
Die klinischen Manifestationen der Hämoglobinopathien reichen von milden bis hin zu lebensbedrohlichen Erkrankungen und können Hämolyse, Infarkte, Organschäden und Entwicklungsverzögerungen umfassen. Die Diagnose erfolgt meist durch Hämoglobinelektrophorese und Gentestung. Die Behandlung kann symptomatisch oder kausal sein und hängt von der Schwere der Erkrankung ab.
Chromosomenkartierung ist ein Verfahren in der Genetik und Molekularbiologie, bei dem die Position von Genen oder anderen DNA-Sequenzen auf Chromosomen genau bestimmt wird. Dabei werden verschiedene molekularbiologische Techniken eingesetzt, wie beispielsweise die FISH (Fluoreszenz-in-situ-Hybridisierung) oder die Gelelektrophorese nach restrictionemfraktionierter DNA (RFLP).
Durch Chromosomenkartierung können genetische Merkmale und Krankheiten, die mit bestimmten Chromosomenabschnitten assoziiert sind, identifiziert werden. Diese Informationen sind von großer Bedeutung für die Erforschung von Vererbungsmechanismen und der Entwicklung gentherapeutischer Ansätze.
Die Chromosomenkartierung hat in den letzten Jahren durch die Fortschritte in der Genomsequenzierung und Bioinformatik an Präzision gewonnen, was zu einer detaillierteren Darstellung der genetischen Struktur von Organismen geführt hat.
Inzucht, auch Inbreeding genannt, ist ein Begriff aus der Genetik und beschreibt die Paarung oder Fortpflanzung zwischen eng miteinander verwandten Organismen wie nahen Verwandten (z.B. Eltern mit ihren Kindern, Geschwistern, Onkeln/Tanten mit Nichten/Neffen). In der Humangenetik wird Inzucht oft als Maß für den Grad der Verwandtschaft zweier Menschen verwendet und wird durch den sogenannten Inzuchtkoeffizienten ausgedrückt.
Inzucht kann zu einer Erhöhung der Wahrscheinlichkeit führen, dass reinerbige oder homozygote Allele (Versionen eines Gens) auftreten, was wiederum das Risiko für genetisch bedingte Krankheiten und Entwicklungsstörungen erhöhen kann. Dies liegt daran, dass bei enger Verwandtschaft die Wahrscheinlichkeit größer ist, dass beide Elternteilen dieselben Allele für ein Gen geerbt haben.
Es ist wichtig zu beachten, dass Inzucht in der menschlichen Population aufgrund sozialer und kultureller Normen selten vorkommt. In einigen Tierpopulationen hingegen kann Inzucht auftreten, wenn die Population klein ist oder wenn es an geeigneten Partnern mangelt.
Population Genetics ist ein Teilgebiet der Genetik, das sich mit der Verteilung und dem Vorkommen von Genen und Allelen in populationsbiologischen Einheiten beschäftigt. Es untersucht die genetische Variation zwischen Individuen einer Population und wie solche Variation durch verschiedene evolutionäre Kräfte wie Mutation, Genfluss, genetische Drift und Selektion beeinflusst wird.
Die Populationsgenetik liefert wichtige Erkenntnisse zur Entstehung und Verbreitung von Krankheiten in Populationen sowie zur Anpassung von Arten an ihre Umwelt. Sie ist daher ein zentrales Forschungsgebiet der theoretischen und angewandten Genetik, Evolutionsbiologie und medizinischen Genetik.
Knockout-Mäuse sind gentechnisch veränderte Mäuse, bei denen ein bestimmtes Gen gezielt ausgeschaltet („geknockt“) wurde, um die Funktion dieses Gens zu untersuchen. Dazu wird in der Regel ein spezifisches Stück der DNA, das für das Gen codiert, durch ein anderes Stück DNA ersetzt, welches ein selektives Merkmal trägt und es ermöglicht, die knockout-Zellen zu identifizieren. Durch diesen Prozess können Forscher die Auswirkungen des Fehlens eines bestimmten Gens auf die Physiologie, Entwicklung und Verhaltensweisen der Maus untersuchen. Knockout-Mäuse sind ein wichtiges Werkzeug in der biomedizinischen Forschung, um Krankheitsmechanismen zu verstehen und neue Therapeutika zu entwickeln.
Die Beta-Thalassämie ist eine erbliche Störung des Hämoglobinstoffwechsels, die durch eine verminderte oder fehlende Synthese der beta-Kette der Hämoglobinmoleküle in den Erythrozyten (rote Blutkörperchen) gekennzeichnet ist. Es gibt verschiedene Schweregrade dieser Erkrankung, von der thalassämielike Anämie bis hin zu schweren, transfusionsabhängigen Krankheitsbildern, die als Beta-Thalassämia major oder Cooley-Anämie bekannt sind.
Die Krankheit wird autosomal rezessiv vererbt und ist in mediterranen, südasiatischen, arabischen und afrikanischen Bevölkerungsgruppen häufiger anzutreffen. Die Symptome der Beta-Thalassämia major treten normalerweise im ersten Lebensjahr auf und umfassen Anämie, Erbrechen, Fettleibigkeit, Gelbsucht, Knochenveränderungen und eine vergrößerte Milz. Ohne Behandlung kann die Krankheit zu Wachstumsverzögerung, Knochenbrüchen, Infektionen und im Extremfall zum Tod führen.
Die Diagnose der Beta-Thalassämia major erfolgt durch Hämoglobin-Elektrophorese und Gentest. Die Behandlung umfasst Bluttransfusionen, Eisentherapie zur Vorbeugung von Eisenüberladung und möglicherweise eine Stammzelltransplantation.
Glukosephosphat-Dehydrogenase (G6PD) ist ein Enzym, das in roten Blutkörperchen vorkommt und bei der Produktion von NADPH hilft, einem wichtigen Antioxidans im Körper. Ein Mangel an diesem Enzym wird als Glukosephosphat-Dehydrogenase-Mangel bezeichnet.
Dieser genetisch bedingte Zustand tritt auf, wenn eine Person zwei defekte Gene für G6PD erbt, eines von jedem Elternteil. Das Fehlen oder Mangel an funktionsfähigem G6PD-Enzym kann dazu führen, dass rote Blutkörperchen bei Belastungen wie Infektionen, Medikamenteneinnahme oder dem Verzehr von Favabohnen und anderen Lebensmitteln mit hohem Oxidationspotenzial beschädigt werden.
Die Symptome des G6PD-Mangels können leicht bis schwerwiegend sein und umfassen Anämie, Gelbsucht, dunklen Urin und Kurzatmigkeit. In schweren Fällen kann ein G6PD-Mangel zu einer hämolytischen Anämie führen, die sofortige medizinische Behandlung erfordert.
Es ist wichtig zu beachten, dass der Mangel an G6PD nicht behandelbar ist, aber viele Menschen mit dieser Erkrankung können ein normales Leben führen, indem sie vermeiden, Medikamente und Lebensmittel einzunehmen, die einen Anfall auslösen können. Betroffene Personen sollten auch ärztliche Hilfe suchen, wenn sie Symptome wie Müdigkeit, Kurzatmigkeit oder Gelbfärbung der Haut oder Augen bemerken.
DNA, oder Desoxyribonukleinsäure, ist ein Molekül, das die genetische Information in allen Lebewesen und vielen Viren enthält. Es besteht aus zwei langen, sich wiederholenden Ketten von Nukleotiden, die durch Wasserstoffbrückenbindungen miteinander verbunden sind und eine Doppelhelix bilden.
Jeder Nukleotidstrang in der DNA besteht aus einem Zucker (Desoxyribose), einem Phosphatmolekül und einer von vier Nukleobasen: Adenin, Thymin, Guanin oder Cytosin. Die Reihenfolge dieser Basen entlang des Moleküls bildet den genetischen Code, der für die Synthese von Proteinen und anderen wichtigen Molekülen in der Zelle verantwortlich ist.
DNA wird oft als "Blaupause des Lebens" bezeichnet, da sie die Anweisungen enthält, die für das Wachstum, die Entwicklung und die Funktion von Lebewesen erforderlich sind. Die DNA in den Zellen eines Organismus wird in Chromosomen organisiert, die sich im Zellkern befinden.
Genetische Marker sind bestimmte Abschnitte der DNA, die mit einer bekanntermaßen variablen Position in der Genomsequenz eines Individuums assoziiert werden. Sie können in Form von einzelnen Nukleotiden (SNPs - Single Nucleotide Polymorphisms), Variationen in der Wiederholungszahl kurzer Sequenzen (VNTRs - Variable Number Tandem Repeats) oder Insertionen/Deletionen (InDels) auftreten.
Genetische Marker haben keine bekannte Funktion in sich selbst, aber sie können eng mit Genen verbunden sein, die für bestimmte Krankheiten prädisponieren oder Merkmale kontrollieren. Daher werden genetische Marker häufig bei der Kartierung von Krankheitsgenen und zur Abstammungstracing eingesetzt.
Es ist wichtig anzumerken, dass die Entdeckung und Nutzung genetischer Marker ein aktives Feld der Genforschung ist und neue Technologien wie Next-Generation Sequencing zu einer Explosion des verfügbaren Datenmaterials und möglicher neuer Anwendungen führen.
Angeborene Störungen des Aminosäurenstoffwechsels sind eine Gruppe von genetischen Erkrankungen, die aufgrund einer Mutation in den Genen auftreten, die für Enzyme codieren, die am Stoffwechsel von Aminosäuren beteiligt sind. Aminosäuren sind die Bausteine von Proteinen und spielen eine wichtige Rolle im Wachstum, der Reparatur und dem Ersatz von Körpergeweben sowie in anderen biochemischen Prozessen.
Es gibt mehr als 50 verschiedene Arten von angeborenen Störungen des Aminosäurenstoffwechsels, die sich in den Symptomen und der Schwere der Erkrankung unterscheiden können. Die häufigsten Symptome sind Erbrechen, Durchfall, Dehydration, Lethargie, Entwicklungsverzögerungen, neurologische Schäden und geistige Behinderungen.
Die Diagnose von angeborenen Störungen des Aminosäurenstoffwechsels erfolgt häufig durch die Analyse von Blut- oder Urinproben, um eine erhöhte Konzentration bestimmter Aminosäuren nachzuweisen. Die Behandlung besteht in der Regel aus einer speziellen Diät, die den Verzehr von Lebensmitteln mit hohem Gehalt an bestimmten Aminosäuren einschränkt oder vermeidet, sowie der Gabe von Nahrungsergänzungsmitteln und Medikamenten, um den Stoffwechsel zu unterstützen.
Die Prognose hängt von der Art und Schwere der Erkrankung ab, aber viele Kinder mit angeborenen Störungen des Aminosäurenstoffwechsels können ein normales Leben führen, wenn sie eine angemessene Behandlung erhalten. Einige schwere Formen der Krankheit können jedoch zu lebenslangen Behinderungen oder zum Tod führen.
Microsatellite Repeats, auch bekannt als Short Tandem Repeats (STRs), sind wiederholende DNA-Sequenzen, die aus 1-6 Basenpaaren bestehen und in der Regel weniger als 10 Wiederholungen aufweisen. Diese Regionen sind über das Genom verteilt und neigen dazu, instabil zu sein, was zu Variationen in der Anzahl der Wiederholungen zwischen Individuen führt. Microsatellite Repeats werden häufig in der Forensik und Humangenetik zur Identifizierung von Individuen oder zur Erkennung von Verwandtschaftsbeziehungen eingesetzt, da die Wahrscheinlichkeit, dass zwei Personen zufällig die gleiche Anzahl an Wiederholungen aufweisen, sehr gering ist. Mutationen in Microsatellite Repeats können auch mit verschiedenen Krankheiten wie neurologischen Erkrankungen und Krebs assoziiert sein.
Globine sind ein Teil von Proteinen, die als Globuline bezeichnet werden und in den Erythrozyten (roten Blutkörperchen) vorkommen. Die bekannteste und am besten untersuchte Gruppe der Globine sind die Hämoglobine, welche den Sauerstofftransport im Blut ermöglichen.
Hämoglobin besteht aus vier Proteinketten: zwei identische alpha-Globine und zwei identische beta-Globine. Diese Globin-Ketten sind mit einem Häm-Gruppen verbunden, die jeweils ein Eisen-Ion enthalten. Das Eisen im Hämoglobin bindet reversibel an Sauerstoff, was es ermöglicht, Sauerstoff von den Lungen zu den Geweben des Körpers zu transportieren.
Abnormalitäten in der Struktur oder Funktion von Globinen können zu verschiedenen Erkrankungen führen, wie z.B. Sichelzellanämie (eine Krankheit, die durch eine Mutation im beta-Globin-Gen verursacht wird) und Thalassämien (eine Gruppe von Erbkrankheiten, die durch eine Störung in der alpha- oder beta-Globin-Produktion gekennzeichnet sind).
Eine Frameshift-Mutation ist ein genetischer Defekt, bei dem sich die Anzahl der Basenpaare in einem Stück Nukleotidsequenz ändert, was dazu führt, dass das Leseraster des Gens verschoben wird. Dies geschieht durch Hinzufügen oder Weglassen von Basenpaaren in dem genetischen Code.
Wenn beispielsweise ein Basenpaar hinzugefügt oder entfernt wird, verschiebt sich der Rest des Codes um eine Position, was dazu führt, dass die nachfolgenden Aminosäuren nicht mehr korrekt codiert werden. Die resultierende Proteinsequenz kann stark verändert sein und möglicherweise zu einer nichtfunktionellen Proteinversion führen.
Frameshift-Mutationen können durch Fehler während der DNA-Replikation, Reparatur oder Rekombination entstehen und sind oft mit schwerwiegenden genetischen Erkrankungen verbunden.
Angeborene Störungen des Lipidstoffwechsels sind eine Gruppe von Erkrankungen, die aufgrund genetischer Mutationen auftreten und zu einer Stoffwechselstörung von Fetten (Lipiden) führen. Diese Erkrankungen können in der Leber, im Gehirn oder in anderen Organen Schäden verursachen und sind oft mit erhöhten Konzentrationen von Cholesterin oder Triglyceriden im Blut verbunden.
Es gibt verschiedene Arten von angeborenen Störungen des Lipidstoffwechsels, wie zum Beispiel:
* Hypercholesterinämien: Hierbei handelt es sich um Erkrankungen, die zu einem Anstieg des Cholesterinspiegels im Blut führen. Eine bekannte Form ist die familiäre Hypercholesterinämie, bei der eine Genmutation dazu führt, dass der Körper nicht in der Lage ist, überschüssiges Cholesterin aus dem Blut zu entfernen.
* Hypertriglyceridämien: Diese Erkrankungen verursachen einen Anstieg des Triglyceridspiegels im Blut. Eine bekannte Form ist die familiäre Hypertriglyceridämie, bei der eine Genmutation dazu führt, dass der Körper nicht in der Lage ist, überschüssige Triglyceride aus dem Blut zu entfernen.
* Lipoproteinlipase-Mangel: Dies ist eine seltene Erkrankung, bei der ein Enzymmangel dazu führt, dass Fette aus der Nahrung nicht richtig verdaut und aufgenommen werden können.
* Abetalipoproteinämie: Hierbei handelt es sich um eine seltene Erkrankung, bei der der Körper nicht in der Lage ist, Lipoproteine zu bilden, die für den Transport von Fetten im Blut erforderlich sind.
Die Behandlung dieser Erkrankungen hängt von der Art und Schwere der Erkrankung ab. In einigen Fällen kann eine Ernährungsumstellung oder Medikamente helfen, den Cholesterin- oder Triglyceridspiegel im Blut zu senken. In schwereren Fällen kann eine Operation erforderlich sein. Es ist wichtig, dass Menschen mit diesen Erkrankungen regelmäßig von einem Arzt untersucht werden, um sicherzustellen, dass sie richtig behandelt werden und Komplikationen vermieden werden.
Eine Fall-Kontroll-Studie ist eine beobachtende Studie in der Epidemiologie, bei der die Exposition gegenüber einem potenziellen Risikofaktor für eine bestimmte Erkrankung zwischen den „Fällen“ (Personen mit der Erkrankung) und einer Kontrollgruppe ohne die Erkrankung verglichen wird. Die Kontrollgruppen werden üblicherweise so ausgewählt, dass sie dem Fall-Kollektiv hinsichtlich Alter, Geschlecht und anderen potentiell konfundierenden Variablen ähnlich sind. Anschließend wird die Häufigkeit der Exposition zu dem potenziellen Risikofaktor in beiden Gruppen verglichen. Fall-Kontroll-Studien eignen sich besonders gut, um seltene Erkrankungen zu untersuchen oder wenn eine langfristige Beobachtung nicht möglich ist.
Ein Nonsense-Codon ist ein genetischer Code, der während der Proteinsynthese die vorzeitige Beendigung der Translation veranlasst und somit zu einer verkürzten, in der Regel funktionsunfähigen Polypeptidkette führt. Das am häufigsten vorkommende Nonsense-Codon ist UAG (Ambigsus-Code: *), gefolgt von UAA (Ambigsus-Code: * ) und UGA (Ambigsus-Code: *). Diese Codons werden auch als "Stoppcodons" oder "Terminationscodons" bezeichnet. Mutationen, die Nonsense-Codons in normalerweise codierende Sequenzen einführen, können zu schwerwiegenden Erbkrankheiten führen, da sie die Produktion von vollständigen und funktionsfähigen Proteinen verhindern.
Die Fabry-Krankheit ist eine seltene, genetisch bedingte lysosomale Speichererkrankung, die durch einen Mangel des Enzyms alpha-Galaktosidase A verursacht wird. Diese Stoffwechselstörung führt zu einer Anhäufung von Lipiden (insbesondere Gb3 oder Globotriaosylceramid) in verschiedenen Zellen und Geweben des Körpers, was zu progressiven Organschäden führen kann.
Typische Symptome der Fabry-Krankheit umfassen neuropathische Schmerzen, Hautausschläge (Angiokeratome), verminderte Schweißsekretion (Hypohidrose), reduzierte Nierenfunktion, Herzprobleme (wie Herzhypertrophie) und zerebrovaskuläre Ereignisse. Darüber hinaus können Augenbeteiligung (korneale Trübungen) und Hörverlust auftreten.
Die Fabry-Krankheit wird X-chromosomal rezessiv vererbt, was bedeutet, dass Männer mit nur einer Kopie des mutierten Gens typischerweise stärker betroffen sind als Frauen, die in der Regel mildere Symptome aufweisen. Die Diagnose erfolgt durch Biochemische oder genetische Tests, und die Behandlung umfasst Enzymersatztherapie (ERT) und supportive Pflege, um die Symptome zu verwalten und Komplikationen vorzubeugen.
Hypolipoproteinämie ist ein medizinischer Zustand, der durch niedrige Konzentrationen von Lipoproteinen im Blut gekennzeichnet ist. Lipoproteine sind komplexe Partikel, die Fette (Lipide) und Proteine umfassen und für den Transport von Fetten in unserem Körper wesentlich sind. Es gibt verschiedene Arten von Lipoproteinen, wie Chylomikronen, VLDL (very low-density lipoproteins), LDL (low-density lipoproteins) und HDL (high-density lipoproteins). Jede Art von Lipoprotein hat eine bestimmte Funktion im Transport von Fetten.
Eine Hypolipoproteinämie kann auftreten, wenn die Leber nicht in der Lage ist, ausreichende Mengen an Lipoproteinen zu produzieren oder wenn es genetische Störungen gibt, die den Abbau von Lipoproteinen beeinträchtigen. Diese Bedingung kann sich auf verschiedene Weise manifestieren, einschließlich niedriger Cholesterin- und Triglyceridspiegel im Blut.
Es ist wichtig zu beachten, dass eine niedrige Konzentration von Lipoproteinen im Blut nicht unbedingt schädlich sein muss. Einige Formen der Hypolipoproteinämie können jedoch mit einem erhöhten Risiko für neurologische Erkrankungen verbunden sein, wie beispielsweise spinale Muskelatrophie oder periphere Neuropathie. Darüber hinaus kann eine schwere Hypolipoproteinämie zu Mangelernährung führen, da Fette eine wichtige Energiequelle darstellen.
DNA-Schäden beziehen sich auf jede Art von Veränderung in der Struktur oder Sequenz der DNA, die entweder spontan auftreten kann oder als Folge externer oder interner Faktoren, wie ionisierende Strahlung, chemische Substanzen oder Fehler während des Replikationsprozesses. Diese Schäden können verschiedene Formen annehmen, einschließlich Basenschäden, DNA-Strangbrüche, Kreuzvernetzungen und DNA-Addukte. Unreparierte oder fehlerhaft reparierte DNA-Schäden können zum Zelltod führen oder mutagene Ereignisse verursachen, die mit der Entstehung von Krankheiten wie Krebs in Verbindung gebracht werden.
Hybrid Vigor, auch bekannt als Heterosis, ist ein Begriff aus der Genetik und beschreibt die verbesserte Leistungsfähigkeit oder Widerstandsfähigkeit gegenüber Krankheiten, die bei der Kreuzung zweier verschiedener Elternrassen oder -linien auftreten kann.
Diese verbesserte Leistungsfähigkeit und Widerstandsfähigkeit können sich auf verschiedene Merkmale beziehen, wie zum Beispiel höhere Erträge, bessere Qualität der Produkte, stärkeres Wachstum oder größere Vitalität.
Hybrid Vigor tritt auf, wenn die Elternrassen unterschiedliche Allele (Versionen desselben Gens) für bestimmte Merkmale besitzen und diese sich in der hybridisierten Nachkommenschaft kombinieren. Die kombinierten Allele können dann zu einer besseren Leistungsfähigkeit oder Widerstandsfähigkeit führen als die einzelnen Elternrassen.
Es ist wichtig zu beachten, dass Hybrid Vigor nicht immer auftritt und dass es von den spezifischen Merkmalen und Elternrassen abhängt. In manchen Fällen kann es auch zu einer verringerten Leistungsfähigkeit oder Widerstandsfähigkeit kommen, die als Outbreeding Depression bekannt ist.
Hexosaminidase A ist ein Enzym, das im menschlichen Körper vorkommt und eine wichtige Rolle bei dem Abbau von bestimmten Fettsäuren spielt, die an Proteine gebunden sind (Glykolipide und Glykoproteine). Diese Fettsäuren werden als GlcNAc bezeichnet und sind ein Bestandteil von Lipid-Komplexen in den Zellmembranen. Das Fehlen oder eine verminderte Aktivität des Enzyms Hexosaminidase A führt zu einem Abbauprodukt, das sich im Gehirn ansammelt und dort neurotoxische Wirkungen entfaltet, was letztendlich zur Entstehung der Tay-Sachs-Krankheit führt. Diese Erbkrankheit geht mit einer fortschreitenden Zerstörung von Nervenzellen einher und betrifft vor allem Kleinkinder.
'Gene Deletion' ist ein Begriff aus der Genetik und bezeichnet den Verlust eines bestimmten Abschnitts oder sogar eines gesamten Gens auf einer DNA-Molekülstrangseite. Diese Mutation kann auftreten, wenn ein Stück Chromosomenmaterial herausgeschnitten wird oder durch fehlerhafte DNA-Reparaturmechanismen während der Zellteilung.
Die Folgen einer Gendeletion hängen davon ab, welches Gen betroffen ist und wie groß der gelöschte Abschnitt ist. In einigen Fällen kann eine Gendeletion zu keinen oder nur sehr milden Symptomen führen, während sie in anderen Fällen schwerwiegende Entwicklungsstörungen, Erkrankungen oder Behinderungen verursachen kann.
Es ist wichtig zu beachten, dass Gendeletionen bei der genetischen Beratung und Diagnostik eine große Rolle spielen, insbesondere wenn es um erbliche Krankheiten geht. Durch die Analyse von Chromosomen und Genen können Ärzte und Forscher feststellen, ob ein bestimmtes Gen fehlt oder ob es Veränderungen in der DNA-Sequenz gibt, die mit einer Erkrankung verbunden sind.
Homocystinurie ist eine seltene, genetisch bedingte Stoffwechselstörung, bei der der Körper den Abbau der Aminosäure Methionin nicht richtig durchführen kann. Dies führt zu einem Anstieg des Homocysteinspiegels im Blut und im Urin. Homocystinurie wird autosomal rezessiv vererbt und ist mit Mutationen in den Genen CBS, MTHFR oder anderen betroffen, die für Enzyme kodieren, die an der Homocystein-Methionin-Homöostase beteiligt sind.
Die Symptome von Homocystinurie können variieren und umfassen Entwicklungsverzögerungen, kognitive Beeinträchtigungen, Verhaltensauffälligkeiten, Muskelhypertonie, Marfanoid-ähnliche Erscheinungsbilder (hohe und schlanke Gestalt, langgestreckte Gliedmaßen, Brustbein- und Linsenluxation), Osteoporose und Thromboembolien. Die Behandlung von Homocystinurie umfasst in der Regel eine methioninarme Diät, die Ergänzung mit Vitamin B6, B9 (Folsäure) und B12 sowie gegebenenfalls die Gabe von Betain. Eine frühzeitige Diagnose und Behandlung sind entscheidend für die Verhinderung von Komplikationen und die Verbesserung der Prognose.
Der Inzuchtstamm C57BL (C57 Black 6) ist ein spezifischer Stamm von Labormäusen, der durch enge Verwandtschaftspaarungen über mehrere Generationen hinweg gezüchtet wurde. Dieser Prozess, bekannt als Inzucht, dient dazu, eine genetisch homogene Population zu schaffen, bei der die meisten Tiere nahezu identische Genotypen aufweisen.
Die Mäuse des C57BL-Stammes sind für biomedizinische Forschungen sehr beliebt, da sie eine Reihe von vorteilhaften Eigenschaften besitzen. Dazu gehören:
1. Genetische Homogenität: Die enge Verwandtschaftspaarung führt dazu, dass die Tiere des C57BL-Stammes ein sehr ähnliches genetisches Profil aufweisen. Dies erleichtert die Reproduzierbarkeit von Experimenten und die Interpretation der Ergebnisse.
2. Robuste Gesundheit: Die Tiere des C57BL-Stammes gelten als gesund und leben im Allgemeinen lange. Sie sind anfällig für bestimmte Krankheiten, was sie zu einem geeigneten Modell für die Erforschung dieser Krankheiten macht.
3. Anfälligkeit für Krankheiten: C57BL-Mäuse sind anfällig für eine Reihe von Krankheiten, wie zum Beispiel Diabetes, Krebs, neurologische Erkrankungen und Immunerkrankungen. Dies macht sie zu einem wertvollen Modellorganismus für die Erforschung dieser Krankheiten und zur Entwicklung neuer Therapeutika.
4. Verfügbarkeit von genetisch veränderten Linien: Da der C57BL-Stamm seit langem in der Forschung eingesetzt wird, stehen zahlreiche genetisch veränderte Linien zur Verfügung. Diese Linien können für die Untersuchung spezifischer biologischer Prozesse oder Krankheiten eingesetzt werden.
5. Eignung für verschiedene experimentelle Ansätze: C57BL-Mäuse sind aufgrund ihrer Größe, Lebensdauer und Robustheit für eine Vielzahl von experimentellen Ansätzen geeignet, wie zum Beispiel Verhaltensstudien, Biochemie, Zellbiologie, Genetik und Immunologie.
Es ist wichtig zu beachten, dass C57BL-Mäuse nicht für jede Art von Forschung geeignet sind. Ihre Anfälligkeit für bestimmte Krankheiten kann sie als Modellorganismus ungeeignet machen, wenn das Ziel der Studie die Untersuchung einer anderen Krankheit ist. Darüber hinaus können genetische und Umweltfaktoren die Ergebnisse von Experimenten beeinflussen, was die Notwendigkeit einer sorgfältigen Planung und Durchführung von Experimenten unterstreicht.
Lipodystrophie ist eine seltene Erkrankung, bei der es zu einem Verlust oder einer Anomalie des Fettgewebes kommt. Dies kann zu verschiedenen Symptomen führen, wie beispielsweise ein vermindertes Körperfett, erhöhte Fette im Blut (Hypertriglyceridämie), Insulinresistenz und Stoffwechselstörungen.
Es gibt mehrere Arten von Lipodystrophien, die sich in Bezug auf das Ausmaß des Fettgewebesverlusts und die betroffenen Körperregionen unterscheiden. Kongenitale generalisierte Lipodystrophie ist eine angeborene Form, bei der das Fettgewebe bereits von Geburt an oder in den ersten Lebensmonaten fehlt oder abnorm ist. Bei der familiären partiellen Lipodystrophie tritt der Fettverlust erst im Kindes- oder Jugendalter auf und betrifft vor allem die Extremitäten und den Rumpf, während das Gesicht und der Hals area normal bleiben.
Lipodystrophien können auch erworben sein und durch bestimmte Erkrankungen, Medikamente oder Infektionen verursacht werden. Beispiele hierfür sind HIV-infizierte Menschen, die bestimmte Medikamente einnehmen, oder Menschen mit Autoimmunerkrankungen.
Die Behandlung von Lipodystrophien hängt von der zugrunde liegenden Ursache ab und kann Ernährungsänderungen, Medikamente zur Senkung des Blutzuckerspiegels und Cholesterinspiegels sowie Operationen umfassen.
Lezithin-Azyltransferasemangel ist ein seltener Stoffwechseldefekt, bei dem das Enzym Lezithin-Azyltransferase (LAT) nicht oder nur in geringen Mengen vorhanden ist. Dieses Enzym ist für die Umwandlung des Lipids Lezithin in andere Verbindungen notwendig, insbesondere in Plasmalogen, eine Art von Phospholipid, das ein wichtiger Bestandteil der Zellmembranen ist.
Der Mangel an LAT führt zu einem Anstieg des Lezithinspiegels im Blut und zu einem Rückgang des Plasmalogenspiegels in den Zellmembranen, insbesondere in denen des Gehirns. Dies kann zu einer Reihe von neurologischen Symptomen führen, wie z.B. verzögerte Entwicklung, Muskelhypotonie (schlaffe Muskeln), Ataxie (koordinationsstörungen), Epilepsie und in schweren Fällen geistiger Behinderung.
Der Lezithin-Azyltransferasemangel wird autosomal-rezessiv vererbt, was bedeutet, dass ein Betroffener zwei Kopien des defekten Gens haben muss, um die Krankheit zu entwickeln. Die Diagnose erfolgt meist durch die Bestimmung der Aktivität der Lezithin-Azyltransferase im Blut oder in Fibroblasten (Hautzellen) und kann durch genetische Tests bestätigt werden.
Apolipoprotein B (ApoB) ist ein großes, essentielles Protein, das bei der Bildung und Transport von Lipoproteinen im Körper beteiligt ist. Es gibt zwei Hauptformen von ApoB: ApoB-48 und ApoB-100.
ApoB-48 wird hauptsächlich in der Darmschleimhaut produziert und ist ein wichtiger Bestandteil von Chylomikronen, den Lipoproteinen, die Lipide aus der Nahrung transportieren.
ApoB-100 hingegen wird in der Leber synthetisiert und ist das Strukturprotein der sehr niedrig density Lipoproteine (VLDL), die während des Fettstoffwechsels Triglyceride aus der Leber zu den peripheren Geweben transportieren. Im Laufe des Stoffwechsels werden VLDL in Low-Density-Lipoproteine (LDL) umgewandelt, die hauptsächlich Cholesterin transportieren. ApoB-100 ist der einzige Apolipoprotein-Bestandteil von LDL und wird daher manchmal auch als "LDL-Rezeptor-Ligand" bezeichnet.
Erhöhte Konzentrationen von ApoB im Blutplasma sind mit einem erhöhten Risiko für kardiovaskuläre Erkrankungen verbunden, da sie auf eine erhöhte Anzahl von atherogenen Lipoproteinpartikeln hinweisen.
Crossing over, genetisch, ist ein Prozess, der während der Meiose auf Chromosomenebene auftritt und zu genetischer Rekombination führt. Es ist der Austausch von genetischem Material zwischen homologen Chromosomen, die eine genetische Vielfalt bei Nachkommen schafft. Während der Prophase I der Meiose nähern sich die homologen Chromosomenpaare einander an und bilden Chiasmen, an denen die Crossing over stattfindet. Die Ergebnisse von Crossing over sind neue Kombinationen von Allelen auf den Tochterchromosomen, was zu genetischer Vielfalt führt.
Stärkegel-Elektrophorese ist ein Labortest, bei dem Proteine in einer Probe aufgrund ihrer elektrischen Ladung und Größe getrennt werden. Im Gegensatz zu anderen Elektrophorese-Methoden verwendet Stärkegel-Elektrophorese eine Gelmatrix mit bekannter Porengröße, um die Migration der Proteine während des Tests besser zu kontrollieren und zu standardisieren.
Das Gel wird in ein Elektrophoresegerät eingesetzt, das eine elektrische Spannung appliziert, wodurch die Proteine in Richtung des Gegenions migrieren. Die Proteine bewegen sich durch das Gel mit einer Geschwindigkeit, die von ihrer Ladung und Größe abhängt. Große oder stark geladene Proteine migrieren schneller als kleinere oder weniger geladene Proteine.
Nach Abschluss der Elektrophorese wird das Gel typischerweise gefärbt, um die Position der Proteine sichtbar zu machen. Die Färbung kann durch verschiedene Methoden erfolgen, wie z.B. mit Coomassie-Brillantblau oder Silbernitrat. Das resultierende Muster der Proteinbanden wird dann analysiert, um Informationen über die Art und Menge der vorhandenen Proteine zu gewinnen.
Stärkegel-Elektrophorese ist ein wichtiges Werkzeug in der klinischen Diagnostik und Forschung, insbesondere in der Proteomik und Klinischen Biochemie. Es wird häufig eingesetzt, um Veränderungen im Proteinmuster bei verschiedenen Krankheiten wie Krebs, Stoffwechselstörungen und neurologischen Erkrankungen zu identifizieren.
Das Lesch-Nyhan-Syndrom ist ein seltenes, X-chromosomal-rezessiv vererbtes Stoffwechseldefekt, welches durch einen Mangel des Enzyms Hypoxanthin-Guanin-Phosphoribosyltransferase (HGPRT) gekennzeichnet ist. Dies führt zu einem Anstieg von Harnsäure im Blut und im Urin (Hyperurikämie und Hyperurikosurie), was wiederum zu Gicht, Nierensteinen und Nierenschäden führen kann.
Die Erkrankung manifestiert sich typischerweise in den ersten Lebensmonaten mit motorischen Auffälligkeiten wie Hypotonie (Muskelschwäche) und Tremor (Zittern). Später kommen dann auch noch Choreoathetose (unwillkürliche, wellenförmige Muskelbewegungen), Dysarthrie (Sprechstörung) und Spastik (erhöhter Muskeltonus) hinzu.
Die bekannteste und auffälligste Symptomatik des Lesch-Nyhan-Syndroms ist jedoch das selbstverletzende Verhalten, welches bei den meisten Betroffenen auftritt. Dazu gehören beispielsweise sich ins Gesicht schlagen, sich in die Mundwinkel beißen oder Fingernägel kauen.
Die Behandlung des Lesch-Nyhan-Syndroms ist bisher symptomatisch und unterstützend, eine ursächliche Therapie existiert nicht.
Ein DNA-Primer ist ein kurzes, einzelsträngiges Stück DNA oder RNA, das spezifisch an die Template-Stränge einer DNA-Sequenz bindet und die Replikation oder Amplifikation der DNA durch Polymerasen ermöglicht. Primers sind notwendig, da Polymerasen nur in 5'-3' Richtung synthetisieren können und deshalb an den Startpunkt der Synthese binden müssen. In der PCR (Polymerase Chain Reaction) sind DNA-Primer entscheidend, um die exakte Amplifikation bestimmter DNA-Sequenzen zu gewährleisten. Sie werden spezifisch an die Sequenz vor und nach der Zielregion designed und erlauben so eine gezielte Vermehrung des gewünschten DNA-Abschnitts.
Lipoproteinlipase (LPL) ist ein Enzym, das am Abbau von Lipoproteinen beteiligt ist, indem es die Triglyceride in den Lipoproteinmolekülen hydrolysiert und so freie Fettsäuren und Glycerin abspaltet. Dieses Enzym spielt eine wichtige Rolle bei der Regulation des Lipidstoffwechsels, indem es die Aufnahme von Fettsäuren in peripheren Geweben wie Muskeln und Fettgewebe fördert.
LPL ist an der Oberfläche von Endothelzellen in Blutgefäßen lokalisiert und wird durch verschiedene Hormone und Enzyme reguliert, darunter Insulin, das seine Aktivität steigert, und Lipoproteinlipaseinhibitoren wie Apolipoprotein C-III, die seine Aktivität hemmen.
Genetische Variationen im LPL-Gen wurden mit einem erhöhten Risiko für Erkrankungen wie Familiäre Hypercholesterinämie und koronare Herzkrankheit in Verbindung gebracht.
Ethylnitrosoharnstoff ist ein chemisches Medikament, das früher zur Behandlung von Angina pectoris eingesetzt wurde. Es wirkt als Vasodilatator, der die glatte Muskulatur in den Blutgefäßen entspannt und so den Blutfluss verbessert. Ethylnitrosoharnstoff ist ein Ester von Nitrosoharnstoff und wird nach der oralen Einnahme schnell vom Körper metabolisiert.
Aufgrund seiner unerwünschten Wirkungen, wie zum Beispiel Kopfschmerzen, niedriger Blutdruck, Übelkeit und Erbrechen, wird Ethylnitrosoharnstoff heutzutage nur noch selten eingesetzt. Stattdessen werden andere Medikamente bevorzugt, die ähnliche Wirkungen haben, aber besser verträglich sind.
Apolipoprotein B-100 (ApoB-100) ist ein großes, essentielles Protein, das bei der Bildung von Lipoproteinen beteiligt ist, insbesondere des Low-Density-Lipoproteins (LDL), auch als "schlechtes Cholesterin" bekannt. ApoB-100 ist die Hauptproteinkomponente von LDL und spielt eine wichtige Rolle bei der Bindung von LDL an seine Rezeptoren auf Leberzellen, was den Clearance-Prozess des LDL-Cholesterins aus dem Blutkreislauf einleitet.
Eine Erhöhung der ApoB-100-Konzentration im Blutplasma ist mit einem erhöhten Risiko für kardiovaskuläre Erkrankungen verbunden, da es zu einer Anhäufung von LDL-Cholesterin in den Blutgefäßen führt und somit die Entstehung von Atherosklerose begünstigt. Daher ist die Bestimmung der ApoB-100-Konzentration ein wichtiger Marker für das kardiovaskuläre Risiko und kann bei der Beurteilung des individuellen Risikos sowie bei der Überwachung der Therapieeffektivität hilfreich sein.
In der Medizin bezieht sich "Genes" auf den Prozess der Wiederherstellung der normalen Funktion und des Wohlbefindens nach einer Krankheit, Verletzung oder Operation. Dieser Begriff beschreibt den Zustand, in dem ein Patient die Symptome seiner Erkrankung überwunden hat und wieder in der Lage ist, seine täglichen Aktivitäten ohne Beeinträchtigung auszuführen.
Es ist wichtig zu beachten, dass "Genes" nicht immer bedeutet, dass eine Person vollständig geheilt ist oder keine Spuren der Erkrankung mehr aufweist. Manchmal kann es sich lediglich um eine Verbesserung des Zustands handeln, bei der die Symptome abgeklungen sind und das Risiko einer erneuten Verschlechterung minimiert wurde.
Die Dauer des Genesungsprozesses hängt von verschiedenen Faktoren ab, wie Art und Schwere der Erkrankung, dem Alter und Gesundheitszustand des Patienten sowie der Qualität der medizinischen Versorgung und Nachsorge. In einigen Fällen kann die Genesung schnell und vollständig sein, während sie in anderen Fällen langwierig und möglicherweise unvollständig sein kann.
Haploidie ist ein Genetik-Begriff, der sich auf die Situation bezieht, in der eine Zelle nur einen vollständigen Satz von Chromosomen enthält. Im Gegensatz dazu besitzen normale diploide Zellen zwei komplette Sätze von Chromosomen, einen vom Vater geerbten und einen von der Mutter geerbten.
In der menschlichen Genetik ist ein normaler diploider Körperzelltyp ein 2N-Zustand, was bedeutet, dass er 46 Chromosomen enthält (23 Paare). Haploide Zellen hingegen, wie die Geschlechtszellen oder Gameten (Eizelle und Spermium), enthalten nur einen einzelnen Satz von 23 ungepaarten Chromosomen, was als N-Zustand bezeichnet wird.
Die Reduktion der Chromosomenzahl von diploid auf haploid erfolgt während des Meiotischen Prozesses (Reifeteilung) in den Keimdrüsen (Gonaden), wobei die Anzahl der Chromosomen durch eine spezielle Art der Zellteilung, die Meiose, halbiert wird. Diese Halbierung ist notwendig, um während der Befruchtung oder Verschmelzung von zwei haploiden Gameten (Eizelle und Spermium) wieder auf die normale diploide Anzahl von Chromosomen zu kommen.
Alpha-1-Antitrypsinmangel ist ein genetisch bedingter Mangel an dem Protein Alpha-1-Antitrypsin (AAT), das normalerweise in der Leber produziert wird und im Blut zirkuliert. Das Protein gehört zur Gruppe der Serpin-Proteine und hat die Funktion, andere Enzyme zu inhibieren, insbesondere das Enzym Elastase, das von weißen Blutkörperchen (Neutrophilen) produziert wird.
Elastase ist ein Protein, das normalerweise hilft, Bakterien im Körper zu zerstören. Wenn es jedoch nicht durch AAT gehemmt wird, kann es Gewebe schädigen, insbesondere in den Lungen. Bei Menschen mit Alpha-1-Antitrypsinmangel ist das Protein defekt und kann seine Funktion nicht richtig ausüben, was zu einem Übermaß an Elastase führt und die Lunge angreift.
Die Krankheit wird autosomal-rezessiv vererbt, was bedeutet, dass eine Person das Gen nur dann von ihren Eltern geerbt haben muss, wenn beide Elternteile Träger des Gens sind. Wenn ein Mensch zwei Kopien des defekten Gens hat, führt dies zu einem schweren Mangel an Alpha-1-Antitrypsin und einem erhöhten Risiko für Lungen- und Lebererkrankungen.
Die Symptome von Alpha-1-Antitrypsinmangel können variieren, aber die meisten Menschen mit dieser Krankheit entwickeln im Erwachsenenalter eine chronisch obstruktive Lungenerkrankung (COPD), insbesondere wenn sie rauchen oder anderen schädlichen Umweltfaktoren ausgesetzt sind. Andere Symptome können Lebererkrankungen, Hautausschläge und Blutgerinnungsstörungen sein.
Die Behandlung von Alpha-1-Antitrypsinmangel umfasst in der Regel die Verwendung von Ersatzpräparaten aus menschlichem Plasma, die das fehlende Protein ersetzen. Andere Behandlungen können auch die Einnahme von Medikamenten zur Linderung der Symptome und die Änderung des Lebensstils umfassen, wie zum Beispiel das Aufhören des Rauchens.
Albinismus ist eine genetisch bedingte Erkrankung, die durch einen Mangel oder vollständiges Fehlen des Enzyms Tyrosinase verursacht wird, das für die Produktion des Pigments Melanin in Haut, Haaren und Augen notwendig ist. Dies führt zu einer stark verminderten oder völlig fehlenden Pigmentierung der Haut, Haare und Augen. Menschen mit Albinismus haben oft weiße Haare, sehr helle Haut und hellblaue oder rosafarbene Augen. Darüber hinaus können sie eine verminderte Sehschärfe aufweisen, da die reduzierte Pigmentierung in der Augenrückseite (Retina) die normale Funktion des Sehnervs beeinträchtigen kann. Albinismus wird autosomal rezessiv vererbt, was bedeutet, dass ein Betroffener das defekte Gen von beiden Elternteilen erben muss, um an der Erkrankung zu leiden.
Eine Chromosomenumkehr (oder Chromosomainversion) ist eine genetische Veränderung, bei der ein Teil eines Chromosoms seine ursprüngliche Orientierung relativ zu dem Telomer am Ende des Chromosoms umgekehrt hat. Dabei bleiben die Reihenfolge und Orientierung der Gene entlang des invertierten Abschnitts erhalten, aber der gesamte Abschnitt ist nun in entgegengesetzter Richtung relativ zu den umliegenden Chromosomenabschnitten angeordnet.
Chromosomeninversionen können während der Meiose auftreten, wenn ein Chromosom bricht und die Bruchstücke anschließend in verkehrter Orientierung wieder zusammengefügt werden. Sie können entweder perizentrische oder parazentrische Inversionen sein, je nachdem, ob der zentromerische Bereich des Chromosoms betroffen ist oder nicht.
Obwohl die genetische Information im Allgemeinen intakt bleibt, kann eine Chromosomenumkehr zu genetischen Unregelmäßigkeiten führen, wenn sie während der Meiose mit einem nicht invertierten Homologen kombiniert wird. Dies kann zu ungleicher Crossing-over und anormaler Segregation der Chromosomen führen, was wiederum zu verschiedenen genetischen Erkrankungen oder Fehlgeburten führen kann.
Hämoglobin, Sichelzell- ist eine Variante des Hämoglobins, einem Proteins, das in den roten Blutkörperchen vorkommt und den Sauerstoff transportiert. Bei dieser Variante liegt eine genetisch bedingte Veränderung der Aminosäuresequenz vor, die dazu führt, dass sich die Hämoglobinmoleküle unter bestimmten Bedingungen verformen und zu einer Sichelbildung der roten Blutkörperchen führen. Diese Verformung kann zu einer Verstopfung kleiner Blutgefäße führen, was eine Vielzahl von Komplikationen wie Schmerzen, Infektionen, Organschäden und Blutgerinnsel verursachen kann. Sichelzell-Hämoglobin ist eine erbliche Erkrankung, die hauptsächlich bei Menschen afrikanischer Abstammung auftritt.
Beta-N-Acetylhexosaminidase ist ein Enzym, das eine wichtige Rolle in der Degradation von Glykoproteinen und Lipochininoiden spielt. Es besteht aus zwei Untereinheiten, α und β, die sich zu drei Isoformen des Enzyms zusammensetzen: αβ, αα und ββ. Das β-Untereinheit ist das defekte Protein bei der seltenen genetischen Erkrankung Tay-Sachs-Krankheit. Diese Krankheit führt zu einem Anstieg von GM2-Gangliosiden in den Neuronen des Gehirns und Nervensystems, was schließlich zu deren Zerstörung und zu einer fortschreitenden neurodegenerativen Erkrankung führt.
Fibroblasten sind Zellen des Bindegewebes, die für die Synthese und Aufrechterhaltung der Extrazellularmatrix verantwortlich sind. Sie produzieren Kollagen, Elastin und proteoglykane, die dem Gewebe Struktur und Elastizität verleihen. Fibroblasten spielen eine wichtige Rolle bei Wundheilungsprozessen, indem sie das Granulationsgewebe bilden, das für die Narbenbildung notwendig ist. Darüber hinaus sind Fibroblasten an der Regulation von Entzündungsreaktionen beteiligt und können verschiedene Wachstumsfaktoren und Zytokine produzieren, die das Verhalten anderer Zellen im Gewebe beeinflussen.
Hyperlipoproteinämie ist ein medizinischer Zustand, der durch einen Anstieg der Blutfette (Lipide) gekennzeichnet ist, die an Proteine (Lipoproteine) gebunden sind. Es gibt verschiedene Arten von Hyperlipoproteinämien, abhängig von dem Typ des beteiligten Lipoproteins. Die zwei häufigsten Formen sind:
1. Hypercholesterolemie: Hierbei liegt ein Anstieg des Cholesterins im Blut vor, hauptsächlich in Form von Low-Density-Lipoprotein (LDL), auch als "schlechtes Cholesterin" bekannt.
2. Hypertriglyceridämie: Hierbei liegt ein Anstieg der Triglyzeride im Blut vor, hauptsächlich in Form von Very-Low-Density-Lipoprotein (VLDL).
Hyperlipoproteinämien können genetisch bedingt sein oder aufgrund von Lebensstilfaktoren wie unausgewogener Ernährung, Bewegungsmangel, Adipositas und Rauchen auftreten. Sie können auch als Nebenwirkungen bestimmter Medikamente auftreten.
Hyperlipoproteinämien sind ein wichtiger Risikofaktor für die Entwicklung von Atherosklerose und kardiovaskulären Erkrankungen, einschließlich Herzinfarkt und Schlaganfall. Daher ist es wichtig, Hyperlipoproteinämien frühzeitig zu diagnostizieren und zu behandeln, um das Risiko von Komplikationen zu minimieren.
2-Aminopurin, auch bekannt als Theophyllin, ist ein methyliertes Xanthin-Alkaloid, das in Teeblättern und anderen Pflanzen vorkommt. Es wird als Bronchodilatator und Stimulans des Zentralnervensystems (CNS) eingesetzt. Theophyllin wirkt durch Hemmung der Phosphodiesterase und damit Erhöhung des cAMP-Spiegels in den glatten Muskelzellen der Atemwege, wodurch diese entspannt werden. Es wird bei der Behandlung von Asthma bronchiale und anderen obstruktiven Atemwegserkrankungen eingesetzt. Theophyllin ist auch ein nicht-selektiver Adenosinrezeptorantagonist, was zu seiner Wirkung als CNS-Stimulans beiträgt.
Eine Aminosäuresequenz ist die genau festgelegte Reihenfolge der verschiedenen Aminosäuren, aus denen ein Proteinmolekül aufgebaut ist. Sie wird direkt durch die Nukleotidsequenz des entsprechenden Gens bestimmt und spielt eine zentrale Rolle bei der Funktion eines Proteins.
Die Aminosäuren sind über Peptidbindungen miteinander verknüpft, wobei die Carboxylgruppe (-COOH) einer Aminosäure mit der Aminogruppe (-NH2) der nächsten reagiert, wodurch eine neue Peptidbindung entsteht und Wasser abgespalten wird. Diese Reaktion wiederholt sich, bis die gesamte Kette der Proteinsequenz synthetisiert ist.
Die Aminosäuresequenz eines Proteins ist einzigartig und dient als wichtiges Merkmal zur Klassifizierung und Identifizierung von Proteinen. Sie bestimmt auch die räumliche Struktur des Proteins, indem sie hydrophobe und hydrophile Bereiche voneinander trennt und so die Sekundär- und Tertiärstruktur beeinflusst.
Abweichungen in der Aminosäuresequenz können zu Veränderungen in der Proteinstruktur und -funktion führen, was wiederum mit verschiedenen Krankheiten assoziiert sein kann. Daher ist die Bestimmung der Aminosäuresequenz von großer Bedeutung für das Verständnis der Funktion von Proteinen und deren Rolle bei Erkrankungen.
Linkage Disequilibrium (LD) ist ein Konzept in der Genetik, das die nicht zufällige Assoziation zwischen Allelen verschiedener Gene oder unterschiedlicher Varianten eines Gens beschreibt. Normalerweise würden man erwarten, dass sich Allele unabhängig voneinander vererben, aber in bestimmten Situationen können zwei Allele häufiger gemeinsam vorkommen als es durch Zufall zu erwarten wäre.
Dies tritt auf, wenn diese Allele nahe beieinander auf demselben Chromosom liegen und nicht unabhängig sortiert werden können, da sie während der Meiose gemeinsam vererbt werden. Die Stärke des Linkage Disequilibrium hängt von der genetischen Distanz zwischen den Allelen ab - je näher sie beieinander liegen, desto stärker ist die Assoziation.
Linkage Disequilibrium wird oft in populationsgenetischen Studien untersucht, um Informationen über die Lage von Genen auf Chromosomen zu gewinnen und um genetische Varianten zu identifizieren, die mit bestimmten Krankheiten assoziiert sind.
Es ist wichtig zu klären, dass es keine medizinische Definition der Gruppe "asiatischer Abstammung" gibt, da Rasse ein soziokulturelles Konstrukt und kein biologisches Merkmal ist. Die Klassifizierung von Menschen nach rassischen Kategorien ist umstritten und wird von vielen Experten abgelehnt, weil sie dazu beitragen kann, diskriminierende Praktiken zu rechtfertigen und Ungleichheiten aufrechtzuerhalten.
Im Gesundheitswesen werden jedoch häufig bestimmte Bevölkerungsgruppen identifiziert, die auf der Grundlage gemeinsamer kultureller, sprachlicher, geografischer oder historischer Merkmale zusammengefasst sind. Diese Gruppen können als "Asiatische Amerikaner und Inselbewohner des Pazifik" bezeichnet werden, wie es in den USA üblich ist. Diese Kategorie umfasst eine sehr heterogene Gruppe von Menschen mit unterschiedlichen ethnischen Hintergründen, Sprachen, Kulturen und Erfahrungen.
Es ist wichtig zu beachten, dass die Verwendung dieser Kategorien nicht bedeutet, dass es biologische Unterschiede zwischen den Gruppen gibt, sondern dass sie verwendet werden, um bestimmte Bevölkerungsgruppen zu identifizieren, die aufgrund historischer und soziokultureller Faktoren möglicherweise unterschiedliche Erfahrungen mit dem Gesundheitssystem machen oder unterschiedlichen Risikofaktoren für bestimmte Krankheiten ausgesetzt sind.
Faktor V, auch bekannt als Proakzelerin oder Labile Faktor, ist ein Protein das im menschlichen Plasma vorkommt und eine wichtige Rolle in der Blutgerinnung spielt. Es wird von Leberzellen produziert und dient als Co-Faktor für die Aktivierung von Faktor X zu Faktor Xa während der Gerinnungskaskade.
Wenn Faktor V durch die Thrombin-aktivierte Faktor V-Protease in seine aktive Form Faktor Va umgewandelt wird, beschleunigt es die Umwandlung von Prothrombin zu Thrombin, was wiederum zur Bildung von Fibrin führt und so zur Blutgerinnung beiträgt.
Eine Fehlfunktion des Faktors V kann zu Gerinnungsstörungen führen, wie zum Beispiel die vererbte Thromboembolie, bei der das Risiko für venöse Thromboembolien (Blutgerinnsel in den Venen) erhöht ist.
Aminosäuresubstitution bezieht sich auf den Prozess, bei dem ein anderes Aminosäurerestmolekül in einen Proteinstrukturstrang eingebaut wird, anstelle des ursprünglichen Aminosäurerests an einer bestimmten Position. Dies tritt auf, wenn es eine genetische Variante oder Mutation gibt, die dazu führt, dass ein anderes Aminosäure codiert wird, was zu einer Veränderung der Aminosäurensequenz im Protein führt. Die Fähigkeit eines Proteins, seine Funktion aufrechtzuerhalten, nachdem eine Aminosäuresubstitution stattgefunden hat, hängt von der Art und Position der substituierten Aminosäure ab. Manche Substitutionen können die Proteinstruktur und -funktion beeinträchtigen oder sogar zerstören, während andere möglicherweise keine Auswirkungen haben.
In der Medizin bezieht sich die Haarfarbe auf die natürliche oder durch Färbemittel veränderte Farbpigmentierung der Haare, die durch zwei Arten von Pigmenten bestimmt wird: Eumelanin und Phäomelanin. Die Menge und Art dieser Pigmente in den Haaren determinieren die Haarfarbe eines Menschen.
Eumelanin ist das dunklere Pigment, das in braunen und schwarzen Haaren vorkommt. Wenn nur wenig Eumelanin vorhanden ist, erscheinen die Haare blond. Phäomelanin hingegen ist das hellere Pigment, das in roten Haaren am stärksten konzentriert ist, aber auch in braunen und schwarzen Haaren vorkommen kann.
Es ist wichtig zu beachten, dass die Haarfarbe eines Menschen im Laufe des Lebens aufgrund von Alterung, Hormonänderungen und anderen Faktoren variieren kann.
Ein Hemizygot ist ein Individuum, das nur einen funktionalen Allel eines Gens besitzt, während das andere Allel auf einem Geschlechtschromosom fehlt oder nicht-funktional ist. Dies tritt normalerweise bei Genen auf, die auf den Geschlechtschromosomen X und Y liegen.
Bei Frauen (XX) sind beide X-Chromosomen normalerweise aktiv und exprimieren das entsprechende Gen. Bei Männern (XY) ist jedoch nur ein X-Chromosom vorhanden, wodurch sie für X-chromosomale Gene hemizygot sind. Wenn das verbleibende Allel auf dem X-Chromosom eines Mannes mutiert oder fehlt, kann dies zu genetischen Erkrankungen führen, da es kein zweites, funktionales Allel gibt, um die Funktion des Gens zu kompensieren.
Es ist wichtig anzumerken, dass Hemizygotie nicht nur auf Geschlechtschromosomen beschränkt ist. Seltene Fälle von Autosomal-Hemizygotie können auftreten, wenn ein Individuum ein großes genetisches Defekt oder Deletion auf einem autosomalen Chromosom aufweist, wodurch sie nur ein funktionales Allel für bestimmte Gene besitzen.
Chromosomen sind im Zellkern befindliche Strukturen, die die Erbinformationen in Form von Desoxyribonukleinsäure (DNA) enthalten. Sie sind bei der Zellteilung und -vermehrung von großer Bedeutung, da sie sich verdoppeln und dann zwischen den Tochterzellen gleichmäßig verteilen, um so eine genetisch identische Kopie der Elternzelle zu erzeugen.
Ein Chromosom besteht aus zwei Chromatiden, die durch einen Zentromer miteinander verbunden sind. Die Chromatiden enthalten jeweils ein lineares DNA-Molekül, das mit Proteinen assoziiert ist und in bestimmten Abschnitten (den Genen) die Erbinformationen kodiert.
Im Menschen gibt es 23 verschiedene Chromosomenpaare, von denen 22 Paare als Autosomen bezeichnet werden und ein Paar Geschlechtschromosomen (XX bei Frauen, XY bei Männern) bildet. Die Gesamtzahl der Chromosomen in einer menschlichen Zelle beträgt daher 46.
Diploidie ist ein Zustand der Chromosomenzahl in den Zellen eines Organismus, bei dem sich das Genom aus zwei vollständigen Sets von Chromosomen zusammensetzt, die jeweils als Homologe bezeichnet werden. In der Regel besteht ein Satz aus einem autosomalen oder nicht-geschlechtsspezifischen Chromosomensatz und einem Geschlechtschromosomensatz. Somit enthält eine diploide Zelle gewöhnlich das doppelte der haploiden Anzahl an Chromosomen, die in den reiferen Gameten (Eizellen und Spermien) vorhanden sind. Bei Menschen beträgt die normale diploide Anzahl von Chromosomen 46 (2N), bestehend aus 23 paarweise Homologe Chromosomen, davon 22 Paare autosomaler Chromosomen und ein Paar Geschlechtschromosomen (XX bei Weibchen oder XY bei Männern). Diploidie ist die übliche Kondition für die Mehrheit der Zellen in den Körpern von vielzelligen Organismen und spielt eine wichtige Rolle bei der genetischen Vielfalt, da sie während der Meiose und Befruchtung zur Rekombination führt. Abweichungen von der normalen Diploidie, wie Aneuploidien (zusätzliche oder fehlende Chromosomen), können zu genetischen Erkrankungen führen.
Die Glucosephosphat-Dehydrogenase (GPD, Gen name: GPI) ist ein Enzym, das im Stoffwechsel eine zentrale Rolle spielt. Es ist beteiligt am ersten Schritt der Glykolyse und an der Pentosephosphat-Pathway (HEX-PATH). Das Enzym katalysiert die Umwandlung von Glucose-6-phosphat in 6-Phosphoglucono-δ-Lacton unter Verbrauch von NADP+ und Freisetzung von Dihydroxyacetonphosphat (DHAP). Diese Reaktion ist ein wichtiger Schritt bei der Regulation des Stoffwechsels, da sie die Menge an reduziertem Nicotinamidadenindinukleotidphosphat (NADPH) kontrolliert, das für den Abbau und die Synthese von Fettsäuren sowie für den Schutz der Zellen vor oxidativem Stress benötigt wird.
Eine genetische Mutation des GPD-Gens kann zu einem Mangel an Glucosephosphat-Dehydrogenase führen, was als GPD-Mangel oder G6PD-Mangel bezeichnet wird. Diese Erkrankung ist eine der häufigsten enzymatischen Stoffwechselstörungen und betrifft vor allem Männer. Symptome eines GPD-Mangels können anfallsartige Hämolyse (Zerstörung der roten Blutkörperchen), Gelbsucht, dunkler Urin und Anämie sein. Diese Symptome treten häufig nach Infektionen oder dem Verzehr bestimmter Medikamente auf.
Der Founder Effekt ist ein Begriff in der Populationsgenetik, der beschreibt, wie sich genetische Variationen in einer Population verändern können, wenn sie von einer kleinen Gruppe Gründungsmitglieder (der "Founder") gegründet wird.
Wenn eine neue Population durch eine kleine Gruppe Gründer entsteht, kann es sein, dass nur bestimmte Allele (Formen eines Gens) in der neuen Population vorhanden sind, da die Gründer möglicherweise nur einige dieser Varianten tragen. Diese reduzierte genetische Vielfalt kann dazu führen, dass bestimmte Merkmale oder Krankheiten häufiger auftreten als in der ursprünglichen Population.
Der Founder Effekt kann auch dazu führen, dass sich die Häufigkeit seltener genetischer Varianten in der neuen Population erhöht, was als "Gründerflaschen-Effekt" bezeichnet wird. Diese Veränderungen in der genetischen Zusammensetzung können das Auftreten von Erbkrankheiten und anderen genetisch bedingten Merkmalen in der neuen Population beeinflussen.
Es ist wichtig zu beachten, dass der Founder Effekt nur eine mögliche Ursache für Veränderungen in der genetischen Zusammensetzung einer Population ist und dass andere Faktoren wie Selektion, Genfluss und Gendrift ebenfalls eine Rolle spielen können.
Familiäres Mittelmeerfieber ist eine autosomal-rezessiv vererbte autoinflammatory Krankheit, die gekennzeichnet ist durch periodische Episoden von Fieber, Bauchschmerzen, Pleuritis, Perikarditis, Arthralgien und Hautausschlägen. Die Krankheit wird durch Mutationen im MEFV-Gen verursacht, das für das Protein Pyrin kodiert. Das familiäre Mittelmeerfieber ist am häufigsten in mediterranen Bevölkerungsgruppen wie Türken, Arabern, Juden und Italienern zu finden. Die Diagnose wird durch charakteristische klinische Merkmale und genetische Tests gestellt. Die Behandlung umfasst kolchizinhaltige Medikamente wie Colchicin zur Vorbeugung von Krankheitsepisoden und Entzündungshemmer wie nicht-steroidale Antirheumatika (NSAIDs) oder Glukokortikoide zur Linderung der Symptome während einer Episode.
In der Medizin wird ein Haar als ein keratinhaltiges Filament definiert, das aus der Haut eines Menschen oder Tieres wächst. Es ist Teil des Haarfollikels und besteht aus einer proteinreichen Substanz namens Keratin. Jedes Haar hat eine Wurzel, die in der Haut tief verwurzelt ist und von Blutgefäßen und Nerven versorgt wird. Die sichtbare, äußere Partie des Haares wird als Stamm bezeichnet.
Die Funktionen von Haaren sind thermoregulierung, Schutz der Haut vor UV-Strahlung, mechanischer Schutz sowie soziale und kulturelle Bedeutungen. Die Anzahl, Verteilung und Beschaffenheit der Körperhaare können je nach Alter, Geschlecht, genetischen Faktoren und Erkrankungen variieren.
Eisenüberlastung, auch bekannt als Hämochromatose, ist ein Zustand, bei dem es zu einer übermäßigen Ansammlung von Eisen in verschiedenen Organen und Geweben des Körpers kommt. Normalerweise wird Eisen aus der Nahrung im Darm aufgenommen und an Transferrin gebunden, um dann zu den Zellen transportiert zu werden, wo es für die Bildung von Hämoglobin und anderen Proteinen benötigt wird.
Bei einer Eisenüberlastung ist dieser Regulationsmechanismus gestört, was dazu führt, dass zu viel Eisen in den Körper aufgenommen und nicht ausreichend ausgeschieden wird. Die übermäßige Menge an Eisen sammelt sich im Laufe der Zeit in Organen wie Leber, Herz, Bauchspeicheldrüse und Hormondrüsen an, was zu Organschäden und verschiedenen Symptomen führen kann, wie zum Beispiel:
* Müdigkeit und Schwäche
* Braune oder graue Verfärbung der Haut (Bronzedermatose)
* Schmerzen in Gelenken und Knochen
* Herzrhythmusstörungen
* Lebererkrankungen, wie Leberzirrhose oder Leberkrebs
* Diabetes mellitus
* Impotenz bei Männern
Eisenüberlastung kann angeboren sein und auf genetischen Defekten beruhen, die die Eisenaufnahme im Darm erhöhen. Sie kann aber auch erworben werden, zum Beispiel durch wiederholte Bluttransfusionen oder übermäßigen Alkoholkonsum.
Die Behandlung von Eisenüberlastung besteht in der Regel darin, überschüssiges Eisen aus dem Körper zu entfernen, was durch regelmäßige Aderlässe (Phlebotomie) erreicht wird. In einigen Fällen kann auch eine medikamentöse Therapie eingesetzt werden, um das Eisen aus den Organen zu entfernen.
'Drosophila melanogaster' ist keine medizinische Bezeichnung, sondern die wissenschaftliche Bezeichnung für die Taufliege oder Fruchtfliege. Es handelt sich um ein kleines Insekt, das häufig in der biologischen und genetischen Forschung eingesetzt wird, da es eine kurze Generationszeit hat, leicht zu züchten und zu manipulieren ist, und sein Genom gut erforscht und verstanden ist. Die Entschlüsselung des Genoms von Drosophila melanogaster hat wertvolle Einblicke in die Funktionsweise von Genen bei verschiedenen Tierarten geliefert, einschließlich Menschen.
"Gene Dosage" bezieht sich auf die Anzahl der Kopien eines Gens, die in einem Genom vorhanden sind. Normalerweise hat ein Mensch zwei Kopien jedes Gens, eine von jedem Elternteil. Eine Veränderung in der Gene Dosage, sei es durch Duplikation oder Deletion eines Gens, kann zu Veränderungen im Proteinspiegel führen und verschiedene Krankheiten oder Fehlbildungen verursachen. Zum Beispiel können zusätzliche Kopien bestimmter Gene mit Erkrankungen wie Down-Syndrom assoziiert sein, während das Fehlen einer Kopie bestimmter Gene mit Krankheiten wie dem Turner-Syndrom einhergehen kann. Die Untersuchung der Gene Dosage ist ein wichtiger Bestandteil der Humangenetik und Genomforschung.
Es scheint, dass Sie nach einer medizinischen Bedeutung oder Verwendung des Begriffs "Italien" suchen, aber der Begriff an sich ist nicht mit Medizin verbunden. Italien ist ein Land in Südeuropa. Wenn Sie nach etwas Bestimmten suchen, wie einer italienischen Bezeichnung für eine Krankheit oder medizinische Organisation in Italien, lassen Sie es mich bitte wissen und ich werde mein Bestes tun, um Ihnen zu helfen.
Erythrozyten, auch als rote Blutkörperchen bekannt, sind die häufigsten Zellen im Blutkreislauf der Wirbeltiere. Laut medizinischer Definition handelt es sich um bikonkave, un nucleierte Zellen, die hauptsächlich den Sauerstofftransport vom Atmungsorgan zu den Geweben ermöglichen. Die rote Farbe der Erythrozyten resultiert aus dem darin enthaltenen Protein Hämoglobin. Inaktive Erythrozyten werden in Milz und Leber abgebaut, während die Bildung neuer Zellen hauptsächlich in Knochenmark stattfindet.
Introns sind nicht-kodierende Sequenzen in einem DNA-Molekül, die während der Transkription in mRNA (messenger RNA) umgeschrieben werden. Sie werden als "inneres" Segment zwischen zwei kodierenden Abschnitten oder Exons definiert.
Geschlechtschromosomen sind ein Typ von Chromosomen, die bei der Bestimmung des biologischen Geschlechts eines Organismus eine wichtige Rolle spielen. Bei Menschen und vielen anderen Säugetieren gibt es zwei Geschlechtschromosomen: X und Y. Die meisten weiblichen Individuen haben zwei X-Chromosomen (XX), während die meisten männlichen Individuen ein X- und ein Y-Chromosom (XY) besitzen.
Die Geschlechtschromosomen enthalten Gene, die das Geschlecht eines Organismus bestimmen und auch an der Entwicklung der primären und sekundären Geschlechtsmerkmale beteiligt sind. Abweichungen in der Anzahl oder Struktur der Geschlechtschromosomen können zu genetisch bedingten Geschlechtsvarianten oder Erkrankungen führen, wie zum Beispiel das Klinefelter-Syndrom (XXY) oder das Turner-Syndrom (X0).
Membranproteine sind Proteine, die sich in der Lipidbilayer-Membran von Zellen oder intrazellulären Organellen befinden. Sie durchdringen oder sind mit der Hydrophobischen Membran verbunden und spielen eine wichtige Rolle bei zellulären Funktionen, wie dem Transport von Molekülen, Signaltransduktion, Zell-Zell-Kommunikation und Erkennung. Membranproteine können in integral (dauerhaft eingebettet) oder peripher (vorübergehend assoziiert) eingeteilt werden, je nachdem, ob sie die Membran direkt durch eine hydrophobe Domäne stabilisieren oder über Wechselwirkungen mit anderen Proteinen assoziiert sind.
Gen-Targeting bezieht sich auf die gezielte Manipulation oder Modulation spezifischer Gene innerhalb von Zellen zur Behandlung oder Erforschung von Krankheiten. Dies wird in der Regel durch Techniken wie Geneditierung (z.B. CRISPR-Cas9), RNA-Interferenz oder Antisense-Oligonukleotide erreicht. Das Ziel ist es, die Funktion eines defekten Gens zu korrigieren, die Expression eines überaktiven Gens zu reduzieren oder gezielt therapeutische Proteine in den Zellen zu produzieren. Diese Techniken haben das Potenzial, neue Behandlungsmöglichkeiten für eine Vielzahl von Erkrankungen wie genetisch bedingte Krankheiten, Krebs und Virusinfektionen zu eröffnen.
HLA-Antigene, auch bekannt als Human Leukocyte Antigens, sind eine Klasse von Proteinen, die auf der Oberfläche der meisten Zellen im menschlichen Körper vorkommen. Sie spielen eine wichtige Rolle im Immunsystem, indem sie dem Körper helfen, zwischen "selbst" und "nicht-selbst" zu unterscheiden.
HLA-Antigene präsentieren kurze Proteinstücke, die aus both inneren und äußeren Quellen stammen, an T-Zellen des Immunsystems. Auf dieser Basis entscheidet das Immunsystem, ob eine Zelle als "normal" oder "krankhaft" eingestuft wird. Wenn beispielsweise ein Virus in eine Zelle eindringt und sich dort vermehrt, werden virale Proteinstücke von HLA-Antigenen präsentiert, was dazu führt, dass das Immunsystem die infizierte Zelle zerstört.
Es gibt drei Hauptklassen von HLA-Antigenen: Klasse I (A, B und C), Klasse II (DP, DQ und DR) und Klasse III. Jede Klasse hat eine unterschiedliche Funktion im Immunsystem. Unterschiede in HLA-Genen können die Anfälligkeit für bestimmte Krankheiten beeinflussen, einschließlich Autoimmunerkrankungen und Infektionskrankheiten. Darüber hinaus werden HLA-Typen zur Übereinstimmung bei Organtransplantationen verwendet, um die Wahrscheinlichkeit von Abstoßungsreaktionen zu minimieren.
Die 'Familiengesundheit' bezieht sich auf den Zustand der Gesundheit und des Wohlbefindens aller Familienmitglieder sowie auf die Fähigkeit der Familie als Einheit, Unterstützung und Ressourcen zur Bewältigung von Krankheiten, Verletzungen und Stressfaktoren bereitzustellen. Es umfasst auch die Förderung einer gesunden Entwicklung und des Wohlbefindens durch die Schaffung einer positiven, unterstützenden und sicheren Umgebung.
Die Familienmitglieder können unterschiedliche Altersgruppen, ethnische Hintergründe, kulturelle Identitäten und sozioökonomische Status haben, was die Komplexität der Familien Gesundheitszustand weiter erhöht. Faktoren wie soziale Determinanten der Gesundheit, genetische Veranlagung, Lebensstilfaktoren und Umweltfaktoren können sich auf den Gesundheitszustand der Familie auswirken.
Daher ist es wichtig, dass die Familien einen Zugang zu hochwertigen Gesundheitsversorgungs- und Präventionsdiensten haben, um chronische Krankheiten, Behinderungen und Verletzungen zu vermeiden oder zu verwalten. Darüber hinaus können Bildungsprogramme und Unterstützungsdienste für Familien dazu beitragen, die Fähigkeiten und Ressourcen der Familie zu stärken, um eine positive Gesundheit und ein positives Wohlbefinden aufrechtzuerhalten.
Kongenitale Adrenale Hyperplasie (CAH) ist ein Sammelbegriff für eine Gruppe seltener erblicher Störungen, die die Entwicklung der Nebennierenrinde betreffen. Die Nebennieren sind endokrine Drüsen, die Hormone produzieren, einschließlich Cortisol, Aldosteron und Androgene.
CAH wird durch Mutationen in den Genen verursacht, die für das Enzym 21-Hydroxylase codieren, das an der Synthese von Cortisol beteiligt ist. Aufgrund dieser Störung kann das Enzym nicht richtig arbeiten, was zu einem Anstieg des Androgenspiegels führt. Androgene sind männliche Sexualhormone, die bei Frauen und Mädchen eine übermäßige männliche Entwicklung verursachen können.
Es gibt verschiedene Formen von CAH, aber die häufigste ist das 21-Hydroxylase-Mangel-Syndrom, das in zwei Hauptformen unterteilt wird: die klassische und die nichtklassische Form. Die klassische Form kann weiter in ein salz-verschwendendes und ein nicht-salz-verschwendendes Untertyp unterteilt werden.
Die Symptome der CAH hängen von der Art und Schwere der Erkrankung ab. Bei Mädchen mit salz-verschwendendem Typ kann es zu einer Virilisierung (maskulinisierende Veränderungen) der Genitalien vor der Geburt kommen, was zu einer ungewöhnlichen Anatomie der Genitalien führt. Bei Jungen und Mädchen können Symptome wie verfrühtes Wachstum, frühzeitige Pubertät, Akne, erhöhte Körperbehaarung und Menstruationsstörungen auftreten.
Die Behandlung von CAH umfasst in der Regel eine lebenslange Hormonersatztherapie mit Glukokortikoiden, um den Hormonspiegel im Körper zu normalisieren und das Wachstum und die Entwicklung des Kindes zu fördern. Eine frühzeitige Diagnose und Behandlung sind wichtig, um Komplikationen wie Wachstumsverzögerung, Knochenschwäche und Bluthochdruck zu vermeiden.
Geschlechtschromosomale Aberrationen sind Abweichungen von der normalerweise vorkommenden Anzahl oder Struktur der Geschlechtschromosomen. Bei Menschen ist das Geschlecht in der Regel durch die Kombination von Chromosomen X und Y bestimmt, wobei Weibchen zwei X-Chromosomen (46, XX) und Männchen ein X- und ein Y-Chromosom (46, XY) besitzen.
Abweichungen in der Anzahl oder Struktur der Geschlechtschromosomen können zu genetischen Störungen führen, die sich auf die physische Entwicklung, das Hormonsystem und die Fruchtbarkeit auswirken können. Beispiele für geschlechtschromosomale Aberrationen sind:
* Klinefelter-Syndrom (47, XXY): Männer mit diesem Syndrom haben ein extra X-Chromosom. Sie können unterentwickelte Hoden, Brustgewebewachstum, reduzierte Fruchtbarkeit und Lernschwierigkeiten haben.
* Turner-Syndrom (45, X): Frauen mit diesem Syndrom fehlt ein X-Chromosom. Sie können kurz sein, breite Hälse und Schultern haben, kein Menstruationszyklus entwickeln und Unfruchtbarkeit aufweisen.
* Triple-X-Syndrom (47, XXX): Frauen mit diesem Syndrom haben ein zusätzliches X-Chromosom. Sie können normal groß sein, aber manchmal geistige Behinderungen oder Lernschwierigkeiten haben.
* Jacob-Syndrom (47, XYY): Männer mit diesem Syndrom haben ein zusätzliches Y-Chromosom. Sie können größer als der Durchschnitt sein, aber oft keine anderen Symptome haben.
Es ist wichtig zu beachten, dass nicht alle Menschen mit diesen Chromosomenanomalien Symptome haben und dass die Auswirkungen sehr unterschiedlich sein können. Ein Gentest kann durchgeführt werden, um festzustellen, ob eine Person ein zusätzliches oder fehlendes X- oder Y-Chromosom hat.
Eine Chromosomendeletion ist ein genetischer Defekt, bei dem ein Teil eines Chromosoms fehlt oder verloren gegangen ist. Dies kann durch Fehler während der Zellteilung (Mitose oder Meiose) verursacht werden und führt zu einer Veränderung der Anzahl oder Struktur des Chromosoms.
Die Größe der Deletion kann variieren, von einem kleinen Fragment bis hin zu einem großen Teil eines Chromosoms. Die Folgen dieser Deletion hängen davon ab, welcher Bereich des Chromosoms betroffen ist und wie viele Gene darin enthalten sind.
Eine Chromosomendeletion kann zu verschiedenen genetischen Erkrankungen führen, je nachdem, welches Chromosom und welcher Bereich betroffen sind. Ein Beispiel für eine genetische Erkrankung, die durch eine Chromosomendeletion verursacht wird, ist das cri du chat-Syndrom, bei dem ein Teil des kurzen Arms von Chromosom 5 fehlt. Diese Erkrankung ist mit charakteristischen Gesichtsmerkmalen, Entwicklungsverzögerungen und geistiger Beeinträchtigung verbunden.
Hyperargininämie ist eine seltene, autosomal-rezessiv vererbte Stoffwechselstörung, bei der es zu einem Anstieg des Argininspiegels im Blut und im Urin kommt. Dies wird durch eine Mutation im Gen für die Arginosuccinatlyase (ASL) verursacht, das ein Enzym kodiert, das für den Abbau von Arginosuccinat notwendig ist.
Der Mangel an funktionsfähiger Arginosuccinatlyase führt zu einem Anstieg des Arginins und der Aminosäure Prolin im Körper. Hyperargininämie kann zu einer Reihe von neurologischen Symptomen führen, darunter Entwicklungsverzögerungen, Krampfanfälle, geistige Behinderung und in schweren Fällen sogar zum Tod im Kindesalter. Es gibt derzeit keine kurative Behandlung für Hyperargininämie, aber eine kohlenhydratarme Diät und die Ergänzung mit essentiellen Aminosäuren können dazu beitragen, einige der Symptome zu lindern.
Elektroretinographie (ERG) ist ein diagnostisches Verfahren in der Augenheilkunde, bei dem die elektrische Antwort der Netzhaut auf Lichtreize gemessen wird. Dabei werden Elektroden an der Außenseite des Auges oder auf der Haut neben dem Auge angebracht, die die sehr kleinen elektrischen Signale erfassen, die von den Photorezeptoren und anderen Zellen in der Netzhaut generiert werden.
Die ERG-Messung liefert Informationen über die Funktion der verschiedenen Zelltypen in der Netzhaut, einschließlich Stäbchen und Zapfen, und kann bei der Diagnose und Überwachung von Erkrankungen wie Retinitis Pigmentosa, Makuladegeneration, diabetischer Retinopathie und anderen Netzhauterkrankungen hilfreich sein.
Die ERG-Untersuchung ist schmerzlos und dauert in der Regel nur wenige Minuten. Der Patient muss während der Untersuchung die Augen offen halten und auf ein Licht oder Muster fixieren, während die verschiedenen Lichtreize präsentiert werden. Die Ergebnisse der ERG-Messung werden dann vom Arzt ausgewertet, um den Zustand der Netzhaut zu beurteilen und gegebenenfalls weitere Behandlungsschritte einzuleiten.
Mukopolysaccharidose Typ II, auch bekannt als Hunter-Syndrom, ist eine seltene, aber schwere erbliche Stoffwechselerkrankung, die durch einen Mangel des Enzyms Iduronat-2-Sulfatase verursacht wird. Diese Erkrankung führt zu einer Anhäufung von Glykosaminoglykanen (auch als Mukopolysaccharide bekannt) in verschiedenen Körpergeweben und Organen, was zu Gewebeschäden und Organdysfunktionen führt.
Die Symptome des Hunter-Syndroms können variieren, aber die häufigsten Anzeichen sind: vergrößerte Leber und Milz, Atemwegsprobleme, Herzklappenerkrankungen, Hörverlust, Veränderungen im Gesicht (dickes Nasenbein, prominente Wangenknochen, große Zunge), Gelenksteifigkeit, eingeschränkte Beweglichkeit und geistige Behinderung.
Das Hunter-Syndrom tritt hauptsächlich bei Jungen auf, da es auf dem X-Chromosom vererbt wird und Mädchen ein zweites intaktes X-Chromosom haben, das die Krankheit kompensieren kann. Es gibt zwei Formen des Hunter-Syndroms: eine schwere, fortschreitende Form und eine mildere, langsam fortschreitende Form. Die schwere Form ist am häufigsten und führt in der Regel vor dem 10. Lebensjahr zum Tod. Die milde Form kann später im Leben auftreten und kann zu einer kürzeren Lebenserwartung führen, aber die Betroffenen können ein normales Erwachsenenalter erreichen.
Die Diagnose des Hunter-Syndroms erfolgt durch eine Blutuntersuchung, bei der das fehlende Enzym Iduronat-2-Sulfatase nachgewiesen wird. Eine frühzeitige Diagnose ist wichtig, um die Behandlung so schnell wie möglich einzuleiten und den Krankheitsverlauf zu verlangsamen.
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) ist ein Protein, das als Chloridkanal in der Membran von Epithelzellen dient. Es spielt eine wichtige Rolle bei der Flüssigkeitsregulation in verschiedenen Organen, wie beispielsweise den Atemwegen, dem Verdauungstrakt und den Schweißdrüsen. Mutationen in dem Gen, das für CFTR codiert, können zu cystischer Fibrose führen, einer erblich bedingten Krankheit, die durch eine übermäßige Produktion von zähem Schleim in verschiedenen Organen gekennzeichnet ist. Diese Mutationen können dazu führen, dass das CFTR-Protein nicht richtig funktioniert oder gar nicht erst gebildet wird, was zu Störungen der Chlorid- und Flüssigkeitstransportmechanismen führt.
Alpha-Galactosidase ist ein Enzym, das den Abbau von Alpha-Galactosiden, einer Klasse von Kohlenhydraten, katalysiert. Genauer gesagt spaltet dieses Enzym die α(1→6)- und α(1→3)-glykosidische Bindungen zwischen Galactose und einem terminalen Galactose-Rest in bestimmten Glykolipiden und Glycoproteinen auf. Das Enzym ist wichtig für den Stoffwechsel von Substanzen, die Alpha-Galactose enthalten, wie beispielsweise Ceramidtrihexoside (auch Globotriaosylceramid oder Gb3 genannt).
Eine verminderte Aktivität der Alpha-Galactosidase kann zu einer Anreicherung von Gb3 in verschiedenen Organen und Geweben führen, was eine Erkrankung namens Fabry-Syndrom verursacht. Dieses ist eine genetisch bedingte lysosomale Speicherkrankheit, die mit verschiedenen Symptomen einhergeht, wie Schmerzen in Händen und Füßen (Acroparesthesien), Hautausschlägen, Herz-Kreislauf-Erkrankungen und Nierenversagen. Die Behandlung des Fabry-Syndroms beinhaltet die Enzymersatztherapie mit rekombinanter Alpha-Galactosidase (z. B. Agalsidase alfa oder Agalsidase beta), um den Mangel an diesem Enzym zu kompensieren und den Fortschritt der Krankheit zu verlangsamen.
In der medizinischen Forschung wird Alpha-Galactosidase auch als potenzielles Therapeutikum zur Behandlung von alpha-Galactose-induzierten Allergien untersucht, wie beispielsweise die sogenannte 'Alpha-Gal-Allergie', bei der Menschen nach dem Verzehr von rotem Fleisch (Rind, Schwein, Lamm) oder bestimmten Milchprodukten allergische Reaktionen entwickeln.
HLA-DRB3 Chain bezieht sich auf eine Untergruppe von Proteinen, die als HLA-DRB3-Moleküle bezeichnet werden und Teil des Humanen Leukozytenantigens (HLA) sind, einem komplexen System aus Molekülen, das an der Regulation der Immunantwort beteiligt ist.
Die HLA-Moleküle sind auf der Oberfläche von Zellen lokalisiert und spielen eine wichtige Rolle bei der Präsentation von Peptiden (kleine Proteinstücke) an T-Zellen, die wiederum eine zentrale Rolle bei der Erkennung und Bekämpfung von Krankheitserregern im Körper spielen.
Die HLA-DRB3-Moleküle sind spezifische Varianten des HLA-DR-Komplexes, die aus zwei Ketten bestehen: einer α-Kette und einer β-Kette. Die HLA-DRB3 Chains sind eine Untergruppe von β-Ketten, die durch genetische Variationen in der DNA codiert werden.
Es ist wichtig zu beachten, dass das Vorhandensein oder Fehlen bestimmter HLA-Typen, einschließlich HLA-DRB3 Chains, mit verschiedenen Krankheiten und Erkrankungen assoziiert sein kann, obwohl die genauen Mechanismen dieser Assoziationen noch nicht vollständig verstanden sind.
Alpha-1-Antitrypsin (AAT) ist ein Protein, das im Blutkreislauf vorkommt und als Serpin-Protease-Inhibitor wirkt. Es schützt Gewebe, insbesondere in der Lunge, vor Zerstörung durch proteolytische Enzyme wie Elastase, die von neutrophilen Granulozyten während Entzündungsprozessen sezerniert werden.
Eine genetisch bedingte Störung im AAT-Gen kann zu einer verminderten Produktion oder Funktionsunfähigkeit des Proteins führen, was als Alpha-1-Antitrypsin-Mangel bezeichnet wird. Dieser Mangel erhöht das Risiko für Lungenkrankheiten wie COPD (Chronisch Obstruktive Lungenerkrankung) und Emphysem sowie für Lebererkrankungen, da AAT auch Leberzellen schützt.
Die Behandlung eines Alpha-1-Antitrypsin-Mangels kann eine wöchentliche Infusion von AAT-Protein umfassen, um die Konzentration des Proteins im Blutkreislauf zu erhöhen und das Fortschreiten der Lungenerkrankung zu verlangsamen. Es ist auch wichtig, andere Risikofaktoren für COPD wie Rauchen zu vermeiden.
Die Ornithin carbamoyltransferase (OCT) ist ein Enzym, das eine wichtige Rolle in dem Stoffwechselprozess der Umwandlung von Ammoniak zu Harnstoff spielt, einem Endprodukt des Eiweißstoffwechsels. Bei einer Ornithin carbamoyltransferase-Mangelkrankheit ist die Aktivität dieses Enzyms reduziert oder fehlt ganz, was zu einer Anreicherung von Ammoniak im Blut und einem damit einhergehenden Anstieg des Blut pH-Werts (Alkalose) führt. Diese Erkrankung wird auch als Hyperammonämie bezeichnet und kann zu schweren neurologischen Schäden führen, wenn sie nicht frühzeitig erkannt und behandelt wird.
Die Symptome einer Ornithin carbamoyltransferase-Mangelkrankheit können variieren, aber typischerweise treten sie bereits im Säuglingsalter auf und umfassen Erbrechen, Lethargie, Reizbarkeit, Hyperaktivität, Krampfanfälle und Entwicklungsverzögerungen. Langfristig kann die Erkrankung zu geistiger Behinderung, Wachstumsverzögerung und Schädigungen des Nervensystems führen.
Die Diagnose einer Ornithin carbamoyltransferase-Mangelkrankheit erfolgt in der Regel durch eine Kombination aus klinischen Untersuchungen, Bluttests und genetischen Tests. Die Behandlung umfasst in der Regel eine Ernährungsumstellung, die Reduzierung des Proteinanteils in der Nahrung, die Gabe von Medikamenten zur Senkung des Ammoniakspiegels im Blut und gegebenenfalls auch eine Lebertransplantation.
Ein Neugeborenes ist ein Kind, das in den ersten 28 Tagen nach der Geburt steht. Dieser Zeitraum wird als neonatale Periode bezeichnet und ist klinisch wichtig, da die meisten Komplikationen und Probleme des Neugeborenen in den ersten Tagen oder Wochen auftreten. Die Betreuung von Neugeborenen erfordert spezielle Kenntnisse und Fähigkeiten, einschließlich der Erkennung und Behandlung von angeborenen Anomalien, Infektionen, Frühgeburtlichkeit und anderen potenziellen Komplikationen. Neugeborene werden oft in spezialisierten Einheiten wie einer Neonatologie oder Neugeboreneneinheit betreut, insbesondere wenn sie vorzeitig geboren sind oder medizinische Probleme haben.
Ein Codon ist ein dreibasiger Nukleotidabschnitt in der DNA oder RNA, der für die genetische Information zur Synthese eines spezifischen Aminosäurerestes in einem Protein kodiert. Es gibt 64 mögliche Codone (inklusive Start- und Stoppcodons), die sich aus den vier Nukleotiden Adenin, Thymin/Uracil, Guanin und Cytosin zusammensetzen. Jedes Codon wird von einem korrespondierenden tRNA-Molekül erkannt und decodiert, um die entsprechende Aminosäure während der Proteinsynthese zu transportieren.
Ferritin ist ein Proteinkomplex, der in allen Zellen des menschlichen Körpers vorkommt und als Speicherprotein für Eisen dient. Es ist vor allem in Leber, Knochenmark, Milz und Muskeln konzentriert. Ferritin kann im Blutserum gemessen werden und dient als Maß für den Gesamtkörper-Eisenspeicher. Niedrige Serumferritinspiegel können auf einen Eisenmangel hinweisen, während hohe Spiegel auf eine übermäßige Eisenakkumulation oder Entzündungen hindeuten können.
Leukozyten, auch weiße Blutkörperchen genannt, sind ein wichtiger Bestandteil des menschlichen Immunsystems. Es handelt sich um spezialisierte Zellen, die im Blutkreislauf zirkulieren und den Körper bei der Abwehr von Infektionen und Krankheiten unterstützen. Leukozyten sind in der Lage, krankheitserregende Mikroorganismen wie Bakterien, Viren und Pilze zu erkennen, zu umhüllen und zu zerstören.
Es gibt verschiedene Arten von Leukozyten, die sich in ihrer Form, Funktion und Herkunft unterscheiden. Dazu gehören Neutrophile, Lymphozyten, Monozyten, Eosinophile und Basophile. Jede dieser Untergruppen hat eine spezifische Rolle bei der Immunabwehr.
Neutrophile sind die häufigsten Leukozyten und spielen eine wichtige Rolle bei der Bekämpfung bakterieller Infektionen, indem sie die Bakterien durch Phagozytose (Einschließung und Zerstörung) eliminieren.
Lymphozyten sind an der zellulären und humoralen Immunantwort beteiligt. Sie produzieren Antikörper, um Krankheitserreger zu neutralisieren, und können infizierte Zellen durch direkte Lyse (Zerstörung) eliminieren.
Monozyten sind große Leukozyten, die sich in Gewebe differenzieren und als Makrophagen oder dendritische Zellen fungieren. Sie spielen eine wichtige Rolle bei der Phagozytose und Präsentation von Antigenen an andere Immunzellen.
Eosinophile sind an der Bekämpfung von Parasiten wie Würmern beteiligt und spielen auch eine Rolle bei allergischen Reaktionen, indem sie die Freisetzung von Histamin aus Mastzellen regulieren.
Basophile sind seltene Leukozyten, die an der Entstehung von Entzündungen beteiligt sind, indem sie Histamin und andere Mediatoren freisetzen, um Immunreaktionen zu verstärken.
Eine Erhöhung oder Verminderung der Anzahl bestimmter Leukozyten kann auf eine Infektion, Entzündung oder eine Erkrankung des blutbildenden Systems hinweisen. Die Analyse von Blutuntersuchungen ist ein wichtiges Instrument zur Diagnose und Überwachung von Krankheiten.
Genotyping techniques are a group of laboratory methods used to identify and analyze the genetic makeup (genotype) of an individual, organism, or virus. These techniques can be used to detect differences or variations in the DNA sequence, gene structure, or number of chromosomes between individuals. Genotyping is used in various fields such as medical research, forensic science, and ancestry testing.
There are several types of genotyping techniques, including:
1. Polymerase Chain Reaction (PCR) based methods: These methods use PCR to amplify specific regions of the DNA and then analyze them for variations. Examples include Amplification Refractory Mutation System (ARMS), TaqMan assays, and High-Resolution Melting (HRM) analysis.
2. Sequencing-based methods: These methods determine the order of nucleotides in a specific region of DNA. Examples include Sanger sequencing, Next-Generation Sequencing (NGS), and Whole Genome Sequencing (WGS).
3. Microarray-based methods: These methods use microarrays to analyze multiple genetic markers simultaneously. Examples include Single Nucleotide Polymorphism (SNP) arrays and Comparative Genomic Hybridization (CGH) arrays.
4. Fragment analysis-based methods: These methods use capillary electrophoresis or gel electrophoresis to separate DNA fragments based on their size, followed by detection using fluorescence or radioactivity. Examples include Short Tandem Repeat (STR) analysis and Restriction Fragment Length Polymorphism (RFLP).
Genotyping techniques have numerous applications in medicine, including diagnosis of genetic disorders, identification of disease-associated genes, pharmacogenomics, and infectious disease surveillance.
Augenproteine, auch als ophthalmologische Proteine bekannt, beziehen sich auf die verschiedenen Proteine, die in den unterschiedlichen Geweben des Auges gefunden werden und an wichtigen physiologischen Prozessen beteiligt sind. Dazu gehören Enzyme, Strukturproteine, Signalproteine und Transportproteine. Einige Beispiele für Augenproteine sind:
* Krystalline: Diese Proteine bilden den größten Teil der Linsenmasse und sind für die Transparenz und Brechung des einfallenden Lichts verantwortlich.
* Opsine: Diese Proteine sind in den Stäbchen und Zapfen der Netzhaut lokalisiert und spielen eine wichtige Rolle bei der visuellen Wahrnehmung, indem sie Licht in elektrische Signale umwandeln.
* Transportproteine: Diese Proteine, wie z.B. Glukose-Transporter und Ionenkanäle, sind für den Transport von Nährstoffen und Metaboliten in das Auge notwendig.
* Enzyme: Verschiedene Enzyme sind im Auge vorhanden und beteiligt an Stoffwechselprozessen, wie z.B. Katalase und Superoxiddismutase, die antioxidative Eigenschaften haben und das Auge vor oxidativen Schäden schützen.
* Strukturproteine: Diese Proteine, wie z.B. Kollagen und Elastin, sind für die Stabilität und Elastizität der verschiedenen Gewebe des Auges verantwortlich.
Störungen in der Funktion oder Regulation dieser Proteine können zu verschiedenen Augenerkrankungen führen, wie z.B. Katarakt, Makuladegeneration und Netzhautdegenerationen.
Ein genetischer Komplementaritätstest ist ein molekularbiologisches Verfahren, bei dem die genetische Kompatibilität zwischen zwei potenziellen Spenderschaften (z.B. Knochenmark oder Nierenspende) untersucht wird. Dabei wird die Histokompatibilität der Gewebemerkmale, insbesondere der humanen Leukozytenantigene (HLA), zwischen Spender und Empfänger bestimmt.
Der Test zielt darauf ab, das Risiko einer Abstoßungsreaktion nach der Transplantation zu minimieren, indem die Übereinstimmung der Gewebemerkmale zwischen Spender und Empfänger so hoch wie möglich ist. Das Verfahren umfasst in der Regel die Analyse von HLA-Proteinen oder -DNA-Sequenzen an mehreren Genloci, um eine genaue Beurteilung der Kompatibilität zu ermöglichen.
Ein höheres Maß an Übereinstimmung in den HLA-Merkmalen zwischen Spender und Empfänger kann die Wahrscheinlichkeit einer erfolgreichen Transplantation erhöhen, indem das Risiko von Abstoßungsreaktionen und transplantatassoziierten Komplikationen reduziert wird.
Die Glykogenose Typ V, auch bekannt als McArdle-Krankheit, ist ein seltenes genetisch bedingtes metabolisches Myopathie-Syndrom. Es ist durch einen Mangel an der Muskelphosphorylase verursacht, einem Enzym, das für den Abbau von Glykogen zu Glukose-1-Phosphat in der Skelettmuskulatur benötigt wird.
Dieser Enzymdefekt führt dazu, dass die Muskeln nicht in der Lage sind, Glykogen effizient als Energiequelle zu nutzen, insbesondere während anaerober Aktivitäten. Als Folge können Betroffene Anzeichen und Symptome wie vorzeitige Ermüdung, Muskelkrämpfe, Muskelsteifigkeit und Myoglobinurie (die Ausscheidung von Myoglobin im Urin, was auf Muskelschäden hindeutet) erfahren, insbesondere nach Phasen ununterbrochener oder intensiver körperlicher Anstrengung.
Die Diagnose der Glykogenose Typ V kann durch klinische Untersuchungen, Blutuntersuchungen und Muskelbiopsie mit anschließender Enzymaktivitätsanalyse gestellt werden. Die Behandlung umfasst in der Regel eine Anpassung des Lebensstils, einschließlich der Vermeidung von Aktivitäten, die Krämpfe oder Myoglobinurie auslösen können, sowie die Einbeziehung von Ausdauertraining in den Alltag, um die Fähigkeit der Muskeln zu verbessern, auf andere Energiequellen zurückzugreifen. In einigen Fällen kann eine Ernährungsumstellung mit einer Erhöhung der Kohlenhydratzufuhr hilfreich sein.
Epistasis ist ein Begriff aus der Genetik und beschreibt ein Phänomen, bei dem die Auswirkungen eines Gens durch die Variation eines oder mehrerer anderer Gene beeinflusst werden. In anderen Worten, das Erscheinungsbild oder die Expression eines Gens wird durch die Interaktion mit einem anderen Gen verändert.
Es gibt verschiedene Arten von Epistasis, aber eine häufige Form ist die sogenannte rezessive Epistasis. Hierbei maskiert ein rezessives Allel (die weniger aktive Variante) eines Gens die Wirkung eines dominanten Allels (die aktivere Variante) eines anderen Gens.
Epistasis spielt eine wichtige Rolle bei der Ausprägung komplexer Merkmale und Krankheiten, wie beispielsweise Stoffwechselstörungen oder Erkrankungen des Immunsystems. Daher ist ein Verständnis von Epistasis für die humane Genetik und personalisierte Medizin von großer Bedeutung.
Augenanomalien sind Abweichungen oder Abweichungen von der normalen Struktur, Funktion oder Positionierung des Auges oder seiner Anhänge. Diese Abweichungen können bei Geburt vorhanden sein (anlagemäßig) oder im Laufe des Lebens erworben werden. Sie können einzeln oder in Kombination mit anderen Gesundheitsproblemen auftreten.
Es gibt viele Arten von Augenanomalien, die von mild bis schwer reichen und eine Vielzahl von Symptomen verursachen können, wie verschwommenes Sehen, Doppeltsehen, Schmerzen, Lichtempfindlichkeit, rote Augen oder einfach nur kosmetische Beeinträchtigungen.
Beispiele für häufige Augenanomalien sind Kurzsichtigkeit (Myopie), Weitsichtigkeit (Hyperopie), Astigmatismus, Grauer Star (Katarakt), Grüner Star (Glaukom), Schielen (Strabismus) und Fehlbildungen des Augapfels oder der Augenlider.
Die Behandlung von Augenanomalien hängt von der Art und Schwere der Anomalie ab. Mildere Fälle können mit Brillen oder Kontaktlinsen korrigiert werden, während schwerwiegendere Fälle möglicherweise eine chirurgische Korrektur erfordern. Regelmäßige augenärztliche Untersuchungen sind wichtig, um Augenanomalien frühzeitig zu erkennen und zu behandeln, bevor sie irreversible Schäden verursachen.
Das Manifestationsalter ist ein Begriff aus der Medizin, der das typische Alter beschreibt, in dem die Symptome (die „Manifestationen“) einer bestimmten Erkrankung erstmals auftreten. Es handelt sich also um das durchschnittliche Alter, in dem die Krankheit klinisch sichtbar wird und vom Patienten oder von einem Arzt bemerkt werden kann.
Das Manifestationsalter ist ein wichtiger Faktor bei der Diagnose vieler Krankheiten, insbesondere bei Entwicklungsstörungen und genetischen Erkrankungen. Zum Beispiel manifestiert sich die spinale Muskelatrophie (SMA) typischerweise im Säuglings- oder Kleinkindalter, während die Alzheimer-Krankheit üblicherweise erst im höheren Erwachsenenalter auftritt.
Es ist jedoch wichtig zu beachten, dass das Manifestationsalter variieren kann und nicht bei allen Patienten mit derselben Erkrankung gleich ist. Einige Menschen können die Symptome einer Krankheit früher oder später im Leben entwickeln als üblich, was als atypische Manifestation oder variable Manifestation bezeichnet wird.
Transgenic Mice sind gentechnisch veränderte Mauslinien, bei denen Fremd-DNA (auch Transgen) in ihr Genom eingebracht wurde, um das genetische Material der Mäuse gezielt zu verändern. Das Ziel ist es, das Verständnis von Genfunktionen und krankheitsverursachenden Genmutationen zu verbessern.
Die Einführung des Transgens kann durch verschiedene Techniken erfolgen, wie beispielsweise per Mikroinjektion in die Keimzellen (Eizelle oder Spermien), durch Nukleofugierung in embryonale Stammzellen oder mithilfe von Virenvektoren.
Die transgenen Mäuse exprimieren das fremde Gen und können so als Modellorganismus für die Erforschung menschlicher Krankheiten dienen, um beispielsweise Krankheitsmechanismen besser zu verstehen oder neue Therapien zu entwickeln. Die Veränderungen im Genom der Tiere werden oft so gestaltet, dass sie die humane Krankheit nachahmen und somit ein geeignetes Modell für Forschungszwecke darstellen.
Apolipoprotein A-I (ApoA-I) ist ein Protein, das hauptsächlich in der Hülle von High-Density-Lipoproteinen (HDL) vorkommt, die allgemein als "gutes Cholesterin" bekannt sind. Es wird hauptsächlich in der Leber und im Dünndarm synthetisiert. ApoA-I spielt eine wichtige Rolle bei der Umwandlung von Cholesterin in HDL, was zur Bildung von so genanntem "umgekehrtem Cholesterintransport" führt, einem Prozess, bei dem überschüssiges Cholesterin aus Geweben (einschließlich Arterienwänden) zu Leber und Darm transportiert wird, wo es metabolisiert oder ausgeschieden werden kann. ApoA-I hat auch entzündungshemmende Eigenschaften und spielt möglicherweise eine Rolle bei der endothelialen Funktion und der Immunantwort.
Embryonenverlust ist ein medizinischer Begriff, der den Verlust einer Schwangerschaft in den frühen Stadien bezeichnet, bevor das Fötus ein Herz hat geschlagen oder eine Größe von mehr als 5 mm erreicht hat. Dies tritt normalerweise vor der 8. Schwangerschaftswoche auf und wird manchmal auch als "früher Fruchtverlust" oder "fehlgeschlagene Empfängnis" bezeichnet.
Die Ursachen für Embryonenverlust können vielfältig sein, dazu gehören Chromosomenanomalien, Hormonstörungen, anatomische Probleme der Gebärmutter oder andere Erkrankungen bei der Mutter. In vielen Fällen ist die Ursache jedoch unbekannt und der Verlust passiert spontan und unerwartet.
Embryonenverlust kann ein emotional belastendes Ereignis sein, und es wird empfohlen, dass Frauen, die einen solchen Verlust erleiden, medizinische und psychologische Unterstützung suchen.
Southern Blotting ist eine Labor-Technik in der Molekularbiologie und Genetik, die verwendet wird, um spezifische DNA-Sequenzen in einer DNA-Probe zu erkennen und zu analysieren. Die Methode wurde nach dem Entwickler des Verfahrens, dem britischen Wissenschaftler Edwin Southern, benannt.
Das Southern Blotting-Verfahren umfasst mehrere Schritte:
1. Zuerst wird die DNA-Probe mit Restriktionsenzymen verdaut, die das DNA-Molekül in bestimmten Sequenzen schneiden.
2. Die resultierenden DNA-Fragmente werden dann auf ein Nitrozellulose- oder PVDF-Membran übertragen und an der Membran fixiert.
3. Als Nächstes wird die Membran in eine Lösung mit markierten DNA-Sonden getaucht, die komplementär zu den gesuchten DNA-Sequenzen sind. Die Markierung erfolgt meistens durch radioaktive Isotope (z.B. 32P) oder fluoreszierende Farbstoffe.
4. Die markierten Sonden binden an die entsprechenden DNA-Fragmente auf der Membran, und die ungebundenen Sonden werden weggespült.
5. Schließlich wird die Membran mit einem Film oder durch direkte Fluoreszenzdetektion ausgewertet, um die Lokalisation und Intensität der markierten DNA-Fragmente zu bestimmen.
Southern Blotting ist eine empfindliche und spezifische Methode zur Analyse von DNA-Sequenzen und wird häufig in der Forschung eingesetzt, um Genexpression, Genmutationen, Genomorganisation und andere genetische Phänomene zu untersuchen.
Inzuchtstämme bei Mäusen sind eng verwandte Populationen von Labor-Nagetieren, die über viele Generationen hinweg durch Paarungen zwischen nahen Verwandten gezüchtet wurden. Diese wiederholten Inzuchtaffen ermöglichen die prädiktive und konsistente genetische Zusammensetzung der Stämme, wodurch sich ihre Phänotypen und Genotypen systematisch von wildlebenden Mäusen unterscheiden.
Die Inzucht führt dazu, dass rezessive Allele einer bestimmten Eigenschaft geäußert werden, was Forscher nutzen, um die genetischen Grundlagen von Krankheiten und anderen Merkmalen zu erforschen. Einige der bekanntesten Inzuchtstämme sind C57BL/6J, BALB/cByJ und DBA/2J. Diese Stämme werden oft für biomedizinische Forschungen verwendet, um Krankheiten wie Krebs, Diabetes, neurologische Erkrankungen und Infektionskrankheiten zu verstehen und Behandlungsansätze zu entwickeln.
Die Heteroduplex-Analyse ist ein Verfahren in der Molekularbiologie, bei dem die komplementären Stränge zweier DNA-Moleküle, die sich in ihrer Sequenz unterscheiden können, paarweise angeordnet (denaturiert und anschließend renaliert) werden, um die Unterschiede zwischen den beiden DNA-Strängen zu identifizieren.
Heteroduplex-Moleküle sind hybride Moleküle, die aus einem komplementären Strang eines wildtypischen DNA-Moleküls und einem komplementären Strang einer mutierten DNA-Sequenz bestehen. Diese Heteroduplex-Moleküle weisen häufig ein ungepaartes, schlecht gepaartes oder unstabiles Segment auf, das als "Bulle" bezeichnet wird und die Lokalisierung der Mutation ermöglicht.
Die Heteroduplex-Analyse ist eine nützliche Methode zur Identifizierung von DNA-Sequenzvarianten, einschließlich Punktmutationen, Insertionen und Deletionen. Sie wird oft in der Genetik, in der Mutationsforschung und in der Entwicklung molekularer Diagnosemethoden eingesetzt.
Lipoproteine sind komplexe Partikel, die sich im Blutplasma befinden und hauptsächlich aus Proteinen (Apolipoproteinen) und Lipiden (Fetten und Cholesterin) bestehen. Ihre Hauptfunktion ist der Transport von Lipiden zwischen den Zellen des Körpers.
Lipoproteine werden in verschiedene Klassen eingeteilt, je nach ihrer Dichte:
- Chylomikronen: die leichtesten und größten Lipoproteine, die Lipide aus der Nahrung transportieren
- VLDL (very low density lipoproteins): sie transportieren Triglyceride von der Leber zu den peripheren Geweben
- IDL (intermediate density lipoproteins): sie sind ein Zwischenprodukt bei der Umwandlung von VLDL in LDL
- LDL (low density lipoproteins): sie werden oft als "schlechtes Cholesterin" bezeichnet, da hohe Konzentrationen im Blutplasma mit einem erhöhten Risiko für Herzkrankheiten verbunden sind
- HDL (high density lipoproteins): sie werden oft als "gutes Cholesterin" bezeichnet, da sie Cholesterin von den Zellen zu Leber transportieren und so das Risiko von Herzkrankheiten verringern können.
Eine ausgewogene Ernährung und ein gesunder Lebensstil können dazu beitragen, die Konzentrationen der verschiedenen Lipoproteine im Blutplasma zu optimieren und so das Risiko von Herz-Kreislauf-Erkrankungen zu verringern.
Die Gaucher-Krankheit ist eine seltene, genetisch bedingte lysosomale Speichererkrankung, die durch einen Mangel an dem Enzym Glukozerebrosidase gekennzeichnet ist. Dieser Enzymmangel führt zu einem Anstieg des Lipids Glukozerebrosid in den Lysosomen von bestimmten Zellen, vor allem Makrophagen, was zur Bildung von Gaucher-Zellen führt. Diese Zellen sammeln sich hauptsächlich in Knochenmark, Leber und Milz an, wodurch diese Organe vergrößert werden können.
Es gibt drei Hauptformen der Gaucher-Krankheit: Typ 1, Typ 2 und Typ 3. Typ 1 ist die häufigste Form und verläuft chronisch, mit Symptomen wie Leber- und Milzvergrößerung, Blutarmut, Knochenschmerzen und -brüchen. Typ 2 ist eine akute neuropathische Form, die bei Säuglingen oder Kleinkindern auftritt und oft tödlich verläuft. Typ 3 ist eine langsam fortschreitende neuropathische Form, die im Kindes- oder Jugendalter beginnt und zu neurologischen Symptomen wie Sprachstörungen, Ataxie und Epilepsie führt.
Die Gaucher-Krankheit wird autosomal rezessiv vererbt, was bedeutet, dass ein Betroffener zwei Kopien des defekten Gens haben muss, um die Krankheit auszubilden. Die Diagnose erfolgt durch Bestimmung der Aktivität der Glukozerebrosidase im Blut oder in Gewebeproben und kann durch genetische Tests bestätigt werden. Die Behandlung umfasst Enzymersatztherapie, Substratreduktionstherapie und symptomatische Therapien.
Iduronat-Sulfatase ist ein Enzym, das für den Abbau von certain Glykosaminoglykanen (GAGs), auch Mukopolysaccharide genannt, in der Zelle verantwortlich ist. Genauer gesagt spaltet Iduronat-Sulfatase die Sulfatgruppen von Dermatan-Sulfat und Heparan-Sulfat ab, zwei Arten von GAGs, die in verschiedenen Geweben des Körpers vorkommen.
Dieses Enzym ist ein essentieller Bestandteil des Lysosoms, eines membranumschlossenen Kompartiments innerhalb der Zelle, in dem verschiedene Stoffwechselvorgänge stattfinden, einschließlich des Abbaus von Biomolekülen. Ohne funktionsfähige Iduronat-Sulfatase können sich GAGs im Lysosom ansammeln und zu einer lysosomalen Speicherkrankheit führen, bekannt als Mukopolysaccharidose Typ II oder Hunter-Syndrom.
Das Hunter-Syndrom ist eine X-chromosomal vererbte Erkrankung, die bei Jungen auftritt und durch eine Vielzahl von Symptomen gekennzeichnet ist, wie z.B. Entwicklungsverzögerung, geistige Behinderung, Gesichtsauffälligkeiten, Vergrößerung von Leber und Milz, Herz- und Atemprobleme sowie Gelenksteifigkeit.
Sichelzellänämie ist ein genetisch bedingtes Krankheitsbild, bei dem die roten Blutkörperchen (Erythrozyten) eine abnorme, sichelartige Form annehmen und damit weniger flexibel sind als normale, rundliche Erythrozyten. Diese Verformung führt dazu, dass die Sichelzellen in kleinen Blutgefäßen stecken bleiben und verstopfen können.
Die Sichelzellänämie wird durch eine Mutation im Gen für das Hämoglobin-Beta-Kettenprotein (HBB) verursacht, die dazu führt, dass anstelle des normalen Hämoglobins (HbA) ein abnormes Hämoglobin S (HbS) gebildet wird. Wenn Sauerstoff aus den Blutkörperchen entweicht, neigt das HbS dazu, sich zusammenzuklumpen und die Zelle in eine sichelartige Form zu verformen.
Die Sichelzellänämie kann zu einer Vielzahl von Symptomen führen, darunter Anämie (Mangel an roten Blutkörperchen), Jaundice (Gelbfärbung der Haut und Augen), Schmerzen in den Extremitäten, Atemnot, Organversagen und einem erhöhten Risiko für Infektionen. Die Krankheit ist unheilbar, aber es gibt Behandlungen, die dazu beitragen können, Symptome zu lindern und Komplikationen zu vermeiden.
Genetische Hybridisierung bezieht sich auf die Kreuzung zweier verschiedener Arten oder Stämme von Organismen durch künstliche Befruchtung (Kreuzungsversuch), wodurch ein neuer Organismus mit genetischem Material aus beiden Elternarten entsteht. Das resultierende Hybrid-Genom kann eine Kombination der Merkmale und Eigenschaften beider Elternorganismen aufweisen, was zu neuen Phänotypen führen kann. Die Fähigkeit zur Fortpflanzung des Hybriden hängt von der Kompatibilität der genetischen Materialien ab; manchmal können Hybride fruchtbar sein und sich fortpflanzen, während sie in anderen Fällen steril oder unfruchtbar sind. Diese Technik wird in verschiedenen Bereichen wie Landwirtschaft, Biotechnologie und Forschung eingesetzt, um neue Sorten mit verbesserten Eigenschaften zu erzeugen.
Eisen ist ein essentielles Spurenelement, das für den Sauerstofftransport im Körper unerlässlich ist. Es ist ein Hauptbestandteil des Hämoglobins in den roten Blutkörperchen und des Myoglobins in den Muskeln. Hämoglobin bindet Eisen, um Sauerstoff aus der Lunge aufzunehmen und zu den Geweben des Körpers zu transportieren, während Myoglobin Eisen verwendet, um Sauerstoff in den Muskeln zu speichern.
Cholesterin ist ein fettartiger, wachsartiger Alkohol, der in den Membranen von Zellen im Körper vorkommt und für die Produktion von Hormonen, Vitamin D und Gallensäuren unerlässlich ist. Es wird hauptsächlich vom Körper selbst produziert, aber es kann auch mit der Nahrung aufgenommen werden, insbesondere durch den Verzehr von tierischen Produkten.
Cholesterin wird im Blutkreislauf durch Lipoproteine transportiert, die als "gutes Cholesterin" (High-Density-Lipoprotein, HDL) und "schlechtes Cholesterin" (Low-Density-Lipoprotein, LDL) bezeichnet werden. Ein hoher Spiegel von LDL-Cholesterin im Blutkreislauf kann zu Ablagerungen in den Arterienwänden führen und das Risiko für Herzkrankheiten und Schlaganfälle erhöhen. Ein niedriger HDL-Spiegel ist ebenfalls mit einem höheren Risiko für Herzkrankheiten verbunden.
Es ist wichtig, einen ausgewogenen Cholesterinspiegel im Blut aufrechtzuerhalten, um das Risiko von Herz-Kreislauf-Erkrankungen zu minimieren. Eine cholesterinarme Ernährung, regelmäßige körperliche Aktivität und gegebenenfalls Medikamente können dazu beitragen, den Cholesterinspiegel im Blut zu kontrollieren.
Neurologische Mutantenmäuse sind genetisch veränderte Labortiere, die für die Erforschung von neurologischen Erkrankungen und Störungen eingesetzt werden. Dabei wird das Erbgut der Mäuse so manipuliert, dass sie Veränderungen aufweisen, die dem menschlichen Krankheitsbild ähneln. Ziel ist es, durch das Studium dieser Tiere mehr über die zugrundeliegenden Mechanismen von neurologischen Erkrankungen wie Parkinson, Alzheimer, Epilepsie oder multipler Sklerose herauszufinden und neue Therapien zu entwickeln.
Es gibt verschiedene Arten von neurologischen Mutantenmäusen, die sich in der Art der genetischen Veränderung unterscheiden. Manche Mäuse tragen zusätzliche Kopien eines Gens, während andere ein Gen gezielt ausschalten (knockout) oder verändern (knock-in). Auch können mehrere Gene gleichzeitig verändert werden, um komplexe Krankheitsbilder abzubilden.
Die Verwendung von neurologischen Mutantenmäusen hat in den letzten Jahren zu wichtigen Erkenntnissen im Bereich der Neurowissenschaften beigetragen und ermöglicht es Forschern, neue Behandlungsansätze für neurologische Erkrankungen zu entwickeln.
Genetic association studies are a type of epidemiological research that aims to identify statistical associations between genetic variations and particular diseases or traits in a population. These studies compare the frequency of specific genetic markers, such as single nucleotide polymorphisms (SNPs) or copy number variants (CNVs), in individuals with a given disease or trait to those without it.
By identifying these associations, researchers can gain insights into the underlying genetic architecture of complex diseases and traits, which may ultimately lead to a better understanding of disease mechanisms, improved diagnostics, and the development of novel therapeutic strategies. It is important to note that while genetic association studies can identify statistical associations between genetic markers and diseases or traits, they do not necessarily imply causation, and further functional validation studies are often required to confirm the role of these genetic variants in disease pathogenesis.
Fetales Tod, auch als intrauteriner Fruchttod oder stiller Tod bekannt, ist die Beendigung einer Schwangerschaft nach der 20. Schwangerschaftswoche mit dem Nachweis eines toten Fetus. In den USA wird es häufig definiert als Tod eines Fetus nach der 20. Schwangerschaftswoche mit einem Gewicht von mehr als 500 Gramm oder einer Länge von mehr als 20 cm. Die Ursachen des fetalen Todes sind vielfältig und können auf Komplikationen während der Schwangerschaft, genetische Faktoren, Infektionen, Mangelernährung oder Umweltfaktoren zurückzuführen sein. Der Nachweis eines fetalen Todes erfolgt durch Ultraschall oder nach der Entbindung durch klinische Untersuchung des Fetus.
'Developmental Gene Expression Regulation' bezieht sich auf die Prozesse, durch die die Aktivität bestimmter Gene während der Entwicklung eines Organismus kontrolliert und reguliert wird. Dies umfasst komplexe Mechanismen wie Epigenetik, Transkriptionsregulation und posttranskriptionelle Regulation, die sicherstellen, dass Gene zur richtigen Zeit, am richtigen Ort und in der richtigen Menge exprimiert werden.
Während der Entwicklung eines Organismus sind Veränderungen in der Genexpression entscheidend für das Wachstum, die Differenzierung und die Morphogenese von Zellen und Geweben. Fehler in der Regulation der Genexpression können zu einer Reihe von Entwicklungsstörungen und Erkrankungen führen.
Daher ist das Verständnis der molekularen Mechanismen, die der Developmental Gene Expression Regulation zugrunde liegen, ein wichtiger Forschungsbereich in der Biomedizin und hat das Potenzial, zu neuen Therapien und Behandlungen für Entwicklungsstörungen und Erkrankungen beizutragen.
Lymphozyten sind eine Art weißer Blutkörperchen (Leukozyten), die eine wichtige Rolle in dem Immunsystem des menschlichen Körpers spielen. Es gibt zwei Hauptgruppen von Lymphozyten: B-Lymphozyten und T-Lymphozyten, die beide an der Abwehr von Krankheitserregern wie Bakterien, Viren und Parasiten beteiligt sind.
B-Lymphozyten produzieren Antikörper, um Krankheitserreger zu bekämpfen, während T-Lymphozyten direkt mit infizierten Zellen interagieren und diese zerstören oder deren Funktion hemmen können. Eine dritte Gruppe von Lymphozyten sind die natürlichen Killerzellen (NK-Zellen), die ebenfalls in der Lage sind, infizierte Zellen zu zerstören.
Lymphozyten kommen in allen Körpergeweben vor, insbesondere aber im Blut und in den lymphatischen Geweben wie Lymphknoten, Milz und Knochenmark. Ihre Anzahl und Aktivität können bei Infektionen, Autoimmunerkrankungen oder Krebs erhöht oder verringert sein.
Es gibt keine einheitliche oder allgemein anerkannte "medizinische Definition" der Bezeichnung "Gruppe afrikanischer Abstammung". Die Klassifizierung von Menschen nach ihrer ethnischen Zugehörigkeit oder Abstammung ist ein komplexes und kontrovers diskutiertes Thema. In der medizinischen Forschung und Versorgung werden manchmal broadrace-Kategorien wie "afrikanische Abstammung" oder "schwarze Bevölkerung" verwendet, um bestimmte Populationen zu beschreiben, die möglicherweise gemeinsame genetische, soziale oder Umweltfaktoren teilen, die für das Gesundheitsverhalten relevant sein könnten.
Die US-Volkszählung definiert beispielsweise "afroamerikanische" oder "schwarze Bevölkerung" als Personen mit afrikanischen Wurzeln, unabhängig von ihrer ethnischen Zugehörigkeit, Hautfarbe oder nationaler Herkunft.
Es ist wichtig zu beachten, dass diese Klassifizierungen vereinfachende Konstrukte sind und die individuelle Vielfalt innerhalb jeder Gruppe nicht vollständig erfassen. Die Verwendung solcher Kategorien in der medizinischen Forschung und Versorgung sollte sorgfältig abgewogen werden, um sicherzustellen, dass sie nicht zu diskriminierenden Praktiken führen oder die Komplexität und Heterogenität von Bevölkerungsgruppen vereinfachen.
Histokompatibilitätsantigene Klasse I sind Proteinkomplexe, die sich auf der Oberfläche der Zellen aller nucleären Organismen befinden. Sie spielen eine entscheidende Rolle im Immunsystem von Wirbeltieren und bestehen aus drei Komponenten: einem großen, transmem membranösen Protein (heavy chain) und zwei kleineren, nicht kovalent gebundenen Proteinen (light chains). Diese Antigene werden in drei verschiedene Untergruppen eingeteilt: HLA-A, HLA-B und HLA-C beim Menschen.
Histokompatibilitätsantigene Klasse I sind von großer Bedeutung bei der Transplantationsmedizin, da sie eine entscheidende Rolle bei der Abstoßungsreaktion gegenüber transplantierten Organen spielen. Die Übereinstimmung der Histokompatibilitätsantigene zwischen Spender und Empfänger ist ein wichtiger Faktor für das Gelingen einer Transplantation. Unterschiede in den HLA-Antigenen können zu einer Abstoßungsreaktion führen, bei der das Immunsystem des Empfängers die transplantierten Zellen als fremd erkennt und angreift.
Isoenzyme sind Enzyme, die die gleiche katalytische Funktion haben, aber sich in ihrer Aminosäuresequenz und/oder Struktur unterscheiden. Diese Unterschiede können aufgrund von Genexpression aus verschiedenen Genen oder durch Variationen im gleichen Gen entstehen. Isoenzyme werden oft in verschiedenen Geweben oder Entwicklungsstadien einer Organismengruppe gefunden und können zur Unterscheidung und Klassifizierung von Krankheiten sowie zur Beurteilung der biochemischen Funktionen von Organen eingesetzt werden.
Apolipoproteine E (ApoE) sind eine Klasse von Proteinen, die in der Regulation des Lipidstoffwechsels eine wichtige Rolle spielen. Sie sind Hauptbestandteil der Lipoproteinpartikel, wie sehr-low-density-Lipoproteine (VLDL) und high-density-Lipoproteine (HDL), die an der Bindung und Aufnahme von Lipiden in Zellen beteiligt sind.
Es gibt drei verschiedene Isoformen des ApoE-Proteins, ApoE2, ApoE3 und ApoE4, die durch genetische Variationen im APOE-Gen codiert werden. Diese Isoformen unterscheiden sich in ihrer Fähigkeit, Lipide zu binden und zu transportieren, was Auswirkungen auf das Risiko für verschiedene Erkrankungen hat.
Zum Beispiel ist die ApoE4-Isoform mit einem erhöhten Risiko für Alzheimer-Krankheit assoziiert, während ApoE2 mit einem reduzierten Risiko verbunden ist. Darüber hinaus spielt ApoE eine Rolle bei der Entstehung und Progression von Herz-Kreislauf-Erkrankungen, da es an der Clearance von Cholesterin aus dem Blutkreislauf beteiligt ist.
Insgesamt sind Apolipoproteine E wichtige Proteine im Lipidstoffwechsel und haben Auswirkungen auf das Risiko für verschiedene Erkrankungen, insbesondere Herz-Kreislauf-Erkrankungen und Alzheimer-Krankheit.
Lipoproteine sind komplexe Partikel, die aus Lipiden (Fette) und Proteinen bestehen und im Blutplasma zirkulieren. Sie spielen eine wichtige Rolle bei der Fetttransportierung im Körper. Es gibt verschiedene Arten von Lipoproteinen, die sich in ihrer Dichte und Zusammensetzung unterscheiden, wie zum Beispiel Low-Density-Lipoproteine (LDL), Very-Low-Density-Lipoproteine (VLDL) und High-Density-Lipoproteine (HDL).
HDL-Cholesterol, auch bekannt als "gutes Cholesterin", bezieht sich auf das Cholesterin, das mit HDL-Partikeln assoziiert ist. HDL-Partikel sind kleiner und dichter als LDL-Partikel und werden manchmal als "Müllabfuhr des Körpers" bezeichnet, weil sie überschüssiges Cholesterin von Zellen im Körper aufnehmen und zu Leber transportieren, wo es abgebaut oder ausgeschieden werden kann. Hohe HDL-Cholesterinspiegel sind mit einem verringerten Risiko für Herzkrankheiten verbunden, während niedrige HDL-Cholesterinspiegel mit einem erhöhten Risiko assoziiert sind.
Hexosaminidase ist ein Enzymkomplex, der aus mehreren Isoenzymen besteht und eine wichtige Rolle bei intrazellulären Abbauprozessen spielt, insbesondere im Rahmen des Abbaus von Glykoproteinen und Gangliosiden. Die Hexosaminidase-Enzyme sind in der Lage, die terminalen Hexosaminidase-Restgruppen (N-Acetylglucosamin oder N-Acetylgalactosamin) von Oligosacchariden und Glycolipiden abzuspalten.
Es gibt zwei Hauptformen des Enzyms, Hexosaminidase A und Hexosaminidase B, die sich in ihrer Substratspezifität und ihrem Gewebespezifischen Vorkommen unterscheiden. Mutationen im Gen, das für das α-Untereinheit von Hexosaminidase A kodiert (HEXA), können zu einem Mangel oder Fehlen des Enzyms führen und sind die Ursache für eine seltene erbliche Stoffwechselerkrankung namens Tay-Sachs-Krankheit.
Eine verminderte Aktivität von Hexosaminidase kann auch mit anderen Erkrankungen assoziiert sein, wie z.B. Sandhoff-Krankheit und GM2-Gangliosidose.
Apolipoproteine A sind ein Hauptbestandteil der High-Density-Lipoproteine (HDL) oder "guten Cholesterins" im Blutkreislauf. Es gibt mehrere Arten von Apolipoproteinen, aber Apolipoprotein A-1 ist das häufigste und wichtigste Protein in HDL-Partikeln.
Apolipoprotein A-1 spielt eine entscheidende Rolle bei der Umwandlung von Cholesterin in wasserlösliche Partikel, die dann vom Körper ausgeschieden werden können. Es hilft auch, entzündliche Prozesse im Körper zu reduzieren und wirkt als Antioxidans, indem es die Oxidation von Low-Density-Lipoproteinen (LDL) oder "schlechtem Cholesterin" verhindert.
Eine niedrige Konzentration von Apolipoprotein A-1 im Blutkreislauf ist ein Risikofaktor für die Entwicklung von Herz-Kreislauf-Erkrankungen, während höhere Konzentrationen mit einem geringeren Risiko verbunden sind.
Neuralrohrdefekte sind Fehlbildungen des sich entwickelnden Nervensystems während der Embryonalentwicklung. Das Neuralrohr ist die Struktur, aus der das zentrale Nervensystem – das Gehirn und das Rückenmark – hervorgeht. Bei einer gestörten Entwicklung kann es zu Fehlbildungen kommen, bei denen sich das Neuralrohr nicht vollständig schließt.
Es gibt drei Hauptarten von Neuralrohrdefekten:
1. Anenzephalie: Dabei handelt es sich um die schwerwiegendste Form von Neuralrohrdefekten, bei der sich der vordere Teil des Neuralrohrs nicht schließt. Das Gehirn und andere Schädelstrukturen fehlen oder sind nur unvollständig ausgebildet. Diese Fehlbildung ist mit dem Leben nicht vereinbar.
2. Spina bifida occulta: Hierbei handelt es sich um eine leichtere Form von Neuralrohrdefekten, bei der sich der hintere Teil des Neuralrohrs nicht schließt. Meist ist nur ein kleiner Bereich des Rückenmarks betroffen, und die Fehlbildung kann ohne Symptome verlaufen. In manchen Fällen können jedoch neurologische Ausfälle auftreten, wie beispielsweise Lähmungen in den Beinen oder Blasen- und Darminkontinenz.
3. Meningomyelozele: Bei dieser Form von Neuralrohrdefekten ist ein größerer Bereich des Rückenmarks betroffen, der durch eine sackartige Ausbuchtung nach außen hervortritt. Die Fehlbildung kann mit neurologischen Ausfällen verbunden sein, wie Lähmungen in den Beinen, Blasen- und Darminkontinenz sowie Sensibilitätsstörungen.
Die Ursachen von Neuralrohrdefekten sind noch nicht vollständig geklärt, aber es wird angenommen, dass genetische Faktoren und Umweltfaktoren wie eine unzureichende Versorgung mit Folsäure während der Schwangerschaft eine Rolle spielen.
Hämolytische, kongenitale nichtsphärozytäre Anämie ist ein medizinischer Begriff, der eine angeborene Form von Anämie beschreibt, die durch eine verstärkte Zerstörung der roten Blutkörperchen (Hämolyse) gekennzeichnet ist. Im Gegensatz zur hämolytischen Anämie, die auf erworbene Ursachen wie Infektionen oder Autoimmunerkrankungen zurückzuführen ist, ist die kongenitale nichtsphärozytäre hämolytische Anämie angeboren und wird durch genetisch bedingte Defekte in den Membranproteinen der roten Blutkörperchen verursacht.
Der Begriff "nichtsphärozytär" bezieht sich darauf, dass die Membranproteine betroffen sind, im Gegensatz zu sphärozytären Formen der Anämie, bei denen die Hämoglobin-Moleküle selbst defekt sind. Die häufigsten Formen der kongenitalen nichtsphärozytären hämolytischen Anämie sind das hereditäre Akanthozytose-Syndrom und die hereditäre Pyropoikilozytose, die durch Mutationen in den Genen für Membranproteine wie die Band 3-Proteine oder die Anionen-Exchanger verursacht werden.
Diese Defekte führen zu einer gestörten Membranstabilität und -deformierbarkeit, was dazu führt, dass die roten Blutkörperchen im Blutkreislauf leichter beschädigt und zerstört werden. Die Hämolyse führt zu einem verstärkten Abbau von roten Blutkörperchen in der Milz und anderen retikuloendothelialen Geweben, was zu einer Anämie führt. Die Symptome können von mild bis schwer reichen und umfassen Müdigkeit, Atemnot, Gelbsucht, Milzvergrößerung und eine erhöhte Anfälligkeit für Infektionen. Die Behandlung kann symptomatisch sein und umfasst Bluttransfusionen, Splenektomie und Medikamente zur Unterstützung der roten Blutkörperchenproduktion.
Hämoglobin C ist eine genetische Variation des Hämoglobins, einem Proteinkomplex in den roten Blutkörperchen, der für den Transport von Sauerstoff im Körper verantwortlich ist. Diese Variante entsteht durch eine Mutation im Gen, das das Beta-Globin-Protein codiert.
Im Fall von Hämoglobin C handelt es sich um einen Glutamat-zu-Lysin-Austausch an der Position 6 der beta-Globinkette (Beta6Glu→Lys). Diese Veränderung führt dazu, dass das Hämoglobin C unnormal klumpen kann und in den roten Blutkörperchen aggregiert.
Die meisten Menschen mit dieser Mutation sind heterozygot (HbAS) und haben nur leichte oder asymptomatische Symptome. Ein kleinerer Anteil der Betroffenen ist homozygot (HbCC) für diese Mutation, was zu einer schwereren Hämoglobinopathie führt, die als Hämoglobin C-Krankheit bekannt ist. Die Hämoglobin C-Krankheit geht mit milden bis mäßigen Anämiesymptomen einher und kann auch eine Erhöhung des Risikos für rezidivierende bakterielle Infektionen, Gallensteine und Sichelzellanämie-assoziierte Komplikationen umfassen.
Genetic Heterogeneity bezieht sich in der Genetik auf die Situation, in der verschiedene genetische Veränderungen oder Mutationen in unterschiedlichen Genen zu ähnlichen oder identischen Phänotypen (klinischen Erscheinungsbildern) führen können. Dies bedeutet, dass ein bestimmtes Krankheitsbild auf unterschiedliche Weise genetisch bedingt sein kann.
Es gibt zwei Arten von Genetic Heterogeneity:
1. Allelic Heterogeneity: Hierbei treten verschiedene Mutationen im selben Gen auf, die aber alle zu derselben Krankheit führen. Zum Beispiel können unterschiedliche Mutationen im BRCA1-Gen zu einer erhöhten Anfälligkeit für Brustkrebs führen.
2. Locus Heterogeneity: Hierbei treten Mutationen in verschiedenen Genen auf, die aber alle zu derselben Krankheit führen. Zum Beispiel können Mutationen in unterschiedlichen Genen wie CFTR, G551D oder ΔF508 bei Mukoviszidose auftreten.
Genetic Heterogeneity ist wichtig zu verstehen, da sie die Identifizierung von Krankheitsgenen und die Entwicklung genetischer Tests für bestimmte Krankheiten erschweren kann.
Genetic Load bezieht sich auf die Gesamtheit der negativen Effekte, die von weniger optimalen Allelen in einer Population verursacht werden. Diese Allele können sich häufiger rezessiv ausdrücken oder einen geringeren Fitnesswert aufweisen als die optimale Variante. Der Begriff "genetische Belastung" kann auch die Anzahl der leicht schädlichen Mutationen umfassen, die in den Genen einer Population vorhanden sind und sich auf ihre genetische Gesundheit auswirken können.
Es gibt drei Arten von genetischer Belastung: Balancierte Belastung, selektive Belastung und neutraler Belastung. Die balancierte Belastung tritt auf, wenn heterozygote Individuen für ein bestimmtes Allel einen Selektionsvorteil haben, während homozygote Individuen für dasselbe Allel einen Selektionsnachteil haben. Die selektive Belastung bezieht sich auf die Reduzierung der Fitness durch den Einfluss schädlicher rezessiver Allele. Schließlich bezieht sich die neutrale Belastung auf die Auswirkungen neutraler Mutationen, die weder einen Selektionsvorteil noch einen Selektionsnachteil verursachen.
Die genetische Belastung ist ein wichtiges Konzept in der Populationsgenetik und Evolutionsbiologie, da sie die Fähigkeit einer Population beeinflusst, sich an Veränderungen in ihrer Umwelt anzupassen. Eine höhere genetische Belastung kann dazu führen, dass eine Population anfälliger für Krankheiten und anderen negativen Effekten ist, was ihre Überlebens- und Fortpflanzungschancen verringern kann.
Ein Embryo ist in der Medizin und Biologie die Bezeichnung für die frühe Entwicklungsphase eines Organismus vom Zeitpunkt der Befruchtung bis zum Beginn der Ausbildung der Körperorgane (ca. 8. Woche beim Menschen). In dieser Phase finden die Hauptprozesse der Embryogenese statt, wie Zellteilungen, Differenzierungen, Migrationen und Interaktionen, die zur Bildung der drei Keimblätter und der sich daraus differenzierenden Organe führen.
Bei Menschen wird nach der 8. Entwicklungswoche auch vom Fötus gesprochen. Es ist wichtig zu beachten, dass verschiedene Definitionen des Begriffs 'Embryo' in unterschiedlichen Kontexten und Rechtssystemen variieren können, insbesondere im Hinblick auf ethische und rechtliche Fragen der Fortpflanzungsmedizin.
Carrierproteine, auch als Transportproteine bekannt, sind Moleküle, die die Funktion haben, andere Moleküle oder Ionen durch Membranen zu transportieren. Sie spielen eine wichtige Rolle im Stoffwechsel von Zellen und im interzellulären Kommunikationsprozess. Carrierproteine sind in der Lage, Substanzen wie Zucker, Aminosäuren, Ionen und andere Moleküle selektiv zu binden und diese durch die Membran zu transportieren, indem sie einen Konformationswandel durchlaufen.
Es gibt zwei Arten von Carrierproteinen: uniporter und symporter/antiporter. Uniporter transportieren nur eine Art von Substanz in eine Richtung, während Symporter und Antiporter jeweils zwei verschiedene Arten von Substanzen gleichzeitig in die gleiche oder entgegengesetzte Richtung transportieren.
Carrierproteine sind von großer Bedeutung für den Transport von Molekülen durch Zellmembranen, da diese normalerweise nicht-polar und lipophil sind und somit nur unpolare oder lipophile Moleküle passiv durch Diffusion durch die Membran transportieren können. Carrierproteine ermöglichen es so, auch polare und hydrophile Moleküle aktiv zu transportieren.
Es gibt keine allgemein anerkannte medizinische Definition der Bezeichnung "Gruppe europäischer Abstammung". Der Begriff wird manchmal in biomedizinischen Forschungen oder öffentlichen Gesundheitsdaten verwendet, um Menschen mit gemeinsamen geografischen und historischen Ursprüngen in Europa zu beschreiben. Es ist jedoch wichtig anzumerken, dass die Verwendung solcher ethnischen Kategorien komplexe soziale und historische Konstrukte sind und nicht unbedingt biologisch oder genetisch bestimmbare Eigenschaften widerspiegeln. Die Verwendung dieser Begriffe in der Forschung oder klinischen Praxis sollte daher sorgfältig geprüft werden, um sicherzustellen, dass sie angemessen, nützlich und respektvoll sind.
Hereditäre Elliptozytose ist ein genetischer Blutbildungsdefekt, bei dem die roten Blutkörperchen (Erythrozyten) eine elliptische oder ovale Form statt der normalerweise diskusförmigen Gestalt aufweisen. Dieses Zustandsbild wird als Autosomal-dominant vererbt, was bedeutet, dass nur ein Elternteil das genetisch defekte Gen weitergeben muss, um die Erkrankung zu übertragen.
Die elliptische Form der Erythrozyten ist auf eine Beeinträchtigung des Zytoskeletts zurückzuführen, welches die Form und Stabilität der roten Blutkörperchen gewährleistet. Bei hereditärer Elliptozytose liegt in der Regel ein Defekt im Spectrin-Protein vor, das ein wichtiger Bestandteil des Zytoskeletts ist.
Die meisten Menschen mit hereditärer Elliptozytose weisen nur eine leichte bis mäßige Anzahl an elliptischen Erythrozyten auf und sind in der Regel asymptomatisch oder entwickeln nur sehr milde Symptome. In einigen Fällen kann es jedoch zu milden bis mäßigen Hämolyseerscheinungen kommen, die sich in Form von Gelbsucht (Ikterus), erhöhter Retikulozytenzahl und Anämie manifestieren können.
In der Regel ist hereditäre Elliptozytose ein harmloser Zustand, der weder eine Behandlung noch besondere Vorsichtsmaßnahmen erfordert. In seltenen Fällen kann die Erkrankung jedoch mit schwerwiegenderen Symptomen einhergehen, wie beispielsweise schwerer Hämolyse und hämolytischer Anämie.
 Faktor-XIII-Mangel
Faktor-XIII-Mangel
Kollagen-Typ 3α1
HCN-Kanal
Kniest-Dysplasie
Klein-Waardenburg-Syndrom
Morbus Fabry
Selektion (Evolution)
Faktor-XI-Mangel
Bombay-Blutgruppe
Sabino Overo
Fehlfarbe (Hundezucht)
Willebrand-Jürgens-Syndrom
Progerie
Tigerschecken-Komplex
Naxos-Krankheit
Nucleolus
Morbus Krabbe
Fragiles-X-Syndrom
Mukoviszidose
Heterosis-Effekt
ABCC11
Refsum-Syndrom
Liste der betroffenen Merkmale des Gutachtens zur Auslegung des Verbotes von Qualzüchtungen
Arylsulfatase A
Louis-Bar-Syndrom
Genetisches Matching
Frontometaphysäre Dysplasie
Hämoglobinopathie
Heterotaxie
Faktor-V-Leiden-Mutation
Faktor-XIII-Mangel - Wikipedia
 Pharmakogenetics/genomics - Universitätsinstitut für Medizinisch-Chemische Labordiagnostik - Zentrallabor LKH
Pharmakogenetics/genomics - Universitätsinstitut für Medizinisch-Chemische Labordiagnostik - Zentrallabor LKH
 Leukodystrophien des Erwachsenenalters - Klinische Neurologie - eMedpedia
Leukodystrophien des Erwachsenenalters - Klinische Neurologie - eMedpedia
 Familiäre Hypercholesterinämie: Ursachen, Risiken und Diagnose
Familiäre Hypercholesterinämie: Ursachen, Risiken und Diagnose
 Atorvastatin AXiromed 10 mg/ -20 mg/ -40 mg/ -80 mg Filmtabletten: Dosierung, Nebenwirkung & Wirkung
Atorvastatin AXiromed 10 mg/ -20 mg/ -40 mg/ -80 mg Filmtabletten: Dosierung, Nebenwirkung & Wirkung
 Ginkgo biloba
Ginkgo biloba
 Hereditäre Katarakt (HSF4)* : LABOKLIN Bad Kissingen
Hereditäre Katarakt (HSF4)* : LABOKLIN Bad Kissingen
 Genomia: Prüfung von Hunden: PK Defizienz
Genomia: Prüfung von Hunden: PK Defizienz
 Galaktokinase | LADR
Galaktokinase | LADR
 Karl II.: Woran litt der „verhexte König" von Spanien?
Karl II.: Woran litt der „verhexte König" von Spanien?
ERA PerMed
 Pathologie: Leber - Wikibooks, Sammlung freier Lehr-, Sach- und Fachbücher
Pathologie: Leber - Wikibooks, Sammlung freier Lehr-, Sach- und Fachbücher
 Was bedeutet heterozygot bio?
Was bedeutet heterozygot bio?
 Von der Erbse bis zur modernen genetischen Medizin - myPoint
Von der Erbse bis zur modernen genetischen Medizin - myPoint
 Description: Einfluss des Transkriptionsfaktors Snail in der Initiation und Progression von PanINs beim invasiven duktalen...
Description: Einfluss des Transkriptionsfaktors Snail in der Initiation und Progression von PanINs beim invasiven duktalen...
 Glucose-6-Phosphat-Dehydrogenase-Defizienz (Favismus): Labor & Diagnostik
Glucose-6-Phosphat-Dehydrogenase-Defizienz (Favismus): Labor & Diagnostik
 Diploidie - Lexikon der Biologie
Diploidie - Lexikon der Biologie
 MUTYH-assoziierte Polyposis (MAP) - Deutsch
MUTYH-assoziierte Polyposis (MAP) - Deutsch
Studienhandbuch | Diseases caused by gene-environment-interaction
 OPUS 4 | Leukemia inhibitory factor enhances neurogenin's pro-neural effect during mouse cortical development
OPUS 4 | Leukemia inhibitory factor enhances neurogenin's pro-neural effect during mouse cortical development
 Wahre Größe misst sich nicht in Zentimetern ...<...
Wahre Größe misst sich nicht in Zentimetern ...<...
 Golden Retriever Hündin 1 Jahr kaufen - Dezember 2023
Golden Retriever Hündin 1 Jahr kaufen - Dezember 2023
 Tabbys - Tabbys
Tabbys - Tabbys
 Osteochondrodysplasie beim Zwergpudel (OCD) - AnimaLabs©
Osteochondrodysplasie beim Zwergpudel (OCD) - AnimaLabs©
Indikationsverzeichnis - Medizinische Laboratorien Düsseldorf
 Jetzt schlägt's 13! Faktor XIII-Mangel - pin-up-docs - don't panic
Jetzt schlägt's 13! Faktor XIII-Mangel - pin-up-docs - don't panic
Labor Renner . Labor für Molekularbiologische Analytik
 Wie entsteht Familiäre Hypercholesterinämie? - FHchol Austria
Wie entsteht Familiäre Hypercholesterinämie? - FHchol Austria
 Hämoglobinopathien - vielgestaltiges Krankheitsbild und Methodenspektrum
Hämoglobinopathien - vielgestaltiges Krankheitsbild und Methodenspektrum
Homozygote5
- Die heterozygote Form tritt am häufigsten auf, während die homozygote Form sehr selten ist. (medical-tribune.ch)
- Bei einer erblichen Erhöhung des Blutfettgehaltes (Hyperlipidämie Typ IIa und IIb nach Frederickson, homozygote und heterozygote familiäre Hypercholesterolämie) wird Atorvastatin ergänzend zu einer Diät verwendet. (onmeda.de)
- Dies bedeutet, dass sich homozygote Allele stärker auswirken als heterozygote. (of-mysterys-dream.de)
- Häufige Genotypen sind die homozygote HBSS-Sichelzellkrankheit, die compound heterozygote Sichelzellkrankheit, HbS-β-Thal und die HbSC-Krankheit. (onkopedia.com)
- Bei der β-Thalassämia major handelt es sich um die homozygote Form, bei der beide Allele schwerwiegend mutiert sind, so dass oft die β-Kettensynthese völlig ausbleibt, während bei der β-Thalassämia minor die heterozygote Form vorliegt und die Polypeptidsynthese um ca. 20 % erniedrigt ist. (chromsystems.com)
Mutation4
- Heterozygote Hunde (Träger der Mutation) sind gewöhnlich asymptomatisch, obwohl sie um die Hälfte reduzierte Pyrunatkinaseaktivität aufweisen. (genomia.cz)
- Was bedeutet heterozygote Mutation? (alleantworten.de)
- Im beschriebenen Fall liegt eine compound-heterozygote Mutation im CDH3-Gen vor. (hjmd.de)
- Neben ihrem Vorhofflimmern war sie heterozygote Trägerin sowohl für die Faktor-V-Leiden-Mutation als auch für die Punktmutation in Position 677 (C677T) der Methyltetrahydrofolatreduktase (MTHFR). (medscape.com)
Asymptomatisch2
- Heterozygote Personen sind asymptomatisch. (wikipedia.org)
- Heterozygote Patienten sind meistens asymptomatisch. (pin-up-docs.de)
Zeigen4
- kombiniert-heterozygote Frauen mit Varianten auf dem X-Chromosom zeigen den voll ausgeprägten Phänotyp. (medizinische-genetik.de)
- Heterozygote Anlageträgerinnen zeigen in der Regel nur dann Symptome, wenn eine präferenzielle Expression des betroffenen Allels, z.B. aufgrund einer verschobenen X-Inaktivierung , vorliegt. (medizinische-genetik.de)
- Heterozygote zeigen keine Symptome. (animalabs.com)
- Heterozygote Merkmalsträger zeigen überwiegend Neutrophile, die über nur 2 Kernsegmente verfügen (bisegmentiert). (mtdialog.de)
Personen1
- Die heterozygote Familiäre Hypercholesterinämie betrifft etwa 1 von 250 Personen. (fhchol.at)
Homozygote3
- ehemals Typ I Hyperlipidämie), die in der Kindheit oder Jugend auftritt und häufig durch homozygote oder kombiniert-heterozygote pathogene Varianten im LPL -Gen oder dessen Cofaktoren APOC2 , APOA5 , GPIHBP1 oder LMF1 verursacht wird. (medizinische-genetik.de)
- Ursache für eine schwere Hypertriglyzeridämie sind homozygote oder gemischt heterozygote pathogene Varianten im LPL -Gen ( LPL-Defizienz ). (medizinische-genetik.de)
- Homozygote (ca. 0,3% der dunkelhäutigen USA Bevölkerung) haben eine Sichelzellanämie, Heterozygote (8-13% der Dunkelhäutigen) sind typischerweise nicht anämisch, besitzen aber ein erhöhtes Risiko für andere Komplikationen. (msdmanuals.com)
Pathogene1
- Mittels Sanger Sequenzierung konnte im TBX5 Gen eine heterozygote, pathogene Variante detektiert werden. (herzmedizin.de)
Variante1
- Die Anwendung des Arzneimittels ist zusätzlich zu einer Diät angezeigt zur Senkung erhöhter Gesamtcholesterin-, LDL-Cholesterin-, Apolipoprotein-B- und Triglyceridspiegel bei Erwachsenen, Jugendlichen und Kinder ab 10 Jahren mit Primärer Hypercholesterinämie, einschließlich Familiärer Hypercholesterinämie (heterozygote Variante) oder Kombinierter (Gemischter) Hyperlipidämie (entsprechend Typ II a und II b nach Fredrickson), wenn Diät und andere nicht-pharmakologische Maßnahmen keine ausreichende Wirkung erbringen. (ouiiou.com)
Genotyp1
- Kombiniert-heterozygote Träger (Genotyp m/s) scheinen die Krankheit nicht auszuprägen. (schwarzzucht.de)
Defekten1
- Weibliche Heterozygote wurden fälschlicherweise als „Trägerinnen des defekten Gens" bezeichnet, die von der Entwicklung von Krankheitsmanifestationen unbetroffen sind. (rethinkfabry.de)
Katzen1
- Aufgrund der unvollständigen Penetranz bei Fd fd - Tieren können in einer Faltohrzucht auch heterozygote, aber normalohrige Katzen das Fd- Gen vererben. (welt-der-katzen.de)