Quantitative Structure-Activity Relationship
Structure-Activity Relationship
Molecular Structure
Models, Molecular
Mescaline
Models, Chemical
Drug Design
The carboxy terminus of the herpesvirus saimiri ORF 57 gene contains domains that are required for transactivation and transrepression. (1/699)
Herpesvirus saimiri (HVS) ORF 57 is homologous to genes identified in all classes of herpesviruses. We have previously shown that ORF 57 encodes a multifunctional protein, responsible for both transactivation and repression of viral gene expression at a post-transcriptional level. This suggests that the ORF 57 protein shares some functional similarities with the herpes simplex virus IE63/ICP27 and Epstein-Barr virus Mta proteins. However, little is known about the functional domains responsible for the properties of ORF 57 due to the limited homology shared between these proteins. In this report, we have identified the functional domains responsible for transactivation and repression by the ORF 57 protein. We demonstrate that the carboxy terminus is required for ORF 57 transactivation, repression and an intense SC-35 nuclear spotting. This region contains two highly conserved motifs amongst its homologues, a zinc finger-like motif and a highly hydrophobic domain. We further show that the hydrophobic domain is required for transactivation and is also involved in nuclear localization of the ORF 57 protein, whereas the zinc finger-like domain is required for transactivation, repression and the intense SC-35 nuclear spotting. (+info)Self-organizing neural network for modeling 3D QSAR of colchicinoids. (2/699)
A novel scheme for modeling 3D QSAR has been developed. A method involving multiple self-organizing neural network adjusted to be analyzed by the PLS (partial least squares) analysis was used to model 3D QSAR of the selected colchicinoids. The model obtained allows the identification of some structural determinants of the biological activity of compounds. (+info)MIPSIM: similarity analysis of molecular interaction potentials. (3/699)
SUMMARY: MIPSIM is a computational package designed to analyse and compare 3D distributions of molecular interaction potentials (MIP) of series of biomolecules. (+info)The UL34 gene product of herpes simplex virus type 2 is a tail-anchored type II membrane protein that is significant for virus envelopment. (4/699)
The UL34 gene of herpes simplex virus type 2 (HSV-2) is highly conserved in the herpesvirus family. The UL34 gene product was identified In lysates of HSV-2-infected cells as protein species with molecular masses of 31 and 32.5 kDa, the latter being a phosphorylated product. Synthesis of these proteins occurred at late times post-infection and was highly dependent on viral DNA synthesis. Immunofluorescence assays revealed that the UL34 protein was localized in the cytoplasm in a continuous net-like structure, closely resembling the staining pattern of the endoplasmic reticulum (ER), in both HSV-2-infected cells and in cells transiently expressing UL34 protein. Deletion mutant analysis showed that this colocalization required the C terminus of the UL34 protein. The UL34 protein associated with virions but not with A, B or C capsids. We treated virions, HSV-2-infected cells and cells expressing the UL34 protein with a protease in order to examine the topology of the UL34 protein. In addition, we constructed UL34 deletion mutant proteins and examined their intracellular localization. Our data strongly support the hypothesis that the UL34 protein is inserted into the viral envelope as a tail-anchored type II membrane protein and is significant for virus envelopment. (+info)Recombinant human monoclonal antibodies against different conformational epitopes of the E2 envelope glycoprotein of hepatitis C virus that inhibit its interaction with CD81. (5/699)
The antibody response to the envelope proteins of hepatitis C virus (HCV) may play an important role in controlling the infection. To allow molecular analyses of protective antibodies, we isolated human monoclonal antibodies to the E2 envelope glycoprotein of HCV from a combinatorial Fab library established from bone marrow of a chronically HCV-infected patient. Anti-E2 reactive clones were selected using recombinant E2 protein. The bone marrow donor carried HCV genotype 2b, and E2 used for selection was of genotype 1a. The antibody clones were expressed as Fab fragments in E. coli, and as Fab fragments and IgG1 in CHO cells. Seven different antibody clones were characterized, and shown to have high affinity for E2, genotype 1a. Three clones also had high affinity for E2 of genotype 1b. They all bind to conformation-dependent epitopes. Five clones compete for the same or overlapping binding sites, while two bind to one or two other epitopes of E2. Four clones corresponding to the different epitopes were tested as purified IgG1 for blocking the CD81-E2 interaction in vitro; all four were positive at 0.3-0.5 microg/ml. Thus, the present results suggest the existence of at least two conserved epitopes in E2 that mediate inhibition of the E2-CD81 interaction, of which one appeared immunodominant in this donor. (+info)Isolation of a Spodoptera exigua baculovirus recombinant with a 10.6 kbp genome deletion that retains biological activity. (6/699)
When Spodoptera exigua multicapsid nucleopolyhedrovirus (SeMNPV) is grown in insect cell culture, defective viruses are generated. These viruses lack about 25 kbp of sequence information and are no longer infectious for insects. This makes the engineering of SeMNPV for improved insecticidal activity or as expression vectors difficult to achieve. Recombinants of Autographa californica MNPV have been generated in insects after lipofection with viral DNA and a transfer vector into the haemocoel. In the present study a novel procedure to isolate SeMNPV recombinants was adopted by alternate cloning between insect larvae and cultured cells. The S. exigua cell line Se301 was used to select the putative recombinants by following a green fluorescent protein marker inserted in the p10 locus of SeMNPV. Polyhedra from individual plaques were fed to larvae to select for biological activity. In this way an SeMNPV recombinant (SeXD1) was obtained with the speed of kill improved by about 25%. This recombinant lacked 10593 bp of sequence information, located between 13.7 and 21.6 map units of SeMNPV and including ecdysteroid UDP glucosyl transferase, gp37, chitinase and cathepsin genes, as well as several genes unique to SeMNPV. The result indicated, however, that these genes are dispensable for virus replication both in vitro and in vivo. A mutant with a similar deletion was identified by PCR in the parental wild-type SeMNPV isolate, suggesting that genotypes with differential biological activities exist in field isolates of baculoviruses. The generation of recombinants in vivo, combined with the alternate cloning between insects and insect cells, is likely to be applicable to many baculovirus species in order to obtain biologically active recombinants. (+info)Expression of unglycosylated mutated prion protein facilitates PrP(Sc) formation in neuroblastoma cells infected with different prion strains. (7/699)
Prion replication involves conversion of the normal, host-encoded prion protein PrP(C), which is a sialoglycoprotein bound to the plasma membrane by a glycophosphatidylinositol anchor, into a pathogenic isoform, PrP(Sc). In earlier studies, tunicamycin prevented glycosylation of PrP(C) in scrapie-infected mouse neuroblastoma (ScN2a) cells but it was still expressed on the cell surface and converted into PrP(Sc); mutation of PrP(C) at glycosylation consensus sites (T182A, T198A) produced low steady-state levels of PrP that were insufficient to propagate prions in transgenic mice. By mutating asparagines to glutamines at the consensus sites, we obtained expression of unglycosylated, epitope-tagged MHM2PrP(N180Q,N196Q), which was converted into PrP(Sc) in ScN2a cells. Cultures of uninfected neuroblastoma (N2a) cells transiently expressing mutated PrP were exposed to brain homogenates prepared from mice infected with the RML, Me7 or 301V prion strains. In each case, mutated PrP was converted into PrP(Sc) as judged by Western blotting. These findings raise the possibility that the N2a cell line can support replication of different strains of prions. (+info)Metabonomics: evaluation of nuclear magnetic resonance (NMR) and pattern recognition technology for rapid in vivo screening of liver and kidney toxicants. (8/699)
The purpose of this study was to evaluate the feasibility of metabonomics technology for developing a rapid-throughput toxicity screen using 2 known hepatotoxicants: carbon tetrachloride (CCl(4)) and alpha-naphthylisothiocyanate (ANIT) and 2 known nephrotoxicants: 2-bromoethylamine (BEA) and 4-aminophenol (PAP). In addition, the diuretic furosemide (FURO) was also studied. Single doses of CCl(4) (0.1 and 0.5 ml/kg), ANIT (10 and 100 mg/kg), BEA (15 and 150 mg/kg), PAP (15 and 150 mg/kg) and FURO (1 and 5 mg) were administered as single IP or oral doses to groups of 4 male Wistar rats/dose. Twenty-four-h urine samples were collected pretest, daily through Day 4, and on Day 10 (high dose CCl(4) and BEA only). Blood samples were taken on Days 1, 2, and 4 or 1, 4, and 10 for clinical chemistry assessment, and the appropriate target organ was examined microscopically. NMR spectra of urine were acquired and the data processed and subjected to principal component analyses (PCA). The results demonstrated that the metabonomic approach could readily distinguish the onset and reversal of toxicity with good agreement between clinical chemistry and PCA data. In at least 2 instances (ANIT and BEA), PCA analysis suggested effects at low doses, which were not as evident by clinical chemistry or microscopic analysis. Furosemide, which had no effect at the doses employed, did not produce any changes in PCA patterns. These data support the contention that the metabonomic approach represents a promising new technology for the development of a rapid throughput in vivo toxicity screen. (+info)Quantitative Structure-Activity Relationship (QSAR) is a method used in toxicology and medicinal chemistry that attempts to establish mathematical relationships between the chemical structure of a compound and its biological activity. QSAR models are developed using statistical methods to analyze a set of compounds with known biological activities and their structural properties, which are represented as numerical or categorical descriptors. These models can then be used to predict the biological activity of new, structurally similar compounds.
QSAR models have been widely used in drug discovery and development, as well as in chemical risk assessment, to predict the potential toxicity of chemicals based on their structural properties. The accuracy and reliability of QSAR predictions depend on various factors, including the quality and diversity of the data used to develop the models, the choice of descriptors and statistical methods, and the applicability domain of the models.
In summary, QSAR is a quantitative method that uses mathematical relationships between chemical structure and biological activity to predict the potential toxicity or efficacy of new compounds based on their structural properties.
A Structure-Activity Relationship (SAR) in the context of medicinal chemistry and pharmacology refers to the relationship between the chemical structure of a drug or molecule and its biological activity or effect on a target protein, cell, or organism. SAR studies aim to identify patterns and correlations between structural features of a compound and its ability to interact with a specific biological target, leading to a desired therapeutic response or undesired side effects.
By analyzing the SAR, researchers can optimize the chemical structure of lead compounds to enhance their potency, selectivity, safety, and pharmacokinetic properties, ultimately guiding the design and development of novel drugs with improved efficacy and reduced toxicity.
Molecular structure, in the context of biochemistry and molecular biology, refers to the arrangement and organization of atoms and chemical bonds within a molecule. It describes the three-dimensional layout of the constituent elements, including their spatial relationships, bond lengths, and angles. Understanding molecular structure is crucial for elucidating the functions and reactivities of biological macromolecules such as proteins, nucleic acids, lipids, and carbohydrates. Various experimental techniques, like X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and cryo-electron microscopy (cryo-EM), are employed to determine molecular structures at atomic resolution, providing valuable insights into their biological roles and potential therapeutic targets.
Molecular models are three-dimensional representations of molecular structures that are used in the field of molecular biology and chemistry to visualize and understand the spatial arrangement of atoms and bonds within a molecule. These models can be physical or computer-generated and allow researchers to study the shape, size, and behavior of molecules, which is crucial for understanding their function and interactions with other molecules.
Physical molecular models are often made up of balls (representing atoms) connected by rods or sticks (representing bonds). These models can be constructed manually using materials such as plastic or wooden balls and rods, or they can be created using 3D printing technology.
Computer-generated molecular models, on the other hand, are created using specialized software that allows researchers to visualize and manipulate molecular structures in three dimensions. These models can be used to simulate molecular interactions, predict molecular behavior, and design new drugs or chemicals with specific properties. Overall, molecular models play a critical role in advancing our understanding of molecular structures and their functions.
Mescaline is a naturally occurring psychoactive alkaloid that is found in several species of cacti, including the peyote (Lophophora williamsii), San Pedro (Echinopsis pachanoi), and Peruvian torch (Echinopsis peruviana) cacti. It is known for its ability to produce profound changes in consciousness, mood, and perception when ingested.
In a medical context, mescaline is classified as a hallucinogen or psychedelic drug. It works by binding to serotonin receptors in the brain, which leads to altered states of consciousness, including visual hallucinations, distorted perceptions of time and space, and altered emotional states.
It's important to note that while mescaline has been used for centuries in religious and spiritual practices among indigenous communities, its use is not without risks. High doses can lead to unpleasant or even dangerous psychological effects, such as anxiety, panic, and psychosis. Additionally, the legal status of mescaline varies by country and region, so it's important to be aware of local laws and regulations before using it.
'Salvia officinalis', also known as garden sage or common sage, is not a medical condition but an herb that has been used in traditional medicine. Here's the botanical definition:
Salvia officinalis, commonly known as sage, garden sage, or common sage, is a perennial, evergreen subshrub, with woody stems, grayish leaves, and blue to purplish flowers. It belongs to the Lamiaceae family, also known as the mint family. The plant is native to the Mediterranean region and has been cultivated throughout the world for its aromatic leaves, which are used in cooking, cosmetics, and medicinal preparations.
In traditional medicine, sage leaves have been used to treat various conditions, such as sore throats, coughs, colds, and digestive issues. However, it is essential to note that the effectiveness of sage for these uses has not been thoroughly studied in clinical trials, and its use should not replace conventional medical care. Always consult with a healthcare professional before starting any new treatment or therapy.
A chemical model is a simplified representation or description of a chemical system, based on the laws of chemistry and physics. It is used to explain and predict the behavior of chemicals and chemical reactions. Chemical models can take many forms, including mathematical equations, diagrams, and computer simulations. They are often used in research, education, and industry to understand complex chemical processes and develop new products and technologies.
For example, a chemical model might be used to describe the way that atoms and molecules interact in a particular reaction, or to predict the properties of a new material. Chemical models can also be used to study the behavior of chemicals at the molecular level, such as how they bind to each other or how they are affected by changes in temperature or pressure.
It is important to note that chemical models are simplifications of reality and may not always accurately represent every aspect of a chemical system. They should be used with caution and validated against experimental data whenever possible.
"Drug design" is the process of creating and developing a new medication or therapeutic agent to treat or prevent a specific disease or condition. It involves identifying potential targets within the body, such as proteins or enzymes that are involved in the disease process, and then designing small molecules or biologics that can interact with these targets to produce a desired effect.
The drug design process typically involves several stages, including:
1. Target identification: Researchers identify a specific molecular target that is involved in the disease process.
2. Lead identification: Using computational methods and high-throughput screening techniques, researchers identify small molecules or biologics that can interact with the target.
3. Lead optimization: Researchers modify the chemical structure of the lead compound to improve its ability to interact with the target, as well as its safety and pharmacokinetic properties.
4. Preclinical testing: The optimized lead compound is tested in vitro (in a test tube or petri dish) and in vivo (in animals) to evaluate its safety and efficacy.
5. Clinical trials: If the preclinical testing is successful, the drug moves on to clinical trials in humans to further evaluate its safety and efficacy.
The ultimate goal of drug design is to create a new medication that is safe, effective, and can be used to improve the lives of patients with a specific disease or condition.
Molecular conformation, also known as spatial arrangement or configuration, refers to the specific three-dimensional shape and orientation of atoms that make up a molecule. It describes the precise manner in which bonds between atoms are arranged around a molecular framework, taking into account factors such as bond lengths, bond angles, and torsional angles.
Conformational isomers, or conformers, are different spatial arrangements of the same molecule that can interconvert without breaking chemical bonds. These isomers may have varying energies, stability, and reactivity, which can significantly impact a molecule's biological activity and function. Understanding molecular conformation is crucial in fields such as drug design, where small changes in conformation can lead to substantial differences in how a drug interacts with its target.
In the context of medical and biological sciences, a "binding site" refers to a specific location on a protein, molecule, or cell where another molecule can attach or bind. This binding interaction can lead to various functional changes in the original protein or molecule. The other molecule that binds to the binding site is often referred to as a ligand, which can be a small molecule, ion, or even another protein.
The binding between a ligand and its target binding site can be specific and selective, meaning that only certain ligands can bind to particular binding sites with high affinity. This specificity plays a crucial role in various biological processes, such as signal transduction, enzyme catalysis, or drug action.
In the case of drug development, understanding the location and properties of binding sites on target proteins is essential for designing drugs that can selectively bind to these sites and modulate protein function. This knowledge can help create more effective and safer therapeutic options for various diseases.
 Quantitative structure-activity relationship
Quantitative structure-activity relationship IUPAC - three-dimensional quantitative structure-activity relationships (DT06959)
IUPAC - three-dimensional quantitative structure-activity relationships (DT06959) EuroQSAR 2016 Verona Italy, 21st European Symposium on Quantitative Structure-Activity Relationship
EuroQSAR 2016 Verona Italy, 21st European Symposium on Quantitative Structure-Activity Relationship Biocatalytic oxidation of phenolic compounds by bovine methemoglobin in the presence of H2O2: quantitative structure-activity...
Biocatalytic oxidation of phenolic compounds by bovine methemoglobin in the presence of H2O2: quantitative structure-activity... Quantitative Structure Activity Relationship Studies of Sulfamide Derivatives as Carbonic Anhydrase Inhibitor: As Antiglaucoma...
Quantitative Structure Activity Relationship Studies of Sulfamide Derivatives as Carbonic Anhydrase Inhibitor: As Antiglaucoma... Interpretable correlation descriptors for quantitative structure-activity relationships | Journal of Cheminformatics | Full Text
Interpretable correlation descriptors for quantitative structure-activity relationships | Journal of Cheminformatics | Full Text Quantitative Structure Activity Relationship (QSAR) - BioNome
Quantitative Structure Activity Relationship (QSAR) - BioNome Unbalance Quantitative Structure Activity Relationship Problem Reduction in Drug Design
Unbalance Quantitative Structure Activity Relationship Problem Reduction in Drug Design Analysis of a Quantitative Relationship Between the Structure and Analgesic Activity of Meperidin Derivatives Using Semi...
Analysis of a Quantitative Relationship Between the Structure and Analgesic Activity of Meperidin Derivatives Using Semi... Quantitative Structure-Activity Relationship (Qsar) Market Size - Best Online New With Newzbuds
Quantitative Structure-Activity Relationship (Qsar) Market Size - Best Online New With Newzbuds 4D- quantitative structure-activity relationship modeling: making a comeback - NCSU Bioinformatics Research Center
4D- quantitative structure-activity relationship modeling: making a comeback - NCSU Bioinformatics Research Center RI UFLA: Quantitative structure-activity relationship studies for potential rho-associated protein kinase inhibitors
RI UFLA: Quantitative structure-activity relationship studies for potential rho-associated protein kinase inhibitors Bisphenol analogues inhibit human and rat 17ß-hydroxysteroid dehydrogenase 1: 3D-quantitative structure-activity relationship ...
Bisphenol analogues inhibit human and rat 17ß-hydroxysteroid dehydrogenase 1: 3D-quantitative structure-activity relationship ... Acronyms and Abbreviations - OECD
Acronyms and Abbreviations - OECD Prooxidant toxicity of polyphenolic antioxidants to HL-60 cells : description of quantitative structure-activity relationships<...
Prooxidant toxicity of polyphenolic antioxidants to HL-60 cells : description of quantitative structure-activity relationships<... Solution Structural Studies of GTP:Adenosylcobinamide-Phosphateguanylyl Transferase (CobY) from Methanocaldococcus jannaschii
Solution Structural Studies of GTP:Adenosylcobinamide-Phosphateguanylyl Transferase (CobY) from Methanocaldococcus jannaschii Exploring Quantitative Structure-Activity Relationships (QSARs) of Cyclooxygenase-2 (COX-2) Inhibitors by MLR, PLS and PC-ANN.
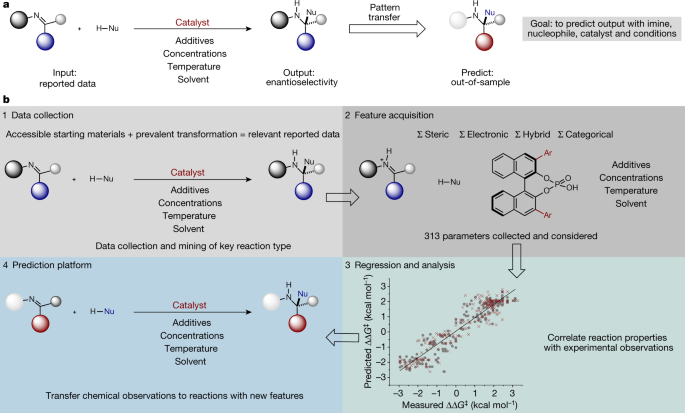
Exploring Quantitative Structure-Activity Relationships (QSARs) of Cyclooxygenase-2 (COX-2) Inhibitors by MLR, PLS and PC-ANN. Holistic prediction of enantioselectivity in asymmetric catalysis | Nature
Holistic prediction of enantioselectivity in asymmetric catalysis | Nature A k-nearest neighbor classification of hERG K+ channel blockers | Journal of Computer-Aided Molecular Design
A k-nearest neighbor classification of hERG K+ channel blockers | Journal of Computer-Aided Molecular Design IJMS | Free Full-Text | Beyond the Flavour: The Potential Druggability of Chemosensory G Protein-Coupled Receptors
IJMS | Free Full-Text | Beyond the Flavour: The Potential Druggability of Chemosensory G Protein-Coupled Receptors